Introduction
Lung cancer continues to be a leading cause of
cancer death both in Japan and worldwide (1) and, despite recent improvements in
chemotherapies and molecular-targeted therapies, the prognosis
remains poor (2–5). Patient selection based on a specific
biomarker is one strategy that could lead to improved lung cancer
treatments. Although some biomarkers predictive of metastasis,
prognosis and drug sensitivity have already been reported in lung
cancer, more sensitive and specific biomarkers could facilitate the
development of novel therapeutic applications (6–8).
Epithelial-mesenchymal transition (EMT) comprises a
complex series of reversible events that can lead to the loss of
epithelial cell adhesion and the induction of a mesenchymal
phenotype (9). Thus, EMT is
characterized by the loss of epithelial differentiation markers
including E-cadherin and the induction of mesenchymal markers such
as vimentin and fibronectin. EMT can be induced by transforming
growth factor-β1 (TGF-β1) (10).
The Smad pathway is a major transducer of TGF-β signaling (11). Smad2 and Smad3 are phosphorylated
by the TGF-β type I receptor and form complexes with Smad4
(11). These complexes accumulate
in the nucleus of the cell, regulating the transcription of target
genes and playing critical roles in the control of cell
proliferation, differentiation, apoptosis and cell migration. In
response to TGF-β, the TGF-β receptors also activate alternative
signaling effectors, such as mitogen-activated protein kinase,
phosphatidylinositol-3 kinase, and Rho-like GTPases (11). It has been recognized that EMT
plays a pivotal role in several diverse processes during embryonic
development, chronic inflammation and fibrosis (12). Recently, several studies
demonstrated that EMT was correlated with carcinogenesis,
metastasis and poor prognosis in various human cancers, including
those of the lung (13–16). Furthermore, EMT has been reported
to be related to reduced sensitivity and acquired resistance to
epidermal growth factor tyrosine kinase inhibitors (EGFR-TKI) in
lung cancer cells (17–19). Taken together, these findings
demonstrate that the suppression of EMT could be used as a
potential target for treatment of lung cancer.
MicroRNA (miRNAs) are a class of short
single-stranded noncoding endogenous RNAs, approximately 18–24
nucleotides in length, which post-transcriptionally modulate gene
expression by either inhibiting translation or inducing mRNA
degradation (20). MiRNAs have
been recognized as a new class of genes involved in human
tumorigenesis (21,22) and recently they have been shown to
be diagnostic, prognostic and therapeutic biomarkers in lung cancer
(22–25). For example, high miR-155 expression
and low let-7a expression, as independent risk factors, have a
negative prognostic impact on outcome in lung adenocarcinoma
patients (23). The miR-17-92
cluster functions as an oncogene, and has been shown to promote
lung cancer carcinogenesis (24).
We previously reported that the inhibition of miR-21, whose
upregulation is associated with EGFR mutations, can be a
therapeutic strategy, either as a monotherapy or in combination
with EGFR-TKI treatment (25).
These findings suggest that miRNA can serve as a novel therapeutic
target as well as diagnostic and prognostic marker in lung
cancer.
A recent study reported that a specific cluster of
miRNA, miR-23a/24/27a, was induced by TGF-β in a Smad-dependent
manner in hepatocellular carcinoma (HCC) cells (26). Upregulation of these miRNAs were
able to suppress TGF-β-induced growth suppressive activities in HCC
cells. In this present study, we analyzed miR-23a/24/27a expression
in non-small cell cancer (NSCLC) cells and evaluated the
correlation between its expression and TGF-β/Smad signaling. We
found that miR-23a could be induced by TGF-β in a Smad-dependent
manner in A549 cells. In addition, overexpression of mature miR-23a
reduced E-cadherin expression and stimulate the EMT phenomenon
which is involved in tumorigenesis. Furthermore, in these A549
cells, inhibition of miR-23a could also partially suppress
TGF-β-induced EMT, while overexpression was associated with the
tendency for EGFR-TKI resistance. We have demonstrated that, in
lung cancer cells, miR-23a is regulated by the TGF-β/Smad pathway
and plays a critical role in EMT through the targeting of
E-cadherin.
Materials and methods
Cell culture
We used 10 NSCLC cell lines: A549, PC9, PC14,
LC2/ad, RERF-LC-KJ, RERF-LC-OK adenocarcinoma (AC) cell lines and
PC1, PC10, LK2 and SQ5 squamous cell carcinoma cell lines (SCC) for
this study. An immortalized tracheal cell line (BET2A) was used as
a normal control cells. A549 and BET2A were purchased from the
American Type Culture Collection (ATCC, Manassas, VA, USA);
RERF-LC-KJ, RERF-LC-OK, LC2-ad, LC2/Ad and SQ5 were obtained from
the RIKEN Cell Bank (Ibaraki, Japan); and PC1, PC9, PC10 and PC14
were obtained from Immuno-Biological laboratories (Gunma, Japan).
NSCLC cell lines were maintained in RPMI-1640 medium (Gibco,
Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS).
BET2A was maintained in RPMI-1640 medium with 5% FBS.
RNA extraction and real-time quantitative
reverse transcription-PCR
Total RNA was extracted from the BET2A and NSCLC
cell lines with Trizol reagent (Invitrogen, Carlsbad, CA, USA). The
miR-23a, miR-24 and miR-27a expression levels were quantified by
quantitative reverse transcription-PCR (qRT-PCR) using
TaqMan® MicroRNA Assay System (Applied Biosystems,
Foster City, CA). RNU66 (PN 4373382) was used as an internal
control (Applied Biosystems). MiRNA expression was quantified and
reported as 2−ΔΔCt value (27).
Antibodies and western blot analysis
Cells were lysed in buffer containing 50 mM
Tris-HCl, pH 7.6, 150 mM NaCl, 0.1% sodium dodecyl sulfate, 1%
Nonidet P-40, and 0.5% sodium-deoxycholate. The lysates were kept
on ice for 30 min, and then centrifuged at 13000 × g for 30 min.
The supernatant was collected and then 10 μg of each of the
proteins was separated by SDS-PAGE on 10% gels and transferred to
nitrocellulose membrane. After being blocked in 5% skimmed milk,
the membrane was incubated with Smad2/3, β-actin (Cell Signaling
Technology, Beverley, MA, USA), E-cadherin, N-cadherin and vimentin
(Santa Cruz Biotechnology, Santa Cruz, CA, USA) antibodies.
Proteins were detected by immunoblotting using ECL-Plus reagents
(GE Healthcare Bio-Science Corp, Piscataway, NJ, USA).
Oligonucleotide transfection
TGF-β1 was purchased from R&D system
(Minneapolis, MN, USA). Cells were treated with 5 ng/ml TGF-β1 for
the indicated time. Small interference RNA (siRNA) targeting
Smad2/3 was purchased from Dharmacon Research Inc. (Lafayette, CO,
USA) and the homologous negative control was obtained from
Invitrogen. MiR-23a inhibitor, its negative control, miR-23a
precursor (Pre-miR-23a) and its cognate negative control
(Pre-miR-ctl) were synthesized by Ambion (Ambion). Pre-miR-23a and
miR-23a inhibitor were transfected using Lipofectamine™ 2000
reagent 24 h after seeding, as per the manufacturer’s instructions
(Invitrogen). Transfections of precursor and inhibitor complexes
were added to cells at a final concentration of 40 nM. Six hours
after the transfection was performed, the transfection medium was
replaced, and after 24 h, 5 ng/ml TGF-β1 was added to the medium
which was then incubated at 37°C for 48 h.
Growth inhibition assay
Gefitinib was purchased from Seleck Chemicals
(Houston, TX, USA). A549 cells (5,000 cells/well) were seeded into
96-well plates for 24 h. After being treated with Pre-miR-ctl or
Pre-miR-23a, at a final concentration of 40 nM for 24 h, the cells
were incubated in the various concentrations of gefitinib for 72 h
at 37°C. Then, MTS was added to each well and the cells were
incubated for a further 2 h at 37°C, after which absorbance was
measured using a microplate reader with a test wavelength of 450
nm. The IC50 value was defined as the concentration
needed for 50% reduction of the growth by treatment with
gefitinib.
Results
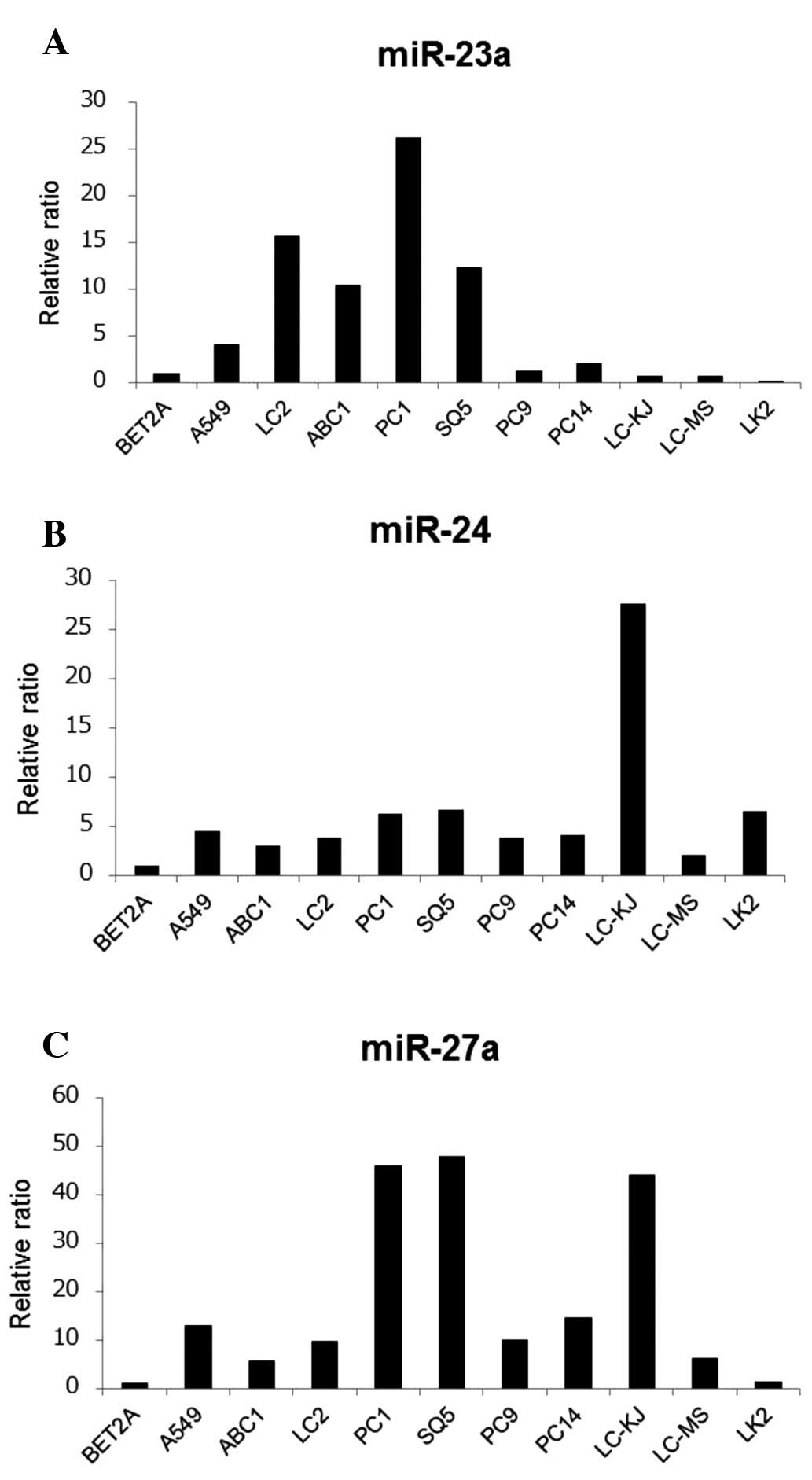
MiR-23a, miR-24 and miR-27a expression in
lung cancer cells
We first investigated miR-23a, miR-24 and miR-27a
expression levels in NSCLC cell lines, including 6 AC cell lines
and 4 SCC cell lines. Expression levels of these mature miRNAs were
examined by qRT-PCR (Fig. 1).
Although these miRNAs belonged to the same miRNA cluster, their
expression levels varied among the 10 cell lines.
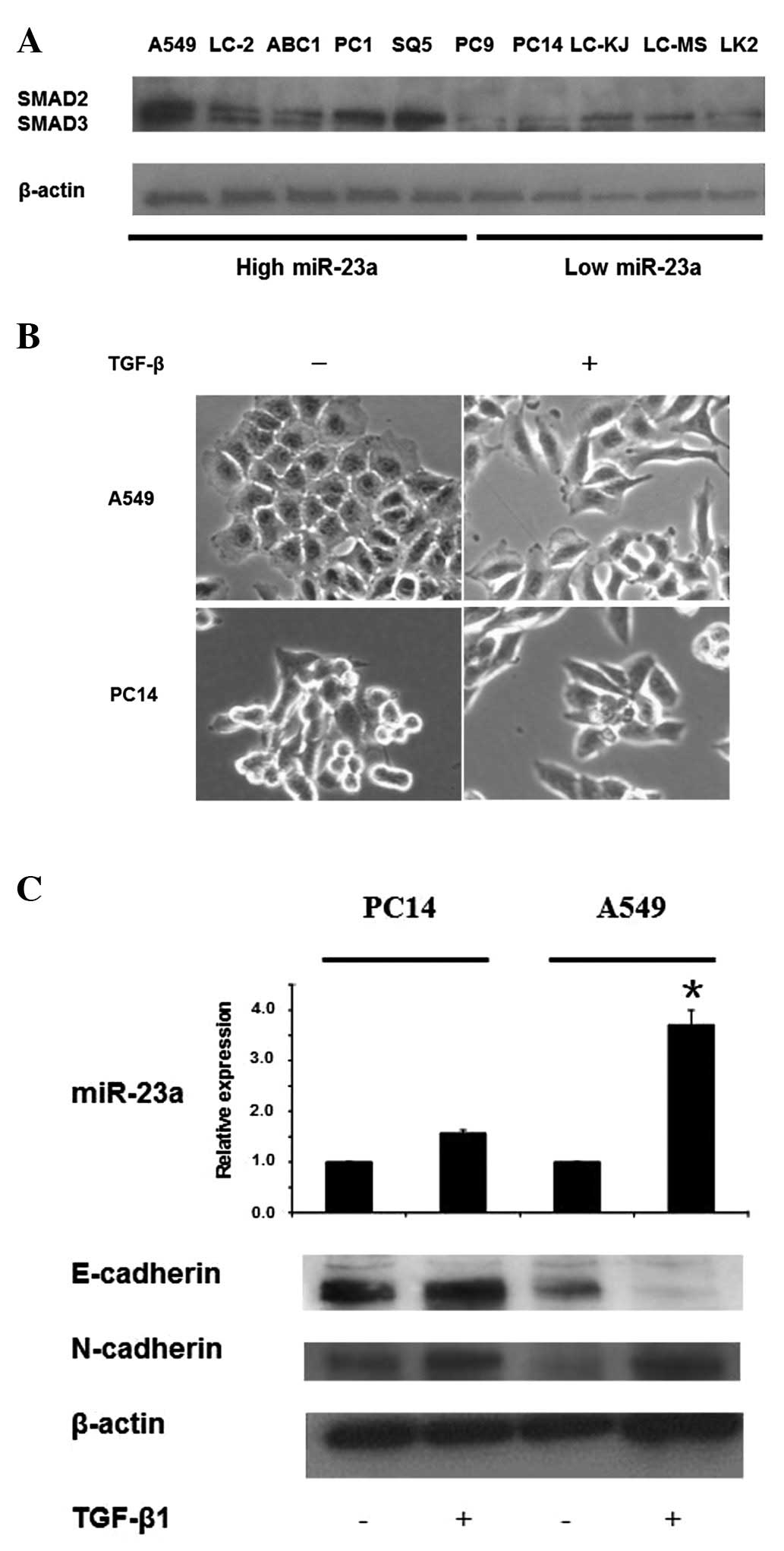
MiR-23a expression is directly induced by
TGF-β1 in a Smad-dependent manner
A recent study reported that miR-23a/24/27a was
induced by TGF-β in a Smad-dependent manner in HCC cells (26). Smad pathway is known as a major
transducer of TGF-β signaling (11). We evaluated the correlation between
miR-23a, miR-24 and miR-27a expression levels and Smad expression.
High expression of miR-23a was observed in those NSCLC cells which
also overexpressed Smad2/3. Thus, 5 cell lines (A549, LC-2/ad,
ABC1, PC1 and SQ5) showed high expression of miR-23a and Smad2/3
(Fig. 2A), while low expression of
miR-23a and Smad2/3 was found in the other 5 cell lines (PC9, PC14,
LC-KJ, LC-MS and LK2) (Fig. 2A).
In contrast, no correlation was observed between miR-24 or miR-27a
and Smad2/3.
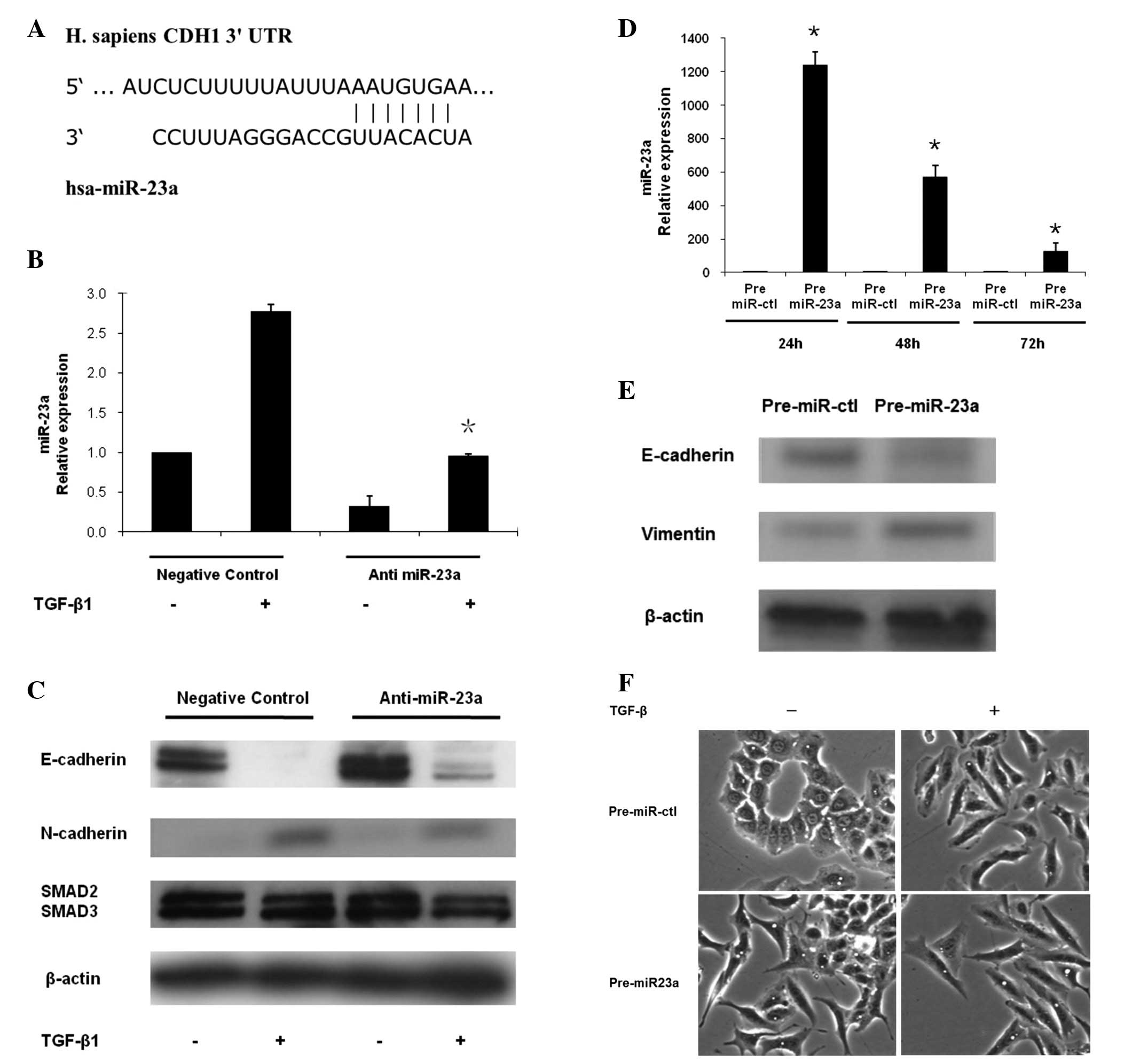
 | Figure 2MiR-23a is directly induced by TGF-β1
in a SMAD-dependent manner and involved in EMT of lung cancer
cells. (A) Five cell lines (A549, LC-2, ABC1, PC1 and SQ5) showed
high expression of both miR-23a and Smad2/3. In contrast, low
expression of miR-23a and Smad2/3 were found in the other five cell
lines (PC9, PC14, LC-KJ, LC-MS and LK2). (B) A549 and PC14 cells
were treated with 5 ng/ml of TGF-β1 for 48 h. The cells were
observed under a light microscope. A549 cells exhibited a classic
epithelial morphology. In contrast, A549 cells after the TGF-β1
exposure appeared to be less uniformly epithelial. (C) MiR-23a and
protein expression of EMT markers in A549 and PC14 cells after
TGF-β1 stimulation. Compared with PC14 cells, high expression of
miR-23a, lost E-cadherin expression and enhanced N-cadherin
expression were observed in A549 cells after TGF-β1 stimulation.
(D) Protein expression of Smad2/3 after treatment of siRNA in A549
cells. A549 cells were treated with control siRNA or siRNA of
Smad2/3 with or without TGF-β1 stimulation. Smad2 or Smad3 specific
siRNA completely diminished Smad2 or Smad3 expression. (E)
Expression of miR-23a after the transfection of Smad2/3 siRNA was
evaluated. MiR-23a expression was significantly decreased after the
treatment of Smad2 or Smad3 siRNAs with TGF-β1 stimulation in A549
cells. (F) MiR-23a expression was unchanged after the transfection
of Smad2 or Smad3 siRNAs with TGF-β1 stimulation in PC14 cells.
Data are mean ± SD from 3 independent experiments.
*p<0.05 when compared with the respective parent
cells. |
It is well known that TGF-β1 stimulates the EMT of
A549 lung cancer cells (10). In
this study, A549 cells which over-expressed miR-23a and Smad2/3 was
treated with 5 ng/ml of TGF-β1 for 48 h. We observed that, while
the parent A549 cells exhibited a classic epithelial morphology
(Fig. 2B), after TGF-β1 exposure
they had a less uniform epithelial appearance (Fig. 2B). In contrast, PC14 cells with low
expression of miR-23a and Smad2/3 retained their epithelial
morphology after TGF-β1 treatment (Fig. 2B). Using western blot analysis, we
evaluated the expression levels of EMT markers in A549 and PC14
cells treated with TGF-β1 in order to confirm the occurrence of
EMT. A549 cells treated with TGF-β1 displayed reduced E-cadherin
expression and increased N-cadherin expression when compared with
A549 cells without TGF-β1 stimulation (Fig. 2C). We also examined whether TGF-β1
stimulated miR-23a expression in these two cells. MiR-23a
expression level was significantly higher in A549 cells, which had
shown the EMT phenomenon after the treatment of the respective
parent cells with TGF-β1 (Fig.
2C). In contrast, miR-23a expression level in PC14 cells, which
had not shown the EMT phenomenon, was unaffected by exposure to
TGF-β1 (Fig. 2C).
Next, we examined whether the Smad signal pathway
directly regulated miR-23a expression. A549 and PC14 cells were
treated with siRNAs of control or Smad2/3 (Fig. 2D). Knockdown of Smad2/3
significantly decreased TGF-β1-induced miR-23a expression in A549
cells in which miR-23a was overexpressed (Fig. 2E). On the other hand, in PC14 cells
with low miR-23a, miR23a expression was unaffected by treatment
with Smad siRNAs (Fig. 2F). These
results suggested that, in A549 lung cancer cells, miR-23a was
directly regulated by TGF-β1/Smad pathway and contributed to the
EMT phenomenon.
MiR-23a regulates TGF-β1-induced EMT by
targeting E-cadherin
Since miR-23a was significantly upregulated in A549
cells after the treatment with TGF-β1 and mediated EMT, we
proceeded to identify potential targets known to play a role in EMT
by using the Target Scan database. Among the candidate miRNAs for
the E-cadherin gene (CDH1), we found that the region of 3′ UTR of
the CDH1 gene may serve as a binding site for miR-23a based on the
prediction of Target Scan database (Fig. 3A).
To examine whether the CDH1 was a target of miR-23a,
we knocked down miR-23a in A549 cells by using a specific
inhibitor. Control or specific miR-23a inhibitor was transfected
into A549 cells for 24 h, which were then treated with or without
TGF-β1 for a further 48 h. We confirmed that miR-23a was
effectively knocked down by miR-23a inhibitor in A549 cells
(Fig. 3B). Using western blot
analysis, we evaluated the expression of EMT markers after the
treatment of miR-23a inhibitor to confirm the occurrence of EMT.
After exposure to TGF-β1, E-cadherin expression in A549 cells was
greatly diminished, resulting in TGF-β1-induced EMT (Fig. 3C). Interestingly, E-cadherin was
still expressed in A549 cells transfected with miR-23a inhibitor
after TGF-β1 exposure (Fig. 3C).
N-cadherin expression was also weak after miR-23a treatment
followed by TGF-β1 exposure (Fig.
3C). These findings demonstrated that miR-23a inhibition
partially suppressed TGF-β-induced EMT phenomenon in A549
cells.
We transiently transfected A549 cells with the
miR-23a precursor (Pre-miR-23a), and the control precursor miR (Pre
miR-ctl). Mature miR-23a was remarkably induced by the miR-23a
precursor in A549 cells between 24 to 72 h (Fig. 3D). After the treatment of precursor
miR-23a, decreased E-cadherin expression and increased levels of
vimentin were observed in A549 cells at 48 h (Fig. 3E). Under a light microscope,
overexpression of miR-23a enhanced the spindle integration,
resulting in an additive effect with TGF-β1-induced EMT in A549
cells (Fig. 3F). These results
suggested that miR-23a may affect EMT by targeting E-cadherin in
lung cancer cells.
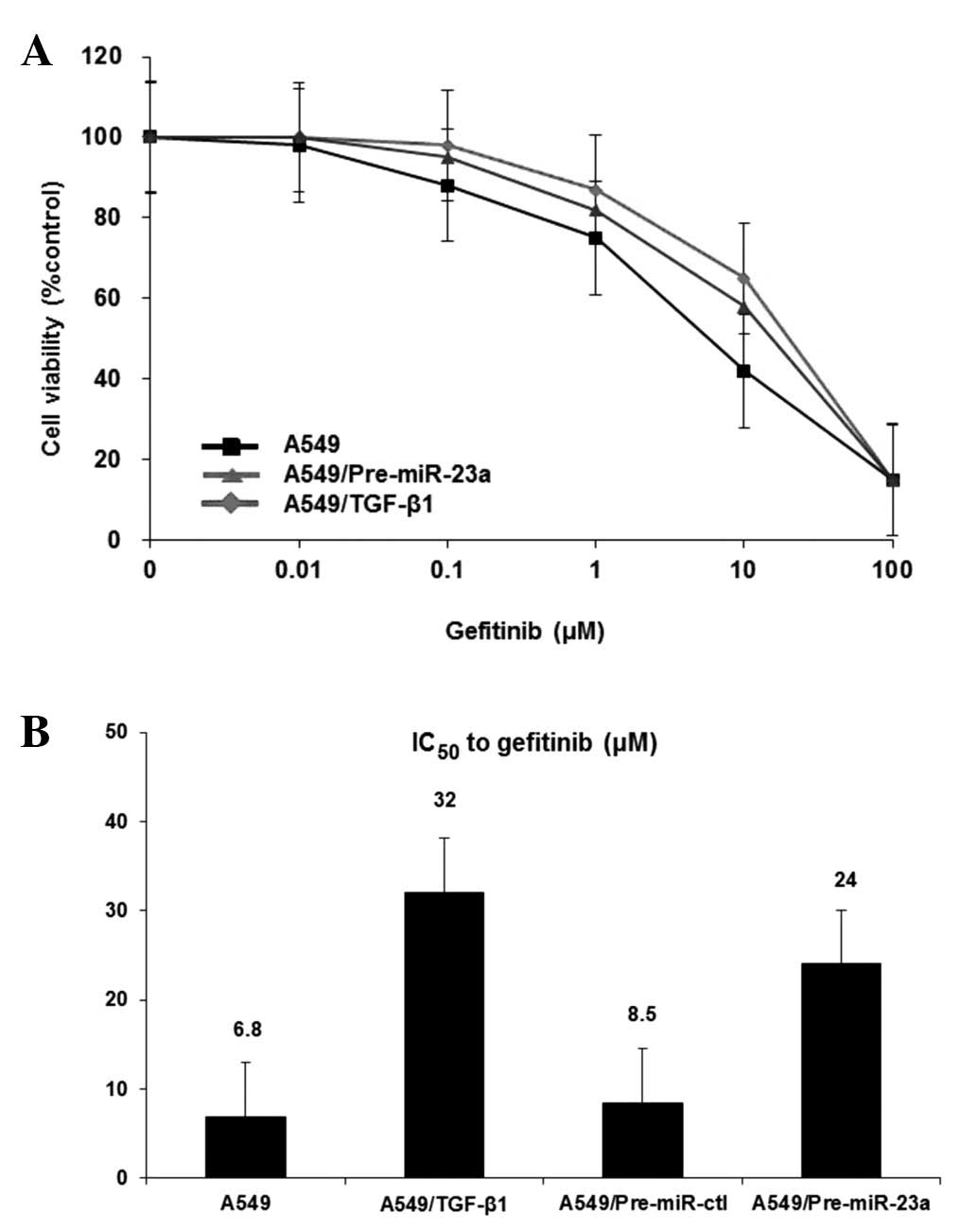
MiR-23a stimulated EMT and induced
resistance to gefitinib
Finally, to evaluate whether EMT leads to resistance
against the EGFR-TKI, gefitinib, we measured the response to
gefitinib after the exposure of A549 cells to TGF-β1 or
Pre-miR-23a. Consistent with previous studies, A549 cells treated
with TGF-β1 were more resistant to gefitinib than were the parental
cells (Fig. 4A). Interestingly,
the resistance to gefitinib was also found in A549 cells after
treatment with Pre-miR-23a only (Fig.
4A). The IC50 values of gefitinib with TGF-β1 and
gefitinib after treatment with Pre-miR-23a were 32 and 24,
respectively, whereas that of gefitinib monotherapy was 6.8
(Fig. 4B). These findings indicate
that induction of EMT affects cellular response to gefitinib and
that miR-23a contributes to the resistance as well as TGF-β1.
Discussion
TGF-β1 has been recognized as a regulator of EMT in
advanced-stage human cancers, a phenomenon which promotes
tumorigenesis, cancer progression and metastasis (10). Previous studies have demonstrated
that EMT plays a key role in lung cancer progression (15,28,29).
In addition, it has been demonstrated that cancer progression might
stimulate EMT in lung cancer, resulting in resistance to anticancer
drugs (17–19). Therefore, the early detection of
EMT development or attenuation of the EMT phenotype in lung cancer
cells may be useful, particularly in helping to improve the
clinical treatment of lung cancer.
Recent reports showed that several miRNAs play a
crucial role in the regulation of EMT of several cancers (30–34).
The miR-200 family and miR-205 have been shown to contribute to EMT
in cancer cells by the direct targeting of transcriptional
repressors of E-cadherin, ZEB1 and ZEB2 (30–32).
More specifically, in breast cancer cells, miR-155 has been shown
to facilitate TGF-β-induced EMT by targeting RhoA (33). Finally, miR-9 activated by MYC/MYCN
mediated E-cadherin down-regulation resulting in the activation of
β-catenin, and VEGF, and metastases in human cancers, including
neuroblastomas and breast tumors (34). However, in lung cancer, the
mechanism by which miRNA contributes to TGF-β-induced EMT is
largely unknown.
MiR-23a/24/27a is a miRNA cluster located in
chromosome 19p13.12 and can be induced by TGF-β (26). This cluster functions as an
oncogenic miRNA in several human cancers, and previous studies have
reported that miR-23a/24/27a was upregulated in human cancers
(26,35). Furthermore, miR-23a/24/27a
functioned as a growth-promoting and anti-apoptotic factor in HCC
cells (26), while miR-23a was
also shown to promote the growth of gastric adenocarcinoma cells
and downregulate interleukin-6 receptor (35). In addition, c-myc suppression of
miR-23a enhances mitochondrial glutamine metabolism and glutaminase
expression (36). The cognate of
glutamine is the major component that catabolizes glutamine to
generate energy and lactate. Plenty of large amounts of glutamine
are aggressively transported into cells to promote cancer cell
proliferation and act as a source of carbon in the carbon cycle.
Taken together, miR-23a/24/27a can be induced by TGF-β and act as
an oncogenic or tumor repressive miRNA in multiple human
malignancies. However, the relation between miR-23a/24/27a and
TGF-β/Smad pathway remains unclear in lung cancer cells.
In this study, we found that expression of miR-23a
was directly induced by the TGF-β1/Smad pathway in A549 lung
adenocarcinoma cells with the EMT phenomenon. In contrast, miR-24
and miR-27a belonging to the same cluster were induced in a
Smad-independent manner in lung adenocarcinoma cells. We suggest
that TGF-β1 mainly regulates the expression of miR-23a in lung
cancer cells. Furthermore, overexpression of miR-23a decreased
E-cadherin expression and increased levels of vimentin, resulting
in the EMT phenomenon in A549 lung cancer cells; silencing of
miR-23a partially restored E-cadherin expression. This is the first
report showing that miR-23a regulated TGF-β1-induced EMT via
E-cadherin suppression in lung cancer cells.
Molecular-targeted therapies have been recently
developed for NSCLC treatment. NSCLC patients with EGFR gene
mutations have shown a dramatic response to EGFR-TKIs such as
gefitinib and erlotinib (4,5). We
have recently reported that first-line gefitinib for advanced NSCLC
patients with EGFR mutations improved progression-free survival
with acceptable toxicity (5,37).
However, it is recognized that, clinically, drug resistance
eventually emerges and this limits the mean duration of response.
Although mechanisms of acquired resistance, such as T790M secondary
mutation and MET amplification, have recently been found, other
mechanisms should be identified to widen the therapeutic strategy
for NSCLC with EGFR mutations (38,39).
EMT has been reported to be correlated with an unfavorable
prognosis for NSCLC patients (40). Some studies showed that the
mesenchymal phenotype was more resistant to EGFR-TKI than the
epithelial phenotype in NSCLC (17–19).
Similarly, restoration of E-cadherin increased the sensitivity to
EGFR-TKI in lung cancer cell lines (41). Consistent with previous findings,
induction of EMT by TGF-β1 in A549 cells led to the acquisition of
resistance to gefitinib. Furthermore, overexpression of miR-23a
induced EMT by suppressing E-cadherin expression and contributed to
the reduced sensitivity to gefitinib in A549 cells. These findings
demonstrated that suppression of EMT by miR-23a inhibition might
overcome the resistance to EGFR-TKIs observed in NSCLC.
In conclusion, our study has provided evidence that
miR-23a regulated TGF-β-induced EMT by suppression of E-cadherin
and contributed to EGFR-TKI resistance in lung cancer cells.
MiR-23a might be a potential prognostic marker and a new
therapeutic target in NSCLC. Further studies should be performed to
clarify the connection between miR-23a and TGF-β/Smad signaling
during the EMT process in NSCLC.
Acknowledgements
This study was supported in part by a
Grant-in-Aid from the Ministry of Education, Culture, Sports,
Science, and Technology (MEXT) of Japan, and MEXT-Supported Program
for the Strategic Research Foundation at Private Universities (to
M.S. and A.G.).
References
|
1
|
Jemal A, Siegel R, Ward E, et al: Cancer
Statistics. CA Cancer J Clin. 59:225–249. 2009.
|
|
2
|
Schiller JH, Harrington D, Belani CP, et
al Eastern Cooperative Oncology Group: Comparison of four
chemotherapy regimens for advanced non-small-cell lung cancer. N
Engl J Med. 346:92–98. 2002. View Article : Google Scholar
|
|
3
|
Ohe Y, Ohashi Y, Kubota K, et al:
Randomized phase III study of cisplatin plus irinotecan versus
carboplatin plus paclitaxel, cisplatin plus gemcitabine, and
cisplatin plus vinorelbine for advanced non-small-cell lung cancer:
Four-Arm Cooperative Study in Japan. Ann Oncol. 18:317–323. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mok TS, Wu YL, Thongprasert S, et al:
Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N
Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maemondo M, Inoue A, Kobayashi K, et al:
Gefitinib or chemotherapy for non-small-cell lung cancer with
mutated EGFR. N Engl J Med. 362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Beer DG, Kardia SL, Huang CC, et al:
Gene-expression profiles predict survival of patients with lung
adenocarcinoma. Nat Med. 8:816–824. 2002.PubMed/NCBI
|
|
7
|
Potti A, Mukherjee S, Petersen R, et al: A
genomic strategy to refine prognosis in early-stage non-small-cell
lung cancer. N Engl J Med. 355:570–580. 2006. View Article : Google Scholar
|
|
8
|
Seike M, Yanaihara N, Bowman ED, et al:
Use of a cytokine gene expression signature in lung adenocarcinoma
and the surrounding tissue as a prognostic classifier. J Natl
Cancer Inst. 99:1257–1269. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zavadil J and Bottinger EP: TGF-β and
epithelial-to-mesenchymal transitions. Oncogene. 24:5764–5774.
2005.
|
|
11
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-β family signalling. Nature.
425:577–584. 2003.
|
|
12
|
Thiery JP: Epithelial-mesenchymal
transitions in development and pathologies. Curr Opin Cell Biol.
15:740–746. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumor
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Singh A, Greninger P, Rhodes D, et al: A
gene expression signature associated with ‘K-Ras addiction’ reveals
regulators of EMT and tumor cell survival. Cancer Cell. 15:489–500.
2009.
|
|
16
|
Mizutani H, Okano T, Minegishi Y, et al:
HSP27 modulates epithelial to mesenchymal transition of lung cancer
cells in a Smad-independent manner. Oncol Lett. 1:1011–1016.
2010.PubMed/NCBI
|
|
17
|
Yauch RL, Januario T, Eberhard DA, et al:
Epithelial versus mesenchymal phenotype determines in vitro
sensitivity and predicts clinical activity of erlotinib in lung
cancer patients. Clin Cancer Res. 11:8686–8698. 2005. View Article : Google Scholar
|
|
18
|
Thomson S, Buck E, Petti F, et al:
Epithelial to mesenchymal transition is a determinant of
sensitivity of non-small-cell lung carcinoma cell lines and
xenografts to epidermal growth factor receptor inhibition. Cancer
Res. 65:9455–9462. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rho JK, Choi YJ, Lee JK, et al: Epithelial
to mesenchymal transition derived from repeated exposure to
gefitinib determines the sensitivity to EGFR inhibitors in A549, a
non-small cell lung cancer cell line. Lung Cancer. 63:219–226.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Johnson SM, Grosshans H, Shingara J, et
al: RAS is regulated by the let-7 microRNA family. Cell.
120:635–647. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lu J, Getz G, Miska EA, et al: MicroRNA
expression profiles classify human cancers. Nature. 435:834–838.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Volinia S, Calin GA, Liu CG, et al: A
microRNA expression signature of human solid tumors defines cancer
gene targets. Proc Natl Acad Sci USA. 103:2257–2261. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yanaihara N, Caplen N, Bowman E, et al:
Unique microRNA molecular profiles in lung cancer diagnosis and
prognosis. Cancer Cell. 9:189–198. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hayashita Y, Osada H, Tatematsu Y, et al:
A polycistronic microRNA cluster, miR-17-92, is overexpressed in
human lung cancers and enhances cell proliferation. Cancer Res.
65:9628–9632. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Seike M, Goto A, Okano T, et al: MiR-21 is
an EGFR-regulated anti-apoptotic factor in lung cancer in
never-smokers. Proc Natl Acad Sci USA. 106:12085–12090. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang S, He X, Ding J, et al: Upregulation
of miR-23a approximately 27a approximately 24 decreases
transforming growth factor-beta-induced tumor-suppressive
activities in human hepatocellular carcinoma cells. Int J Cancer.
123:972–978. 2008. View Article : Google Scholar
|
|
27
|
Bustin SA: Absolute quantification of mRNA
using real-time reverse transcription polymerase chain reaction
assays. J Mol Endocrinol. 25:169–193. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Saito RA, Watabe T, Horiguchi K, et al:
Thyroid transcription factor-1 inhibits transforming growth
factor-beta-mediated epithelial-to-mesenchymal transition in lung
adenocarcinoma cells. Cancer Res. 69:2783–2791. 2009. View Article : Google Scholar
|
|
29
|
Soltermann A, Tischler V, Arbogast S, et
al: Prognostic significance of epithelial-mesenchymal and
mesenchymal-epithelial transition protein expression in non-small
cell lung cancer. Clin Cancer Res. 14:7430–7437. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gregory PA, Bert AG, Paterson EL, et al:
The miR-200 family and miR-205 regulate epithelial to mesenchymal
transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10:593–601.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Korpal M, Lee ES, Hu G, et al: The miR-200
family inhibits epithelial-mesenchymal transition and cancer cell
migration by direct targeting of E-cadherin transcriptional
repressors ZEB1 and ZEB2. J Biol Chem. 283:14910–14914. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tryndyak VP, Beland FA and Pogribny IP:
E-cadherin transcriptional down-regulation by epigenetic and
microRNA-200 family alterations is related to mesenchymal and
drug-resistant phenotypes in human breast cancer cells. Int J
Cancer. 126:2575–2583. 2010.PubMed/NCBI
|
|
33
|
Kong W, Yang H, He L, et al: MicroRNA-155
is regulated by the transforming growth factor beta/Smad pathway
and contributes to epithelial cell plasticity by targeting RhoA.
Mol Cell Biol. 28:6773–6784. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ma L, Young J, Prabhala H, et al: miR-9, a
MYC/MYCN-activated microRNA, regulates E-cadherin and cancer
metastasis. Nat Cell Biol. 12:247–256. 2010.PubMed/NCBI
|
|
35
|
Zhu LH, Liu T, Tang H, et al: MicroRNA-23a
promotes the growth of gastric adenocarcinoma cell line MGC803 and
downregulates interleukin-6 receptor. FEBS J. 277:3726–3734. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gao P, Tchernyshyov I, Chang TC, et al:
c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase
expression and glutamine metabolism. Nature. 458:762–765. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Inoue A, Kobayashi K, Usui K, et al:
First-line gefitinib for patients with advanced non-small-cell lung
cancer harboring epidermal growth factor receptor mutations without
indication for chemotherapy. J Clin Oncol. 27:1394–1400. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kobayashi S, Boggon TJ, Dayaram T, et al:
EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Engelman JA, Zejnullahu K, Mitsudomi T, et
al: MET amplification leads to gefitinib resistance in lung cancer
by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bremnes RM, Veve R, Gabrielson E, et al:
High-throughput tissue microarray analysis used to evaluate biology
and prognostic significance of the E-cadherin pathway in
non-small-cell lung cancer. J Clin Oncol. 20:2417–2428. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Witta SE, Gemmill RM, Hirsch FR, et al:
Restoring E-cadherin expression increases sensitivity to epidermal
growth factor receptor inhibitors in lung cancer cell lines. Cancer
Res. 66:944–945. 2006. View Article : Google Scholar : PubMed/NCBI
|