Introduction
Colorectal cancer (CRC) is one of the most common
malignancies worldwide (1). Other
than surgery, treatment of CRC patients relies primarily on
chemotherapy, especially the patients with advanced CRC. Among the
chemotherapeutic agents for CRC, 5-fluorouracil (5-FU), which is a
classical chemotherapy agent, has been the first line regimen for
treating CRC over several decades (2,3).
However, 5-FU has many disadvantages, for example, poor selectivity
and sensitivity to tumor and high toxicity to bone marrow,
gastrointestinal tract, and skin even at the therapeutic dose
(4). Therefore, combined therapy
with 5-FU, named FOLFOX regimen (5-fluorouracil, leucovorin, and
oxaliplatin), has widely been used to enhance 5-FU efficiency.
However, the problems have been attributed to innate or acquired
resistance, resulting from DNA damage repair, decreased drug import
or enhanced tolerance to platinum adduct accumulation (5). Therefore, development of new
chemotherapeutic agents and strategies are required to improve drug
efficacy, tolerance, and disease-free survival.
Parthenolide (PT) is one of the main sesquiterpene
lactones present in Feverfew, a traditional herbal medicine, that
has been used for the treatment of migraine, fever, and arthritis
in Europe (6). It is well known to
inhibit interleukin-1 (IL-1) and tumor necrosis factor-α-mediated
nuclear factor-κB (NF-κB) activation, which is responsible for its
inflammatory activity (7–9). Recent studies have demonstrated
anti-cancer property of PT through induction of apoptotic cell
death in a number of human cancer cells (10,11).
Multiple pathways might be involved in PT-induced apoptotic cell
death, including oxidative stress, endoplasmic reticulum (ER)
stress, intracellular thiol depletion, caspase activation, and
mitochondrial dysfunction (12,13).
Especially, PT has been demonstrated to activate the caspase
cascade through regulation of Bcl-2 family, mitochondrial damage,
and release of cytochrome c in cholangiocarcinoma and gastric
cancer cells (11,14). Furthermore, it has been shown that
the proapoptotic Bcl-2 family (Bid, BAX, BAK) members are important
mediators relaying the cell death signaling elicited by PT from
caspase 8 downstream in CRC cells (15). Functional role of Bcl-2 family
member in PT-induced apoptosis has been studied well. However,
there is only one report on PT-induced apoptosis in CRC cells.
Especially, PT as a chemotherapeutic agent using a CRC animal model
has not been evaluated.
In this study, we examined whether anti-tumor
effects of PT involves induction of mitochondrial dysfunction and
apoptosis in CRC cell lines and xenograft models. We evaluated the
potential as a new chemotherapeutic drug using mouse models of
cancer.
Materials and methods
Chemicals and reagents
Parthenolide and Z-VAD-FMK were from Calbiochem (San
Diego, CA). Rhodamine-123 (Rh-123) and Annexin V-FITC were
purchased from Invitrogen (Eugene, OR). TUNEL assay kit was from
Promega (Madison, WI). Anti-Bcl-2, anti-Bid, anti-Bax,
anti-cytochrome c, anti-caspase 3 and anti-p53 antibody were from
Santa Cruz (Beverly, MA). Anti-PARP antibody was from Cell
Signaling (Beverly, MA). Anti-actin was from Sigma (St. Louis,
MO).
Cell culture and treatment
Human colorectal cancer cell lines HT-29, LS174T,
and SW480 cells (American Type Culture Collection, Rockville, MD)
were cultured in the RPMI-1640 medium supplemented with 10% FBS,
100 units penicillin and 100 units streptomycin. For the treatment
of cells with PT, cells were sub-cultured in RPMI-1640 medium
without FBS for 12 h. PT was dissolved in DMSO as a stock solution
at 100 mM and diluted with FBS-free medium to achieve designated
concentrations. Same concentration of DMSO was always applied to
cells as a control.
MTT colorimetric survival assay
HT-29, LS174T, and SW480 cells were plated at a
density of 1.0×104 cells per well in 96-well plates.
Cells were treated with various concentrations of PT for 24 h, and
then the medium was removed and 200 μl of fresh medium plus 20 μl
of 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide
(MTT, 2.5 mg dissolved in 50 μl of dimethylsulfoxide, Sigma) were
added to each well. After incubation for 4 h at 37°C, the culture
medium containing MTT was withdrawn and 200 μl of dimethylsulfoxide
(DMSO) was added, followed by shaking until the crystals were
dissolved. Viable cells were detected by measuring absorbance at
570 nm using a microplate reader (Molecular Devices, Sunnyvale,
CA). The cell growth was expressed as a percentage of absorbance in
cells with PT treatment to that in cells without PT treatment
(100%).
Detection of apoptosis
After being incubated with PT for 24 h, the cells
were trypsinized, collected, washed with ice-cold PBS, suspended in
a 500 μl Annexin V binding buffer containing 5 μl of Annexin
V-FITC, and incubated for 15 min at room temperature in the dark.
The fluorescence was measured on a BD LSR flow cytometer (Becton
Dickinson, NY) and processed with Cell Quest software (Becton
Dickinson, NY) for analysis. PT-induced apoptosis in colon cancer
cells was assessed using Hoechst 33258. The cells were treated with
various concentrations of PT for 24 h, and then stained with
Hoechst 33258 (1 μg/ml) at 37°C for 10 min. Nuclear morphology was
examined under a Confocal Laser Scanning Microscope (Carl Zeiss,
Germany) to identify cells undergoing apoptosis.
Mitochondrial transmembrane potential
(ΔΨm)
The mitochondrial membrane was monitored using
Rhodamine-123 fluorescent dye (Ex/Em = 485 nm/535 nm; Sigma), a
cell-permeable cationic dye, which preferentially enters into
mitochondria due to the highly negative mitochondrial membrane
potential (ΔΨm). Depolarization of ΔΨm results in the loss of
Rhodamine-123 from the mitochondria and a decrease in intracellular
fluorescence. In brief, cells were incubated with the designated
doses of PT for 24 h. Cells were washed twice with PBS and
incubated with Rhodamine-123 (0.1 μg/ml) at 37°C for 30 min. The
intensity of Rhodamine-123 staining was determined using a BD LSR
flow cytometer.
Cell extraction and western blotting
Cells were collected, washed twice with PBS, and
then lysed for 30 min on ice in a lysis buffer (50 mM Tris-HCl pH
8.0, 150 mM EDTA, 1% Triton X-100, 0.5% SDS and protease inhibitor
cocktail). The protein concentration in cell lysates was measured
by using Protein Quantification kit from Bio-Rad. Total 30 μg
proteins were loaded onto an SDS-PAGE gel. After transferring and
blocking, the membrane was probed with various antibodies
(anti-Bcl-2, anti-Bax, anti-Bid, anti-cytochrome c, anti-caspase 3,
anti-PARP, anti-p53 and anti-actin, Santa Cruz Biotechnology). The
signal was detected by using enhanced Westone (Intron, Daejeon,
Korea), and captured, analyzed by a Luminescent Image Analyzer
(LAS-3000, Fuji film, Japan).
Xenograft models
HT-29 (6×106) cells were injected into
nude mice. Mice were randomized and assigned to control group and
treatment group and intraperitoneally injected 3 times a week
vehicle (DMSO) and 4 mg/kg PT, respectively. PT or vehicle
treatment was started on 5 days after tumor cell implantation (0.5
mm3 tumor volume). Tumor diameters were measured 3 times
a week, and tumor volumes were also calculated (volume = X x Y x Z
x π/6). The experiment was terminated on 28 days, and the tumors
were harvested for immunohistochemistry.
Immunohistochemistry
Immunohistochemistry was carried out in
paraffin-embedded (5 μm) tissue sections. Apoptosis was measured
quantitatively using the terminal deoxynucleotidyl transferase
(TdT)-mediated dUTP nick end-labeling (TUNEL) assay using a ApopTag
In Situ Apoptosis Detection kit (Chemicon, Temecula, CA)
according to the manufacturer’s instructions. Four fields at ×40
magnification were selected at the proliferation front of each
tumor, and TUNEL-positive cells were counted. For analysis of
expression of CD31 (VEGF), slides were incubated with anti-CD31
(sc-507; Santa Cruz Biotechnology), and incubated with secondary
antibody (goat anti-rabbit; Santa Cruz Biotechnology). Five
equal-sized fields were randomly chosen.
Statistical analysis
The data are presented as the mean ± SE of at least
three independent experiments done in duplicate. Representative
blots are shown. All the data were entered into the Microsoft Excel
5.0, and SPSS software was used to perform the two-tailed t-tests
or the analysis of the variance, where appropriate. P-values
<0.05 were considered significant.
Results
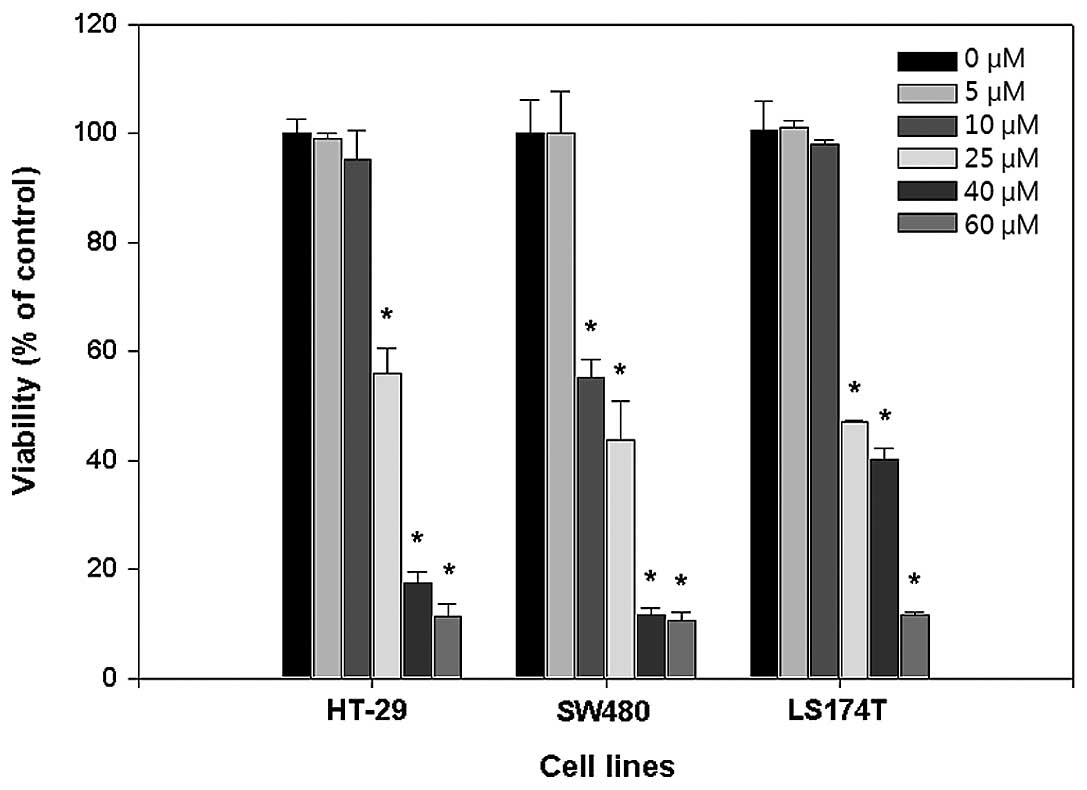
Inhibitory effect of PT on cell
proliferation
Human colorectal cancer cell lines, HT-29, SW620,
and LS174T cells were treated with various concentrations (0, 5,
10, 20, 40, and 60 μM) of PT for 24 h. At 20 μM of PT,
proliferation of these cells was inhibited approximately 50% in all
cell lines (Fig. 1). At 40 μM of
PT, inhibition of proliferation of HT-29 and SW620 cells reached
over 80% whereas proliferation of LS174T cells was inhibited
approximately 60%. At 60 μM, over 90% of cell death was observed
with all cell lines.
Apoptosis induction by PT
HT-29 cells were treated with 0, 5, 10, 20, 40 μM PT
for 24 h. The apoptosis of HT-29 cells was induced in a
dose-dependent manner of PT, reaching approximately 93.38% cell
death at 40 μM of PT. In addition, at 40 μM of PT, induction of
apoptosis in SW620 and LS174T cells were 67.215±3.755% and
72.79±7.824%, respectively (Fig.
2A).
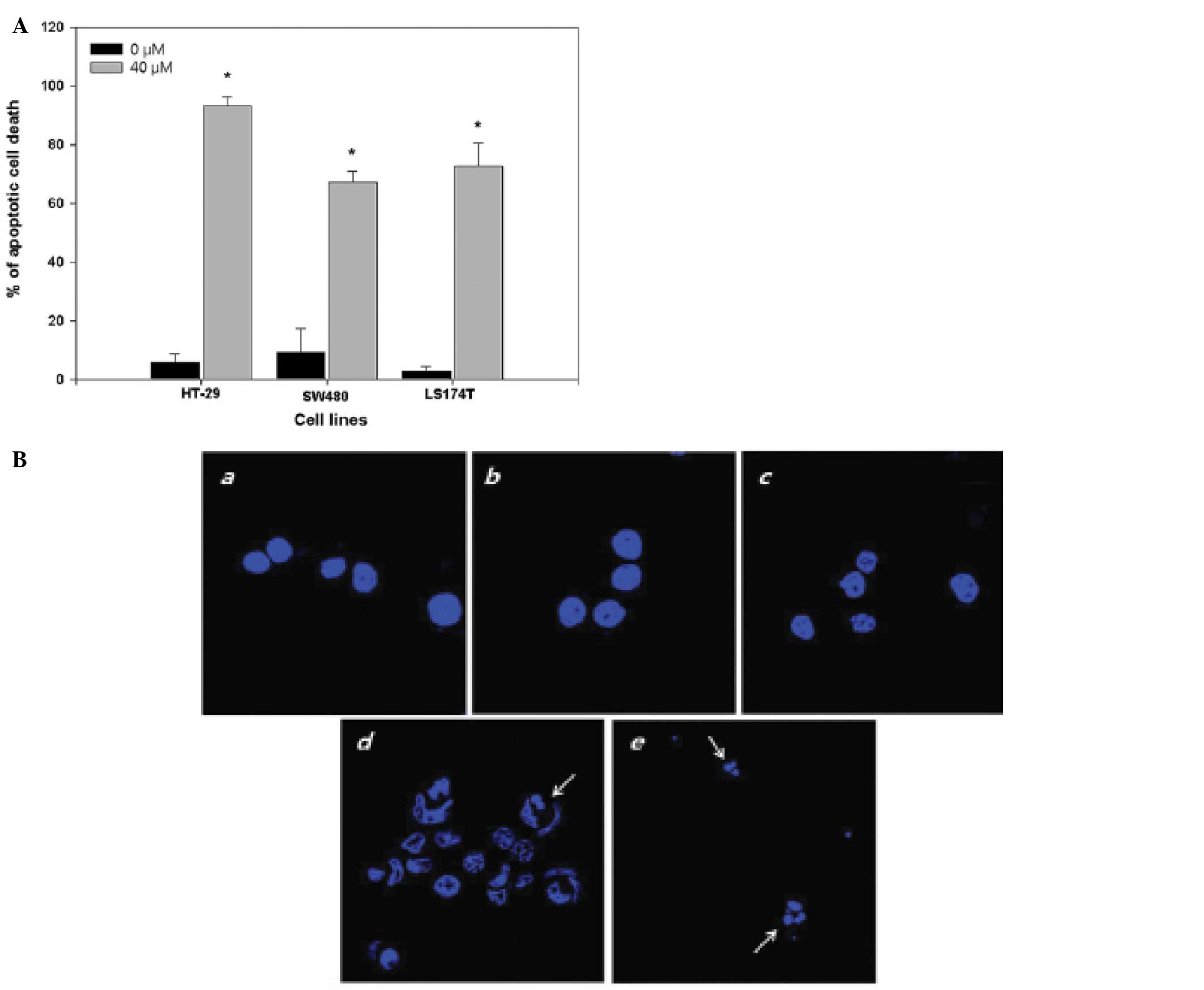 | Figure 2Apoptotic effect of PT on human
colorectal cancer cells. (A), FACS analysis of Annexin V-FITC.
After PT treatment (40 μM) for 24 h, cells were harvested and
stained with Annexin V-FITC. Total 10,000 cells were collected for
each group. The experiments were done at least thrice and the
result of one representative experiment is shown. Columns, means;
bars, ± SE. *P<0.05 compared with control of HT-29,
SW480 or LS174T cells. (B), Hoechst 33258 staining images of the
HT-29 cells. After PT treatment (a, control; b, 5 μM; c, 10 μM; d,
20 μM; e, 40 μM) for 24 h, cells were fixed and stained with
Hoechst 33258 (1 μg/ml). |
Apoptotic nuclear morphology was observed after
Hoechst 33258 staining using fluorescence microscopy. After
treatment with 20 μM of PT for 24 h, HT-29 cells began to exhibit
apoptotic characteristics, such as cell shrinkage, nuclear
condensation, and fragmentation. At 40 μM of PT, DNA condensation
was observed in most of cells, and DNA fragments found on surface
of glass plate. In the control group, the cells were regular in
morphology and grew fully in patches and were confluent, rarely
sloughing off (Fig. 2B).
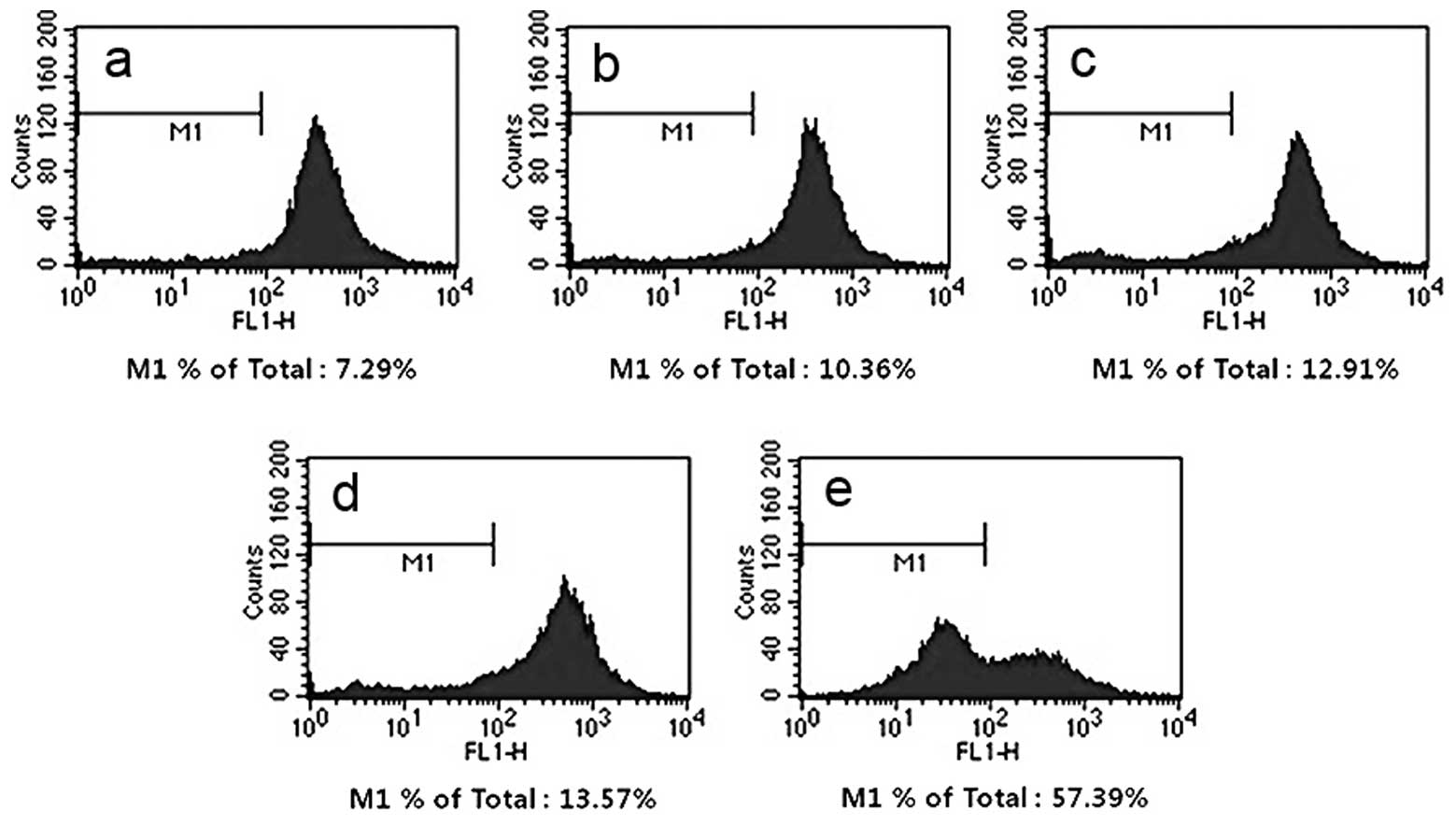
Loss of mitochondrial membrane potential
(ΔΨm)
HT-29 cells were treated with 0, 5, 10, 20, 40 µM PT
for 24 h. Treatment of the cells with PT caused a decrease in ΔΨm
in HT-29 cells in a PT concentration-dependent manner. The
decreased ratio of ΔΨm peak was 7.29% (control), 10.36% (5 µM),
12.91% (10 µM), 13.57% (20 µM) and 57.39% (40 µM) respectively
(Fig. 3).
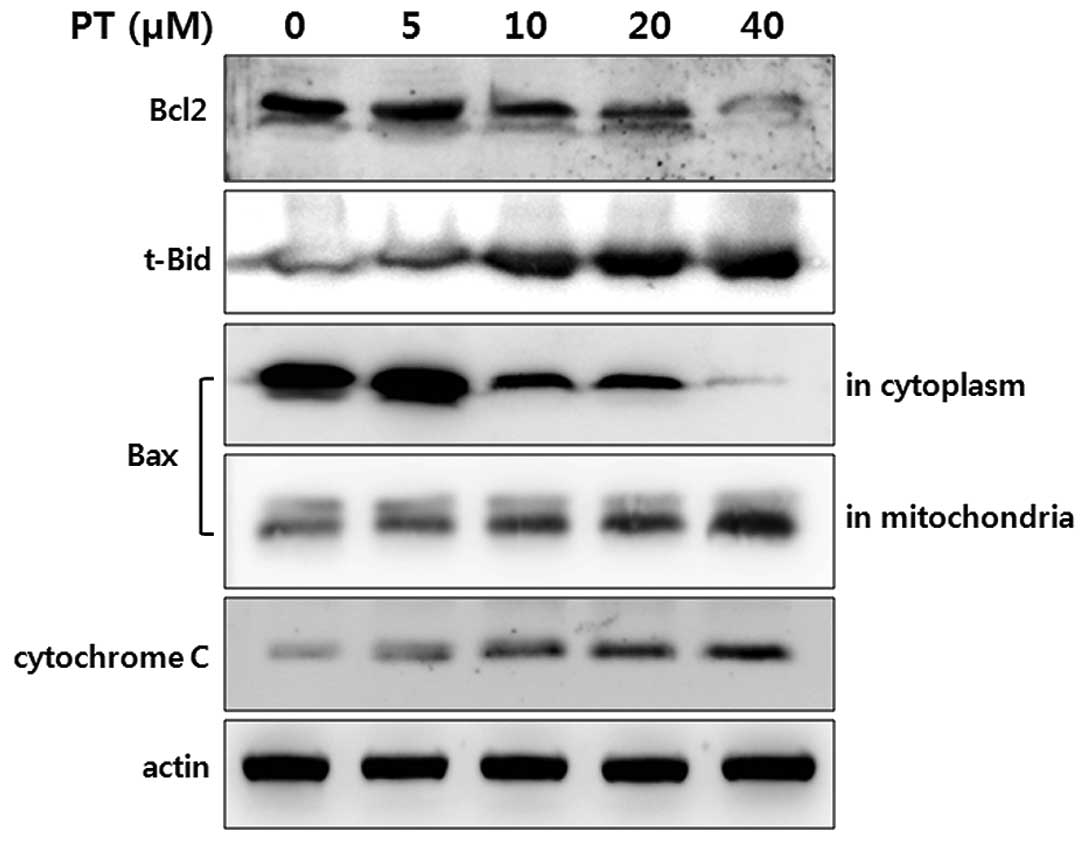
Regulation of Bcl-2 family and cytochrome
c release
The underlying mechanism of PT was also explored in
the study. The activation of several apoptosis-related proteins may
contribute to PT-induced apoptosis. In Bcl-2 family members, the
expression of Bcl-2 and truncated-Bid were detected by Western
blotting in HT-29. The level of Bcl-2 protein in HT-26 was
decreased in a dose-dependent manner of PT (Fig. 4, first panel). In contrast, the
expression of truncated-Bid in HT-29 cells was increased
dramatically in a dose-dependent manner of PT (Fig. 4, second panel).
Following a mitochondria-dependent death signal, Bax
translocates from the cytosol to the mitochondria at which time the
Bax conformation changes. This mitochondrial localization of Bax is
essential for mitochondrial permeabilization and plays an important
role in triggering apoptosis. Therefore, the Bax level was
determined in the cytosol and mitochondrial extracts after PT
treatment. Treatment of HT-29 cells with PT resulted in a decrease
in cytosolic Bax level expression showed a dose-dependent
progressive decrease of Bax, whereas the Bax level in mitochondrial
extracts was increased (Fig. 4,
third and fourth panel).
One of the consequences following the changes of
Bcl-2 family members is a dissipation of mitochondrial poteintial
and release of mitochondrial pro-apoptotic protein, cytochrome c.
After treatment of HT-29 cells with PT, the release of cytochrome c
was increased in a dose-dependent manner (Fig. 4, fifth panel).
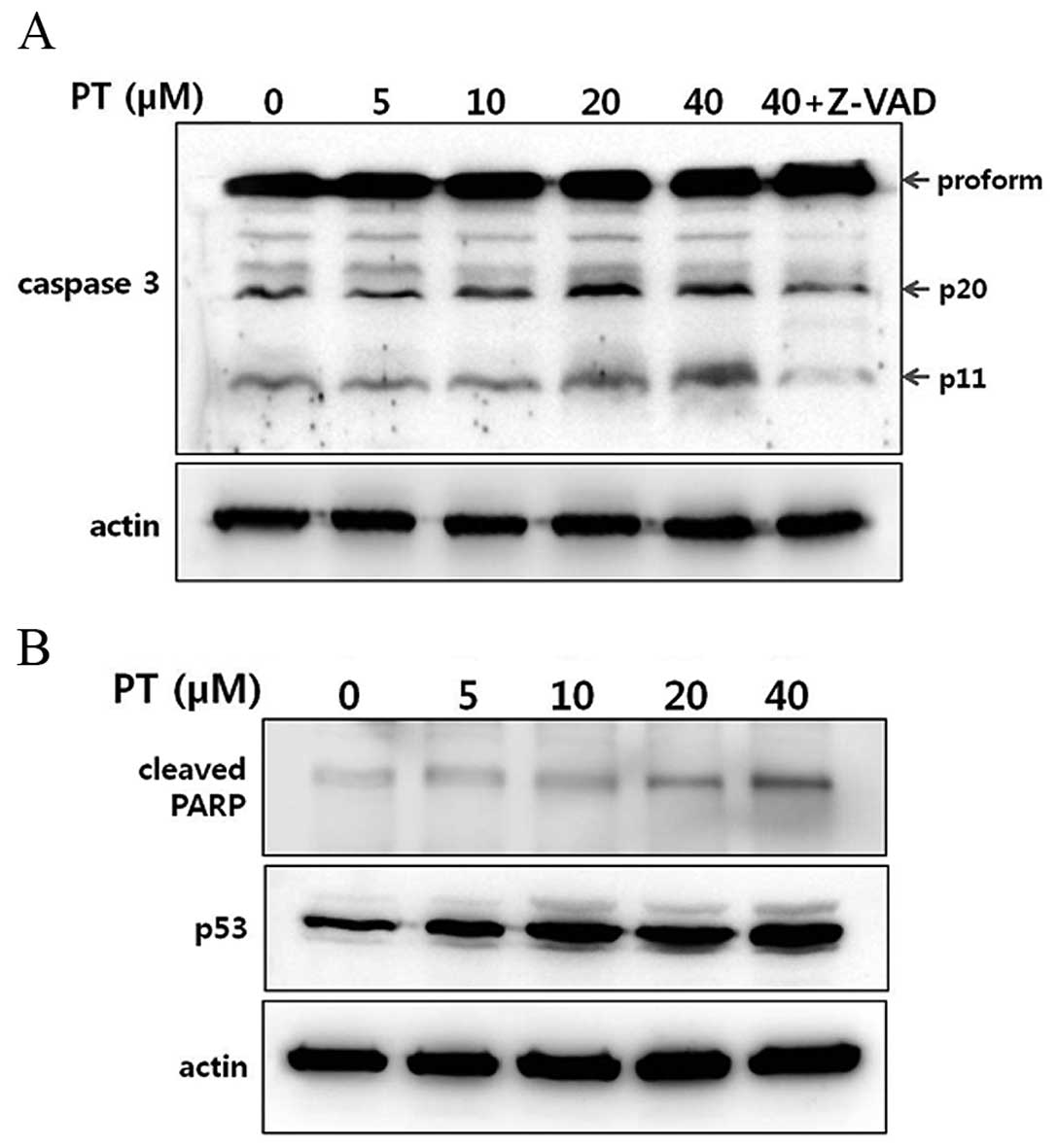
Cleavage of caspase 3 and increase of
apoptotic marker proteins
Caspase 3 is the terminal factor in the enzymatic
cascade reaction related to apoptosis in mammalian cells (16). Here we also tested effects of PT on
the caspase 3 activity in HT-29 cells. While amount of pro-caspase
3 was not changed after treatment with PT, cleavage of caspase 3
was increased in a dose-dependent manner. Furthermore, the cleavage
of caspase 3 was significantly blocked by pretreatment of a general
caspase inhibitor Z-VAD-FMK (Fig.
5A).
The activation of caspase 3 then leads to the
cleavage of their downstream molecular targets including PARP and
p53, a hallmark of apoptosis (17,18).
As shown in Fig. 5B, the levels of
cleaved PARP and p53 were increased by treatment with PT. Taken
together, these results demonstrate that PT-induced apoptosis is
caspase 3-dependent (Fig. 5B).
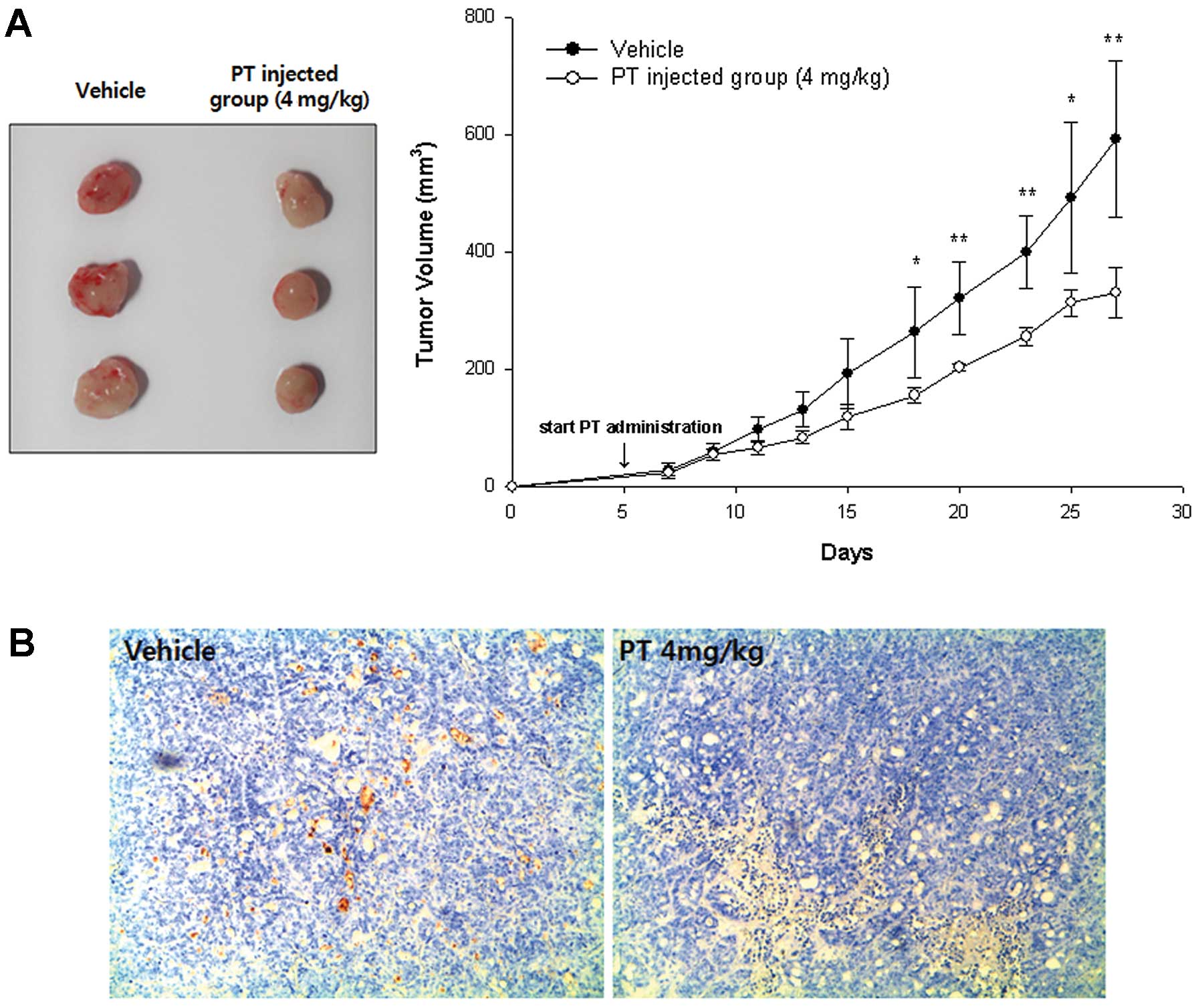
Effect of PT on tumor growth in xenograft
mice
To examine the effects of PT on tumor growth in
vivo, we used a xenograft nude mouse tumor model with
subcutaneously implanted HT-29 cells. About 4 weeks after the start
of treatment with PT, mean tumor volume of PT-treated mice was
330.01±42.09 mm3 and it was significantly smaller than
that in control mice (592.18±132.42 mm3, p= 0.0075,
Fig. 6A). We further examined the
effect of PT on angiogenesis in mice bearing HT-29 cell xenografts.
Immunohistochemical analysis of CD31, a well-established marker for
angiogenesis, revealed that the blood vessel network was well
developed in the tumors from control mice, whereas the development
of the blood vessel network appeared to be inhibited by PT
(Fig. 6B). Representative tumors
were also analyzed by the TUNEL assay to determine apoptotic cells.
Tumor tissues from mice treated with PT displayed drastically more
positively stained apoptotic cells, as compared to those from
control mice (Fig. 6C).
Discussion
Recent reports, including our own, have suggested
the involvement of multiple pathways in PT-induced apoptotic cell
death in human cancer cells, and the pathways include oxidative
stress, inhibition of DNA synthesis, activation of STAT and NF-κB,
and mitochodrial dysfunction (10,11,19–24).
Especially, PT has been demonstrated to regulate the mitochondrial
pathway in vitro. However, there is no report on its ability
to inhibit tumor growth in vivo through induction of
apoptosis via mitochondrial dysfunction. In the present study, we
demonstrated that PT exhibits anti-cancer property, inducing
apoptosis via mitochondrial pathway in vitro and in
vivo.
Many factors mediating apoptosis converge to
activate the critical effector caspase 3, which is considered as
the key protease of caspase family in mammalian cell apoptosis
(25). Caspase-dependent apoptosis
pathway includes mitochondria pathway, death receptor pathway, and
endoplasmic reticulum pathway (26,27).
The mitochondria pathway is controlled and regulated by the Bcl-2
family (27,28), which are divided into two groups,
the anti-apoptotic members (Bcl-2, Bcl-xl) and proapoptotic members
(Bax, BAD, Bid) (29). We
investigated effects of PT on changes in Bcl-2, truncated-Bid and
caspase 3 level. The result of western blot analysis showed that
the levels of truncated-Bid and cleaved caspase 3 were increased by
treatment with PT, while the level of Bcl-2 was decreased. The
cleavage of caspase 3 was prevented by pretreatment with a
pancaspase inhibitor, Z-VAD-FMK. These results have suggested that
apoptosis induced by PT involves the caspase-dependent mitochondria
pathway. Moreover, another important Bcl-2 family member, Bax is
functionally related to truncated-Bid. The direct binding of
truncated-Bid to Bax is a prerequisite for Bax translocation into
mitochondrial outer membrane, then balance of mitochondrial
membrane permeabilization is broken, resulting in cytochrome c
release (30,31). In the present study, a
dose-dependent progressive decrease of Bax in the cytosol fraction
and the increase of Bax in the mitochondrial fraction after PT
treatment were detected, suggesting the translocation of Bax from
cytosol to the mitochondrial membrane. Through the series of
biochemical events, cytochrome c which is located in the
mitochondrial membranes is released into the cytoplasm (32,33).
Downstream of cytochrome c, the activation of caspase 3 then leads
to the cleavage of their downstream molecular targets including
PARP, a hallmark of apoptosis (17,18).
Moreover, p53 induces cell cycle arrest or apoptosis in response to
DNA damage and regulates Bax and Bcl-2 protein expression (34). In this study, we showed that levels
of PARP and p53 were decreased by PT treatment. These observations
indicate that PT induces apoptosis via the mitochondrial pathway,
causing mitochondrial dysfunction in CRC cells.
Many death signals, such as anti-cancer agents,
radiation or ROS could trigger the loss of mitochondrial membrane
potential (ΔΨm), a critical step in the apoptosis processes which
leads to an irreversible apoptosis (35). Bcl-2 families are mainly involved
to apoptosis and mitochondria is the principle site of apoptotic
action (31,36). Correspondingly, treatment with 40
µM PT induced the 57.39% of loss of ΔΨm in HT-29 cells. Notably
there were similar changes of Annexin V positive-stained and
Rhodamine-123 negative-stained cells in 40 µM PT-treated HT-29
cells, suggesting that apoptosis by PT is tightly related to or
dependent on the loss of mitochondrial membrane potential
(ΔΨm).
To assess the effectiveness of PT against CRC in
vivo, we examined whether PT could inhibit the growth of HT-29
tumors in xenograft models. The growth of HT-29 xenografts was
inhibited by treatment with PT and many apoptotic cells were
observed in PT treated mice by TUNEL assay. These findings suggest
that PT has an effect on tumor growth and apoptosis induction in
CRC. The anti-tumor effect of PT by mitochondrial dysfunction has
not been studied in vivo. In the present study, we confirm
that regulation of Bcl-2 family and dissipation of MMP by PT can
lead to inhibit tumor growth in CRC xenografts models.
We also observed reduced blood and blood vessels in
PT-treated mice compared to those in control mice. VEGF plays a
central role in angiogenesis by promoting the growth of vascular
endothelial cells and enhancing vascular permeability (37,38).
In a study of renal cell carcinoma, the production of VEGF was
decreased by PT in vitro and in vivo (39). Moreover, PT inhibits the
proliferation and induces cell cycle arrest at G0/G1 phase in
vascular smooth muscle cells (40). Accumulation of these inhibitory
effects of PT on angiogenic factors may lead to significant tumor
growth inhibition. In this study, we confirm that PT also
participates in regulating angiogenesis and suppressing VEGF, using
immunohistochemistry images for CD31. Moreover, the regulatory
mechanism of angiogenesis by PT requires further study.
In conclusion, the present study demonstrates that
PT inhibits colon cancer development and tumor growth by the
induction of apoptosis through mitochondrial dysfunction. These
results suggest that PT could be a potential chemopreventive and
therapeutic agent of colon cancer.
Acknowledgements
This study was supported by Fund of
Chonbuk National University Hospital Research Institute of Clinical
Medicine.
References
|
1
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J and
Thun MJ: Cancer statistics, 2009. CA Cancer J Clin. 59:225–249.
2009. View Article : Google Scholar
|
|
2
|
Meyerhardt JA and Mayer RJ: Systemic
therapy for colorectal cancer. N Engl J Med. 352:476–487. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tebbutt NC, Cattell E, Midgley R,
Cunningham D and Kerr D: Systemic treatment of colorectal cancer.
Eur J Cancer. 38:1000–1015. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gusella M, Frigo AC, Bolzonella C, et al:
Predictors of survival and toxicity in patients on adjuvant therapy
with 5-fluorouracil for colorectal cancer. Br J Cancer.
100:1549–1557. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Raymond E, Faivre S, Chaney S, Woynarowski
J and Cvitkovic E: Cellular and molecular pharmacology of
oxaliplatin. Mol Cancer Ther. 1:227–235. 2002.
|
|
6
|
Knight DW: Feverfew: chemistry and
biological activity. Nat Prod Rep. 12:271–276. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Murphy JJ, Heptinstall S and Mitchell JR:
Randomised double-blind placebo-controlled trial of feverfew in
migraine prevention. Lancet. 2:189–192. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hehner SP, Heinrich M, Bork PM, et al:
Sesquiterpene lactones specifically inhibit activation of NF-kappa
B by preventing the degradation of I kappa B-alpha and I kappa
B-beta. J Biol Chem. 273:1288–1297. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lyss G, Knorre A, Schmidt TJ, Pahl HL and
Merfort I: The anti-inflammatory sesquiterpene lactone helenalin
inhibits the transcription factor NF-kappaB by directly targeting
p65. J Biol Chem. 273:33508–33516. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang S, Ong CN and Shen HM: Critical
roles of intracellular thiols and calcium in parthenolide-induced
apoptosis in human colorectal cancer cells. Cancer Lett.
208:143–153. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wen J, You KR, Lee SY, Song CH and Kim DG:
Oxidative stress-mediated apoptosis. The anticancer effect of the
sesquiterpene lactone parthenolide J Biol Chem. 277:38954–38964.
2002.PubMed/NCBI
|
|
12
|
Pajak B, Gajkowska B and Orzechowski A:
Molecular basis of parthenolide-dependent proapoptotic activity in
cancer cells. Folia Histochem Cytobiol. 46:129–135. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mathema VB, Koh YS, Thakuri BC and
Sillanpaa M: Parthenolide, a sesquiterpene lactone, expresses
multiple anti-cancer and anti-inflammatory activities.
Inflammation. 35:560–565. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhao LJ, Xu YH and Li Y: Effect of
parthenolide on proliferation and apoptosis in gastric cancer cell
line SGC7901. J Dig Dis. 10:172–180. 2009. View Article : Google Scholar
|
|
15
|
Zhang S, Ong CN and Shen HM: Involvement
of proapoptotic Bcl-2 family members in parthenolide-induced
mitochondrial dysfunction and apoptosis. Cancer Lett. 211:175–188.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen YC, Shen SC, Lee WR, et al: Emodin
induces apoptosis in human promyeloleukemic HL-60 cells accompanied
by activation of caspase 3 cascade but independent of reactive
oxygen species production. Biochem Pharmacol. 64:1713–1724. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Grutter MG: Caspases: key players in
programmed cell death. Curr Opin Struct Biol. 10:649–655. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang JC and Cortopassi GA: Induction of
the mitochondrial permeability transition causes release of the
apoptogenic factor cytochrome c. Free Radic Biol Med. 24:624–631.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tacchini L, De Ponti C, Matteucci E,
Follis R and Desiderio MA: Hepatocyte growth factor-activated
NF-kappaB regulates HIF-1 activity and ODC expression, implicated
in survival, differently in different carcinoma cell lines.
Carcinogenesis. 25:2089–2100. 2004. View Article : Google Scholar
|
|
20
|
Dai Y, Guzman ML, Chen S, et al: The NF
(nuclear factor)-kappaB inhibitor parthenolide interacts with
histone deacetylase inhibitors to induce MKK7/JNK1-dependent
apoptosis in human acute myeloid leukaemia cells. Br J Haematol.
151:70–83. 2010. View Article : Google Scholar
|
|
21
|
Nakshatri H, Rice SE and Bhat-Nakshatri P:
Antitumor agent parthenolide reverses resistance of breast cancer
cells to tumor necrosis factor-related apoptosis-inducing ligand
through sustained activation of c-Jun N-terminal kinase. Oncogene.
23:7330–7344. 2004. View Article : Google Scholar
|
|
22
|
Carlisi D, D’Anneo A, Angileri L, et al:
Parthenolide sensitizes hepatocellular carcinoma cells to TRAIL by
inducing the expression of death receptors through inhibition of
STAT3 activation. J Cell Physiol. 226:1632–1641. 2011. View Article : Google Scholar
|
|
23
|
Chen KF, Tai WT, Liu TH, et al: Sorafenib
overcomes TRAIL resistance of hepatocellular carcinoma cells
through the inhibition of STAT3. Clin Cancer Res. 16:5189–5199.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu C, Chen F, Rushing JW, et al:
Antiproliferative activities of parthenolide and golden feverfew
extract against three human cancer cell lines. J Med Food. 9:55–61.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fernandes-Alnemri T, Litwack G and Alnemri
ES: CPP32, a novel human apoptotic protein with homology to
Caenorhabditis elegans cell death protein Ced-3 and
mammalian interleukin-1 beta-converting enzyme. J Biol Chem.
269:30761–30764. 1994.PubMed/NCBI
|
|
26
|
Mehmet H: Caspases find a new place to
hide. Nature. 403:29–30. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang E and Korsmeyer SJ: Molecular
thanatopsis: a discourse on the BCL2 family and cell death. Blood.
88:386–401. 1996.PubMed/NCBI
|
|
28
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Reed JC: Double identity for proteins of
the Bcl-2 family. Nature. 387:773–776. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Adams JM and Cory S: The Bcl-2 protein
family: arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gross A, McDonnell JM and Korsmeyer SJ:
BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Terrones O, Antonsson B, Yamaguchi H, et
al: Lipidic pore formation by the concerted action of proapoptotic
BAX and tBID. J Biol Chem. 279:30081–30091. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Van Mau N, Kajava AV, Bonfils C, Martinou
JC and Harricane MC: Interactions of Bax and tBid with lipid
monolayers. J Membr Biol. 207:1–9. 2005.PubMed/NCBI
|
|
34
|
Coutts AS and La Thangue N: The p53
response during DNA damage: impact of transcriptional cofactors.
Biochem Soc Symp. 181–189. 2006.PubMed/NCBI
|
|
35
|
Kroemer G and Reed JC: Mitochondrial
control of cell death. Nat Med. 6:513–519. 2000. View Article : Google Scholar
|
|
36
|
Borner C: The Bcl-2 protein family:
sensors and checkpoints for life-or-death decisions. Mol Immunol.
39:615–647. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ferrara N, Hillan KJ, Gerber HP and
Novotny W: Discovery and development of bevacizumab, an anti-VEGF
antibody for treating cancer. Nat Rev Drug Discov. 3:391–400. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang S, Pettaway CA, Uehara H, Bucana CD
and Fidler IJ: Blockade of NF-kappaB activity in human prostate
cancer cells is associated with suppression of angiogenesis,
invasion, and metastasis. Oncogene. 20:4188–4197. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Oka D, Nishimura K, Shiba M, et al:
Sesquiterpene lactone parthenolide suppresses tumor growth in a
xenograft model of renal cell carcinoma by inhibiting the
activation of NF-kappaB. Int J Cancer. 120:2576–2581. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Weng SX, Sui MH, Chen S, et al:
Parthenolide inhibits proliferation of vascular smooth muscle cells
through induction of G0/G1 phase cell cycle arrest. J Zhejiang Univ
Sci B. 10:528–535. 2009. View Article : Google Scholar : PubMed/NCBI
|