Introduction
Cholangiocarcinoma (CCA), caused by liver fluke
infection, is a major public health problem in the Northeastern and
Northern parts of Thailand. It is a slow growing cancer with rapid
metastasis and a high mortality rate (1). The incidence of CCA has increased
worldwide with the highest rate in Thailand where around 93–100 per
100,000 peoples have been diagnosed with this disease (2). The pathogenesis of CCA has been
demonstrated mainly by genetic mutations of bile duct epithelial
cells themselves as the result of chronic inflammation which can
also create a local environment enriched with cytokines and growth
factors (3). In addition to cancer
cells, the role of cancer-associated fibroblasts has been revealed
in cancer pathogenesis by production of several mitogenic and
pro-invasive factors into the tumor microenvironment (4). Several current reports indicate the
crucial involvement of stromal fibroblasts in the progression of
CCA via certain types of secreted tumorigenic substances (5–7).
These substances can prime cancer cells in a paracrine mode to
develop tumorigenic intracellular signaling pathways resulting in
the increased cell proliferation, growth and invasion.
Almost all of the CCA stromal fibroblasts have been
revealed to be α-smooth muscle actin (α-SMA)-positive cells
(5,8). The fibroblasts were transformed into
activated fibroblasts or myofibroblasts and were incorporated into
the tumor and produced extracellular matrix proteins that led to
tumor fibrosis (8). The recent
work from the authors of the present group suggests that levels of
α-SMA positive CCA stromal fibroblasts were correlated with poor
patient survival (5). CCA
fibroblasts isolated from CCA tissues have exhibited the ability to
promote tumor cell proliferation and invasion (5,6). The
whole gene expression analysis of CCA fibroblasts compared to
normal fibroblasts has reported several differential expressed
genes encoded to produce tumorigenic secreted proteins including
periostin (PN) (6). The findings
reported here are in support of several groups of researchers,
wherein PN has been confirmed to express in CCA tissues solely from
stromal fibroblasts (6,9,10).
It has been proposed as a potential marker of poor prognosis in CCA
patients (6). Though in
vitro studies revealed the function of PN in induction of CCA
cell proliferation, growth, and invasion, little is known regarding
the mechanism of PN-induced CCA cell invasion that is a crucial
phenomenon to activate CCA metastasis.
PN is an extracellular matrix (ECM) protein with
multi-functional roles in tumorigenesis and tumor progression at
each step of the transformation of normal into malignant cells and
metastatic tumors (11). It has
been proposed as a marker associated with cancer aggressiveness in
pancreatic cancer (12,13), gastric cancer (14), breast cancer (15), thyroid carcinoma (16) and non-small cell lung cancer
(17). In CCA, the impact of
fibroblast-derived PN is convincing by its ability to activate
cancer cell proliferation and invasion (6). To activate biological functions of
cells, PN has been investigated for its ability to bind to integrin
(ITG) receptors. In epithelial ovarian carcinoma, PN bound with
ITGs αvβ3 and αvβ5 promoted cancer cell motility (18). The interference of these two ITGs
by specific anti-ITG antibodies had an effect on the ability of PN
to mediate cell adhesion in head and neck squamous cell carcinoma
(19). For intracellular
signaling, PN potently promotes metastatic growth of colon cancer
by augmenting cell survival via the AKT/PKB pathway in colon cancer
(20). Similarly, PN from
pancreatic cancer cells activated ITGβ4 and promoted invasiveness
of cancer cells through the PI3K pathway (21). But in vascular smooth muscle cells,
PN was demonstrated to induce cell migration through ITGs αvβ3 and
αvβ5 and the focal adhesion kinase pathway (22). In breast cancer, PN enhanced
angiogenesis, in part, from the up-regulation of the vascular
endothelial growth factor receptor on endothelial cells through the
ITGαvβ3-focal adhesion kinase (FAK)-mediated signaling pathway
(23). In addition, PN could
induce epithelial-mesenchymal transition characteristics resulting
in tumor metastasis through cross-talk between ITGαvβ5 and the
epidermal growth factor receptor signaling pathways (24). It seems reasonable then to conclude
that PN, in the dependent context, can activate specific
ITG-mediated signal pathways and different biological
responses.
Even though, it was found that the invasive property
of CCA cells stimulated by PN was reduced after transient knockdown
ITGα5 (6), little is known
regarding the intracellular signaling pathway activated by PN
through ITGα5-mediated cell invasion. In the present study, the
ITGs expression in CCA cell lines were explored and showed
abundance of ITGs α5β1 and α6β4. The adhesion assay revealed the
propensity of CCA cells to bind PN via ITGα5β1. Using cells with a
low level of functional ITGα5β1 by treating these cells with either
siITGα5β1 or neutralizing anti-ITGα5 antibody, the results
showed that the PI3K/AKT-mediated, but not the ERK-mediated
signaling pathway was involved in PN-stimulated CCA invasion.
Since, PN is abundant in CCA tissues, understanding the role of PN
in induction of cancer cell invasion may provide important
information to attenuate metastasis and help identify the possible
therapeutic targets.
Materials and methods
CCA cell culture
Human CCA cell lines KKU-M055, KKU-100, KKU-M139,
KKU-M156, KKU-M213, KKU-M214 and KKU-OCA17 were kindly donated from
Associate Professor Banchob Sripa, Khon Kaen University, Thailand.
KKU-M213, KKU-M214 and KKU-OCA17 originated from well
differentiated CCA tissues; KKU-M055, KKU-M139 and KKU-M156 from
moderate CCA; and KKU-100 was isolated from poorly differentiated
tissue (25). The non-tumorigenic
immortalized bile duct epithelial cell MMNK1 was kindly provided by
Professor Naoya Kobayashi, Department of Surgery, Okayama
University Graduate School of Medicine and Dentistry, Japan
(26). CCA cells and MMNK1 were
cultured in Ham F-12 medium (Invitrogen, Carlsbad, CA, USA)
supplemented with 10% fetal bovine serum (FBS), 100 U/ml
penicillin, 100 μg/ml streptomycin (Invitrogen) and
anti-fungal agent. Cells were cultured in a humidified 5%
CO2 incubator at 37°C. Cells were passaged by 0.25%
trypsin-EDTA and those of more than 90% viability were used in
further experiments.
Measurement of ITG expression pattern in
CCA cell lines by real-time PCR
Total-RNA was extracted from all CCA cell lines and
MMNK1 using PerfectPure RNA Cultured Cell Kit (5 Prime,
Gaithersburg, MD, USA) according to the manufacturer’s
instructions. The cDNA was synthesized using SuperScript™ III
First-Strand Synthesis System (Invitrogen) according to the
instructions. Expression levels of ITGs αv, α5,
α6, β1, β3, β4 and β5 were
determined by SYBR-Green-based real-time PCR in Light
Cycler® 480 II machine (Roche Applied Sciences,
Indianapolis, IN, USA). The β-actin served as an internal
control to adjust the amount of starting cDNA. The expression of
each ITG was calculated by the
2−ΔCp equation. In this
case, ΔCp=Cp(ITG) −
Cp(β-actin). The sequences of genes used in this
study were retrieved from PubMed (www.ncbi.nln.nih.gov) and primers were designed using
Primer 3 software. A list of primers is summarized (Table I).
 | Table IThe primers used in this study. |
Table I
The primers used in this study.
| Gene | Forward primer
5′-3′ | Reverse primer
5′-3′ | Size (bp) | Accession no. |
|---|
| ITGαv |
TGACTGGTCTTCTACCCGC |
CTCACAGATGCTCCAAACCA | 121 | NM_002210 |
| ITGα5 |
AGTTGCATTTCCGAGTCTGG |
CTCTGGGAGCACCAGATACAA | 223 | NM_002205 |
| ITGα6 |
GGCCTTATGAAGTTGGTGGA |
CTCTGGGAGCACCAGATACAA | 144 | NM_000210 |
| ITGβ1 |
TCCCTGAAAGTCCCAAGTGT |
TTTCCTGCAGTAAGCATCCA | 143 | NM_033666 |
| ITGβ3 |
TGGTCCTGCTCTCAGTGATG |
TGAAGGTAGACGTGGCCTCT | 180 | NM_000212 |
| ITGβ4 |
TCTCCTACCGCACACAGGA |
CTTCACCTGCAGCTCTTTCC | 110 | NM_001005619 |
| ITGβ5 |
CTCCACTCTGGGAAACCTGA |
AGGACGGTCAGGTTGGACTT | 188 | NM_002213 |
| β-actin |
CACACTGTGCCCATCTACGA |
CTCCTTAATGTCACGCACGA | 162 | X00351 |
Flow cytometry analysis of ITG
expression
For detection of the actual ITG α5β1 and α6β4 levels
in biliary epithelial cells, cell pellets of around
1×106 were fixed in 2% formaldehyde for 15 min at room
temperature. The fixed cells were incubated with 1:50 goat
anti-human ITGα5 (Santa Cruz Biotechnology, Santa Cruz, CA, USA)
diluted in a washing solution which was HAM/F-12 containing 2%
(v/v) FBS, 1% (w/v) bovine serum albumin (BSA) and 10 mM
NaN3 for 2 h at room temperature. Cells in the washing
solution were centrifuged at 400 x g for 3 times and 5 min each to
remove the excess primary antibody and this was followed by
staining with 1:2,000 donkey anti-goat IgG-Alexa 488 (Invitrogen)
diluted in the washing solution for 1 h at room temperature with
light protection. For ITGα6β4 detection, 1:100 mouse anti-human
ITGβ4 (Millipore, Temecula, CA, USA) diluted in washing solution
was incubated with cells for 1 h at 4°C and followed by 1:100
rabbit anti-mouse IgG-FITC (Dako, Carpinteria, CA, USA) diluted in
washing solution for 30 min at 4°C with light protection.
The ITGα5 and ITGβ4 signals were determined in the
FL-1 channel of a Becton Dickinson FACSort (Becton Dickinson,
Franklin Lakes, NJ, USA) and data analysis was performed by
CellQuest software (Becton Dickinson). The relative mean
fluorescence intensity (MFI) of CCA cell lines was normalized to
that of the negative control stained with secondary antibody only.
Two independent experiments were performed.
Immunocytochemistry of ITGα5β1 and α6β4
in CCA cell lines
Immunocytochemistry was employed to localize ITGα5β1
and ITGα6β4 on the cell membrane. KKU-M213 (2×104 cells)
were cultured on sterile cover slips placed in a 24-well plate for
48 h. Cells were fixed in 4% paraformaldehyde for 15 min and
blocked with 1% BSA for 30 min at room temperature. Then the cells
were incubated with 1:50 goat anti-human ITGα5 (Santa Cruz
Biotechnology) for 2 h at room temperature and subsequently stained
with 1:500 donkey anti-goat IgG-Alexa 488 (Invitrogen) for 1 h at
room temperature with light protection.
For localization of the ITGβ4, cells were plated
onto glass cover slips at a density of 4×104 cells in
24-well plate. After 48 h, cells were washed twice with 1X PBS and
then blocked with blocking solution (10% FBS containing 1X PBS) for
30 min at room temperature. Cells were further incubated with 1:500
mouse anti-human ITGβ4 (Millipore) for 2 h at room temperature and
then incubated in 1:2,000 goat anti-mouse IgG-Cy3 (Jackson
ImmunoResearch Laboratories Inc., West Grove, PA, USA).
The nuclei were stained with 1:1,000 Hoechst 33258
(Invitrogen) for 30 min at room temperature. The fluorescence
signal was observed under the LSM 510 Meta laser scanning confocal
microscope (Carl Zeiss, Jena, Germany) at the Division of Medical
Molecular Biology, Office for Research and Development, Faculty of
Medicine, Siriraj Hospital, Mahidol University, Bangkok,
Thailand.
Neutralization of ITGα5β1 and ITGβ4 on
CCA cells
To ensure roles of ITGα5β1 and ITGβ4 on PN-mediated
CCA cell adhesion and invasion, neutralizing antibody specific to
the ITGα5β1 heterodimer and ITGβ4 subunits was employed to block
intact ITGα5β1 and ITGβ4 on the cell membrane of CCA cells.
KKU-M213 CCA cells were trypsinized and washed with 1X PBS two
times. Cell pellets of around 1×105 cells were incubated
with 1:200 anti-human ITGα5β1 (Millipore) or 1:200 mouse anti-human
ITGβ4 (Millipore) at 37°C for 1 h. Cells in the antibody solution
were centrifuged at 400 x g for 5 min to remove excess antibody.
The ITG-blocked cells were then collected to explore their
responses to PN-induced invasion and adhesion. The number of either
adhered or invaded cells induced by PN were compared with and
without antibody blocking conditions. Two independent experiments
were performed.
Adhesion assay of CCA cell lines on
PN-coated surface
Recombinant PN (rPN) (1 μg) (Biovendor,
Heidelberg, Germany) was coated on a 96-well plate surface at 37°C
for 2 h. Cells with or without exposure to neutralizing antibodies
against ITGα5β1 or ITGβ4 (Millipore) were then added to each well
and incubated at 37°C for 1 h. Unattached cells were removed by
rinsing twice with serum-free media. The number of adherent cells
was determined by an MTS assay kit (Promega, Madison, WI, USA)
according to the manufacturer’s instructions. The percentage of
PN-induced cell adhesion was normalized to that of cells attached
on 1% BSA-coated wells. Two independent experiments were
performed.
Invasion assay
To investigate the effect of rPN on the invasion of
parental KKU-M213 cells, siITGa5-treated cells, and
ITGα5β1-blocked cells; 2×104 cells of each condition
were suspended in 100 ng/ml rPN containing complete medium and
cultured in the upper chamber of the Matrigel™ invasion chamber (BD
Biosciences, San Jose, CA, USA) for 24 h. Invaded cells were fixed
in 5% (v/v) glutaraldehyde and then hematoxylin and eosin staining
was performed. The number of invaded cells was counted under an
inverted microscope by two independent investigators using ×100
magnification fields. The assays were done in replicates of three
independent experiments. Numbers of invaded cells was compared to
those without rPN treatment.
Western blot analysis of pAKT and
pERK
Cells with or without ITGα5 silencing and cells in
the presence or absence of 100 μM LY294002, a PI3K inhibitor
(Calbiochem, San Diego, CA, USA) or 30 μM U0126, an ERK
inhibitor (Tocris Bioscience, MO) were induced by 100 ng/ml rPN for
30 and 120 min. Then the levels of pAKT and pERK1/2 were determined
using western blot analysis. Cell pellets were collected after
centrifugation of cell suspensions at 400 x g for 5 min in a
refrigerated centrifuge. The cell pellets were rinsed by cold 1X
PBS 2 times before lysed in 1X sample buffer containing 50 mM
Tris-HCl pH 6.8, 2% (w/v) SDS, 10% (v/v) glycerol, 5% (v/v)
β-mercaptoethanol and 0.05% (w/v) bromophenol blue. Cell lysate was
boiled for 10 min and centrifuged to remove the undissolved
proteins and cell debris at 8,000 x g for 1 min. Cell extracts were
then separated in 10% SDS-PAGE and transferred onto PVDF membranes
(Amersham, Buckinghamshire, UK). Membranes were blocked in 5% skim
milk containing TBST for 1 h at room temperature. Rabbit anti-human
pAKT (Thr308) (Santa Cruz Biotechnology) at the dilution of 1:1,000
and rabbit anti-human pERK1/2 (Cell Signaling Technology Inc.,
Danvers, MA, USA) at the dilution of 1:2,000 were used as primary
antibodies for incubation with the membrane for 1 h at room
temperature. The 1:2,000 goat anti-rabbit IgG-HRP (Abcam,
Cambridge, MA, USA) was used as the secondary antibody and
incubated for 1 h at room temperature. The immunoreactive signals
were visualized by enhanced chemiluminescense (Pierce, Rockford,
IL, USA). The β-actin protein level was used as an internal control
to determine the equal amount of loading proteins.
Statistical analysis
The values from different independent experiments
were expressed as mean ± SD. The significance of the different data
sets was determined by the Student’s t-test. A P-value of ≤0.05 was
defined as statistically significant.
Results
Integrin expression profile in CCA cell
lines
ITGs that were previously reported in CCA, and in
the related cancer, hepatocellular carcinoma, and those that have
been revealed as PN receptors including α-subunits: αv, α5 and α6;
and β-subunits: β1, β3, β4 and β5 were explored. Real-time PCR
using 50 ng starting cDNA exhibited different levels of each
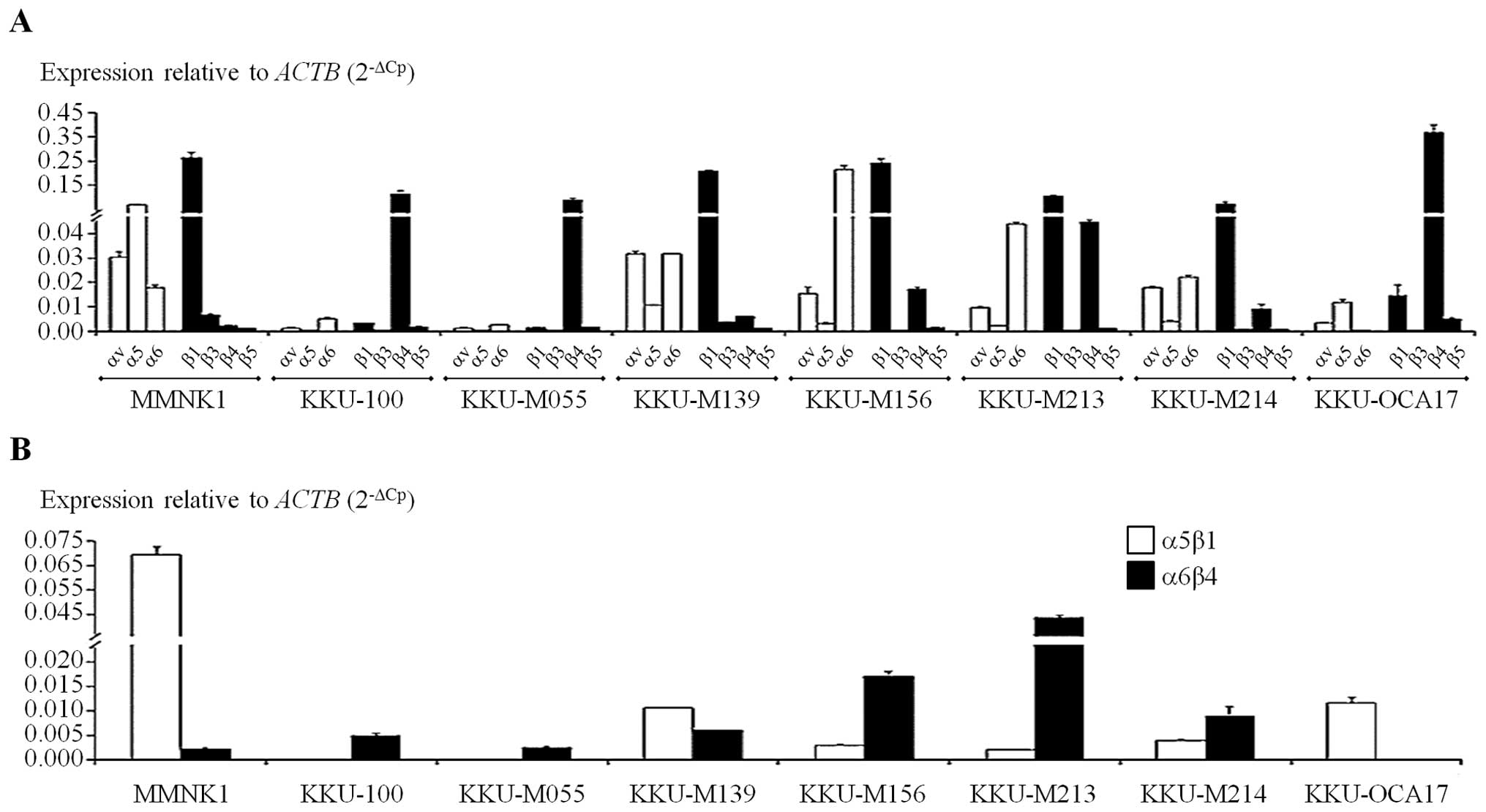
subunit in CCA cell lines and MMNK1 cells. Among α-subunits, α6 was
expressed at the highest level in almost of cell types except
KKU-OCA17 (Fig. 1A). In addition,
α5-subunit showed the highest level among other α-subunits in
MMNK1, but a moderate expression level was found in KKU-M139,
KKU-M213 and KKU-M214. For the β-subunit, the results revealed that
ITGβ1 had the highest expression level in KKU-M139, KKU-M156,
KKU-M213, KKU-M214 and MMNK1, whereas KKU-K100, KKU-M055 and
KKU-OCA17 had highest level of ITGβ4.
Since certain type of α-subunit ITG can be paired
with a specific type of β-subunit, the predicted level of intact
ITGs were presented based on the minimal level of their counterpart
of either α-subunit or β-subunit. It has been shown that ITGα5 can
bind only to β1 while ITGβ4 can only bind with α6 (27). Moreover, ITGαv can bind to several
β-subunits including β1, β3, β5, β6 and β8. So the level of ITGβ3
may determine the maximal level of ITGαvβ3. Using the same concept,
the amount of ITGβ5 would roughly present the possible maximal
level of ITGαvβ5. Whereas, for ITGα5β1 and ITGα6β4, the expression
of α5-subunit and β4-subunit may determine the maximal levels.
Hence, the possible maximal levels of ITGs αvβ3, αvβ5, α5β1 and
α6β4 are shown (Fig. 1B). The
expressions of both α5β1 and α6β4 ITGs were found in all cell
types. Some cell types had ITGα5β1 as the predominant expression
level including MMNK1 and KKU-M139, but KKU-M156, KKU-M213 and
KKU-M214 CCA cells showed predominant ITGα6β4 expression. Cells
with high levels of both ITGα5β1 and ITGα6β4 were KKU-M139,
KKU-M156, KKU-M213 and KKU-M214. Interestingly, KKU-M055 showed
very low levels of ITGα5β1 as well as KKU-100.
Expressions of membrane ITGs α5β1 and
α6β4 on CCA cell lines
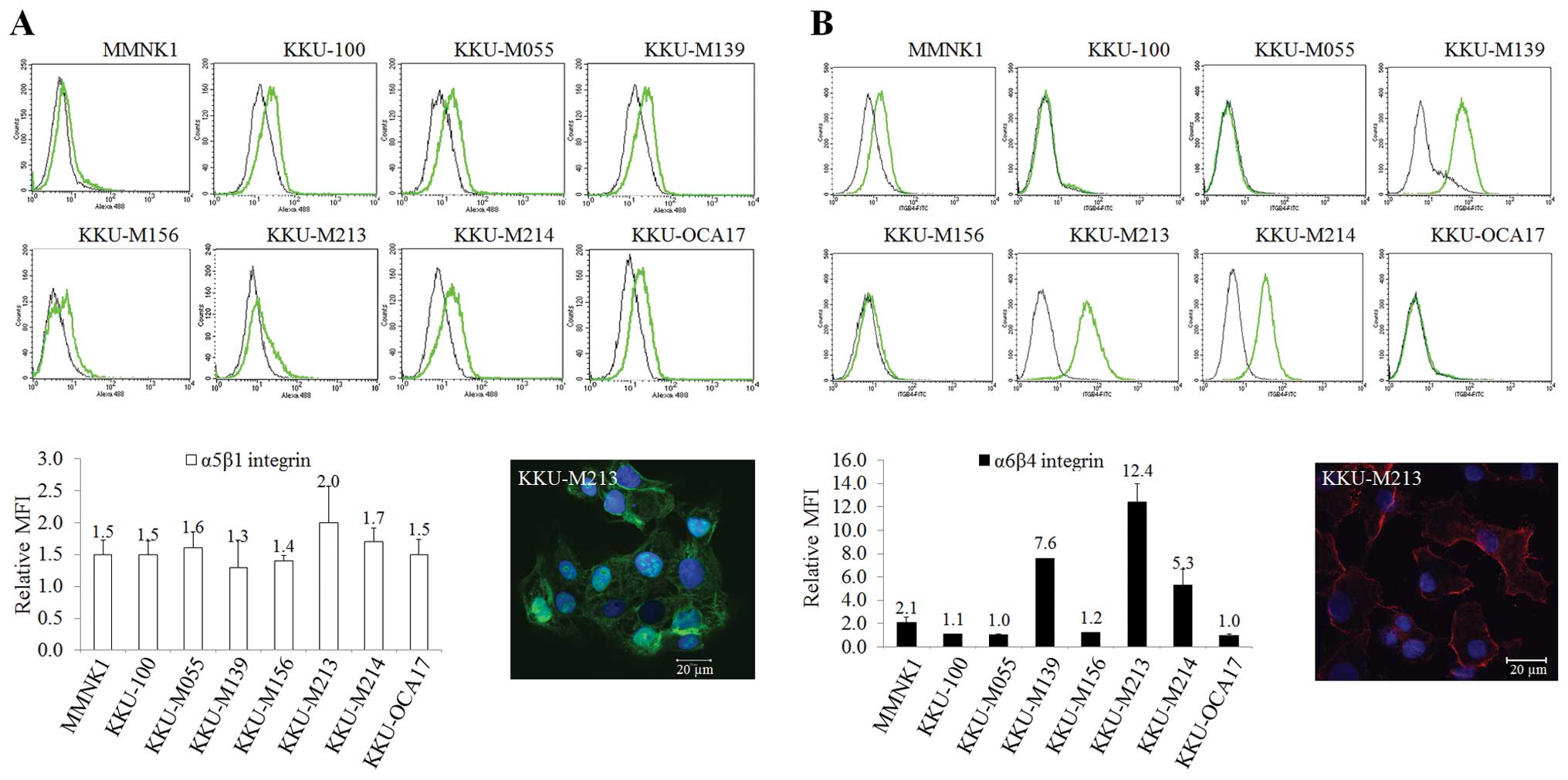
To confirm the actual protein expression levels of
ITG α5β1 and α6β4, FACS analysis was performed. The results
revealed different amounts of these two ITGs among different types
of CCA cell lines. For ITGα5β1, the relative mean fluorescence
intensity (MFI) showed similar levels of expression in all cell
lines with the highest signal in KKU-M213 (Fig. 2A). Most of CCA cells originated
from well differentiated cancers including KKU-M213 and KKU-M214
expressed high levels of ITGα6β4 similar to KKU-M139 which was
derived from moderate differentiation of squamous cell types
(Fig. 2B). Almost all cell lines
derived from moderately differentiated types (KKU-M055 and
KKU-M156) and the poorly differentiated type (KKU-100) had low
expression levels of ITGα6β4. In contrast, the expression of
ITGα6β4 was found higher in KKU-M139, KKU-M213 and KKU-M214 than
other cells, which is concordant to the results of mRNA levels.
Interestingly, the highest level of ITGα6β4 (relative MFI=12.4±1.6)
and ITGα5β1 (relative MFI=2.0±0.57) were detected in KKU-M213
(Fig. 2B).
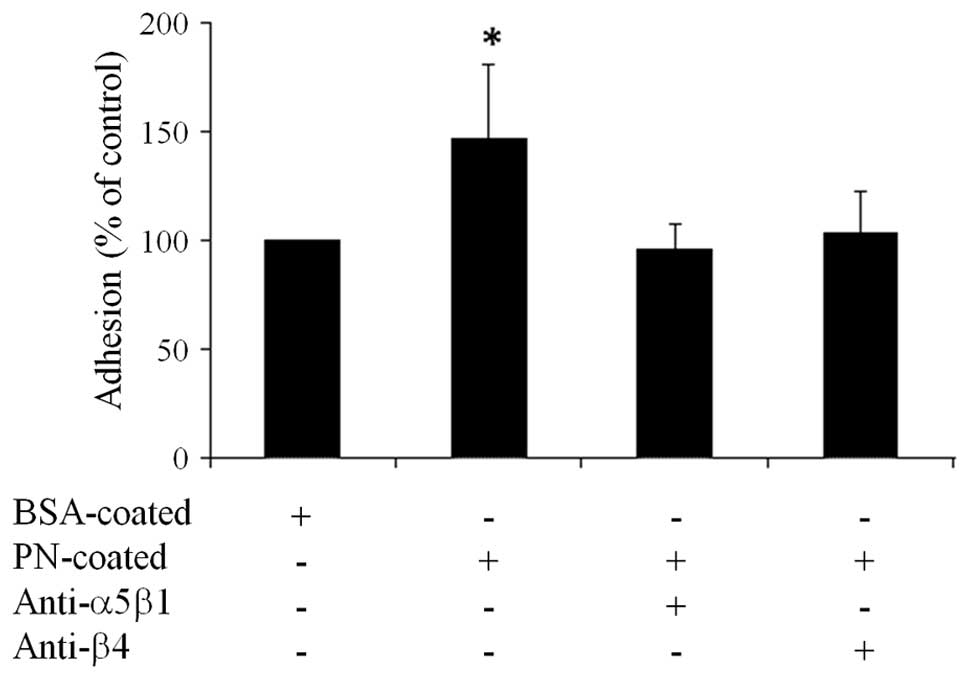
PN mediates cell adhesion through
ITGsα5β1 and α6β4 receptors
To demonstrate the impact of ITGα5β1 and ITGα6β4 in
PN-activated tumorigenic function of CCA cells, KKU-M213 cells
having the highest level of both ITGs were used to perform the
adhesion assay. PN-coated culture plates were utilized to explore
the binding efficiency of the cancer cells with and without
functional ITG receptors on the cell membrane after incubation with
the neutralizing antibodies. The results showed that CCA cells
could intrinsically bind to PN-coated surface more than to
BSA-coated surfaces or negative controls with statistical
significance (Fig. 3). The binding
efficiency was reduced to a similar level of the negative control
when cells were blocked either with the intact ITGα5β1 or ITGα6β4
by specific neutralizing antibodies.
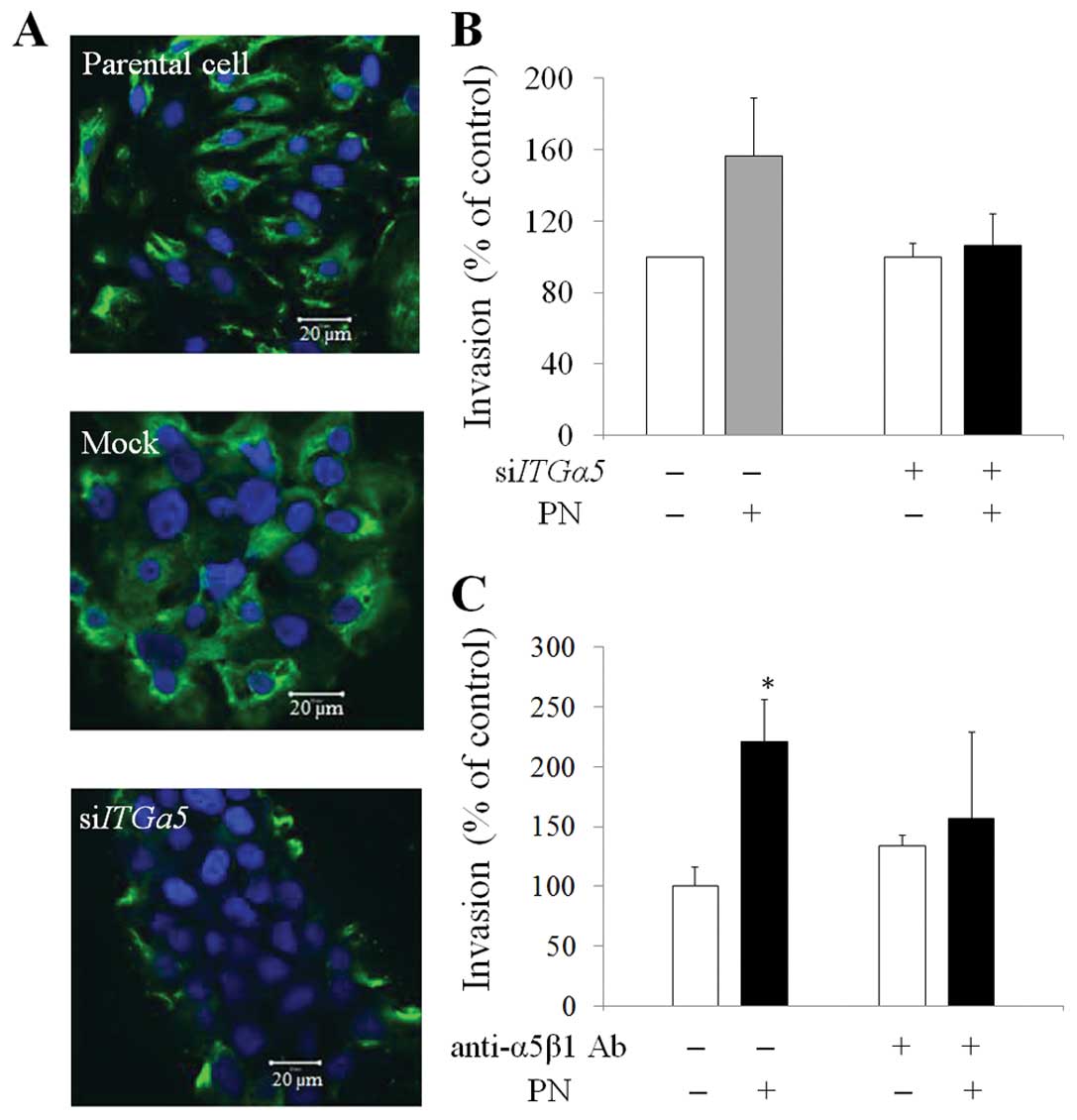
PN-mediated CCA invasion via ITGα5β1
To confirm whether cells with unavailable ITGα5β1
could be induced by PN, siITGα5-treated and anti-ITGα5β1
antibody-treated cells were performed invasion assay with and
without PN treatment. The intact ITGα5β1 receptor on the membrane
of CCA cells was detected by immunocytochemistry and showed that
cells exposed to siITGα5 successfully inhibited the
expression of ITGα5β1 on the membrane of cancer cells as compared
to the intrinsic expression in parental and mock cells (Fig. 4A). The decreased PN-induced
invasive capability of the ITGα5-knockdown cells was
revealed (Fig. 4B). Significant
increases of PN-induced invasion was observed in mock cells
(156±18%) as compared to cells without PN stimulation. The results
revealed that ITGα5-knockdown cells showed decreased
PN-induced invasive capability (106±18% of invaded cells) when
compared to negative controls without PN. Moreover, in cells
blocking ITGα5β1 with neutralizing anti-ITGα5β1 antibody, the
results showed in a similar way that PN could not induce KKU-M213
cell invasion if there was no ITGα5β1 available on the cell
membrane (Fig. 4C).
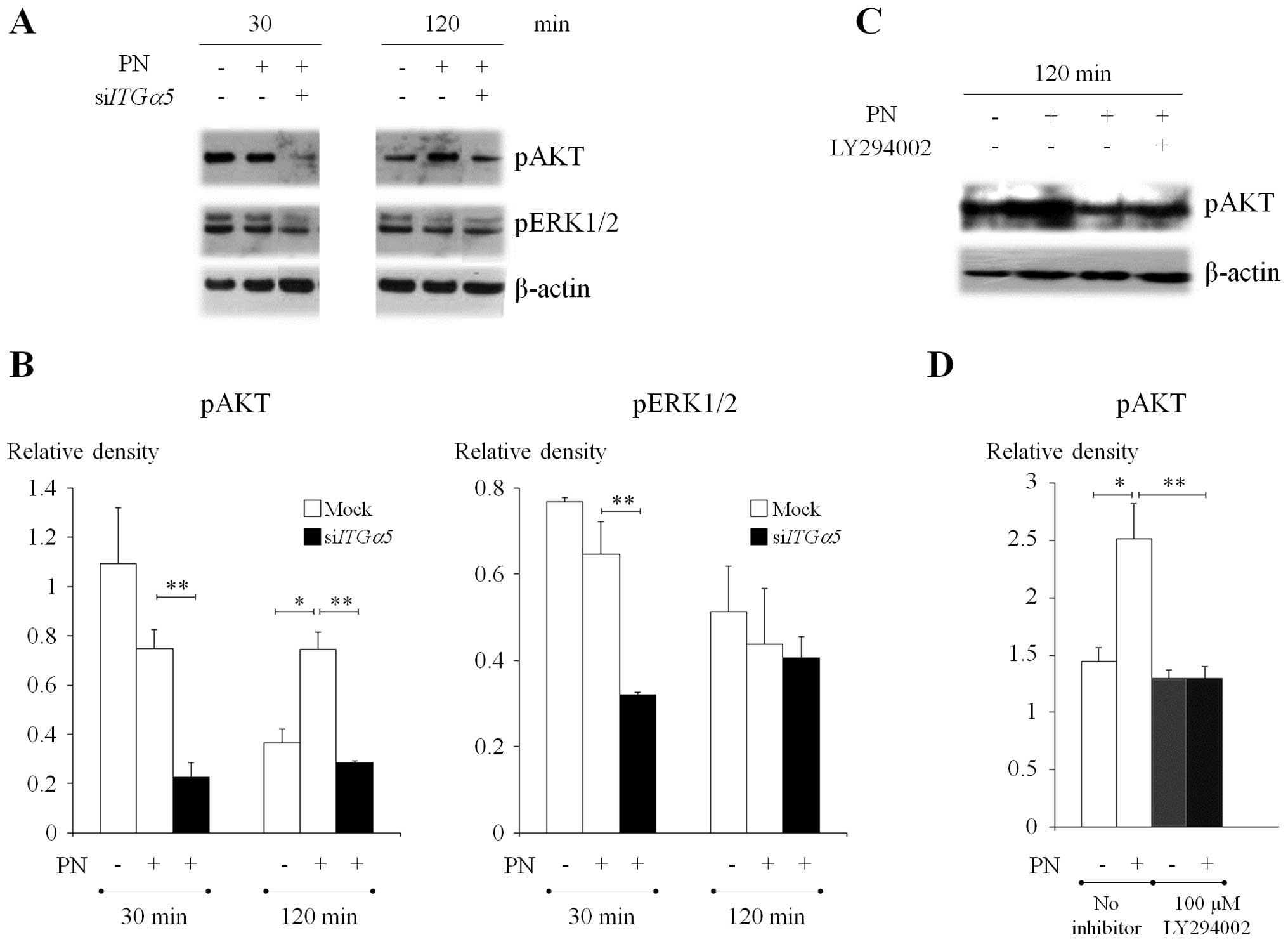
PN induces pAKT through activation of
PI3K pathway in CCA cells
In order to investigate the intracellular signaling
pathway activated by PN via ITGα5β1, cells with normal levels and
ITGα5β1-knockdown cells were treated with recombinant PN. The
results revealed that exogenous PN could significantly induce
phosphorylation of AKT in KKU-M213 parental cells at 120-min
post-treatment compared to control cells without PN treatment
(Fig. 5A and B). PN could not
activate pAKT in cells with transient knockdown of ITGα5, pERK1/2,
however, did not change as a result of PN stimulation when compared
between cells with and without ITGα5β1 (Fig. 5A and B). These results suggested
that upon PN stimulation via ITGα5β1, AKT, but not ERK, was
activated.
To confirm the signaling pathway of PN-induced AKT,
the pharmacological inhibitor of PI3K (LY294002) which is the
upstream molecule of AKT, was applied to KKU-M213 CCA cells and the
level of AKT phosphorylation was determined under the conditions
with and without PN stimulation. The results showed depletion of
pAKT level in responding to LY294002 treatment which confirmed the
antagonist effect of this inhibitor to pAKT (Fig. 5C and D). Interestingly, PN could
not induce pAKT in the PI3K-inhibited cells. Hence, in cells
activated with PN, the pAKT was significantly higher in parental
cells than in cells pretreated with PI3K inhibitor.
Discussion
The progression of cancer is no longer recognized as
an independent aberrant event occurring only in cancer cells.
Substances surrounding the cancer cells secreted from a variety of
cells in the tumor microenvironment and signaling pathways induced
by cancer and other cells, and cancer cell-substance interactions
are thought to play crucial roles. The role of tumor-associated
fibroblasts as the most abundant cell types in the tumor
microenvironment and the major source of growth factors and
extracellular matrix substances in tumorigenesis and tumor
progression have been reported as important contributors (4,28).
In the current situation drug resistance of cancer treatment is a
common phenomenon in cancer patients due to the vulnerability of
cancer cells to undergo genetic changes as the result of their
rapid proliferation rate. Cancer fibroblasts are therefore suitable
to target since they exhibit less genetic instabilities (29). Based on this concept, several
studies have been performed and the obtained information has been
summarized in several review articles to confirm the possibility of
targeting cancer fibroblast as an additional treatment strategy in
cancer patients (4,30–32).
Different cancer fibroblasts have their unique
properties in the production of certain substances (6,33,34).
Fibroblasts isolated from CCA tissues can produce a variety groups
of tumorigenic substances which play important roles in induction
of cancer cell proliferation (5).
Unpublished data from the present research team has revealed
migration induction when CCA cells were treated with the
conditioned-medium from primary cultured-CCA fibroblasts. The gene
expression profile of CCA fibroblasts has been performed by this
group and the increased expression of tumor-related genes in
CCA-derived fibroblasts has been reported (6). Among these genes, PN has been
confirmed with high expression in the microenvironment of CCA
tissues with relation to the short survival time of the patients
together with tumorigenic induction in cancer cells in vitro
including cell proliferation, growth, and invasion (6). Herein, the underlying mechanism of
how CCA cells respond to PN-driven invasion has been explored.
ITGs, as the receptors for PN (18,21,22),
were explored in CCA cells. Adhesion assays indicated that ITGα5β1
and ITGα6β4 were the receptors for PN and the invasion assay
confirmed that ITGα5β1 was involved in PN-induced CCA cell invasion
and PI3K/AKT was the signaling pathway underlying this
mechanism.
Previous reports on ITG expression in CCA cells
indicated that α1 had no expression whereas ITGs α2, α3, α6, β1 and
β4 were expressed in almost all CCA cell lines (35–37).
This is consistent with the present findings that all CCA cell
lines expressed high level of ITGα6, though the synthesis of ITGs
α2 and α3 were not included in the current study. Notably, the
expression of ITGα5 is not uniformly expressed in CCA (36). The result showed the expression of
ITGα5 in some CCA cells; in particular the cells with previously
reported high responses to PN-induced invasion such as KKU-M213 CCA
cell lines (6). Though the
expression of ITGαv has mostly been reported as PN receptor, no
evidence was found in CCA cells. The current results revealed that
ITGαv could be detected in almost all CCA cell lines but at a lower
level than that of ITGs α5 and α6. These results imply that in
addition to ITGs α2, α3, and α6 previously reported, CCA cells
could express αv and α5. It is difficult to predict the level of
ITGαvβ3 and αvβ5 because ITGαv can bind to several β-subunits of
ITG including β1, β3, β5, β6 and β8 (27). ITGα5, however, can form
heterodimers only with ITGβ1 (27,38).
It can then be concluded that ITGα5β1 may be one of the existing
ITGs on the CCA cell membrane and may have an important impact in
cancer progression after the stimulation by stromal PN.
For β-subunit ITGs, the present results are similar
to previous reports that ITGs β1 and β4 were expressed in almost
all CCA cells (35,36). ITGβ4 was found at higher levels in
CCA cells as compared to those in MMNK1 immortalized
non-tumorigenic biliary cells. Though in the previous report
(35), ITGβ4 was detected in
normal and proliferating biliary epithelial cells but was an
inconsistent finding in CCA. This study provides conclusive
evidence of the presence of ITGβ4 in CCA cells. Since ITGβ4 can
bind only to ITG α6, it is likely that the level of ITGβ4 can be
roughly determined by the level of ITGα6β4 presented on CCA cell
membranes. In addition, this study indicated β3 and β5, generally
form heterodimers with ITGαv, as the ITGs with low expression in
CCA cells. These results therefore suggested that CCA cells express
high levels of ITGs α5, α6, β1 and β4. It is suggested that the
heterodimers of ITG α5β1 and α6β4 may be the major ITG receptors
expressed on CCA cell membrane. The flow cytometry analysis and
immunofluorescence staining confirmed the presence of these two
ITGs on the membrane of KKU-M213 cells. The favorable binding of PN
on ITGs α5β1 and α6β4 indicated by the lower numbers of cells bound
onto the PN-coated surface after treatment of cells with the
specific neutralizing antibody. The ITGα6β4 was able to interact
with PN as well as ITGα5β1 at similar levels. Hence, it can be
concluded that PN may influence the progressive tumor behavior of
CCA cells though either ITGα5β1 or ITGα6β4.
PN has been demonstrated to activate cellular
responses via different ITGs depending on cell type context, for
example, ITGαvβ3 in non-small cell lung cancer (39) and ITGα6β4 in pancreatic cancer
(21). Previous work by this group
reports that siITGα5-treated cells have lower PN-induced
cell proliferation and invasion compared to the parental cells
(6) and shows in the first report
presenting the association of PN and ITGα5β1 in tumor promotion of
CCA cells. It is possible that the interaction of PN and ITGα5β1
may be the unique phenomenon of PN in promotion of CCA. Hence, in
this current report, we explored the signaling pathway starting
from ITGα5β1 in the induction of cancer invasion by PN. Similar to
the data on the PN-activated signaling pathway reported previously
(21,39), PN could activate cell invasion via
the stimulation of AKT-dependent, but not ERK, pathway in CCA
cells. It is well known that activation of the PI3K-AKT-dependent
pathway is essential in the regulation of several different
biological functions including cell survival, growth and
proliferation, invasion and migration in a ligand specific manner
(40–42). ITGα5β1 plays an important role in
metastasis, invasion and poor prognosis of some cancers (43). Cancer cells with high expression of
ITGα5β1 showed an increased invasiveness into 3D collagen matrices
through enhancement of the contractile force (44). In addition, the role of ITGα5β1 in
stimulation of cell invasion has been revealed through activation
of matrix metalloproteinase 2 (MMP2) in breast cancer (45). The fact that PN-ITGα5β1 interaction
stimulates the enhancement of cell contractile force and some MMP
expressions is the possible underlying mechanisms of how PN helps
tumor cells to invade and finally metastasize. Unpublished data by
the present authors showed that PN induced MMP9 and MMP13 from CCA
cells and activated cancer cells to migrate (data not shown) which
may be the effect of PN-mediated change of cytoskeletal proteins
through FAK (22). Finally, the
actual downstream signaling pathway after AKT activation is of
particular interest because the proper inhibitor can be proposed to
apply for the attenuation of cancer progression with minimal
side-effects (46).
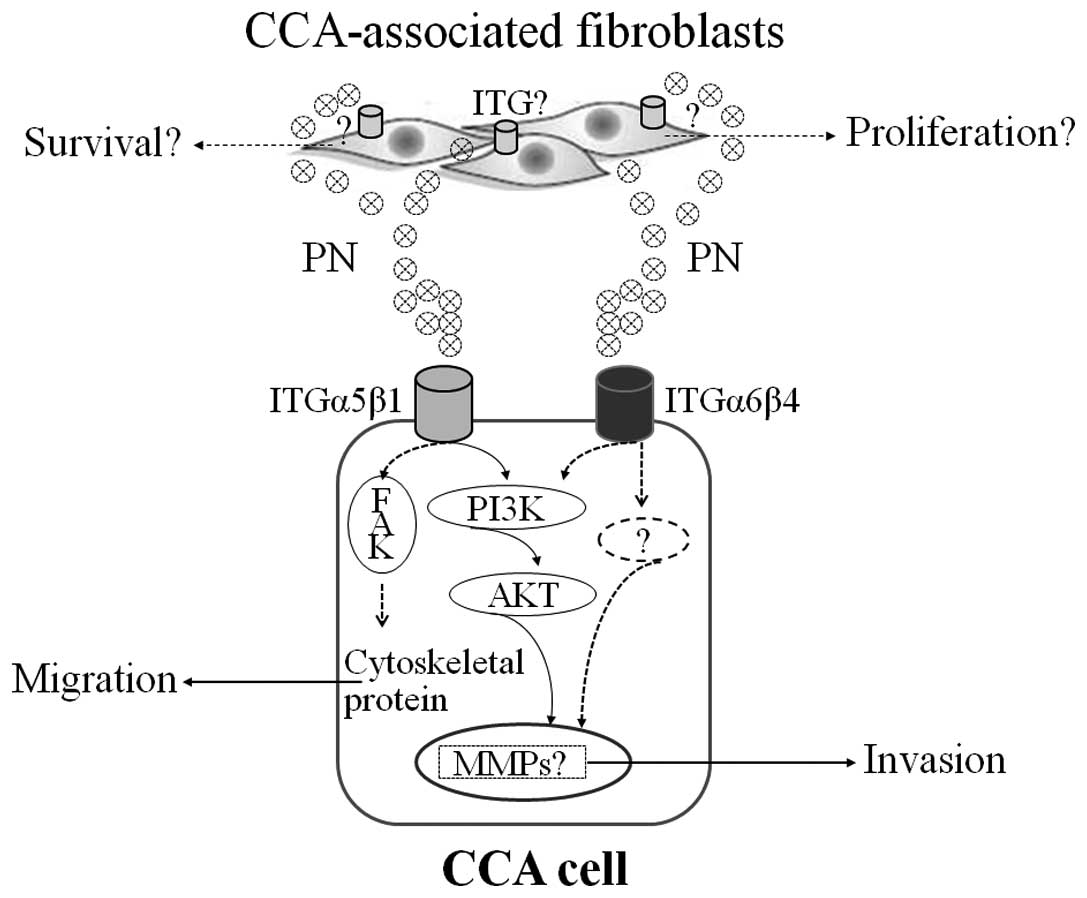
In summary, fibroblasts are not passive bystanders
in tumor environment. The current trend in cancer research is the
inclusion of the cancer fibroblast as a major contributor of
disease progression and suggests the inhibition of
fibroblast-derived tumor-promoting factors as the first line
approach. This study provides evidence for the contribution of
CCA-associated fibroblast-derived PN in the activation of
tumorigenic properties of CCA cells through receptor ITGs (Fig. 6). PN secreted mainly from
fibroblasts binds to either ITGα5β1 or ITGα6β4 and can facilitate
the invasiveness property of CCA cells. The ITG5α5β1-mediated
PI3K-dependent or FAK (22)
signaling pathways are activated eventually regulating several
cellular responses in particular MMP production and cell migration.
The obtained knowledge implies the potential of using an anti-PN
antibody (47), anti-ITGα5β1
antibody (48), and PI3K/AKT
inhibitors (49,50) to attenuate CCA progression driven
by fibroblasts. Understanding the exact mechanisms responsible for
fibroblast-associated cancer progression is a challenge for the
future as the alternative and synergistic cancer-targeted therapy
in CCA patients.
Acknowledgements
This project was co-supported by a
Mid-Career Grant (RMU5080069), Thailand Research Fund (TRF) and the
Research Strengthening Grant 2007 from National Center for Genetic
Engineering and Biotechnology (BIOTEC), National Science and
Technology Development Agency (NSTDA). The editing of this
manuscript was kindly performed by Professor James A. Will,
University of Wisconsin, Madison, WI, USA.
References
|
1.
|
Sirica AE: Cholangiocarcinoma: molecular
targeting strategies for chemoprevention and therapy. Hepatology.
41:5–15. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Sriamporn S, Pisani P, Pipitgool V,
Suwanrungruang K, Kamsaard S and Parkin DM: Prevalence of
Opisthorchis viverrini infection and incidence of
cholangiocarcinoma in Khon Kaen, Northeast Thailand. Trop Med Int
Health. 9:588–594. 2004.
|
|
3.
|
Wise C, Pilanthananond M, Perry BF, Alpini
G, McNeal M and Glaser SS: Mechanisms of biliary carcinogenesis and
growth. World J Gastroenterol. 14:2986–2989. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Franco OE, Shaw AK, Strand DW and Hayward
SW: Cancer associated fibroblasts in cancer pathogenesis. Semin
Cell Dev Biol. 21:33–39. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Chuaysri C, Thuwajit P, Paupairoj A,
Chau-In S, Suthiphongchai T and Thuwajit C: Alpha-smooth muscle
actin-positive fibroblasts promote biliary cell proliferation and
correlate with poor survival in cholangiocarcinoma. Oncol Rep.
21:957–969. 2009.PubMed/NCBI
|
|
6.
|
Utispan K, Thuwajit P, Abiko Y, et al:
Gene expression profiling of cholangiocarcinoma-derived fibroblast
reveals alterations related to tumor progression and indicates
periostin as a poor prognostic marker. Mol Cancer. 9:132010.
View Article : Google Scholar
|
|
7.
|
Sirica AE, Campbell DJ and Dumur CI:
Cancer-associated fibroblasts in intrahepatic cholangiocarcinoma.
Curr Opin Gastroenterol. 27:276–284. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Terada T, Makimoto K, Terayama N, Suzuki Y
and Nakanuma Y: Alpha-smooth muscle actin-positive stromal cells in
cholangiocarcinomas, hepatocellular carcinomas and metastatic liver
carcinomas. J Hepatol. 24:706–712. 1996. View Article : Google Scholar
|
|
9.
|
Darby IA, Vuillier-Devillers K, Pinault E,
et al: Proteomic analysis of differentially expressed proteins in
peripheral cholangiocarcinoma. Cancer Microenviron. 4:73–91. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Dumur CI, Campbell DJ, DeWitt JL, Oyesanya
RA and Sirica AE: Differential gene expression profiling of
cultured neu-transformed versus spontaneously-transformed rat
cholangiocytes and of corresponding cholangiocarcinomas. Exp Mol
Pathol. 89:227–235. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Ruan K, Bao S and Ouyang G: The
multifaceted role of periostin in tumorigenesis. Cell Mol Life Sci.
66:2219–2230. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Erkan M, Kleeff J, Gorbachevski A, et al:
Periostin creates a tumor-supportive microenvironment in the
pancreas by sustaining fibrogenic stellate cell activity.
Gastroenterology. 132:1447–1464. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Fukushima N, Kikuchi Y, Nishiyama T, Kudo
A and Fukayama M: Periostin deposition in the stroma of invasive
and intraductal neoplasms of the pancreas. Mod Pathol.
21:1044–1053. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Li JS, Sun GW, Wei XY and Tang WH:
Expression of periostin and its clinicopathological relevance in
gastric cancer. World J Gastroenterol. 13:5261–5266. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Puglisi F, Puppin C, Pegolo E, et al:
Expression of periostin in human breast cancer. J Clin Pathol.
61:494–498. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Puppin C, Fabbro D, Dima M, et al: High
periostin expression correlates with aggressiveness in papillary
thyroid carcinomas. J Endocrinol. 197:401–408. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Takanami I, Abiko T and Koizumi S:
Expression of periostin in patients with non-small cell lung
cancer: correlation with angiogenesis and lymphangiogenesis. Int J
Biol Markers. 23:182–186. 2008.PubMed/NCBI
|
|
18.
|
Gillan L, Matei D, Fishman DA, Gerbin CS,
Karlan BY and Chang DD: Periostin secreted by epithelial ovarian
carcinoma is a ligand for alpha(V)beta(3) and alpha(V)beta(5)
integrins and promotes cell motility. Cancer Res. 62:5358–5364.
2002.PubMed/NCBI
|
|
19.
|
Kudo Y, Ogawa I, Kitajima S, et al:
Periostin promotes invasion and anchorage-independent growth in the
metastatic process of head and neck cancer. Cancer Res.
66:6928–6935. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Bao S, Ouyang G, Bai X, et al: Periostin
potently promotes metastatic growth of colon cancer by augmenting
cell survival via the Akt/PKB pathway. Cancer Cell. 5:329–339.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Baril P, Gangeswaran R, Mahon PC, et al:
Periostin promotes invasiveness and resistance of pancreatic cancer
cells to hypoxia-induced cell death: role of the beta4 integrin and
the PI3k pathway. Oncogene. 26:2082–2094. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Li G, Jin R, Norris RA, et al: Periostin
mediates vascular smooth muscle cell migration through the
integrins alphavbeta3 and alphavbeta5 and focal adhesion kinase
(FAK) pathway. Atherosclerosis. 208:358–365. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Shao R, Bao S, Bai X, et al: Acquired
expression of periostin by human breast cancers promotes tumor
angiogenesis through up-regulation of vascular endothelial growth
factor receptor 2 expression. Mol Cell Biol. 24:3992–4003. 2004.
View Article : Google Scholar
|
|
24.
|
Yan W and Shao R: Transduction of a
mesenchyme-specific gene periostin into 293T cells induces cell
invasive activity through epithelial-mesenchymal transformation. J
Biol Chem. 281:19700–19708. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Sripa B, Leungwattanawanit S, Nitta T, et
al: Establishment and characterization of an
opisthorchiasis-associated cholangiocarcinoma cell line (KKU-100).
World J Gastroenterol. 11:3392–3397. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Maruyama M, Kobayashi N, Westerman KA, et
al: Establishment of a highly differentiated immortalized human
cholangiocyte cell line with SV40T and hTERT. Transplantation.
77:446–451. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Gahmberg CG, Fagerholm SC, Nurmi SM,
Chavakis T, Marchesan S and Gronholm M: Regulation of integrin
activity and signalling. Biochim Biophys Acta. 1790:431–444. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Angeli F, Koumakis G, Chen MC, Kumar S and
Delinassios JG: Role of stromal fibroblasts in cancer: promoting or
impeding? Tumour Biol. 30:109–120. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Qiu W, Hu M, Sridhar A, et al: No evidence
of clonal somatic genetic alterations in cancer-associated
fibroblasts from human breast and ovarian carcinomas. Nat Genet.
40:650–655. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Pietras K and Ostman A: Hallmarks of
cancer: interactions with the tumor stroma. Exp Cell Res.
316:1324–1331. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Gonda TA, Varro A, Wang TC and Tycko B:
Molecular biology of cancer-associated fibroblasts: can these cells
be targeted in anti-cancer therapy? Semin Cell Dev Biol. 21:2–10.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Rasanen K and Vaheri A: Activation of
fibroblasts in cancer stroma. Exp Cell Res. 316:2713–2722. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Rosenthal E, McCrory A, Talbert M, Young
G, Murphy-Ullrich J and Gladson C: Elevated expression of TGF-beta1
in head and neck cancer-associated fibroblasts. Mol Carcinog.
40:116–121. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Paland N, Kamer I, Kogan-Sakin I, Madar S,
Goldfinger N and Rotter V: Differential influence of normal and
cancer-associated fibroblasts on the growth of human epithelial
cells in an in vitro cocultivation model of prostate cancer. Mol
Cancer Res. 7:1212–1223. 2009. View Article : Google Scholar
|
|
35.
|
Volpes R, van den Oord JJ and Desmet VJ:
Integrins as differential cell lineage markers of primary liver
tumors. Am J Pathol. 142:1483–1492. 1993.PubMed/NCBI
|
|
36.
|
Enjoji M, Sakai H, Nakashima M and Nawata
H: Integrins: utility as cell type- and stage-specific markers for
hepatocellular carcinoma and cholangiocarcinoma. In Vitro Cell Dev
Biol Anim. 34:25–27. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Patsenker E, Wilkens L, Banz V, et al: The
alphavbeta6 integrin is a highly specific immunohistochemical
marker for cholangiocarcinoma. J Hepatol. 52:362–369. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Hynes RO: Integrins: bidirectional,
allosteric signaling machines. Cell. 110:673–687. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Ouyang G, Liu M, Ruan K, Song G, Mao Y and
Bao S: Upregulated expression of periostin by hypoxia in
non-small-cell lung cancer cells promotes cell survival via the
Akt/PKB pathway. Cancer Lett. 281:213–219. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Fresno Vara JA, Casado E, de Castro J,
Cejas P, Belda-Iniesta C and Gonzalez-Baron M: PI3K/Akt signalling
pathway and cancer. Cancer Treat Rev. 30:193–204. 2004.PubMed/NCBI
|
|
41.
|
Sarker D, Reid AH, Yap TA and de Bono JS:
Targeting the PI3K/AKT pathway for the treatment of prostate
cancer. Clin Cancer Res. 15:4799–4805. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Yadav V and Denning MF: Fyn is induced by
Ras/PI3K/Akt signaling and is required for enhanced
invasion/migration. Mol Carcinog. 50:346–352. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Roman J, Ritzenthaler JD, Roser-Page S,
Sun X and Han S: alpha5beta1-integrin expression is essential for
tumor progression in experimental lung cancer. Am J Respir Cell Mol
Biol. 43:684–691. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Mierke CT, Frey B, Fellner M, Herrmann M
and Fabry B: Integrin alpha5beta1 facilitates cancer cell invasion
through enhanced contractile forces. J Cell Sci. 124:369–383. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Morozevich G, Kozlova N, Cheglakov I,
Ushakova N and Berman A: Integrin alpha5beta1 controls invasion of
human breast carcinoma cells by direct and indirect modulation of
MMP-2 collagenase activity. Cell Cycle. 8:2219–2225. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Castillo SS, Brognard J, Petukhov PA, et
al: Preferential inhibition of Akt and killing of Akt-dependent
cancer cells by rationally designed phosphatidylinositol ether
lipid analogues. Cancer Res. 64:2782–2792. 2004. View Article : Google Scholar
|
|
47.
|
Kyutoku M, Taniyama Y, Katsuragi N, et al:
Role of periostin in cancer progression and metastasis: inhibition
of breast cancer progression and metastasis by anti-periostin
antibody in a murine model. Int J Mol Med. 28:181–186.
2011.PubMed/NCBI
|
|
48.
|
Almokadem S and Belani CP: Volociximab in
cancer. Expert Opin Biol Ther. 12:251–257. 2012. View Article : Google Scholar
|
|
49.
|
Li Z, Tan F, Liewehr DJ, Steinberg SM and
Thiele CJ: In vitro and in vivo inhibition of neuroblastoma tumor
cell growth by AKT inhibitor perifosine. J Natl Cancer Inst.
102:758–770. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Pal SK, Reckamp K, Yu H and Figlin RA: Akt
inhibitors in clinical development for the treatment of cancer.
Expert Opin Investig Drugs. 19:1355–1366. 2010. View Article : Google Scholar : PubMed/NCBI
|