Introduction
World Health Organization (WHO) grade II gliomas
(low-grade gliomas) are slow-growing tumors that include several
subtypes, such as diffuse astrocytomas, mixed oligoastrocytomas,
and oligodendrogliomas (1). The
10- and 20-year survival rates for patients with grade II glioma
are reported to be 48 and 22% (2),
reflecting the malignant potential of these tumors in long-term
survival. Radiotherapy is often the treatment of choice for
patients with incompletely resected grade II gliomas. However, the
timing of radiotherapy for patients with these malignancies remains
controversial and no difference in overall survival (OS) between
groups receiving early and delayed radiation has been reported
(3). Moreover, the efficacy of
chemoradiotherapy for grade II gliomas is largely unknown. The
addition of procarbazine, lomustine, and vincristine (PCV) therapy
to radiotherapy for grade II gliomas conferred a significant
increase in OS and progression-free survival (PFS) of >2 years
in the Radiation Therapy Oncology Group (RTOG) 9802 study (4), suggesting that chemoradiotherapy
might be effective for a subset of these patients.
Several studies have attempted to identify
prognostic factors for grade II gliomas. To date, older age,
astrocytic histology, the presence of neurologic deficits before
surgery, larger tumor diameters, and tumors crossing the midline
have been proposed as unfavorable prognostic factors (5–9).
Several genetic markers, such as 1p/19q codeletion or mutations of
the isocitrate dehydrogenase 1 and 2 genes (IDH1/2), have
also been associated with patient survival. Oligodendrogliomas
typically show 1p/19q codeletion (≤70%) (10,11),
and its presence is reported to predict longer survival in
oligodendroglial tumors (12). The
1p/19q codeletion is also a statistically significant predictor of
prolonged survival in patients with astrocytomas (13). Furthermore, 1p/19q codeletion was
associated with longer survival in all types of adult gliomas,
independent of age and diagnosis (14,15).
On the other hand, 1p/19q codeletion did not appear to be a
sensitive prognostic biomarker in patients with either grade II
astrocytic or oligodendroglial tumors who did not receive
radiotherapy or chemotherapy after surgery (16).
Mutations of the IDH1/2 genes are common
events in gliomas (17),
especially among grade II gliomas, where IDH1 mutations are
observed in 70–80% of cases (11,17,18).
Glioblastomas and anaplastic astrocytomas (WHO grade III) with
IDH1/2 mutations have more favorable prognoses than those with a
wild-type phenotype (17). Several
studies have indicated that IDH1/2 mutations are
significantly associated with positive prognosis and
chemosensitivity in low-grade gliomas (19,20),
whereas others have reported that IDH1/2 mutations were not
associated with prognosis (21,22).
Thus, the prognostic or predictive values of these genetic markers
in grade II gliomas remain controversial.
In the present study, the clinicopathological
factors, including age, Karnofsky performance status (KPS),
histology, extent of resection, radiotherapy, chemoradiotherapy,
largest tumor diameter, and MIB-1 staining index, as well as
IDH1/2 mutations and 1p/19q codeletion, were analyzed in
grade II gliomas and correlated with the clinical course of the
patients. Oligodendroglial tumors, age <40 years, initial KPS
≥80, and IDH1/2 mutations were favorable prognostic factors
for PFS and OS. The IDH1/2 mutation was a predictive factor of
response to chemoradiotherapy in grade II gliomas.
Materials and methods
Patients and tissue collections
The data were collected from 72 patients who were
found with WHO grade II gliomas at the first surgery. These
included 49 diffuse astrocytomas and 23 oligodendroglial tumors,
including 4 oligodendrogliomas and 19 oligoastrocytomas
(male-female, 40:32; median age, 39.0 years). These consecutive
cases were diagnosed and treated between 1991 and 2010 at the
National Cancer Center Hospital in Japan. The clinical records of
the patients were reviewed, and the data on the extent of tumor
resection were obtained from the surgical report. Total or subtotal
resection was defined as the removal of 90% or more of the tumor
based on the surgeon’s clinical report. Fifty-eight patients
(80.6%) underwent initial surgeries followed by radiotherapy
(22.2%) or chemoradiotherapy with ACNU (58.3%). Three patients with
total or subtotal removal and 11 with partial resection or biopsy
(19.4%) were followed-up without radiotherapy until progressive
disease. Of the remaining patients, those who underwent initial
treatment between 1991 and 2006 were treated with chemoradiotherapy
and those treated between 2007 and 2010 underwent radiotherapy
alone based on our treatment protocols. The radiation doses were 60
Gy before 2006 and 54 Gy after 2007. The chemotherapy in the
diffuse astrocytoma cases consisted of ACNU administered twice
during radiotherapy and 3 additional doses every 2 months after
radiotherapy. The patients with oligodendroglial tumors received
ACNU + VCR (vincristine) twice during radiotherapy, and thereafter,
PAV [ACNU + VCR + PCZ (procarbazine)] was administered in 3 cycles
every 2 months after radiotherapy. Each patient was worked up by
MRI every 3–4 months until 2 years from the initial treatment and
then every 6 months after 2 years. Progression was determined when
the MRI showed a new enhancing lesion with Gd-DTPA, a new high
intensity lesion or an obvious increased lesion (at least 20%
larger than previous MRI in diameter) on T2/FLAIR images. Clinical
deterioration of a patient was also determined as progression.
The formalin-fixed paraffin-embedded tumor samples
and frozen specimens, when available, were collected from the
primary resection for all the patients who underwent surgery in the
National Cancer Center and whenever possible for those operated at
other hospitals. The samples were examined for IDH1/2
mutations and 1p/19q codeletion only when sufficient material for
DNA extraction was available at either the primary or secondary
resection. The study was approved by the internal review board of
the National Cancer Center. The detailed information for all the 72
patients is listed in Table I.
 | Table ICharacteristics of patients with
grade II gliomas. |
Table I
Characteristics of patients with
grade II gliomas.
| Characteristic | No. of
patients | Years | Percentage (%) |
|---|
| Sex | | | |
| Male | 40 | | 55.6 |
| Female | 32 | | 44.4 |
| Age (years) | | | |
| Median | | 39 | |
| Range | | 21–75 | |
| Histology | | | |
| Astrocytoma | 49 | | 68 |
|
Oligodendroglioma | 4 | | 6 |
|
Oligoastrocytoma | 19 | | 26 |
| Extent of
removal | | | |
| Total
removal | 12 | | 16.7 |
| Subtotal
removal | 2 | | 2.8 |
| Partial
removal | 27 | | 37.5 |
| Biopsy | 31 | | 43.1 |
| Largest diameter of
initial tumor (cm) | | | |
| <6 | 40 | | 55.6 |
| ≥6 | 32 | | 44.4 |
| Initial KPS | | | |
| <80 | 4 | | 5.6 |
| ≥80 | 68 | | 94.4 |
| MIB-1 index
(%) | | | |
| Median | 3 | | |
| IDH
mutation | | | |
| Mutation | 42 | | 58.3 |
| Wild-type | 30 | | 41.7 |
| Loss of 1p/19q | | | |
| 1p/19q
codeletion | 15 | | 25.0 |
| 1p deletion | 24 | | 40.0 |
| 19q deletion | 23 | | 38.3 |
| Initial
radiotherapy | | | |
| + | 58 | | 80.6 |
| − | 14 | | 19.4 |
| Initial
chemotherapy | | | |
| ACNU | 44 | | 61 |
| TMZ | 2 | | 3 |
| None | 26 | | 36 |
| PFS (years) | | | |
| Median | | 5.8 | |
| Overall survival
(years) | | | |
| Median | | 10.3 | |
Hematoxylin and eosin staining and
immunohistochemical staining for MIB-1 and IDH1
The surgical specimens were fixed in 10% formalin
and embedded in paraffin. The hematoxylin and eosin-stained
specimens were examined to determine the histological tumor type.
The multiple serial sections were subjected to immunohistochemical
analyses (IHC) to visualize local staining. Antigen retrieval was
carried out by exposing the tissue sections to 15 min of microwave
heating in 0.1-mol/l sodium citrate (pH 6.0). This was followed
with immunostaining of the specimens with the
streptavidin-biotin-peroxidase complex method (Vectastain, Vector
Laboratories, Inc., Burlingame, CA, USA). The samples were
incubated in human monoclonal antibodies against MIB-1 (Dako,
Tokyo, Japan). Positive immunostaining results were detected with
the diaminobenzidine reaction, and the slides were subsequently
counterstained with hematoxylin, dehydrated, cleared, and
mounted.
Cell counting was performed with the aid of a light
microscope (Olympus Corp., Tokyo, Japan). Cell counting was done at
a magnification of ×400. At least 200 tumor cells were counted, and
the results were expressed as the mean of the counts obtained from
3 different locations within each specimen. The MIB-1-stained cells
were also counted, and the percentage of the MIB-1-stained cells
was calculated within the observed field and expressed as the MIB-1
index.
Human monoclonal antibodies specific against
IDH1-R132H and IDH1-R132S were used to identify these 2 types of
IDH1 mutations (Medical & Biological Laboratories, Tokyo,
Japan). Positive immunostaining results were detected with the
diaminobenzidine reaction, and the slides were subsequently
counterstained with hematoxylin, dehydrated, cleared, and mounted.
The positive granular cytoplasmic staining of the tumor cells was
evaluated for mutant IDH1 (23).
Extraction of nucleic acids
The tumor samples were immediately frozen in liquid
nitrogen and stored at −80°C. A peripheral blood sample was drawn
from each patient and stored at −80°C. Total DNA was extracted from
either frozen tissue samples or paraffin-embedded specimens and
from the patients’ blood with a DNeasy Blood & Tissue kit
(Qiagen Sciences, Germantown, MD, USA) according to the
manufacturer’s instructions.
Sequencing of IDH1/2
A 129-base pair (bp) fragment of IDH1
containing codon 132 or a 150-bp fragment of IDH2 containing
codon 172 was PCR amplified using the forward primer IDH1f
(CGGTCTTCAGAGAAGCCATT) and reverse primer IDH1r
(GCAAAATCACATTATTGCCAAC) for IDH1 and the forward primer
IDH2f (AGCCCATCATCTGCAAAAAC) and reverse primer IDH2r
(CTAGGCGAGGAGCTCCAGT) for IDH2 (18). The thermocycling conditions
consisted of 5 min at 95°C, 35 cycles for 30 sec at 95°C, 40 sec at
56°C, and 50 sec at 72°C, followed by 10 min at 72°C. For
confirmation, the forward primer IDH1fc (ACCAAATGGCACCATACGA) and
reverse primer IDH1rc (TTCATACCTTGCTTAATGGGTGT) generating a 254-bp
fragment and the forward primer IDH2fc (GCTGCAGTGGGACCACTATT) and
reverse primer IDH2rc (TGTGGCCTTGTACTGCAGAG) generating a 293-bp
fragment were used for amplification with the same thermocycling
conditions (24). After the
purification of the PCR products using the QIAquick PCR
Purification kit (Qiagen), DNA sequencing for the IDH1/2
gene was performed with an ABI PRISM 310 Genetic Analyzer (Applied
Biosystems), using the same primers as for PCR.
1p and 19q status by fluorescence in situ
hybridization
For fluorescence in situ hybridization
(FISH), the tumor sections were deparaffinized in Hemo-De (Falma,
Tokyo, Japan), dehydrated with 100% ethanol, and digested using a
Paraffin Pretreatment kit (Vysis-Abbott, Tokyo, Japan) according to
the manufacturer’s protocol. Each section was hybridized with LSI
1p36/1q25 and LSI 19q13/19p13 probes (Vysis-Abbott). The probes and
target DNA were denatured individually at 72°C for 5 min, followed
by 2 overnight incubations at 37°C. Posthybridization washes were
carried out in standard saline solution twice, and the sections
were air-dried. The nuclei were counterstained with
4,6-diamidino-2-phenylindole. The sections were analyzed using a
fluorescence microscope (Biorevo BZ-9000, Keyence, Japan).
The 1p or 19q deletions were considered present when
the population of the cells with single 1p36 or single 19q13 was
<50% of the cells with double 1p36 or double 19p13,
respectively. At least 100 non-overlapping nuclei were counted per
hybridization.
1p and 19q status by multiplex
ligation-dependent probe amplification analysis
We used the SALSA P088 kit (MRC Amsterdam, The
Netherlands) containing 16 1p probes (6 probe at 1p36), 8 19q
probes, and 21 control probes specific to other chromosomes,
including 2 probes for 19p. Information regarding the probe
sequences and ligation sites can be found at http://www.mlpa.com. Multiplex ligation-dependent
probe amplification (MLPA) analysis was performed as described
previously (25,26). The 1p36 or 19q deletions were
considered present when 5 of 6 markers for 1p36 and 5 of 8 markers
for 19q in each chromosome arm had normalized ratios <0.75.
Statistical analysis
All the statistical analyses, including the
Kaplan-Meier survival analysis, were performed using the JMP ver. 8
software (Tokyo, Japan). The multivariate analysis with Cox
regression, which was used to assess the independent prognostic
factors for all the 72 cases, was performed only for the variables
with p<0.1 and which included the data obtained in the
univariate analysis for all the patients. A similar analysis was
performed for 58 cases with radiotherapy or chemoradiotherapy.
Results
Progression-free and OS
The PFS and median OS times for all the 72 grade II
glioma patients were 5.8 and 10.3 years, respectively (male-female,
40:32; median age, 39.0 years; Table
I). These patients were initially treated with surgery followed
by radiotherapy (22.2%) or chemoradiotherapy (58.3%). The median
follow-up time for all the 72 patients was 6.4 years, and it was
7.6 years for the patients treated with chemoradiotherapy (n=42)
and 4.0 years for those who underwent radiotherapy alone
(n=16).
Progression-free and OS times according
to clinical factors
The univariate analysis (Table II) showed that the patients with
oligodendroglial tumors (n=23) had longer OS than those with
diffuse astrocytoma (n=49; p=0.04). The PFS and OS were 3.6 and 8.3
years, respectively, in the patients with diffuse astrocytoma, and
8.3 and 11.7 years, respectively, in the patients with
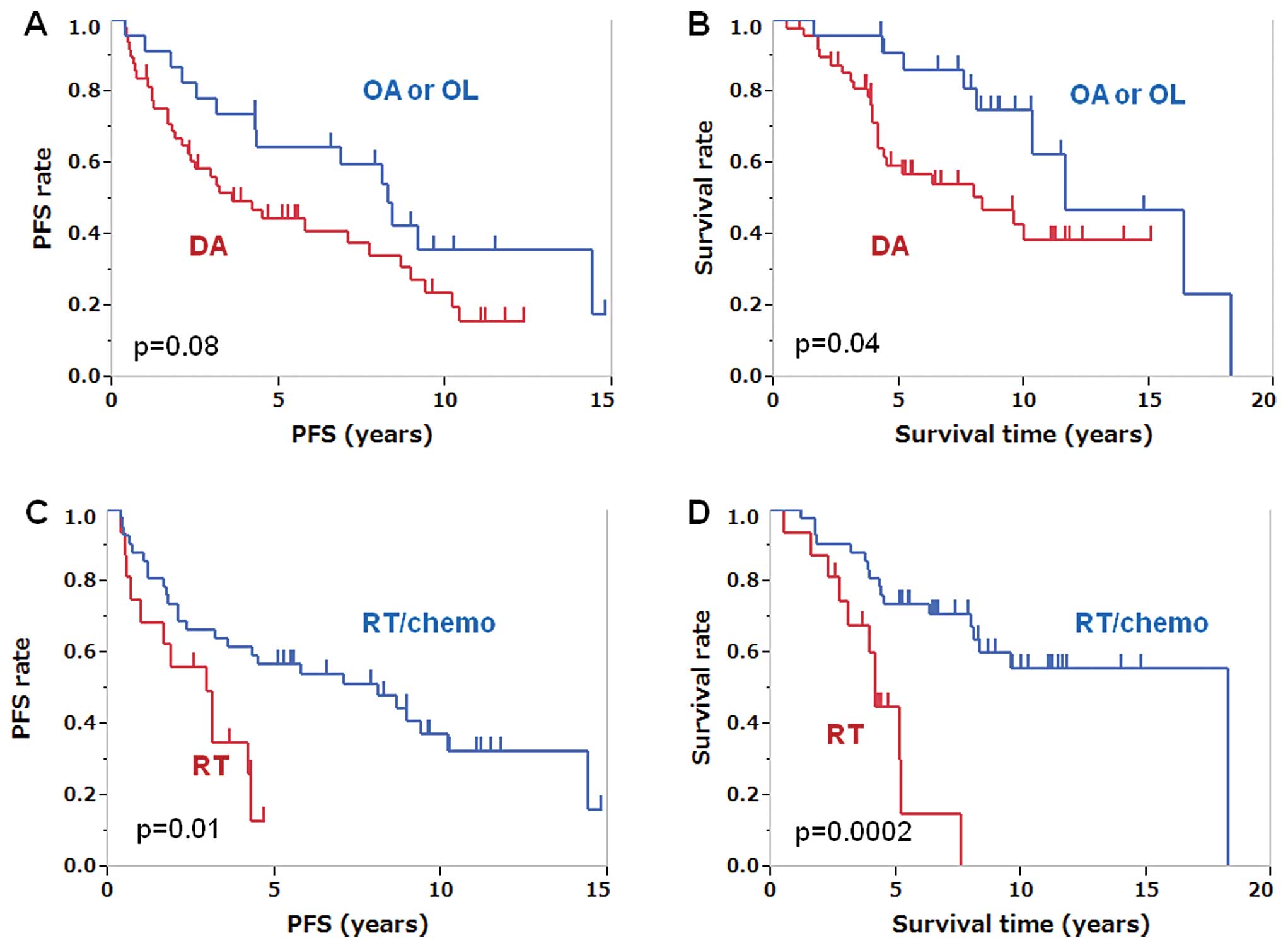
oligodendroglioma or oligoastrocytoma (Fig. 1A and B). The patients younger than
40 years (n=38) had longer OS than those who were 40 years or older
(n=34; p=0.02). The PFS and median survival time of the patients in
the younger age groups were 7.0 years and still not reached,
respectively, whereas the PFS and OS of the patients in the older
age groups were 3.1 and 4.3 years, respectively. The patients with
an initial KPS score ≥80 (n=68) had significantly longer OS
(p=0.0006) and PFS (p=0.01) than those with a KPS score <80
(n=4). The PFS and OS of the patients with a KPS score ≥80 were 6.8
and 11.7 years, respectively, and those of the patients with a KPS
score <80 were 0.6 and 1.7 years, respectively. The patients in
the total or subtotal resection (≥90% removal) groups (n=14; median
age, 34.0 years) tended to have longer OS than those in the partial
(<90%) removal or biopsy groups (n=58; median age, 41.0;
p=0.08). The PFS and OS were 10.4 and 18.3 years, respectively, in
the patients in the total or subtotal resection groups and 4.3 and
10.0 years, respectively, in the patients in the partial resection
or biopsy groups. The patients who were initially treated with
chemoradiotherapy after surgery showed significantly longer PFS (p=
0.01) and OS (p= 0.0002) than those treated with radiotherapy alone
(Fig. 1C and D). The PFS and OS of
the patients who were initially treated with radiotherapy after
surgery (n=16) were 2.9 and 4.2 years, respectively, and the PFS
and OS of the patients who were initially treated with
chemoradiotherapy after surgery (n=42) were 8.1 and 18.2 years,
respectively. According to MIB-1 staining index, there was no
significant difference of survival between groups with cut-off
point at 4, 8 and 15% in our study.
| Table IIUnivariate analyses of
progression-free survival time and overall survival time of
patients with grade II gliomas. |
Table II
Univariate analyses of
progression-free survival time and overall survival time of
patients with grade II gliomas.
| Variable | No.of cases | PFS (95% CI) | p-value
(log-rank) | OS (95% CI) | p-value
(log-rank) |
|---|
| Histology | | | | | |
| Astrocytoma | 49 | 3.6 (2.1–7.7) | 0.08 | 8.3 (4.2-NR) | 0.04 |
|
Oligodendroglioma/oligoastrocytoma | 23 | 8.3 (4.3–14.4) | | 11.7
(8.1–18.2) | |
| Age | | | | | |
| <40 years | 38 | 7.0 (3.6–9.3) | 0.2 | NR (8.0-NR) | 0.02 |
| ≥40 years | 34 | 3.1 (1.8–8.9) | | 4.3 (3.9–16.3) | |
| IDH
mutation | | | | | |
| Mutation | 42 | 8.4 (3.2–10.2) | 0.04 | 16.3
(9.6–18.2) | 0.004 |
| Wild-type | 30 | 3.3 (1.7–7.0) | | 4.5 (3.9–10.0) | |
| Extent of
removal | | | | | |
| Total and
subtotal removal | 14 | 10.4
(2.5–14.4) | 0.1 | 18.3
(4.1–18.3) | 0.08 |
| Partial removal
and biopsy | 58 | 4.3 (2.3–8.3) | | 10.0
(5.2–16.3) | |
| Largest diameter of
initial tumor (cm) | | | | | |
| <6 | 40 | 7.7 (2.3–10.4) | 0.2 | 10.0 (8.0–NR) | 0.7 |
| ≥6 | 32 | 4.3 (2.1–8.3) | | 10.3
(5.1–16.3) | |
| Initial KPS | | | | | |
| <80 | 4 | 0.6 (0.4–8.4) | 0.01 | 1.7 (0.5–10.3) | 0.0006 |
| ≥80 | 68 | 6.8 (3.1–8.9) | | 11.7
(8.0–18.2) | |
| MIB-1 index | | | | | |
| <4% | 33 | 8.1 (2.3–8.9) | 0.6 | 9.6 (5.1-NR) | 0.6 |
| ≥4% | 21 | 4.3 (1.8-NR) | | NR (3.9-NR) | |
| 1p/19q | | | | | |
| 1p/19q codeletion
(+) | 15 | 6.8 (2.2-NR) | 0.4 | 11.7
(4.3–11.7) | 0.2 |
| 1p/19q codeletion
(−) | 45 | 3.6 (2.3–8.4) | | 8.3 (4.4-NR) | |
| 1p | | | | | |
| 1p deletion | 24 | 5.8 (2.5–9.3) | 0.96 | 11.7
(4.2–11.7) | 0.9 |
| Intact | 36 | 4.2 (2.1–10.2) | | 9.6 (4.4-NR) | |
| 19q | | | | | |
| 19q deletion | 23 | 7.0 (4.2–9.3) | 0.5 | 11.7
(4.5–11.7) | 0.5 |
| Intact | 37 | 3.1 (1.9–10.2) | | 8.3 (3.9-NR) | |
| Initial
radiotherapy | | | | | |
| + | 58 | 4.3 (2.9–8.9) | 0.98 | 8.3 (5.1–18.2) | 0.2 |
| − | 14 | 7.7 (2.5–9.1) | | 11.7
(4.2–16.3) | |
| Initial
treatment | | | | | |
| Radiotherapy
alone | 16 | 2.9 (0.7–4.3) | 0.01 | 4.2 (2.7–5.1) | 0.0002 |
|
Chemoradiotherapy | 42 | 8.1 (3.2–10.2) | | 18.2
(8.1–18.2) | |
Presence of 1p/19q codeletion, 1p
deletion, and 19q deletion and survival
The presence of 1p/19q deletions was determined for
25 or 26 primary resections and for 7 or 2 secondary resection
samples by MLPA or FISH, respectively. The 1p/19q codeletion was
observed in 15.9% (7/44) of the astrocytomas and 50% (8/16) of the
oligodendroglial tumors. The OS of the patients with 1p/19q
codeletion was 11.7 years, and the OS of those without 1p/19q
codeletion was 8.3 years (p=0.2; Fig.
1E and F). In the patients with astrocytic tumors, the median
survival time of those with 1p/19q codeletion was not reached and
the OS of those without 1p/19q codeletion was 6.3 years (p=0.5).
The OS of the patients with 1p/19q codeletion was 11.7 years, and
the OS of those without 1p/19q codeletion was 10.3 years in the
oligodendroglial tumors (p=0.5). The presence of 1p/19q codeletion,
1p deletion, or 19q deletion was not correlated with the PFS or OS
time (Table II).
IDH1/2 mutations and survival in the
whole series
IDH1/2 mutations were determined in 55
samples at the primary resection and 17 at the secondary resection
by IHC alone for 32 cases (44.4%) and by direct sequencing in 40
cases (55.6%). IDH1/2 mutations were found in 46.9% (23/49) of the
astrocytomas, 84.2% (16/19) of the oligoastrocytomas, and 75.0%
(3/4) of the oligodendrogliomas (Table
III).
| Table IIIMutation of IDH1/2. |
Table III
Mutation of IDH1/2.
| Diffuse astrocytoma
(%) | Oligodendroglioma
(%) | Oligoastrocytoma
(%) |
|---|
| IDH1/2
mutation by sequence | | | |
| IDH1 R132H | 13 (26.5) | 2 (50.0) | 5 (26.3) |
| IDH1 R132S | 1 (2.0) | 0 (0.0) | 0 (0.0) |
| IDH2 R172K | 1 (2.0) | 0 (0.0) | 0 (0.0) |
| Wild-type | 15 (30.6) | 1 (25.0) | 2 (10.5) |
| IDH mutation by
IHC | | | |
| IDH1 R132H | 8 (16.3) | 1 (25.0) | 11 (57.9) |
| IDH1 R132S | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Mutation (−) | 11 (22.4) | 0 (0.0) | 1 (5.3) |
| Total | 49 (100) | 4 (100) | 19 (100) |
| Mutation | 23 (46.9) | 3 (75.0) | 16 (84.2) |
| Wild-type | 26 (53.1) | 1 (25.0) | 3 (15.8) |
The patients with IDH1/2 mutations (n=42) had
longer PFS (p=0.04) and OS (p=0.004) than those without IDH1/2
mutations (n=30; Table II). The
PFS and OS of the patients with IDH1/2 mutations were 8.4
and 16.3 years, respectively, and the PFS and OS of the patients
without IDH1/2 mutations were 3.3 and 4.5 years,
respectively (Fig. 1G and H). The
diffuse astrocytoma patients with IDH1/2 mutations (n=23)
tended to have longer survival times than those without
IDH1/2 mutations (n=26), although the difference was not
significant (p=0.08). The median survival time of the diffuse
astrocytoma patients with IDH1/2 mutations was not reached
and that of the diffuse astrocytoma patients without IDH1/2
mutations was 4.4 years. The oligodendroglial tumor patients with
IDH1/2 mutations also tended to have longer, though not
significant, survival times (p=0.1).
The survival of the patients with IDH1/2
mutations and 1p/19q codeletion was longer than that of the
patients with neither IDH1/2 mutations nor 1p/19q codeletion
(11.7 vs. 4.4 years, respectively), although the difference did not
reach statistical significance (p=0.1). Furthermore, a combined
IDH1/2 and 1p/19q status did not correlate with the PFS and
OS of the patients who were initially treated with
chemoradiotherapy after surgery regardless of the histological
tumor type.
In the total or subtotal resection group, the
patients with IDH1/2 mutations had longer OS than those
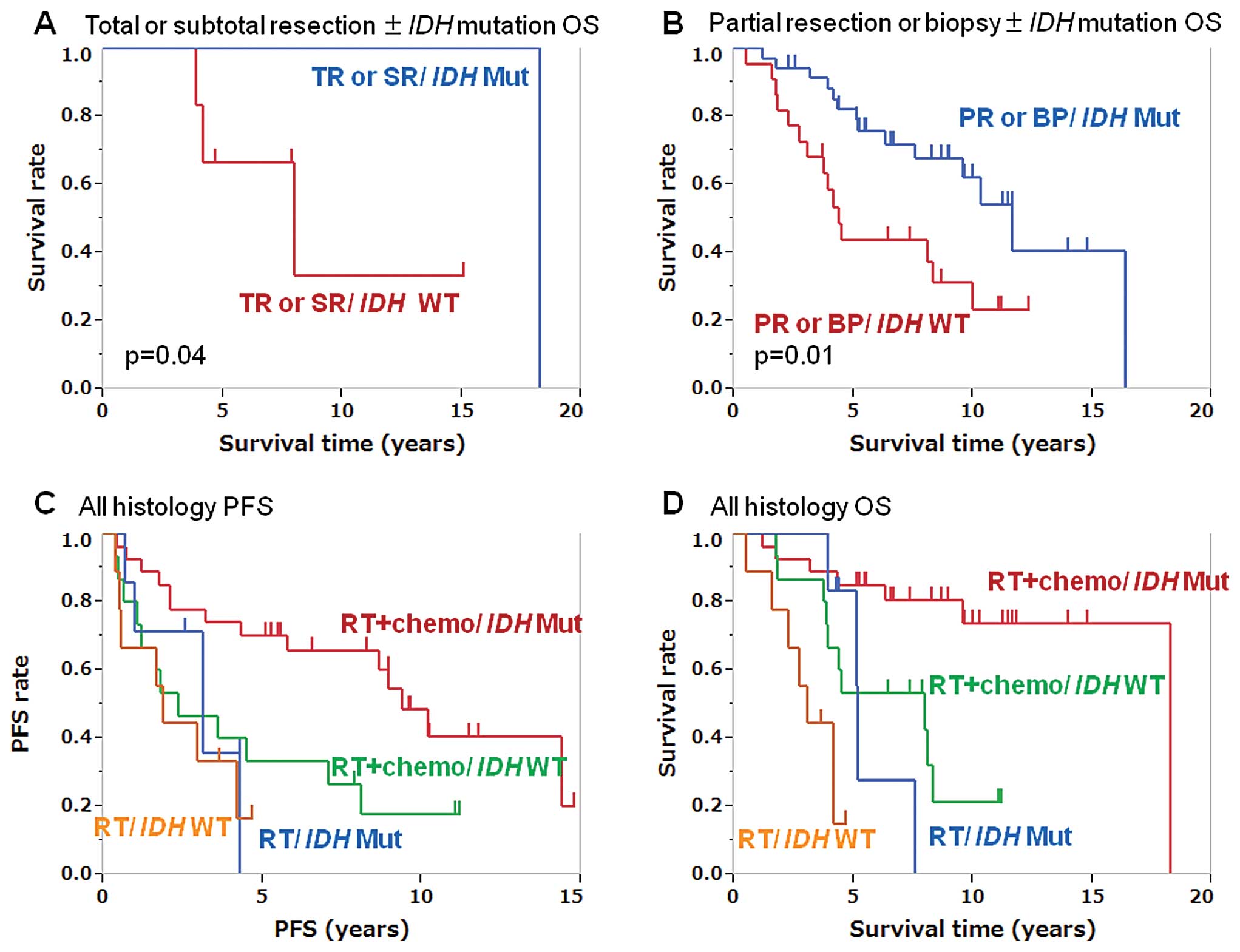
without IDH1/2 mutations (p=0.04; Fig. 2A). The OS of the patients with
IDH1/2 mutations (n=6, 2 diffuse astrocytomas, 3
oligoastrocytomas, and 1 oligodendrogliomas) was 18.2 years; to
date, 5 are still alive and 1 is dead. The OS of the patients
without IDH1/2 mutations (n=8, 7 astrocytomas and 1
oligoastrocytoma) was 8.0 years. In the partial resection or biopsy
group, the patients with IDH1/2 mutations had longer OS than
those without IDH1/2 mutations in the partial resection or
biopsy group (p=0.01; Fig. 2B).
The OS of the patients with IDH1/2 mutations (n=36, 21
diffuse astrocytomas, 13 oligoastrocytomas, and 2
oligodendrogliomas) was 11.7 years, and that of the patients
without IDH1/2 mutations in these groups (n=22, 19 diffuse
astrocytomas, 2 oligoastrocytomas, and 1 oligodendroglioma) was 4.4
years.
IDH1/2 mutations and survival in the
patients who underwent chemoradiotherapy after surgery
Among the grade II glioma patients who were
initially treated with chemoradiotherapy after surgery, those with
IDH1/2 mutations had significantly longer PFS and OS than
those without IDH1/2 mutations (PFS: p=0.02, OS: p=0.004;
Fig. 2C and D; Table IV).
| Table IVPFS and OS in patients with
radiotherapy or chemoradiotherapy according to IDH1/2 status. |
Table IV
PFS and OS in patients with
radiotherapy or chemoradiotherapy according to IDH1/2 status.
| Variable | | No. of cases | PFS (95% CI) | OS (95% CI) |
|---|
| All grade II
gliomas |
| RT+chemo | Mut (+) | 27 | 9.3
(4.3-NA)a,b | 18.2
(9.6–18.2)c,d |
| RT+chemo | Mut (−) | 15 | 2.3
(0.6–7.0)a-,b | 8.0
(3.8–8.3)c,d |
| RT only | Mut (+) | 7 | 3.1
(0.7–4.3)b | 5.1
(3.9–7.5)b,d,e |
| RT only | Mut (−) | 9 | 1.9 (0.4–4.2) | 3.1
(0.5–4.2)b,e |
| Diffuse
astrocytoma |
| RT+chemo | Mut (+) | 16 | 8.6
(2.1–10.2)f | NR
(6.3-NR)g,h |
| RT+chemo | Mut (−) | 13 | 1.8
(0.6–4.5)f | 4.5
(3.8-NR)g,h |
| RT only | Mut (+) | 3 | 3.1
(0.7–3.1)f | 4.5
(3.9–5.1)h |
| RT only | Mut (−) | 8 | 2.4 (0.5-NR) | 3.6 (0.5-NR) |
|
Oligodendroglioma/oligoastrocytoma |
| RT+chemo | Mut (+) | 11 | 14.4
(2.1-NR)e | 18.2 (NR)a |
| RT+chemo | Mut (−) | 2 | 8.1 (NR)e | 8.1 (NR)a |
| RT only | Mut (+) | 4 | 3.7
(1.0–4.3)e,i | 6.3
(5.1–7.5)a,i |
| RT only | Mut (−) | 1 | 0.4 (NR)i | 1.6 (NR)i |
An important finding is that the patients who were
initially treated with chemoradiotherapy after surgery and had
IDH1/2 mutations showed significantly longer PFS and OS than
those treated with radiotherapy alone with IDH1/2 mutations.
The PFS and OS of the patients with IDH1/2 mutations who
were initially treated with chemoradiotherapy after surgery (n=27)
were 9.3 and 18.2 years, respectively, and the PFS and OS of those
treated with radiotherapy alone with IDH1/2 mutations (n=7)
were 3.1 and 5.1 years, respectively (PFS, p=0.01; OS, p=0.008). In
the oligodendroglial tumors, the PFS and OS of the patients with
IDH1/2 mutations who were initially treated with
chemoradiotherapy (n=11) were 14.4 and 18.2 years, respectively,
and the PFS and OS of those treated with radiotherapy alone with
IDH1/2 mutations (n=4) were 3.7 and 6.3 years, respectively
(PFS: p=0.03, OS: p=0.02; Fig. 2G and
H). Similar tendencies, although not reaching statistical
significance, were observed in the astrocytic tumors (PFS: p=0.1,
OS: p=0.07; Fig. 2E and F).
The IDH1/2 status had no impact on the PFS of
all the grade II glioma or diffuse astrocytoma patients who
underwent radiotherapy alone. No significant difference in PFS was
observed between the radiotherapy and chemoradiotherapy groups in
the grade II glioma patients without IDH1/2 mutations.
Chemoradiotherapy did not prolong the PFS of the patients without
IDH1/2 mutations in the astrocytic and oligodendroglial
tumors.
Multivariate analysis
Oligodendroglial tumors (hazard ratio (HR)=0.29,
p=0.02), age <40 years (HR=0.40, p=0.02), initial KPS ≥80
(HR=0.045, p=0.0002), and IDH1/2 mutations (HR=0.37, p=0.01)
were favorable prognostic factors for OS time, as determined by the
multivariate analysis, of the 72 patients included in the study
(Table V). The IDH1/2
mutation status was not a prognostic factor for PFS when all the
patients were considered, including those who did not undergo
initial radiotherapy or chemotherapy (p=0.08). In contrast, total
or subtotal tumor resection (HR=0.36, p=0.03), chemoradiotherapy
(HR=0.41, p=0.04), and IDH1/2 mutations (HR=0.47, p=0.05)
were favorable prognostic factors for PFS, as determined by the
multivariate analysis, of the patients who were initially treated
with radiotherapy or chemoradiotherapy (Table VI). Histological appearance was not
a prognostic marker for PFS in this series (p=0.2) compared with
IDH1/2 mutations (p=0.05).
| Table VMultivariate analyses of PFS and OS
of patients with all grade II gliomas. |
Table V
Multivariate analyses of PFS and OS
of patients with all grade II gliomas.
| Variable | No. of cases | PFS hazard ratio
(95% CI) | PFS p-value
(Cox) | OS hazard ratio
(95% CI) | OS p-value
(Cox) |
|---|
| Histology | | | | | |
| Diffuse
astrocytoma | 49 | 1 | 0.1 | 1 | 0.02 |
|
Oligodendroglioma/oligoastrocytoma | 23 | 0.576
(0.262–1.186) | | 0.290
(0.086–0.815) | |
| IDH
mutation | | | | | |
| Wild-type | 31 | 1 | 0.08 | 1 | 0.01 |
| Mutation | 41 | 0.558
(0.289–1.068) | | 0.365
(0.155–0.819) | |
| Age (years) | | | | | |
| ≥40 | 34 | 1 | 0.5 | 1 | 0.02 |
| <40 | 38 | 0.802
(0.440–1.460) | | 0.400
(0.175–0.877) | |
| Extent of
removal | | | | | |
| Partial removal
and biopsy | 58 | 1 | 0.1 | 1 | 0.2 |
| Total and
subtotal removal | 14 | 0.556
(0.222–1.217) | | 0.463
(0.107–1.403) | |
| Initial KPS | | | | | |
| <80 | 4 | 1 | 0.01 | 1 | 0.0002 |
| ≥80 | 68 | 0.179
(0.063–0.640) | | 0.045
(0.011–0.198) | |
| Table VIMultivariate analyses of PFS and OS
of patients with all grade II gliomas with radiotherapy ±
chemotherapy. |
Table VI
Multivariate analyses of PFS and OS
of patients with all grade II gliomas with radiotherapy ±
chemotherapy.
| Variable | No. of cases | PFS hazard ratio
(95% CI) | PFS p-value
(Cox) | OS hazard ratio
(95% CI) | OS p-value
(Cox) |
|---|
| Histology | | | | | |
| Diffuse
astrocytoma | 40 | 1 | 0.2 | 1 | 0.2 |
|
Oligodendroglioma/Oligoastrocytoma | 18 | 0.549
(0.209–1.290) | | 0.490
(0.133–1.445) | |
| IDH
mutation | | | | | |
| Wild-type | 24 | 1 | 0.05 | 1 | 0.01 |
| Mutation | 34 | 0.467
(0.215–0.999) | | 0.316
(0.117–0.793) | |
| Age (years) | | | | | |
| ≥40 | 28 | 1 | 0.5 | 1 | 0.5 |
| <40 | 30 | 0.758
(0.362–1.559) | | 0.745
(0.300–1.808) | |
| Extent of
removal | | | | | |
| Partial removal
and biopsy | 47 | 1 | 0.03 | 1 | 0.08 |
| Total and
subtotal removal | 11 | 0.364
(0.118–0.918) | | 0.356
(0.080–1.120) | |
| Initial
treatment | | | | | |
| Radiotherapy
alone | 16 | 1 | 0.04 | 1 | 0.002 |
|
Chemoradiotherapy | 42 | 0.408
(0.182–0.948) | | 0.198
(0.073–0.529) | |
Discussion
WHO grade III and IV astrocytomas with IDH1/2
mutations have more favorable prognoses than those with wild-type
IDH1/2 (17). IDH1/2
mutations, 1p/19q codeletion, and MGMT promoter methylation
are pivotal prognostic factors in anaplastic oligodendroglial
tumors treated with radiotherapy or chemoradiotherapy (EORTC 26951)
(27). However, the impact of
IDH1/2 mutations and/or 1p/19q codeletion as biomarkers in
grade II gliomas remains controversial. The present study was
therefore aimed at identifying prognostic and/or predictive factors
in grade II gliomas.
The presence of IDH1/2 mutations is a
favorable prognostic marker for OS
The results of the univariate analysis revealed that
the presence of IDH1/2 mutations was a prognostic factor of
longer OS (p=0.004) and PFS (p=0.04) in the entire patient cohort
and among the patients who underwent with or without radiation
therapy after initial surgery with or without chemotherapy. The
multivariate analysis revealed that the presence of IDH1/2
mutations was associated with prolonged PFS (p=0.05) and OS
(p=0.01) in the patients who initially underwent radiotherapy with
or without chemotherapy. Our results suggest that IDH1/2
mutations may be involved in the response to genotoxic therapy,
such as radiotherapy or chemotherapy, and may act as a prognostic
factor for chemotherapy or radiotherapy in grade II gliomas. There
are currently increasing numbers of reports showing that
IDH1/2 mutations are prognostic markers for several
malignancies, including grade II gliomas. Houillier et al
(19) reported that the presence
of IDH1/2 mutations is a significant prognostic marker for
OS and chemosensitivity in low-grade glioma patients who were
initially treated with temozolomide (TMZ) before any other
treatment except surgery. Hartmann et al (16) reported that the IDH1
mutation was a prognostic factor for PFS and OS in grade II glioma
patients who underwent radiotherapy or chemotherapy after surgery.
In our study, the presence of IDH1/2 mutations was
demonstrated by multivariate analysis to be a favorable prognostic
factor (p=0.01) for OS but not a prognostic marker for PFS (p=0.08)
in whole cohort, which included 14 patients who did not receive
initial radiotherapy. Our finding that IDH1/2 status did not
affect PFS was in line with the findings reported by Hartmann et
al (16) or Houillier et
al (16,19), who showed that IDH1
mutations did not affect the PFS in grade II glioma patients who
did not receive radiotherapy or chemotherapy alone after surgery.
Kim et al (21) and Mukasa
et al (22) reported that
the presence of IDH1/2 mutations was not a prognostic factor
for the survival of patients with low-grade glioma in univariate or
multivariate analyses. The treatment of those patients was not
fully described in their reports.
The presence of IDH1/2 mutations is a
predictive marker for PFS in the grade II glioma patients treated
with chemoradiotherapy
The patients who were initially treated with
chemoradiotherapy after surgery showed significantly longer OS
(p=0.0002) and PFS (p=0.01) than those treated with radiotherapy
alone in our study. Chemoradiotherapy significantly prolonged PFS
and OS compared with radiotherapy alone in all the grade II gliomas
with IDH1/2 mutations (p=0.01 and 0.0008, respectively),
diffuse astrocytoma (p=0.1 and 0.07, respectively), and
oligodendroglial tumors (p=0.03 and 0.02, respectively) in the
univariate analysis. Chemoradiotherapy was shown by multivariate
analysis (p=0.04) to significantly prolong the PFS of grade II
glioma patients carrying IDH1/2 mutations who underwent
radiotherapy with or without concomitant chemotherapy (p=0.04). In
contrast, there were no differences in PFS between the radiotherapy
and chemoradiotherapy groups among the grade II glioma patients
without IDH1/2 mutations in the univariate analysis. PFS did
not differ by IDH1/2 status in the grade II glioma patients
who underwent radiotherapy alone. However, the present study was
limited by the small number of samples and the differences in the
follow-up periods between the radiation and chemoradiotherapy
groups (4 and 7.6 years, respectively). A prospective study
including a larger patient cohort is required to obtain conclusive
evidence that the presence of IDH1/2 mutations is a
predictive marker for chemoradiotherapy in grade II gliomas.
Nonetheless, our results suggest that IDH1/2 mutation is a
predictive marker for chemoradiotherapy in grade II glioma patients
and indicate that these patients may benefit from concurrent
chemotherapy and radiotherapy compared with patients who do not
carry IDH1/2 mutations.
Mutations in IDH1/2 result in the acquisition
of new enzymatic activity that enables the NADPH-dependent
reduction of α-ketoglutarate to 2-hydroxyglutarate, and the
mutation confers oncogenic properties (28). IDH1 mutations are early
events in the development of astrocytomas and oligodendrogliomas
(11). Another possible function
of IDH1/2 mutations is the dominant-negative inhibition of
the oxidative decarboxylation of isocitrate as a result of the
formation of a wild-type/mutant heterodimer (29). Cellular IDH1 levels are associated
with the protection from apoptosis and cell death after exposure to
reactive oxygen species or ultraviolet B-induced phototoxicity and
IDH1/2 functions in cellular defense reactions (30). Glioma cells with IDH1/2
mutations may be vulnerable to irradiation and chemotherapeutic
agents, which might explain why IDH1/2 mutations could be a
predictive and prognostic marker for grade II gliomas in patients
receiving chemoradiotherapy. Our findings warrant a prospective
large-scale clinical study addressing the efficacy of
chemoradiotherapy in grade II glioma patients in association with
IDH1/2 status.
Grade II glioma patients with wild-type
IDH1/2 have poor prognoses even after total resection
The extent of resection of tumors has been reported
to be significantly associated with survival and recurrence of
disease in low-grade glioma patients (9,31).
In our study, the patients in the total or subtotal resection (≥90%
removal) group tended to have longer survival times than the
patients in the partial (<90% removal) or biopsy group (p=0.08).
The patients without IDH1/2 mutations had shorter OS than
those with IDH1/2 mutations in the total and subtotal
resection groups (p=0.04) and in the partial and biopsy groups
(p=0.01). Although the number of patients examined was small, we
believe that this is a very important finding and that it indicates
that patients without IDH1/2 mutations may require more
intensive treatment, such as chemoradiotherapy, even after total
resection of the tumor.
1p/19q codeletion is not a prognostic
factor
In our study, the OS and PFS in the diffuse
astrocytomas with 1p/19q codeletion tended to be longer than those
in the patients without 1p/19q codeletion, but the difference did
not reach statistical significance. Furthermore, no significant
differences were observed between the grade II glioma patients with
regard to 1p/19q status. Prior studies reported that the presence
of the 1p/19q codeletion was significantly associated with longer
OS in low-grade gliomas (12,13,15,21,32).
On the other hand, Houillier et al and Mukasa et al
(19,22) reported that loss of 1p/19q was not
a sensitive prognostic biomarker. Ichimura et al and
Vogazianou et al reported that total 1p/19q loss is rare and
that when present, it is associated with longer survival than other
1p/19q changes in adult gliomas independent of pathological
diagnosis (14,15). Deletion of 1p or 19q was determined
mainly by FISH analysis in our study, and this technique cannot
discriminate between total and partial 1p/19q deletion, which might
explain the discrepancy in the results.
Clinicopathological factors in grade II
gliomas
The multivariate analysis showed that age ≥40 years
(p=0.02), astrocytic tumors (p=0.02), initial KPS <80
(p=0.0002), and wild-type IDH1/2 (p=0.01) were unfavorable
prognostic factors in our series. These results are generally in
line with previous reports showing that older age, astrocytic
histology, presence of neurologic deficits before surgery, largest
tumor diameter, and tumors crossing the midline were important
unfavorable prognostic factors for survival in adult patients with
low-grade gliomas (5–9).
In conclusion, the multivariate analysis showed that
age <40 years, oligodendroglial tumors, initial KPS ≥80, and
IDH1/2 mutations were favorable prognostic factors for
survival of the grade II glioma patients. The presence of
IDH1/2 mutations was a prognostic factor for grade II glioma
patients with radiotherapy. Furthermore, it is a predictive factor
of response to chemoradiotherapy in grade II gliomas. Patients
carrying IDH1/2 mutations may benefit more from concurrent
chemotherapy and radiotherapy compared with those without
IDH1/2 mutations.
Acknowledgements
We thank all the doctors, nurses and
medical staff in National Cancer Center Hospital who attended to
the glioma patients in these 20 years. This study was supported by
National Cancer Center Research and Development Fund no.
23-A-49.
References
|
1
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bauman G, Fisher B, Watling C, Cairncross
JG and Macdonald D: Adult supratentorial low-grade glioma:
long-term experience at a single institution. Int J Radiat Oncol
Biol Phys. 75:1401–1407. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
van den Bent MJ, Afra D, de Witte O, Ben
Hassel M, Schraub S, Hoang-Xuan K, Malmstrom PO, Collette L,
Pierart M, Mirimanoff R and Karim AB: Long-term efficacy of early
versus delayed radiotherapy for low-grade astrocytoma and
oligodendroglioma in adults: the EORTC 22845 randomised trial.
Lancet. 366:985–990. 2005.PubMed/NCBI
|
|
4
|
Shaw EG, Wang M, Coons S, Brachman D,
Buckner JC, Stelzer K, Barger G, Brown PD, Gilbert MR and Mehta MP:
Final report of Radiation Therapy Oncology Group (RTOG) protocol
9802: radiation therapy (RT) versus RT + procarbazine, CCNU, and
vincristine (PCV) chemotherapy for adult low-grade glioma (LGG). J
Clin Oncol (ASCO Meeting abst). 26:20062008.
|
|
5
|
Bauman G, Lote K, Larson D, Stalpers L,
Leighton C, Fisher B, Wara W, MacDonald D, Stitt L and Cairncross
JG: Pretreatment factors predict overall survival for patients with
low-grade glioma: a recursive partitioning analysis. Int J Radiat
Oncol Biol Phys. 45:923–929. 1999. View Article : Google Scholar
|
|
6
|
Chang EF, Smith JS, Chang SM, Lamborn KR,
Prados MD, Butowski N, Barbaro NM, Parsa AT, Berger MS and
McDermott MM: Preoperative prognostic classification system for
hemispheric low-grade gliomas in adults. J Neurosurg. 109:817–824.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pignatti F, van den Bent M, Curran D,
Debruyne C, Sylvester R, Therasse P, Afra D, Cornu P, Bolla M,
Vecht C and Karim AB: Prognostic factors for survival in adult
patients with cerebral low-grade glioma. J Clin Oncol.
20:2076–2084. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schiff D, Brown PD and Giannini C: Outcome
in adult low-grade glioma: the impact of prognostic factors and
treatment. Neurology. 69:1366–1373. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Smith JS, Chang EF, Lamborn KR, Chang SM,
Prados MD, Cha S, Tihan T, Vandenberg S, McDermott MW and Berger
MS: Role of extent of resection in the long-term outcome of
low-grade hemispheric gliomas. J Clin Oncol. 26:1338–1345. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Smith JS, Alderete B, Minn Y, Borell TJ,
Perry A, Mohapatra G, Hosek SM, Kimmel D, O’Fallon J, Yates A, et
al: Localization of common deletion regions on 1p and 19q in human
gliomas and their association with histological subtype. Oncogene.
18:4144–4152. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Watanabe T, Nobusawa S, Kleihues P and
Ohgaki H: IDH1 mutations are early events in the development of
astrocytomas and oligodendrogliomas. Am J Pathol. 174:1149–1153.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Smith JS, Perry A, Borell TJ, Lee HK,
O’Fallon J, Hosek SM, Kimmel D, Yates A, Burger PC, Scheithauer BW
and Jenkins RB: Alterations of chromosome arms 1p and 19q as
predictors of survival in oligodendrogliomas, astrocytomas, and
mixed oligoastrocytomas. J Clin Oncol. 18:636–645. 2000.PubMed/NCBI
|
|
13
|
Mariani L, Deiana G, Vassella E, Fathi AR,
Murtin C, Arnold M, Vajtai I, Weis J, Siegenthaler P, Schobesberger
M and Reinert MM: Loss of heterozygosity 1p36 and 19q13 is a
prognostic factor for overall survival in patients with diffuse WHO
grade 2 gliomas treated without chemotherapy. J Clin Oncol.
24:4758–4763. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ichimura K, Vogazianou AP, Liu L, Pearson
DM, Backlund LM, Plant K, Baird K, Langford CF, Gregory SG and
Collins VP: 1p36 is a preferential target of chromosome 1 deletions
in astrocytic tumours and homozygously deleted in a subset of
glioblastomas. Oncogene. 27:2097–2108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vogazianou AP, Chan R, Backlund LM,
Pearson DM, Liu L, Langford CF, Gregory SG, Collins VP and Ichimura
K: Distinct patterns of 1p and 19q alterations identify subtypes of
human gliomas that have different prognoses. Neurooncology.
12:664–678. 2010.PubMed/NCBI
|
|
16
|
Hartmann C, Hentschel B, Tatagiba M,
Schramm J, Schnell O, Seidel C, Stein R, Reifenberger G, Pietsch T,
von Deimling A, Loeffler M and Weller M: Molecular markers in
low-grade gliomas: predictive or prognostic? Clin Cancer Res.
17:4588–4599. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yan H, Parsons DW, Jin G, McLendon R,
Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ,
et al: IDH1 and IDH2 mutations in gliomas. N Engl J Med.
360:765–773. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Balss J, Meyer J, Mueller W, Korshunov A,
Hartmann C and von Deimling A: Analysis of the IDH1 codon 132
mutation in brain tumors. Acta Neuropathol. 116:597–602. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Houillier C, Wang X, Kaloshi G, Mokhtari
K, Guillevin R, Laffaire J, Paris S, Boisselier B, Idbaih A,
Laigle-Donadey F, et al: IDH1 or IDH2 mutations predict longer
survival and response to temozolomide in low-grade gliomas.
Neurology. 75:1560–1566. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Metellus P, Coulibaly B, Colin C, de Paula
AM, Vasiljevic A, Taieb D, Barlier A, Boisselier B, Mokhtari K,
Wang XW, et al: Absence of IDH mutation identifies a novel
radiologic and molecular subtype of WHO grade II gliomas with
dismal prognosis. Acta Neuropathol. 120:719–729. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim YH, Nobusawa S, Mittelbronn M, Paulus
W, Brokinkel B, Keyvani K, Sure U, Wrede K, Nakazato Y, Tanaka Y,
et al: Molecular classification of low-grade diffuse gliomas. Am J
Pathol. 177:2708–2714. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mukasa A, Takayanagi S, Saito K, Shibahara
J, Tabei Y, Furuya K, Ide T, Narita Y, Nishikawa R, Ueki K and
Saito N: Significance of IDH mutations varies with tumor histology,
grade, and genetics in Japanese glioma patients. Cancer Sci.
103:587–592. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Camelo-Piragua S, Jansen M, Ganguly A, Kim
JC, Louis DN and Nutt CL: Mutant IDH1-specific immunohistochemistry
distinguishes diffuse astrocytoma from astrocytosis. Acta
Neuropathol. 119:509–511. 2010. View Article : Google Scholar
|
|
24
|
Hartmann C, Meyer J, Balss J, Capper D,
Mueller W, Christians A, Felsberg J, Wolter M, Mawrin C, Wick W, et
al: Type and frequency of IDH1 and IDH2 mutations are related to
astrocytic and oligodendroglial differentiation and age: a study of
1,010 diffuse gliomas. Acta Neuropathol. 118:469–474. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Franco-Hernandez C, Martinez-Glez V, de
Campos JM, Isla A, Vaquero J, Gutierrez M, Casartelli C and Rey JA:
Allelic status of 1p and 19q in oligodendrogliomas and
glioblastomas: multiplex ligation-dependent probe amplification
versus loss of heterozygosity. Cancer Genet Cytogenet. 190:93–96.
2009. View Article : Google Scholar
|
|
26
|
Schouten JP, McElgunn CJ, Waaijer R,
Zwijnenburg D, Diepvens F and Pals G: Relative quantification of 40
nucleic acid sequences by multiplex ligation-dependent probe
amplification. Nucleic Acids Res. 30:e572002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
van den Bent MJ, Dubbink HJ, Marie Y,
Brandes AA, Taphoorn MJ, Wesseling P, Frenay M, Tijssen CC, Lacombe
D, Idbaih A, et al: IDH1 and IDH2 mutations are prognostic but not
predictive for outcome in anaplastic oligodendroglial tumors: a
report of the European Organization for Research and Treatment of
Cancer Brain Tumor Group. Clin Cancer Res. 16:1597–1604.
2010.PubMed/NCBI
|
|
28
|
Dang L, White DW, Gross S, Bennett BD,
Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et
al: Cancer-associated IDH1 mutations produce 2-hydroxyglutarate.
Nature. 462:739–744. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang
P, Yu W, Li Z, Gong L, Peng Y, et al: Glioma-derived mutations in
IDH1 dominantly inhibit IDH1 catalytic activity and induce
HIF-1alpha. Science. 324:261–265. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Reitman ZJ and Yan H: Isocitrate
dehydrogenase 1 and 2 mutations in cancer: alterations at a
crossroads of cellular metabolism. J Natl Cancer Inst. 102:932–941.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ahmadi R, Dictus C, Hartmann C, Zurn O,
Edler L, Hartmann M, Combs S, Herold-Mende C, Wirtz CR and
Unterberg A: Long-term outcome and survival of surgically treated
supratentorial low-grade glioma in adult patients. Acta Neurochir.
151:1359–1365. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Daniels TB, Brown PD, Felten SJ, Wu W,
Buckner JC, Arusell RM, Curran WJ, Abrams RA, Schiff D and Shaw EG:
Validation of EORTC prognostic factors for adults with low-grade
glioma: a report using intergroup 86-72-51. Int J Radiat Oncol Biol
Phys. 81:218–224. 2011. View Article : Google Scholar : PubMed/NCBI
|