Introduction
Amyloid precursor-like protein 2 (APLP2) is a
protein that is well conserved between human and mouse, and it has
high homology to another ubiquitously expressed family member,
amyloid precursor protein or APP (1). Similarity between APLP2 and APP
sequences is the primary explanation for a redundancy in some
cellular functions; however, knock-out studies in mice have
demonstrated an essential and unique function in viability for
APLP2 that remains unidentified (2–5). As
shown in various reports, APLP2 and APP are involved in cell
migration, signaling, adhesion, proliferation and healing (1,4,6–12).
As demonstrated by our laboratory, APLP2 can also bind to major
histocompatibility complex class I molecules (receptors that
present tumor and viral antigens to T lymphocytes), and increase
their endocytosis and delivery to lysosomes (13–15).
Higher levels of APLP2 mRNA are present in
the pancreas after partial pancreatectomy, suggesting that APLP2
may have a function in regeneration of pancreas tissue (16). Furthermore, a few studies have
shown increased expression of APLP2 in cancers. For example, in a
screen of tumors, APLP2 was found to be overexpressed (17) and APLP2 was discovered to be
elevated in invasive breast cancer adenocarcinoma compared to
non-invasive adenocarcinoma (18).
Among the many cancer cell lines that we previously examined, APLP2
was expressed at the highest level in the pancreatic cancer cell
lines SUIT-2 and a SUIT-2 subline, S2-013 (19). Regulated intramembrane proteolysis
is a process by which APLP2 or APP C-terminal fragments are
liberated from secreted, extracellular N-terminal fragments
(1,20–23).
This process has been particularly noted in the BxPC3 pancreatic
cancer cell line, which has been reported to exhibit a high level
of APP cleavage; however, the accompanying expression and cleavage
of APLP2 in this cell line was not examined (24). Proteolysis of APLP2 or APP can be
accomplished by the β-site APP cleaving enzyme 1 (BACE1) or BACE2
(22,23,25).
In the context of Alzheimer’s disease, BACE1 and BACE2 cleavage of
APP has been well characterized, and both conserved and unique
cleavage sites on APP have been demonstrated for the two BACE
proteins (26–28). Recently, one BACE1 cleavage site in
APLP2 was identified (23);
however, BACE2 cut site(s) in APLP2 remain(s) unknown. Both BACE
proteins have been reported in pancreatic tissue, but reports
differ on BACE1 and BACE2 expression and activity in pancreatic
ductal and acinar cells (22,23,27,29–32),
which are cell types proposed to give rise to pancreatic cancer
(33).
In our current studies, we have identified increased
APLP2 in human pancreatic cancer tissues, as compared to normal
pancreatic tissues, and have investigated the forms of APLP2
expressed in pancreatic cancer cell lines. We observed high
molecular mass APLP2, at the molecular mass previously shown to be
modified by glycosaminoglycans (GAG) (20,34,35),
in the majority of pancreatic cancer cell lines, as well as
full-length APLP2 without GAG modification and 12–15 kDa C-terminal
fragments generated from secretase cleavage (22,23)
in all these cell lines. C-terminal fragments of APP were only
abundantly observed in the BxPC3 cell line in our panel of
pancreatic cancer cell lines, suggesting that cleavage of APLP2,
rather than APP, is a consistent molecular feature of pancreatic
cancer cell lines. Furthermore, we have shown that transformation
of pancreatic ductal cells by transfected oncogenes induces an
increase in APLP2 expression, with particular enhancement in the
expression of the APLP2 C-terminal fragments. Downregulation of
APLP2 and/or APP in the pancreatic cancer S2-013 cell line, which
displays representatively low expression of APP C-terminal
fragments, decreased cell proliferation, suggesting a role for both
family members in the growth of pancreatic cancer cell lines.
Finally, treatment with inhibitors of β-secretases, enzymes that
cleave APLP2 or APP to release C-terminal fragments, decreased the
growth and viability of the pancreatic cancer cell line S2-013 but
not of a non-transformed pancreatic ductal cell line. Overall,
these studies suggest that APLP2 undergoes extensive modification
and cleavage in pancreatic cancer cell lines, APLP2 (and APP)
facilitate pancreatic cancer cell growth, and treatments that block
APLP2 cleavage can diminish the growth of pancreatic cancer
cells.
Materials and methods
Antibodies and immunostaining
Rabbit polyclonal antibodies against the full-length
form of APLP2, the APLP2 C-terminus and the APP C-terminus were
purchased from EMD Biosciences (San Diego, CA, USA). Mouse
monoclonal anti-actin antibody was purchased from Novus Biologicals
(Littleton, CO, USA). The secondary antibodies used for western
blot analysis were peroxidase-conjugated AffiniPure goat anti-mouse
IgG light chain or peroxidase-conjugated IgG fraction mouse
anti-rabbit IgG light chain (Jackson ImmunoResearch Laboratories,
West Grove, PA, USA).
Tissue sections were obtained from the University of
Nebraska Medical Center (UNMC) Rapid Autopsy Program, according to
a protocol approved by the UNMC Institutional Review Board. All
tissue donors had provided written consent. For immunostaining,
paraffin-fixed sections were stained with anti-APLP2 antibody
before evaluation in a blinded manner, with scoring for APLP2
expression (− for negative; weak for low expression; + for moderate
expression; ++ for strong expression).
Cell lines and culturing conditions
The pancreatic cancer cell lines used in these
studies were BxPC3, Capan-2, Hs766T, SUIT-2 and S2-013 (36–41).
The S2-013 cell line is a cloned subline of the SUIT-2 human
pancreatic tumor cell line (which was derived from a liver
metastasis) (37,39). The hTERT-HPNE cell line is a line
of telomerase-immortalized cells from normal human pancreatic
ducts. This cell line lacks cancer-associated changes, has a normal
karyotype, and can serve as the progenitor of pancreatic ductal
cells (42,43). The hTERT-HPNE cell line and
transfectants have been previously used in studies of pancreatic
ductal cell transformation (44–46).
All the human pancreatic cancer cell lines used in
these studies were cultured in Roswell Park Memorial Institute
(RPMI)-1640 medium that was supplemented with 10 or 15% (vol/vol)
fetal bovine serum, 1 mM sodium pyruvate, 2 mM L-glutamine, 100
U/ml penicillin and 100 μg/ml streptomycin. The hTERT-HPNE cells
were grown in Medium D (as described in reference 43) or in Dulbecco’s modified Eagle’s
medium supplemented with 10% v/v fetal bovine serum, 1 mM sodium
pyruvate, 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml
streptomycin. Basal media and additives were purchased from
Invitrogen (Carlsbad, CA, USA) and fetal bovine serum was obtained
from Atlanta Biologicals (Lawrenceville, GA, USA).
Downregulation of target proteins in culture was
achieved by transient transfections of short interfering RNA
(siRNA). ON-TARGETplus SMARTpool siRNA against human APLP2 or APP
was obtained from Thermo Scientific Dharmacon (Lafayette, CO, USA).
ON-TARGETplus control, non-targeting pool was used as a negative
control (Thermo Scientific Dharmacon). Transfections were performed
following the manufacturer’s instructions for cells in base
maintenance medium. Briefly, cells were seeded at 1×105
cells/well in a 6-well plate the day before transfection and the
medium was exchanged on the day of transfection. DharmaFECT
transfection reagent no. 1 (Thermo Scientific Dharmacon) was
incubated with 0.4 pmol of siRNA for 20 min at room temperature and
the mixture was added drop-wise to wells. Reduced expression of
target proteins was confirmed by western blot analysis of cell
lysates.
Preparation of cell lysates
Cells (1×107) were harvested, washed
three times in 20 mM iodoacetamide (with centrifugation for 5 min
at 450 x g each time), and resuspended in cell lysis buffer (150 mM
NaCl, 5 mM EDTA, 20 mM Tris-HCl pH 7.5, 0.5% Triton X-100).
Resuspended cells were incubated on ice for 1 h with occasional
mixing, and then stored at −80°C overnight. The following day, the
cell lysates were thawed on ice, and then centrifuged at top speed
in a desktop microcentrifuge for 30 min at 4°C. The supernatants
were transferred to new tubes and stored at −80°C. Aliquots of
lysates were mixed with 5X sodium dodecyl sulfate loading dye (250
mM Tris-HCl pH 6.8, 10% w/v sodium dodecyl sulfate, 30% v/v
glycerol, 5% v/v β-mercaptoethanol, 0.02% w/v bromophenol blue) and
boiled for 5 min prior to gel loading.
Western blot analysis
Cell lysate samples were boiled for 5 min and then
loaded on 4–20% Tris-glycine pre-cast gels (Invitrogen).
Electrophoresis was performed at 125 V at room temperature. The
proteins were transferred at 30 V for 2 h at room temperature to
polyvinylidene difluoride Immobilon-P membranes (Millipore,
Billerica, MA, USA). After overnight blocking in a 5% w/v solution
of nonfat dry milk, the membranes were incubated with primary
antibodies (diluted with 5% nonfat dry milk). Incubation with
anti-actin antibody (1:2,000) was for 1 h and incubation with
anti-full-length APLP2 antibody (1:1,000), anti-APLP2 C-terminus
antibody (1:1,000) or anti-APP C-terminus antibody (1:1,000) was
for 1.5 h. After primary antibody incubation, 3 washes with 0.05%
Tween-20 in phosphate-buffered saline (PBS) for 10 min were
performed. The membranes were subsequently incubated for 1 h in
secondary antibodies, diluted 1:10,000 in 0.05% Tween-20 in PBS,
and washed 3 times for 10 min with 0.3% Tween-20 in PBS. For
protein visualization, the membranes were incubated for 1 min in
Pierce ECL Western Blotting Substrate, or (for anti-APLP2
C-terminus) in Pierce SuperSignal West Pico Chemiluminescent
Substrate (Thermo Scientific, Rockford, IL, USA) and exposed to
Kodak BioMax MR film (Rochester, NY, USA). For densitometry of
protein bands, quantification was done with the Molecular Imager
ChemiDoc XRS system with Quantity One 1-D analysis software
(BioRad, Hercules, CA, USA).
Assessment of cellular growth
Growth of cells was assessed using thiazol blue
(3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide, MTT)
purchased from Sigma (St. Louis, MO, USA), by the MTT assay.
Briefly, cells were seeded in 6-well plates at 1×105
cells per well 24 h prior to the start of any cell culture
treatments. The addition of β-secretase inhibitors to S2-013 or
hTERT-HPNE cells signified the start of the time course. For siRNA
studies, S2-013 cells were transfected with siRNA for 48 h to
reduce expression of the target protein and re-seeded at
5×104 cells per well, which then served as the start of
the MTT time course. At the time point indicated, culture medium
was removed and cells were incubated in 2.5 ml/well of 1 mg/ml MTT
reagent (dissolved in base maintenance medium lacking phenol red)
for 3 h at 37°C in 5% CO2. Suspension cells were
collected pre- and post-incubation in MTT by centrifugation. The
MTT solution was then removed and the metabolized MTT reagent was
dissolved in isopropanol. Aliquots of the isopropanol-MTT solution
were transferred a 96-well microtiter plate in replicate, and
absorbances at 570 nm and 690 nm were taken on a SpectraMax M5e
instrument (kindly provided by Dr Amar Natarajan, University of
Nebraska Medical Center) using SoftMax Pro software (Molecular
Devices, Sunnyvale, CA, USA). To determine MTT-specific absorbance,
A690 was subtracted from A570.
β-secretase inhibition experiments
The cells were seeded at low density in 6-well
plates and allowed to adhere for 24 h prior to treatment with
β-secretase inhibitors (signifying 0 h). The inhibitors used were
NB2-755, −281, −418, −897 (donated by Novartis, Basel,
Switzerland), or β-secretase inhibitor IV (purchased from
Calbiochem/EMD Biosciences). The inhibitors were added at 2 μM and
untreated wells were used as controls. Cells were assayed at 0, 24,
48 and 72 h by the MTT assay (described above) or by mixing
aliquots of the cells with 0.4% trypan blue stain (Invitrogen) and
counting the cells on a hemocytometer. The percentage of viable
cells was calculated as unstained cell number divided by total cell
number x 100. At the 24 h time point, samples of S2-013 cells
treated with the Novartis inhibitors were also collected for
preparation of cell lysates and western blot analysis (described
above).
Statistical analysis of data
To determine statistical differences in results
obtained in the MTT growth assay, the analysis of variance (ANOVA)
test was applied with the criterion for significance set at
p<0.05. Statistical differences in the results from experiments
utilizing the Calbiochem β-secretase inhibitor were determined by
the Student’s t-test and the criterion for significance was set at
p<0.05.
Results
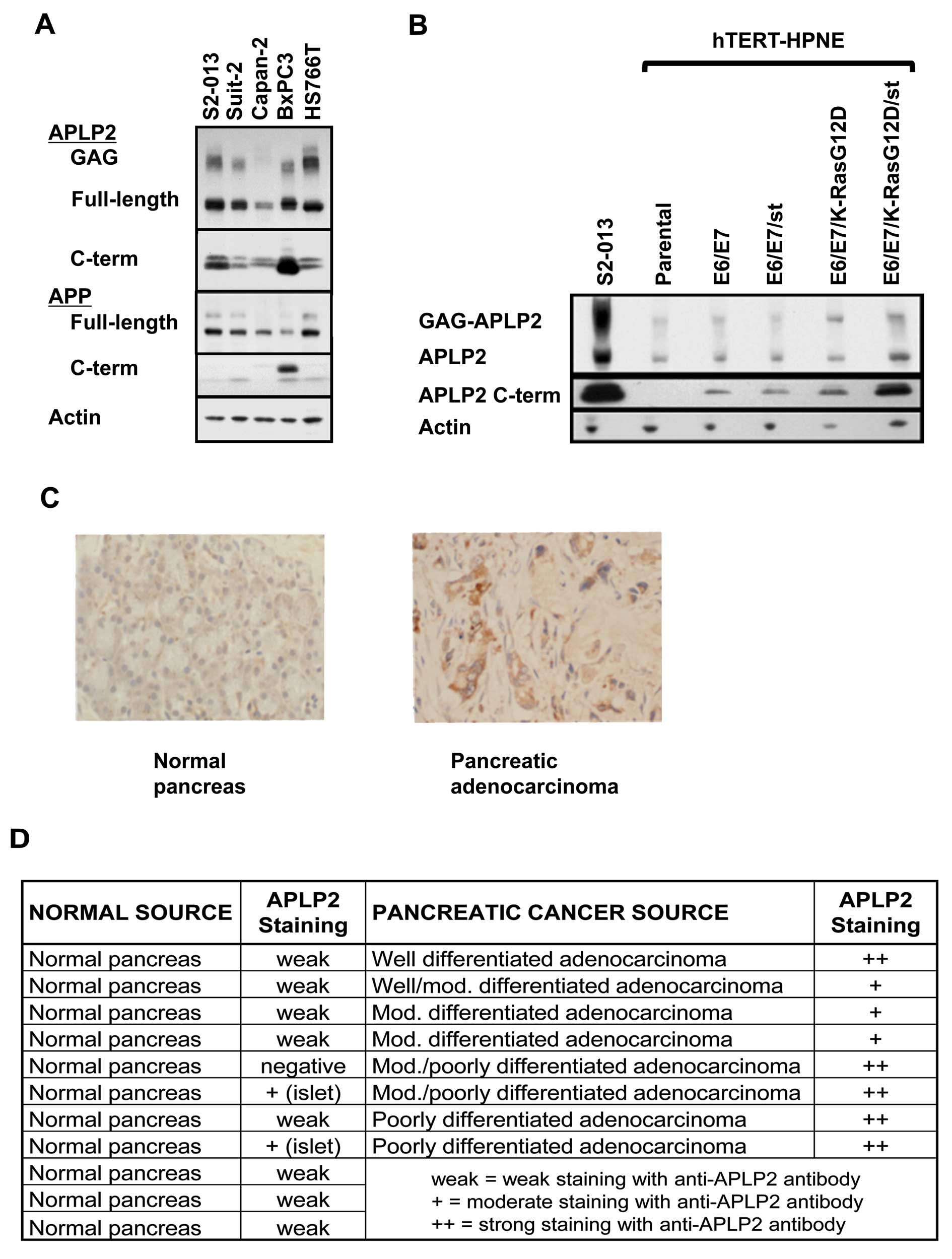
APLP2 is expressed, modified by GAG, and
cleaved in human pancreatic cancer cell lines
Previously, we found high levels of APLP2 expression
in several cancer cell lines, including pancreatic cancer lines
(S2-013, SUIT2 and Hs766T), prostate cancer lines and a melanoma
line (19). We have confirmed the
high expression of APLP2 in pancreatic cancer cell lines, using
additional pancreatic cancer lines (BxPC3 and Capan-2) and observed
additional bands in the molecular mass range previously shown to be
GAG-modified APLP2 (20,34,35; Fig.
1A). Notably, earlier studies by others (using corneal
epithelium and Chinese hamster ovary cells) have shown that
expression of GAG-modified APLP2 correlates with increased cellular
migration (7,8). Thus, the extensive GAG modification
of APLP2 in pancreatic cancer cells may have a pro-migratory
influence.
In addition to APLP2, APP was expressed in each of
the pancreatic cancer cell lines (Fig.
1A). The two bands of APP represent immature (bottom) and
mature (upper) full-length protein, with the higher molecular mass
of the mature form due to glycosylation (47). APP has been shown to promote
proliferation of the BxPC3 pancreatic cancer cell line, conferring
this activity through the soluble N-terminal fragment (24,48),
and soluble APLP2 has been identified in the secretome of another
pancreatic cancer cell line, SUIT-2 (49). Liberation of soluble APP and
soluble APLP2 occurs following secretase cleavage of the
full-length proteins, simultaneously creating 12–15 kDa
intracellular C-terminal fragments (1,20–23).
We determined the expression of C-terminal fragments for APP and
APLP2 in our panel of pancreatic cancer cell lines by western blot
analysis. APLP2 C-terminal fragments were present in all cell lines
examined, whereas APP C-terminal fragments were only substantially
present in the BxPC3 cell line (Fig.
1A). In some cell types, APLP2 or APP C-terminal fragments have
been shown to be additionally be cleaved by γ-secretase, generating
smaller, 4 kDa C-terminal fragments (1,2,20).
These smaller fragments of APLP2 or APP were not observed in our
panel of pancreatic cancer cell lines, even when samples were
electrophoresed on 18% Tris-glycine gels and the film was exposed
overnight (data not shown). These data implicate APLP2 as the
primary cleavage target of secretases in the majority of pancreatic
cancer cells, rather than APP. We therefore pursued the function of
APLP2 expression and cleavage in transformed pancreatic cells.
Oncogene expression increases the
presence of APLP2 and of APLP2 C-terminal fragments
Our identification of GAG-modified APLP2 and APLP2
C-terminal fragment expression in pancreatic cancer cell lines did
not clarify whether these forms of APLP2 occur only in the
transformed state, or if GAG-APLP2 and APLP2 C-terminal fragments
are also endogenously present in untransformed pancreatic ductal
cells. To address this question, we determined the expression of
APLP2 in a series of cell lines generated from the hTERTHPNE cell
line, which has been well characterized and used previously in
several pancreatic cancer studies (42–46).
The hTERT-HPNE cell line is an immortalized, but non-transformed,
cell line derived from human pancreatic ductal epithelium (42,43),
and with oncogene-transfected derivatives of this line it has been
demonstrated that expression of the human papillomavirus E6 and E7
oncogenes (which block p53 and Rb function, respectively) and SV40
small t antigen (st) can permit mutant K-Ras transformation
(44,45). With these cell lines we have been
able to compare expression of full-length APLP2, GAG-modified APLP2
and APLP2 C-terminal fragments in transformed versus matched,
non-transformed pancreatic cells. By western blot analysis, we
demonstrated that all APLP2 forms examined are over-expressed in
the transformed hTERT-HPNE E6/E7/K-RasG12D/st cells, compared to
the related, untransformed cell lines (Fig. 1B). The pancreatic cancer cell line
S2-013 was used as a positive control for APLP2 forms observed in
pancreatic cancer cell lines. The most dramatic enhancement in
APLP2 expression during oncogene-induced transformation of
pancreatic ductal cells was detected in the APLP2 C-terminal
cleavage fragments (Fig. 1B,
middle panel). These data indicate that the presence of APLP2, and
even more so the presence of APLP2 C-terminal fragments, is
increased by oncogene expression in pancreatic ductal cells, and
especially by oncogene-mediated pancreatic ductal cell
transformation.
Expression of APLP2 in human pancreatic
cancer tissue
By immunostaining, we also examined whether APLP2
expression was increased in human pancreatic cancer tissue,
relative to normal tissues. As indicated in Fig. 1 (C and D), APLP2 was weakly
expressed in 8 out of 11 normal pancreas samples, and not detected
at all in 1 normal pancreas sample. In 2 other normal pancreas
tissue samples, APLP2 was detected at a moderate level, but only in
the islets. Higher APLP2 expression was found throughout all 8
pancreatic cancer samples: in 5 samples, APLP2 staining was
detected at a strong level, and in 3 samples at a moderate level.
Among the 5 samples with strong APLP2 staining, 4 were either
moderate/poorly differentiated or poorly differentiated pancreatic
adenocarcinoma, confirming our observation from the hTERT-HPNE cell
lines that APLP2 expression may increase as pancreatic cancer cells
diverge further from normal morphology.
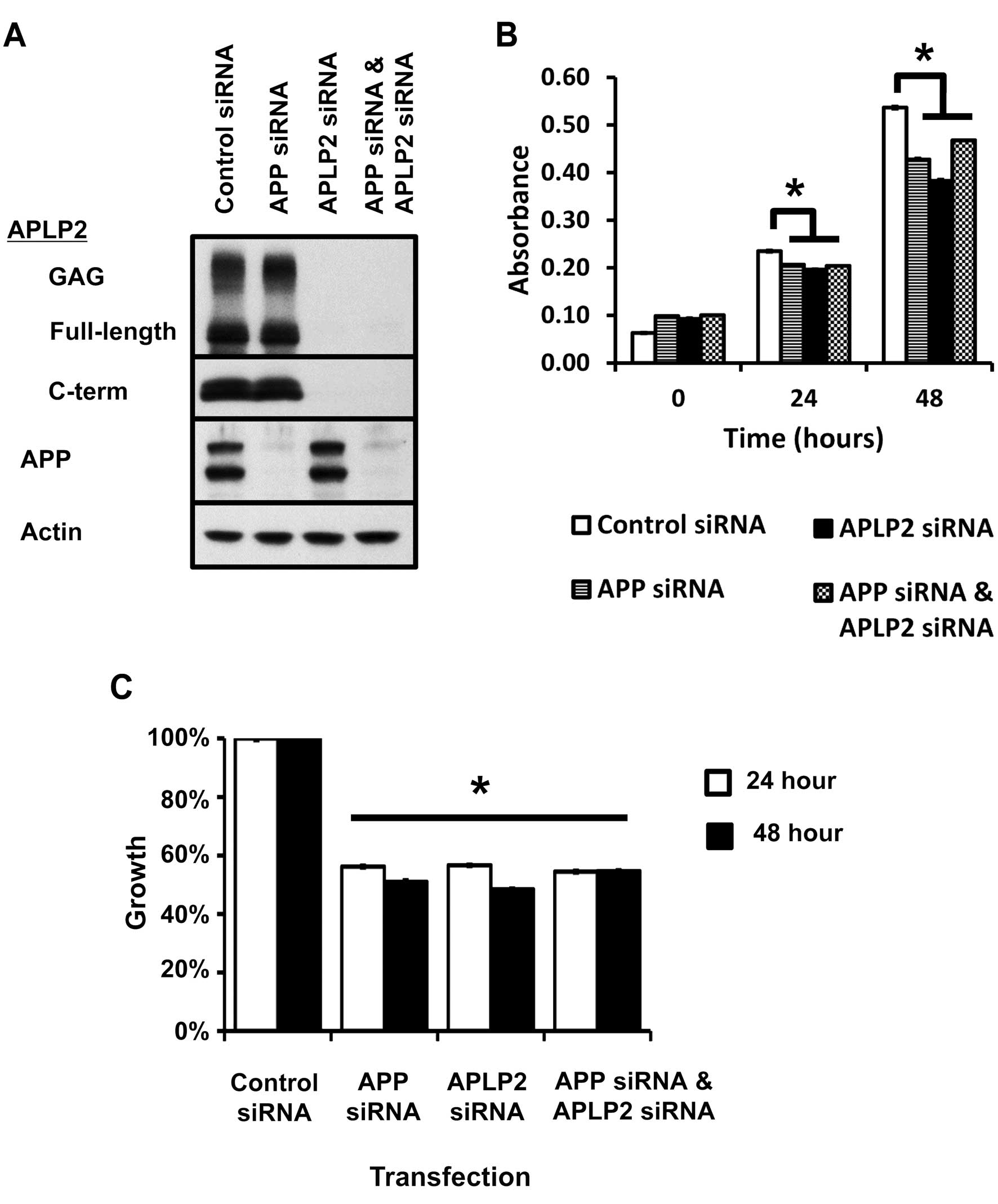
Loss of APLP2 and/or APP impairs the
growth of S2-013 pancreatic cancer cells
Downregulation of APP by siRNA has been shown to
impair the growth of BxPC3 cells (11, 29), which we found have enhanced
processing of APP compared to other pancreatic cancer cell lines
(Fig. 1A). However, the
contribution of APP to the growth of pancreatic cancer cells with
minimal expression of APP C-terminal fragments has not been
assessed previously. To determine the effect on such cells, the
S2-013 cell line (which we identified as having relatively low APP
C-terminal fragment expression, Fig.
1A) was transfected with siRNA against APP. Immature and mature
(glycosylated) full-length APP (47) expression was reduced by 48-h
post-transfection, without affecting APLP2 expression (Fig. 2A), and growth of cells was
determined by the MTT assay. S2-013 cells transfected with APP
siRNA demonstrated significantly reduced growth, compared to
control siRNA (Fig. 2B and C),
suggesting that full-length APP, rather than secretase cleavage of
APP, contributes to the growth of pancreatic cancer cell lines.
Since we had observed increasing APLP2 expression
with transformation of pancreatic cancer cells (Fig. 1), we then determined the
contribution of APLP2 to the growth of S2-013 cells. Western blot
analysis of S2-013 cells transfected with siRNA against APLP2 for
48 h revealed downregulation of all APLP2 forms: full-length APLP2,
GAG-modified APLP2 and APLP2 C-terminal fragments (Fig. 2A). Growth of S2-013 cells was
significantly impaired following transfection with siRNA against
APLP2, compared to control (Fig. 2B
and C). Downregulation of APLP2 had a growth-inhibitory effect
on S2-013, despite maintained expression of APP (Fig. 2). Knockdown of APLP2 or APP
comparably inhibited the growth of S2-013 cells (Fig. 2), demonstrating that loss of either
protein had a deleterious effect on cell growth. We then reduced
expression of APLP2 and APP by co-transfection with both siRNAs to
determine if loss of both proteins would further restrict the
growth of pancreatic cancer cells. Reductions in expression of all
forms of APLP2 and APP at 48-h post-transfection were confirmed by
western blot analysis (Fig. 2A).
APLP2 siRNA and APP siRNA co-transfected S2-013 cells displayed an
inhibition in growth compared to cells treated with control siRNA,
but not compared to S2-013 cells that were transfected with either
APLP2 siRNA or APP siRNA alone (Fig.
2B and C). These data demonstrate that both APLP2 and APP
contribute to the growth of pancreatic cancer cells, and
simultaneous loss of both proteins does not further enhance the
growth inhibition of pancreatic cancer cells. Therefore, it is
probable that APLP2 and APP act through the same pathway to promote
the growth of pancreatic cancer cells. Because loss of one protein
still reduced pancreatic cancer cell growth, we can conclude that
the remaining protein cannot compensate for the loss of the other.
These data suggest that APLP2 and APP have unique roles within the
same pathway.
Blocking β-secretase activity reduced
pancreatic cancer cell viability
The data shown in Fig.
1B suggest a relationship between oncogene expression and the
cleavage of APLP2, and APLP2 C-terminal fragments were observed in
all pancreatic cancer cell lines examined (Fig. 1A). In order to test the impact of
blocking APLP2 cleavage on the growth of pancreatic cancer cells,
we incubated S2-013 cells with chemical inhibitors of the
β-secretase enzymes, which subsequently reduced the production of
APLP2 C-terminal fragments within 24 h (Fig. 3A). By the MTT assay, we observed
that the presence of a β-secretase inhibitor impaired the growth of
S2-013 cells compared to mock-treated cells (Fig. 3B). The reduced growth of S2-013 was
accompanied by a reduction in the number of viable cells over time
(Fig. 3C and D). In contrast,
culturing the non-transformed hTERT-HPNE pancreatic cells with the
β-secretase inhibitors did not influence the growth and survival of
the cells (Fig. 3C and D).
Although β-secretases are known to cleave molecules in addition to
APLP2 (50–52), and cleavage of other proteins may
contribute to the effect of β-secretase inhibitors on pancreatic
cancer cell viability, these results are consistent with the notion
that APLP2 cleavage fragments influence the survival of pancreatic
cancer cells, and they suggest that further investigation of the
efficacy and mechanism of β-secretase inhibitors as potential
therapies for pancreatic cancer is warranted.
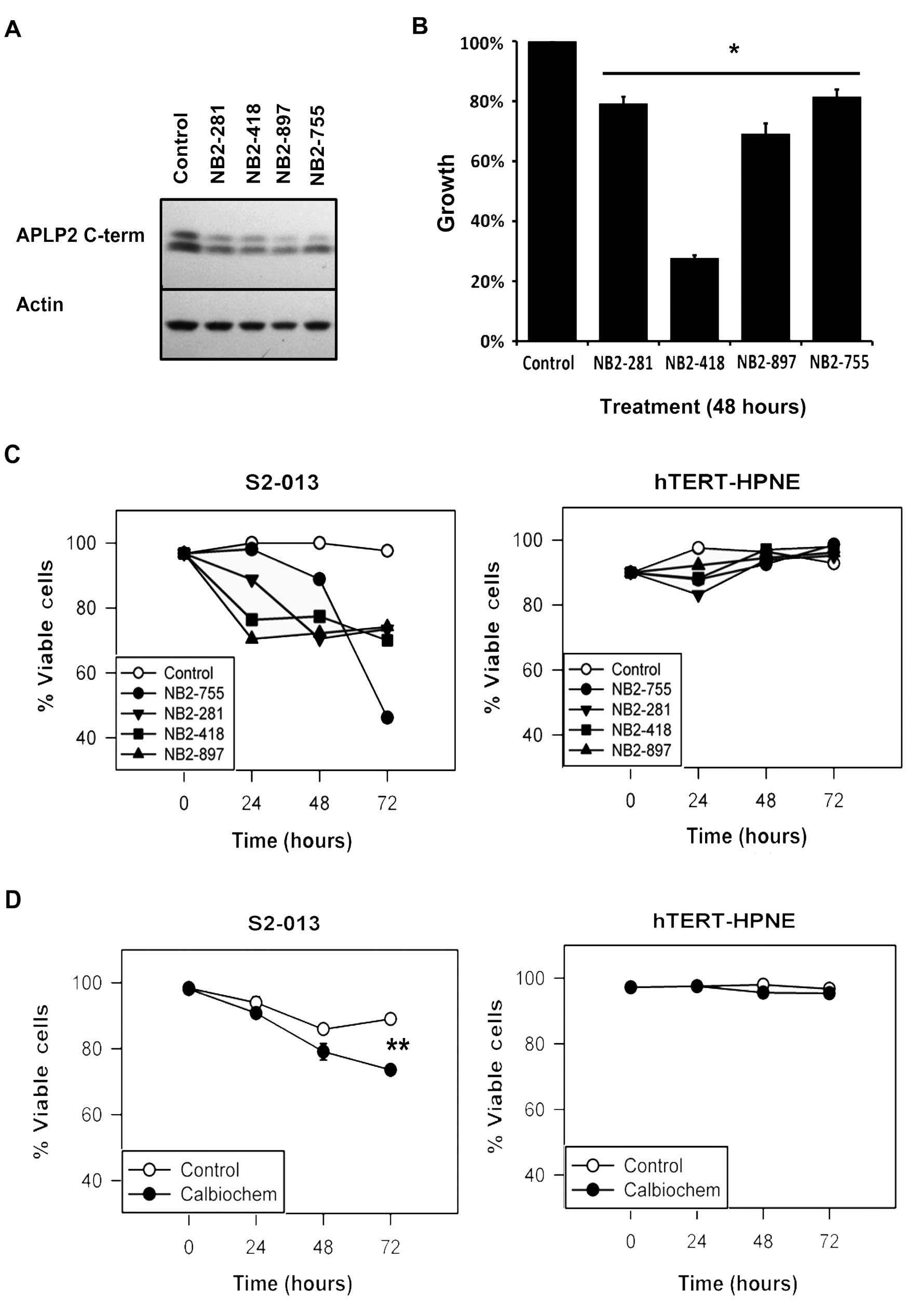 | Figure 3Chemical inhibition of β-secretase
reduced APLP2 C-terminal fragment expression and pancreatic cancer
cell viability. (A) Western blot analysis showed reduced expression
of APLP2 C-terminal fragments in S2-013 cell lysates following 24-h
incubation with 2 µM of each of four Novartis β-secretase
inhibitors (NB2-755, −281, −418 and −897). Mock-treated cells and
actin were respectively used as controls for β-secretase inhibitors
and loading. (B) Novartis β-secretase inhibitors impaired the
growth of S2-013 cells. Cell growth was determined by the MTT assay
and values were compared to respective control cells, which were
set as 100% (asterisk denotes p<0.005 by the Student’s t-test;
n=6, error bars indicate standard error of the mean). (C) The
viability of S2-013 cells in culture was reduced over time by
β-secretase inhibition, as demonstrated by trypan blue staining
following treatment with 2 µM NB2-755, −281, −418 or −897. In
contrast, the viability of the hTERT-HPNE pancreatic ductal cells
was not adversely affected by the same treatments with β-secretase
inhibitors. Viability is expressed on the graph as the percentage
of live cells, with the 0 h time point for control cells set at
100%. (D) S2-013 cells treated with 2 µM Calbiochem β-secretase
inhibitor also exhibited impaired viability. For S2-013, the
asterisk denotes p<0.001 at 72 h by the Student’s t-test. The
viability of the control hTERT-HPNE cells was not reduced by
incubation of the cells with the Calbiochem β-secretase inhibitor
(p<0.05 by the Student’s t-test). The data shown are
representative of the findings from two separate experiments, each
with n=3 and the error bars represent the standard error of the
mean. |
Discussion
APLP2, like APP, has been implicated in Alzheimer’s
disease, although APLP2’s sequence does not include the β-amyloid
peptide found within APP (1,53).
In research to develop improved treatments for Alzheimer’s disease,
there has been considerable investigation of the cleavage of APP
and APLP2, and inhibitors of β-secretases have been developed that
block the cleavage of these proteins (1,54–56).
Two β-secretases, β-site APP-cleaving enzyme (BACE)1 and BACE2,
have been implicated in the cleavage of APP family members
(50,56). BACE2 is expressed in the pancreas
(30) and specifically in
pancreatic ductal cells (31),
although other reports place BACE2 expression within islet cells
(32). The relative contribution
of BACE1 and BACE2 to the overall activity of β-secretase in
pancreatic cancer cell lines and tissues remains an area of active
research. The results shown in Fig.
3 include data derived with four β-secretase inhibitors
produced by Novartis (NB2-755, −281, −418 and −897) and one from
Calbiochem, all with activity towards BACE1 and BACE2. The
commercially available Calbiochem β-secretase inhibitor has 15
times greater ability to inhibit BACE1 compared to BACE2 (according
to the manufacturer) and it required prolonged time in culture to
inhibit pancreatic cancer cell survival (Fig. 3D). Ultimately, increased
understanding of BACE1 and BACE2 expression and activity in
pancreatic cancer will allow optimal selection of β-secretase
inhibitors that have greater efficacy in reducing the viability of
pancreatic cancer cells.
APLP2 and APP are capable of influencing growth
signaling pathways through phosphorylation, subcellular
localization and interactions with protein binding partners, which
are in turn regulated by secretase cleavage and post-translational
modifications (1,2,20,21).
In order to interpret Fig. 2 and
to clarify the pathway relationship of APLP2 and APP in pancreatic
cancer cell proliferation, Table I
was constructed exploring various pathway relationships between
APLP2 and APP. The anticipated outcome on cell proliferation
following downregulation of APLP2 and/or APP was used as the
functional readout (fourth and fifth columns, Table I). Three basic questions were
considered for the construction of the table. First, are APLP2 and
APP in the same pathway? If APLP2 and APP are not in the same
pathway, parallel pathways and unrelated pathways should be
considered. As shown in Fig. 2,
simultaneous downregulation of both APLP2 and APP did not enhance
growth inhibition beyond down-regulation of APLP2 or APP alone,
signifying that APLP2 and APP likely act upon the same pathway in
pancreatic cancer cell growth (eliminating the scenarios in the
bottom half of Table I). Second,
what is the relationship between APLP2 and APP within the pathway?
In the same pathway, APLP2 and APP could act at the same point, or
sequentially to one another, or in a parallel (or switchboard)
relationship. The last question considered was whether APP and
APLP2 have the same role. If APLP2 and APP cannot exchange for one
another, they evidently have unique roles. Because growth
inhibition was still observed when either APLP2 or APP was
downregulated, APLP2 expression does not compensate for loss of APP
in pancreatic cancer cell growth, and vice versa. Therefore,
pathway relationships outlined in rows 1 and 4 from Table I do not match the experimental data
(Fig. 2). Consequently, two models
were devised (rows 2 and 3) that match the experimental data from
Fig. 2.
 | Table IPhenotypic outcomes of postulated
pathways including APP and/or APLP2. |
Table I
Phenotypic outcomes of postulated
pathways including APP and/or APLP2.
| Pathway type | APP and APLP2
pathway relationship | APP and APLP2
role | Growth of cells
expressing APP siRNA or APLP2 siRNA | Growth of cells
expressing APP siRNA and APLP2 siRNA |
|---|
| Same | Same | Same | No effect | ↓ |
| Same | Same | Unique | ↓ | ↓ (to same extent
as cells expressing either siRNA) |
| Same | Sequential | Unique | ↓ | ↓ (to same extent
as cells expressing either siRNA) |
| Same | Parallel | Unique | No effect | ↓ |
| Parallel | + and + | Unique | ↓ | ↓ |
| Parallel | + and inhibits
inhibitor | Unique | ↓ | ↓ |
| Parallel | Inhibits inhibitor
and inhibits inhibitor | Unique | ↓ | ↓ |
| Unrelated | Separate | Unique | ↓ | ↓ |
The second row of Table
I indicates that APLP2 and APP could be involved at the same
point within the same pathway, but yet serve unique roles; an
example of this scenario would be a functional heterodimer of APLP2
and APP. Heterodimers of APLP2 and APP are known to form (57). In this context, a homodimer of
APLP2 or APP could not recapitulate the function of the APLP2-APP
heterodimer, and equal growth inhibition would occur with loss of
one or both proteins. The scenario in the third row also matches
our experimental data. Instead of APLP2 or APP acting at the same
point in a pathway, one protein would be upstream of the other, and
both would be required for a growth signal to be conducted. In both
devised scenarios matching the experimental data, APLP2 and APP
would have unique roles in the growth of pancreatic cancer cells.
Ultimately, the conclusion best fitting the data is that APLP2 and
APP serve unique roles in the growth of pancreatic cancer cell
lines.
Downregulation of APLP2 C-terminal fragments (by
siRNA or β-secretase inhibitors) resulted in growth inhibition of
S2-013 cells, despite maintained expression of APP full-length
protein (Fig. 2A and B and data
not shown); this is noteworthy since β-secretase inhibitors are
also capable of reducing cleavage of APP. However, APP appears to
be a poor target for secretases expressed in pancreatic cancer cell
lines, as evidenced by minimal expression of APP C-terminal
fragments in the majority of pancreatic cancer cell lines (Fig. 1A) and failure of APLP2 reduction
(by siRNA or β-secretase inhibitors) to enhance APP C-terminal
cleavage (data not shown). Selective cleavage of APLP2 over APP can
arise through secretase specificity, intracellular
compartmentalization, APLP2-APP homo- or hetero-oligomerization
and/or isoforms of APLP2 or APP expressed (58–63).
Full-length APLP2 and APP are capable of forming homo- or
hetero-oligomers, which may be disrupted upon soluble APLP2 or
soluble APP binding (57,65–67).
The transmembrane-soluble complexes formed by APLP2 and APP are
significant because the activity of APLP2 and APP receptors depends
upon the complex formed. For example, dimers of full-length APP
have been proposed to activate cell death in neuronal cells, where
full-length APP-soluble APP dimers disrupt the cytotoxic signal
(67). While in our experiments
protein expression of APLP2 or APP was not altered following loss
of the other family member, alterations to additional regulatory
mechanisms of APLP2 and/or APP may occur. Future investigations are
required to explore the possibilities of altered APLP2 or APP
regulatory mechanisms and to dissect functional identities of APLP2
and APP in pancreatic cancer growth and viability.
Treatment with β-secretase inhibitors caused not
only a decrease in APLP2 C-terminal fragments, but also a reduction
in S2-013 cell viability (Fig. 3).
Our data indicate that inhibitory therapies that target APLP2, APP
and BACE may be promising for the treatment of pancreatic cancer.
Notably, tolfenamic acid, which is currently under investigation
for use in pancreatic cancer (68–71),
has been shown to impair expression of APP and BACE (72). Reduced cleavage of other proteins
(in addition to APLP2) might also contribute to the ability of the
β-secretase inhibitors to affect pancreatic cancer cell viability.
The β-secretase inhibitors target β-secretases that cleave APP as
well as APLP2. However, our analysis has not shown a very high
expression of APP C-terminal fragments in pancreatic cancer cell
lines except BxPC3 (Fig. 1).
Cleavage of additional proteins (not in the APP/APLP2 family) able
to influence viability may be affected by the β-secretase
inhibitors. BACE2 substrate proteins are not well defined, although
there is somewhat more information on BACE1 substrates, which
include heregulins (73–75). Heregulin proteins have been noted
to be over-expressed in pancreatic cancer cells and to influence
their growth (76). Thus,
heregulins might have a role in β-secretase inhibitor-mediated
reduction of pancreatic cancer cell viability. Additional credence
for β-secretase inhibitors as a therapy for pancreatic cancer could
be obtained by conducting future studies to test β-secretase
inhibitors in xenograft models of pancreatic cancer, alone or in
combination with gemcitabine (used in clinical treatment for
pancreatic cancer patients) (77).
The available treatments for pancreatic cancer are
unable to decrease morbidity and mortality effectively, and only by
attaining a better understanding of pancreatic cancer cell biology
and testing novel therapies can we hope to increase the odds for
patients with this disease. In this study, our findings have
revealed several new aspects of the relationship between APLP2 and
pancreatic cancer. First, APLP2 is highly expressed in human
pancreatic cancer cell lines and clinical tissue samples, relative
to normal pancreatic tissues. Second, C-terminal fragments are more
consistently generated from APLP2 rather than APP in pancreatic
cancer cell lines. Third, the presence of APLP2 and APLP2
C-terminal fragments is increased by pancreatic ductal cell
transformation. Fourth, down-regulation of APLP2 and/or APP impairs
the growth of a pancreatic cancer cell line, and each protein
likely contributes to the same growth pathway, but in a unique
manner. Finally, inhibitors of β-secretase reduce APLP2 cleavage
and pancreatic cancer cell viability. Overall, our results suggest
that β-secretase, APLP2 (and notably its C-terminal cleavage
fragments), and APP influence the survival of pancreatic cancer
cells.
Abbreviations:
|
APLP2
|
amyloid precursor-like protein 2;
|
|
APP
|
amyloid precursor protein;
|
|
DMSO
|
dimethyl sulfoxide;
|
|
MTT
|
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide;
|
|
PBS
|
phosphate-buffered saline;
|
|
siRNA
|
short interfering RNA
|
Acknowledgements
β-secretase inhibitors NB2-755,
NB2-281, NB2-418 and NB2-897 were kindly provided by Novartis
Pharma AG, Basel, Switzerland. We gratefully acknowledge Dr Amar
Natarajan for providing necessary instrumentation, and the
assistance of the personnel of the University of Nebraska Medical
Center Rapid Autopsy Program and the UNMC Tissue Sciences Facility.
This study received support from the NIH SPORE (P50CA127297) (to
M.A.H. and M.M.O), an NIH SPORE (P50CA127297) Developmental
Research Program Grant, an NIH/NCRR COBRE grant from the National
Center for Research Resources (5P20RR018759-10) and the National
Institute of General Medicine Sciences (8P20GM103489-10) for the
Nebraska Center for Cellular Signaling through an ARRA pilot
project grant (to J.C.S. and R.G.M.), NIH Grant R01 GM57428, a
Nebraska Department of Health and Human Services Award (to J.C.S.),
University of Nebraska Medical Center Graduate Studies Emley and
Regents Tuition Fellowships, an NIH Training Grant T32 CA009476
Fellowship, a Graduate Assistance in Areas of National Need
Fellowship (to H.P.), and a University of Nebraska Graduate Studies
Assistantship (to A.T.). A.T. is supported by the Ramanujan
Fellowship instituted by the Department of Science and Technology,
Government of India. Core facilities at the University of Nebraska
Medical Center receive support from NIH Cancer Center Support Grant
P30CA036727. The funders of this research had no role in study
design, data collection and analysis, decision to publish, or
preparation of the manuscript.
References
|
1
|
Walsh DM, Minogue AM, Frigerio CS, Fadeeva
JV, Wasco W and Selkoe DJ: The APP family of proteins: similarities
and differences. Biochem Soc Trans. 35:416–420. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Orcholski ME, Zhang Q and Bredesen DE:
Signaling via amyloid precursor-like proteins APLP1 and APLP2. J
Alzheimers Dis. 23:689–699. 2011.PubMed/NCBI
|
|
3
|
Needham BE, Wlodek ME, Ciccotosto GD, Fam
BC, Masters CL, Proietto J, Andrikopoulos S and Cappai R:
Identification of the Alzheimer’s disease amyloid precursor protein
(APP) and its homologue APLP2 as essential modulators of glucose
and insulin homeostasis and growth. J Pathol. 215:155–163.
2008.
|
|
4
|
Korte M, Herrmann U, Zhang X and Draguhn
A: The role of APP and APLP for synaptic transmission, plasticity,
and network function: lessons from genetic mouse models. Exp Brain
Res. 217:435–440. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
von Koch CS, Zheng H, Chen H, Trumbauer M,
Thinakaran G, van der Ploeg LH, Price DL and Sisodia SS: Generation
of APLP2 KO mice and early postnatal lethality in APLP2/APP double
KO mice. Neurobiol Aging. 18:661–669. 1997.PubMed/NCBI
|
|
6
|
Cappai R, Mok SS, Galatis D, et al:
Recombinant human amyloid precursor-like protein 2 (APLP2)
expressed in the yeast Pichia pastoris can stimulate neurite
outgrowth. FEBS Lett. 442:95–98. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guo J, Thinakaran G, Guo Y, Sisodia SS and
Yu FX: A role for amyloid precursor-like protein 2 in corneal
epithelial wound healing. Invest Ophthalmol Vis Sci. 39:292–300.
1998.PubMed/NCBI
|
|
8
|
Li XF, Thinakaran G, Sisodia SS and Yu FS:
Amyloid precursor-like protein 2 promotes cell migration toward
fibronectin and collagen IV. J Biol Chem. 274:27249–27256. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rassoulzadegan M, Yang Y and Cuzin F:
APLP2, a member of the Alzheimer precursor protein family, is
required for correct genomic segregation in dividing mouse cells.
EMBO J. 17:4647–4656. 1998. View Article : Google Scholar
|
|
10
|
Thinakaran G, Kitt CA, Roskams AJ, et al:
Distribution of an APP homolog, APLP2, in the mouse olfactory
system: a potential role for APLP2 in axogenesis. J Neurosci.
15:6314–6326. 1995.PubMed/NCBI
|
|
11
|
Kummer C, Wehner S, Quast T, Werner S and
Herzog V: Expression and potential function of beta-amyloid
precursor proteins during cutaneous wound repair. Exp Cell Res.
280:222–232. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
McLoughlin DM and Miller CCJ: The FE65
proteins and Alzheimer’s disease. J Neurosci Res. 86:744–754.
2008.
|
|
13
|
Tuli A, Sharma M, Naslavsky N, Caplan S
and Solheim JC: Specificity of amyloid precursor-like protein 2
interactions with MHC class I molecules. Immunogenetics.
60:303–313. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tuli A, Sharma M, McIlhaney MM, Talmadge
JE, Naslavsky N, Caplan S and Solheim JC: Amyloid precursor-like
protein 2 increases the endocytosis, instability, and turnover of
the H2-Kd MHC class I molecule. J Immunol.
181:1978–1987. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tuli A, Sharma M, Capek HL, Naslavsky N,
Caplan S and Solheim JC: Mechanism for amyloid precursor-like
protein 2 enhancement of major histocompatibility complex class I
molecule degradation. J Biol Chem. 284:34296–34307. 2009.
View Article : Google Scholar
|
|
16
|
Choi JH, Lee MY, Kim Y, et al: Isolation
of genes involved in pancreas regeneration by subtractive
hybridization. Biol Chem. 391:1019–1029. 2010.PubMed/NCBI
|
|
17
|
Covell DG, Wallqvist A, Rabow AA and
Thanki N: Molecular classification of cancer: unsupervised
self-organizing map analysis of gene expression microarray data.
Mol Cancer Therap. 2:317–332. 2003.PubMed/NCBI
|
|
18
|
Abba MC, Drake JA, Hawkins KA, et al:
Transcriptomic changes in human breast cancer progression as
determined by serial analysis of gene expression. Breast Cancer
Res. 6:499–513. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tuli A, Sharma M, Wang X, et al: Amyloid
precursor-like protein 2 association with HLA class I molecules.
Cancer Immunol Immunother. 58:1419–1431. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Eggert S, Paliga K, Soba P, Evin G,
Masters CL, Weidemann A and Beyreuther K: The proteolytic
processing of the amyloid precursor protein gene family members
APLP-2 and APLP-2 involves alpha-, beta-, gamma-, and epsilon-like
cleavages: modulation of APLP-1 processing by N-glycosylation. J
Biol Chem. 279:18146–18156. 2004. View Article : Google Scholar
|
|
21
|
Sisodia SS, Thinakaran G, Slunt HH, Kitt
CA, Von Koch CS, Reed RR, Zheng H and Price DL: Studies on the
metabolism and biological function of APLP2. Ann NY Acad Sci.
777:77–81. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pastorino L, Ikin AF, Lamprianou S,
Vacaresse N, Revelli JP, Platt K, Paganetti P, Mathews PM, Harroch
S and Buxbaum JD: BACE (beta-secretase) modulates the processing of
APLP2 in vivo. Mol Cell Neurosci. 25:642–649. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hogl S, Kuhn PH, Colombo A and
Lichtenthaler SF: Determination of the proteolytic cleavage sites
of the amyloid precursor-like protein 2 by the proteases ADAM10,
BACE1 and γ-secretase. PLoS One. 6:e213372011.PubMed/NCBI
|
|
24
|
Hansel DE, Rahman A, Wehner S, Herzog V,
Yeo CJ and Maitra A: Increased expression and processing of the
Alzheimer amyloid precursor protein in pancreatic cancer may
influence cellular proliferation. Cancer Res. 63:7032–7037.
2003.
|
|
25
|
Vassar R, Bennett BD, Babu-Khan S, Kahn S,
Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo
Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL,
Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G and
Citron M: Beta-secretase cleavage of Alzheimer’s amyloid precursor
protein by the transmembrane aspartic protease BACE. Science.
286:735–741. 1999.
|
|
26
|
Farzan M, Schnitzler CE, Vasilieva N,
Leung D and Choe H: BACE2, a beta-secretase homolog, cleaves at the
beta site and within the amyloid-beta region of the amyloid-beta
precursor protein. Proc Natl Acad Sci USA. 97:9712–9717. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Basi G, Frigon N, Barbour R, Doan T,
Gordon G, McConlogue L, Sinha S and Zeller M: Antagonistic effects
of beta-site amyloid precursor protein-cleaving enzymes 1 and 2 on
beta-amyloid peptide production in cells. J Biol Chem.
278:31512–31520. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun X, Wang Y, Qing H, Christensen MA, Liu
Y, Zhou W, Tong Y, Xiao C, Huang Y, Zhang S, Liu X and Song W:
Distinct transcriptional regulation and function of the human BACE2
and BACE1 genes. FASEB J. 19:739–49. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bodendorf U, Fischer F, Bodian D, Multhaup
G and Paganetti P: A splice variant of beta-secretase deficient in
the amyloidogenic processing of the amyloid precursor protein. J
Biol Chem. 276:12019–12023. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bennett BD, Babu-Khan S, Loeloff R, Louis
JC, Curran E, Citron M and Vassar R: Expression analysis of BACE2
in brain and peripheral tissues. J Biol Chem. 275:20647–20651.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Figueroa DJ, Shi XP, Gardell SJ and Austin
CP: Abetapp secretases are co-expressed with Abetapp in the
pancreatic islets. J Alzheimers Dis. 3:393–396. 2001.PubMed/NCBI
|
|
32
|
Casas S, Casini P, Piquer S, Altirriba J,
Soty M, Cadavez L, Gomis R and Novials A: BACE2 plays a role in the
insulin receptor trafficking in pancreatic β-cells. Am J Physiol
Endocrinol Metab. 299:E1087–E1095. 2010.PubMed/NCBI
|
|
33
|
Rooman I and Real FX: Pancreatic ductal
adenocarcinoma and acinar cells: a matter of differentiation and
development? Gut. 61:449–458. 2012. View Article : Google Scholar
|
|
34
|
Thinakaran G and Sisodia SS: Amyloid
precursor-like protein 2 (APLP2) is modified by the addition of
chondroitin sulfate glycosaminoglycan at a single site. J Biol
Chem. 269:22099–22104. 1994.PubMed/NCBI
|
|
35
|
Thinakaran G, Slunt HH and Sisodia SS:
Novel regulation of chondroitin sulfate glycosaminoglycan
modification of amyloid precursor protein and its homologue, APLP2.
J Biol Chem. 270:16522–16525. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Owens RB, Smith HS, Nelson-Rees WA and
Springer EL: Epithelial cell cultures from normal and cancerous
human tissues. J Natl Cancer Inst. 56:843–849. 1976.PubMed/NCBI
|
|
37
|
Taniguchi S, Iwamura T and Katsuki T:
Correlation between spontaneous metastatic potential and type I
collagenolytic activity in a human pancreatic cancer cell line
(SUIT-2) and sublines. Clin Exp Metastasis. 10:259–266. 1992.
View Article : Google Scholar
|
|
38
|
Iwamura T, Katsuki T and Ide K:
Establishment and characterization of a human pancreatic cancer
cell line (SUIT-2) producing carcinoembryonic antigen and
carbohydrate antigen 19-9. Jpn J Cancer Res. 78:54–62.
1987.PubMed/NCBI
|
|
39
|
Chin J, Miller F and Lane BP: Detection of
human pancreatic adenocarcinomas by histochemical staining with
monoclonal antibody AR1-28. Diagn Immunol. 3:99–105.
1985.PubMed/NCBI
|
|
40
|
Tan MH, Nowak NJ, Loor R, Ochi H, Sandberg
AA, Lopez C, Pickren JW, Berjian R, Douglass HO Jr and Chu TM:
Characterization of a new primary human pancreatic tumor line.
Cancer Invest. 4:15–23. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kyriazis AA, Kyriazis AP, Sternberg CN,
Sloane NH and Loveless JD: Morphological, biological, biochemical,
and karyotypic characteristics of human pancreatic ductal
adenocarcinoma Capan-2 in tissue culture and the nude mouse. Cancer
Res. 46:5810–5815. 1986.
|
|
42
|
Lee KM, Nguyen C, Ulrich AB, Pour PM and
Ouellette MM: Immortalization with telomerase of the
Nestin-positive cells of the human pancreas. Biochem Biophys Res
Commun. 301:1038–1044. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lee KM, Yasuda H, Hollingsworth MA and
Ouellette MM: Notch 2-positive progenitors with the intrinsic
ability to give rise to pancreatic ductal cells. Lab Invest.
85:1003–1012. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Campbell PM, Groehler AL, Lee KM,
Ouellette MM, Khazak V and Der CJ: K-Ras promotes growth
transformation and invasion of immortalized human pancreatic cells
by Raf and phosphatidylinositol 3-kinase signaling. Cancer Res.
67:2098–2106. 2007. View Article : Google Scholar
|
|
45
|
Campbell PM, Lee KM, Ouellette MM, Kim HJ,
Groehler AL, Khazak V and Der CJ: Ras-driven transformation of
human nestin-positive pancreatic epithelial cells. Methods Enzymol.
439:451–465. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Matsuo Y, Campbell PM, Brekken RA, et al:
K-Ras promotes angiogenesis mediated by immortalized human
pancreatic epithelial cells through mitogen-activated protein
kinase signaling pathways. Mol Cancer Res. 7:799–808. 2009.
View Article : Google Scholar
|
|
47
|
Weidemann A, König G, Bunke D, Fischer P,
Salbaum JM, Masters CL and Beyreuther K: Identification,
biogenesis, and localization of precursors of Alzheimer’s disease
A4 amyloid protein. Cell. 57:115–126. 1989.PubMed/NCBI
|
|
48
|
Venkataramani V, Rossner C, Iffland L,
Schweyer S, Tamboli IY, Walter J, Wirths O and Bayer TA: Histone
deacetylase inhibitor valproic acid inhibits cancer cell
proliferation via down-regulation of the alzheimer amyloid
precursor protein. J Biol Chem. 285:10678–10689. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mauri P, Scarpa A, Nascimbeni AC, Benazzi
L, Parmagnani E, Mafficini A, Della Peruta M, Bassi C, Miyazaki K
and Sorio C: Identification of proteins released by pancreatic
cancer cells by multidimensional protein identification technology:
a strategy for identification of novel cancer markers. FASEB J.
19:1125–1127. 2005.
|
|
50
|
Li Q and Südhof TC: Cleavage of
amyloid-beta precursor protein and amyloid-beta precursor-like
protein by BACE 1. J Biol Chem. 279:10542–10550. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sugimoto I, Futakawa S, Oka R, et al:
Beta-galactoside alpha2,6-sialyltransferase I cleavage by BACE1
enhances the sialylation of soluble glycoproteins. A novel
regulatory mechanism for alpha2,6-sialylation. J Biol Chem.
282:34896–34903. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kuhn PH, Marjaux E, Imhof A, De Strooper
B, Haass C and Lichtenthaler SF: Regulated intramembrane
proteolysis of the interleukin-1 receptor II by alpha-, beta-, and
gamma-secretase. J Biol Chem. 282:11982–11995. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zheng H and Koo E: The amyloid precursor
protein: beyond amyloid. Mol Neurodegener. 1:52006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wolfe MS: Selective amyloid-β lowering
agents. BMC Neurosci. 9(Suppl 2): S42008.
|
|
55
|
Ostermann N, Eder J, Eidhoff U, et al:
Crystal structure of human BACE2 in complex with a
hydroxyethylamine transition-state inhibitor. J Mol Biol.
355:249–261. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Stockley JH and O’Neill C: The proteins
BACE1 and BACE2 and β-secretase activity in normal and Alzheimer’s
disease brain. Biochem Soc Trans. 35:574–576. 2007.
|
|
57
|
Jacobsen KT and Iverfeldt K: Amyloid
precursor protein and its homologues: a family of
proteolysis-dependent receptors. Cell Mol Life Sci. 66:2299–2318.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Jacobsen KT, Adlerz L, Multhaup G and
Iverfeldt K: Insulin-like growth factor-1 (IGF-1)-induced
processing of amyloid-beta precursor protein (APP) and APP-like
protein 2 is mediated by different metalloproteinases. J Biol Chem.
285:10223–10231. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Vassar R: Beta-secretase (BACE) as a drug
target for Alzheimer’s disease. Adv Drug Deliv Rev. 54:1589–1602.
2002.
|
|
60
|
Gandhi S, Refolo LM and Sambamurti K:
Amyloid precursor protein compartmentalization restricts
beta-amyloid production: therapeutic targets based on BACE
compartmentalization. J Mol Neurosci. 24:137–143. 2004. View Article : Google Scholar
|
|
61
|
Zou L, Wang Z, Shen L, Bao GB, Wang T,
Kang JH and Pei G: Receptor tyrosine kinases positively regulate
BACE activity and Amyloid-beta production through enhancing BACE
internalization. Cell Res. 17:389–401. 2007.PubMed/NCBI
|
|
62
|
Eggert S, Midthune B, Cottrell B and Koo
EH: Induced dimerization of the amyloid precursor protein leads to
decreased amyloid-beta protein production. J Biol Chem.
284:28943–28952. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Isbert S, Wagner K, Eggert S, Schweitzer
A, Multhaup G, Weggen S, Kins S and Pietrzik CU: APP dimer
formation is initiated in the endoplasmic reticulum and differs
between APP isoforms. Cell Mol Life Sci. 69:1353–1375. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Baumkötter F, Wagner K, Eggert S, Wild K
and Kins S: Structural aspects and physiological consequences of
APP/APLP transdimerization. Exp Brain Res. 217:389–395.
2012.PubMed/NCBI
|
|
65
|
Soba P, Eggert S, Wagner K, Zentgraf H,
Siehl K, Kreger S, Löwer A, Langer A, Merdes G, Paro R, Masters CL,
Müller U, Kins S and Beyreuther K: Homo- and heterodimerization of
APP family members promotes intercellular adhesion. EMBO J.
24:3624–3634. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kaden D, Munter LM, Reif B and Multhaup G:
The amyloid precursor protein and its homologues: structural and
functional aspects of native and pathogenic oligomerization. Eur J
Cell Biol. 91:234–239. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Gralle M, Botelho MG and Wouters FS:
Neuroprotective secreted amyloid precursor protein acts by
disrupting amyloid precursor protein dimers. J Biol Chem.
284:15016–15025. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Abdelrahim M, Baker CH, Abbruzzese JL and
Safe S: Tolfenamic acid and pancreatic cancer growth, angiogenesis,
and Sp protein degradation. J Natl Cancer Inst. 98:855–868. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Konduri S, Colon J, Baker CH, Safe S,
Abbruzzese JL, Abudayyeh A, Basha MR and Abdelrahim M: Tolfenamic
acid enhances pancreatic cancer cell and tumor response to
radiation therapy by inhibiting survivin protein expression. Mol
Cancer Ther. 8:533–542. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Jia Z, Gao Y, Wang L, Li Q, Zhang J, Le X,
Wei D, Yao JC, Chang DZ, Huang S and Xie K: Combined treatment of
pancreatic cancer with mithramycin A and tolfenamic acid promotes
Sp1 degradation and synergistic antitumor activity. Cancer Res.
70:1111–1119. 2010. View Article : Google Scholar
|
|
71
|
Basha R, Baker CH, Sankpal UT, Ahmad S,
Safe S, Abbruzzese JL and Abdelrahim M: Therapeutic applications of
NSAIDS in cancer: special emphasis on tolfenamic acid. Front Biosci
(Schol Ed). 3:797–805. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
72
|
Adwan LI, Basha R, Abdelrahim M, Subaiea
GM and Zawia NH: Tolfenamic acid interrupts the de novo synthesis
of the β-amyloid precursor protein and lowers amyloid beta via a
transcriptional pathway. Curr Alzheimer Res. 8:385–92.
2011.PubMed/NCBI
|
|
73
|
Turner RT III, Loy JA, Nguyen C,
Devasamudram T, Ghosh AK, Koelsch G and Tang J: Specificity of
memapsin 1 and its implications on the design of memapsin 2
(beta-secretase) inhibitor selectivity. Biochemistry. 41:8742–8746.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Hemming ML, Elias JE, Gygi SP and Selkoe
DJ: Identification of beta-secretase (BACE1) substrates using
quantitative proteomics. PLoS One. 4:e84772009. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Vassar R, Kovacs DM, Yan R and Wong PC:
The beta-secretase enzyme BACE in health and Alzheimer’s disease:
regulation, cell biology, function, and therapeutic potential. J
Neuroscience. 29:12787–12794. 2009.
|
|
76
|
Kolb A, Kleeff J, Arnold N, Giese NA,
Giese T, Korc M and Friess H: Expression and differential signaling
of heregulins in pancreatic cancer cells. Int J Cancer.
120:514–523. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Berlin J and Benson AB III: Chemotherapy:
gemcitabine remains the standard of care for pancreatic cancer. Nat
Rev Clin Oncol. 7:135–137. 2010. View Article : Google Scholar : PubMed/NCBI
|