Introduction
Lung cancer is the most common cause of
cancer-related death, and the worldwide annual death by lung cancer
was estimated to be 1.3 million (1). Esophageal squamous cell carcinoma
(ESCC) is one of the most common gastrointestinal tract cancers in
Asian countries (2). Although a
huge body of knowledge about the biology of lung or esophageal
carcinogenesis has been accumulated, the development of novel
cancer therapeutics remains inefficient to improve patients with
these cancers (3). In fact, in
spite of development of various molecular targeted therapies, a
limited proportion of patients can receive clinical benefit from
them (4).
Through genome-wide gene expression analysis of lung
and esophageal cancers, we have isolated a number of oncogenes that
were involved in the development and/or progression of cancer
(5–41). Among the genes upregulated in these
cancers, we focused on MMS22L (methyl
methanesulfonate-sensitivity protein 22-like) which is highly
expressed in the majority of clinical lung and esophageal cancers.
Our original gene expression profile database also revealed that
this gene is highly expressed in clinical cervical cancers, but
scarcely expressed in normal tissues except testis, suggesting that
MMS22L encodes a cancer-testis antigen that can be defined
by predominant expression in various types of cancer and
undetectable expression in normal tissues except germ cells in
testis or ovary (4). Cancer-testis
antigens are considered to be good candidate molecular targets for
developing new therapeutic strategies for cancers.
Constitutive activation of the NFKB pathway is
involved in some forms of cancer such as leukemia, lymphoma, colon
cancer and ovarian cancer as well as inflammatory diseases
(42–45). The main mechanism of this pathway
is reported to be the inactivation of IκB proteins by mutations as
well as amplifications and rearrangements of genes encoding the
NFKB transcription factor subunits (42–45).
However, more commonly it is thought that changes in the upstream
pathways that lead to NFKB activation are likely to be aberrantly
upregulated in cancer cells (45).
Recently some reports suggested that MMS22L-NFKBIL2 interaction
could be essential for genomic stability and homologous
recombination in immortalized cell lines, suggesting MMS22L to be a
new regulator of DNA replication in human cells (46–49).
However, no study has indicated critical roles of activation of
MMS22L and NFKBIL2 in clinical cancers and investigated their
functional importance in carcinogenesis. Here, we report that
MMS22L is involved in NFKB pathway in cancer cells through its
interaction with NFKBIL2 and might be a promising target for
development of novel cancer therapy.
Materials and methods
Cell lines and clinical samples
The 12 human lung-cancer cell lines used for in this
study included nine NSCLC cell lines (A549, NCI-H1373, LC319,
NCI-H1781, PC-14, NCI-H358, NCI-H2170, NCI-H520 and LU61) and three
small-cell lung cancer (SCLC) cell lines (SBC-3, SBC-5 and DMS114).
The 9 human esophageal carcinoma cell lines used in this study were
as follows: eight SCC cell lines (TE1, TE3, TE8, TE9, TE10, TE12,
TE13 and TE15) and one adenocarcinoma (ADC) cell line (TE7). A
cervical cancer cell line HeLa was also included in the study. All
cells were grown in monolayers in appropriate media supplemented
with 10% fetal calf serum (FCS) and were maintained at 37°C in an
atmosphere of humidified air with 5% CO2. Human airway
epithelial cells, SAEC (Cambrex Bio Science Inc.), were also
included in the panel of the cells used in this study. Primary lung
and esophageal cancer samples had been obtained earlier with
informed consent (5–10). This study and the use of all
clinical materials mentioned were approved by individual
institutional ethics committees.
Semiquantitative RT-PCR
We prepared appropriate dilutions of each
single-stranded cDNA prepared from mRNAs of clinical lung and
esophageal cancer samples, taking the level of β-actin
(ACTB) expression as a quantitative control. The primer sets
for amplification were as follows: ACTB-F
(5′-GAGGTGATAGCATTGCTTTCG-3′) and ACTB-R
(5′-CAAGTCAGTGTACAGGTAAGC-3′) for ACTB, MMS22L-F
(5′-GTCTCACCTTGGACAGATGG-3′) and MMS22L-R
(5′-CCAAGGATCCTATTACACAGTTGC-3′) for MMS22L. All reactions
involved initial denaturation at 95°C for 5 min followed by 22 (for
ACTB) or 30 (for MMS22L) cycles of 95°C for 30 sec,
56°C for 30 sec, and 72°C for 60 sec on a GeneAmp PCR system 9700
(Applied Biosystems).
Northern blot analysis
Human multiple-tissue northern blots (16 normal
tissues including heart, brain, placenta, lung, liver, skeletal
muscle, kidney, pancreas, spleen, thymus, prostate, testis, ovary,
small intestine, colon, leukocyte; BD Biosciences Clontech) were
hybridized with a 32P-labeled PCR product of
MMS22L. The partial-length cDNA of MMS22L was
prepared by RT-PCR using primers MMS22L-F1
(CTGGAAGAGGCAGTTGAAAA) and MMS22L-R1 (ATCGCCCAATATACTGCTCA).
Prehybridization, hybridization, and washing were performed
according to the supplier’s recommendations. The blots were
autoradiographed with intensifying screens at −80°C for 7 days.
Anti-MMS22L antibody
Synthesized peptide with the amino acids sequence of
CLGQMGQDEMQRLENDNT [1227–1243] (Cysteine was added to the
N-terminal) was inoculated into rabbits; the immune sera were
purified on affinity columns according to standard methodology.
Affinity-purified anti-MMS22L antibodies were used for western blot
as well as immunocytochemical analyses. We confirmed that the
antibody was specific to MMS22L on western blots using lysates from
cell lines that had been transfected with MMS22L expression vector
as well as those from lung and esophageal cancer cell lines that
endogenously expressed MMS22L or not.
Western blot analysis
Cells were lysed in lysis buffer; 50 mM Tris-HCl (pH
8.0), 150 mM NaCl, 0.5% NP-40, 0.5% deoxycholate-Na, 0.1% SDS, plus
protease inhibitor (Protease Inhibitor Cocktail Set III;
Calbiochem). We used ECL western blot analysis system (GE
Healthcare Bio-Sciences), as described previously (11).
Immunocytochemical analysis
Cultured cells were washed twice with PBS(-), fixed
and rendered permeable in 1:1 acetone: methanol solution for 10 min
at −20°C. Prior to the primary antibody reaction, cells were
covered with blocking solution [5% bovine serum albumin in PBS(-)]
for 10 min to block non-specific antibody binding. After the cells
were incubated with a rabbit polyclonal antibody to human MMS22L
(generated to synthesized peptide MMS22L; please see above) or a
mouse monoclonal antibody to human NFKBIL2 (Abnova), the Alexa
Fluor 488-labelled donkey anti-rabbit secondary antibody (Molecular
Probes) or Alexa Fluor 594-labbelled donkey anti-mouse secondary
antibody (Molecular Probes) was added to detect endogenous MMS22L
or NFKBIL2, individually. Nuclei were stained with
4′,6-diamidino-2-phenylindole (DAPI). The antibody-stained cells
were viewed with a laser-confocal microscope (TSC SP2 AOBS; Leica
Microsystems).
RNA interference assay
Two independent siRNA oligonucleotides against
MMS22L were designed using the MMS22L sequences
(GenBank accession no: NM198468). Each siRNA (600 pM) was
transfected into two NSCLC cell lines, LC319 and A549 or a cervical
cancer cell line HeLa using 30 μl of lipofectamine 2,000
(Invitrogen) following the manufacturer’s protocol. The transfected
cells were cultured for seven days. Cell numbers and viability were
measured by Giemsa staining and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay in triplicate (cell counting kit-8 solution; Dojindo
Laboratories). The siRNA sequences used were as follows: control-1
(si-LUC: luciferase gene from Photinus pyralis),
5′-CGUACGCGGAAUACUUCGA-3′; control-2 (CNT: On-TARGETplus siControl
non-targeting siRNAs of a pool of four oligosnucleotides:
5′-UGGUUUACAUGUCGACUAA-3′; 5′-UGGUUUACAUGUUUUCUGA-3′;
5′-UGGUUUACAUGUUUUCCUA-3′; and 5′-UGGUUUACAUGUUGUGUGA-3′);
siRNA-MMS22L-#1 (si-MMS22L-#1: 5′-CCGCCAAUAUCAUCUCUAAUU-3′);
siRNA-MMS22L-#2 (si-MMS22L-#2: 5′-GAACCUGCAAUACAUGGUAUU-3′).
Downregulation of endogenous MMS22L expression in the cell lines by
siRNAs for MMS22L, but not by controls, was confirmed by
semiquantitative RT-PCR and western blot analyses.
Cell growth assay
COS-7 or HEK293 cells that express endogenous
MMS22L at a very low level were transfected with mock or
MMS22L-expressing vectors (pCAGGSn-3xFlag-MMS22L) using
lipofectamine 2,000 transfection reagent (Roche). Transfected cells
were incubated in the culture medium containing 0.8 mg/ml neomycin
(Geneticin, Invitrogen) for 7 days. Expression of MMS22L as well as
viability and colony numbers of cells were evaluated by western
blot analysis, and MTT and colony-formation assays at day 7.
Flow cytometric analysis
Cells transfected with siRNA oligonucleotides
against MMS22L or control siRNAs were plated at densities of
5x105 per 60-mm dish. Cells were collected in PBS, and
fixed in 70% cold ethanol for 30 min. After treatment with 100
μg/ml RNase (Sigma-Aldrich), the cells were stained with 50
μg/ml propidium iodide (Sigma-Aldrich) in PBS. Flow
cytometric analysis was done on a Cell Lab Quanta SC (Beckman
Coulter) and analyzed by CXP Analysis software (Beckman Coulter).
The cells selected from at least 10,000 ungated cells were analyzed
for DNA content.
Results
Expression of MMS22L in lung and
esophageal cancers
We previously performed genome-wide expression
profile analysis of 120 lung cancer cases using microarray
consisting of 27,648 cDNAs or ESTs (5–10).
Among the genes upregulated in lung and esophageal cancers, we
identified MMS22L transcript to be frequently overexpressed
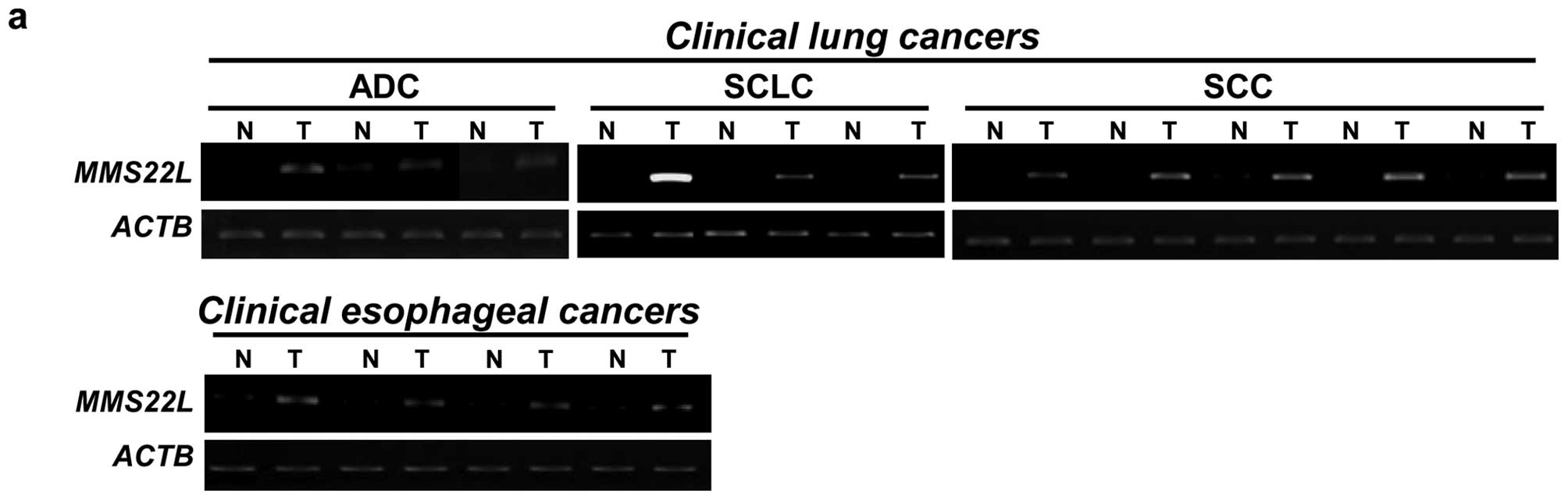
in lung and esophageal cancers, and confirmed by semiquantitative
RT-PCR experiments its elevated expression in all of eleven
clinical lung cancers and in four clinical esophageal cancers,
although its expression was not detectable in adjacent normal lung
and esophagus tissues (Fig. 1a).
We further confirmed by western blot analysis high levels of
endogenous MMS22L protein in 11 of 12 lung cancer cell lines and in
all of 9 esophageal cancer cell lines using anti-MMS22L antibody
(Fig. 1b). Northern blot analysis
of 16 normal tissues confirmed that MMS22L was hardly
detectable in normal tissues except the testis (Fig. 1c).
Growth effect of MMS22L
To investigate the relevance of MMS22L to the growth
and/or survival of cancer cells, we knocked down the expression of
endogenous MMS22L in two lung cancer cell lines, LC319 and
A549, by means of the RNAi technique using siRNA oligonucreotide
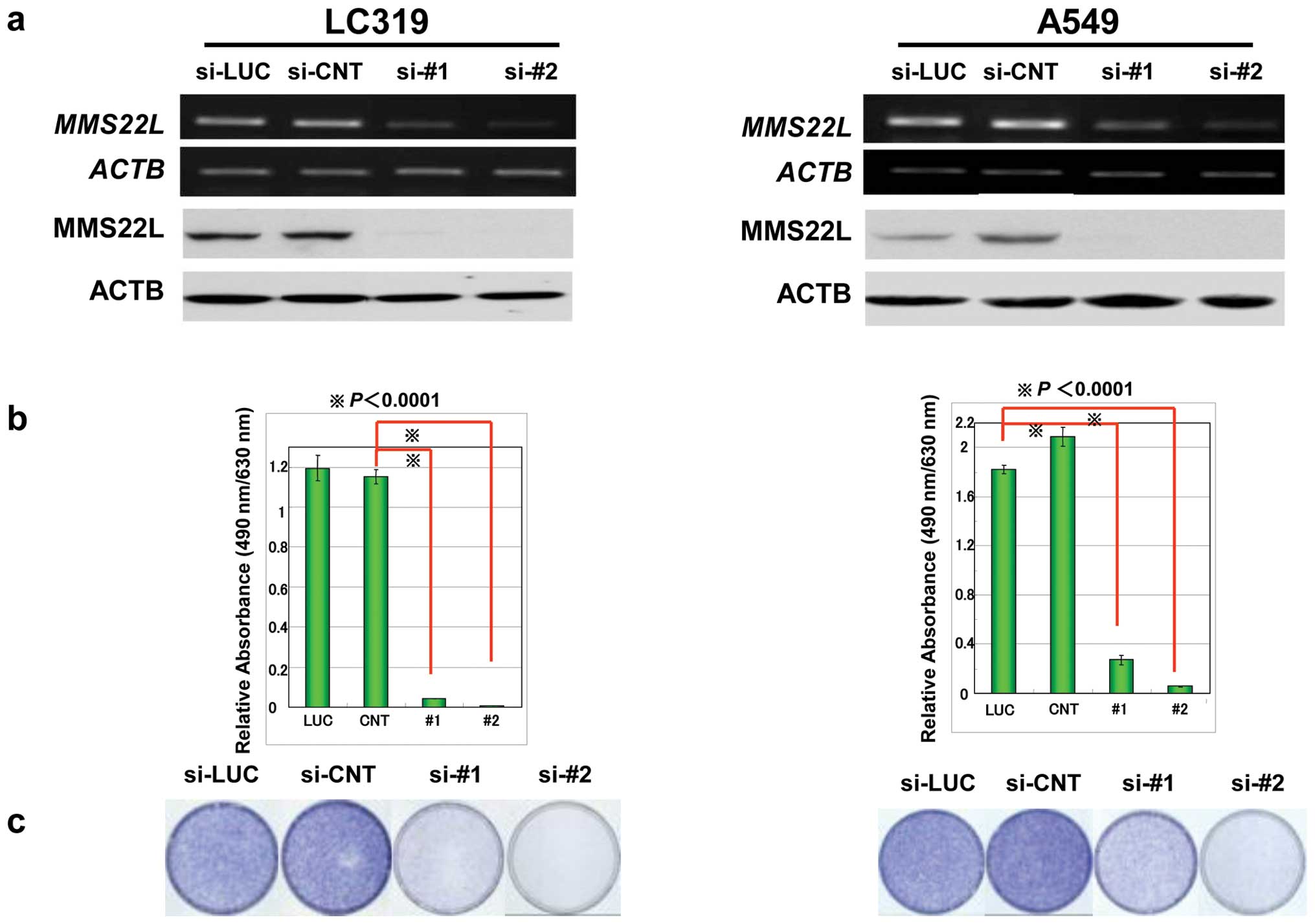
for MMS22L. Semiquantitative RT-PCR experiments detected
significant reduction of MMS22L expression in the cells transfected
with siRNAs against MMS22L (si-#1 and si-#2), but not in
those with control siRNAs (si-LUC and si-CNT) (Fig. 2a). Colony formation and MTT assays
clearly demonstrated that the viability of lung cancer cells
transfected with two effective siRNAs for MMS22L (si-#1 and
si-#2) were reduced in correlation with the reduction of MMS22L
expression level, implying essential role of MMS22L in the growth
of cancer cells (Fig. 2b and c).
Since our original gene expression profile database also revealed
its high level of expression in clinical cervical cancers, we also
knocked down the expression of MMS22L by siRNAs in a
cervical cancer cell line, HeLa, and observed the growth
suppressive effect by siRNAs for MMS22L.
To further examine the effect of MMS22L
overexpression on the growth of mammalian cells, we transiently
transfected plasmid designed to express Flag-tagged MMS22L
(pCAGGSn-3xFlag-MMS22L) or mock plasmid into COS-7 or HEK293 cells
that expressed endogenous MMS22L at very low level. The significant
growth promoting effect was observed in the cells transfected with
the MMS22L expressing vector compared to those transfected with the
mock vector (Fig. 2d).
NFKBIL2 controls the nuclear localization
and stability of MMS22L protein
To investigate the biological function of MMS22L
protein, we screened MMS22L-interacting proteins in lung cancer
cells using mass spectrometric analysis and identified the
interaction between MMS22L and NFKBIL2 [nuclear factor of kappa
(NFKB) light polypeptide gene enhancer in B-cells inhibitor-like
2]. Previous reports independently suggested the roles of
MMS22L-NFKBIL2 interaction in genomic stability and DNA replication
in immortalized cell lines (46–49),
however, no study has indicated critical roles of activation of
MMS22L and NFKBIL2 in clinical cancers and investigated their
functional importance in carcinogenesis. Western blot analysis
using cell lines derived from lung cancers and antibodies to MMS22L
and NFKBIL2 revealed the co-expression of these two proteins (data
not shown), suggesting some functional roles of their interaction
in human carcinogenesis. Therefore, we next performed
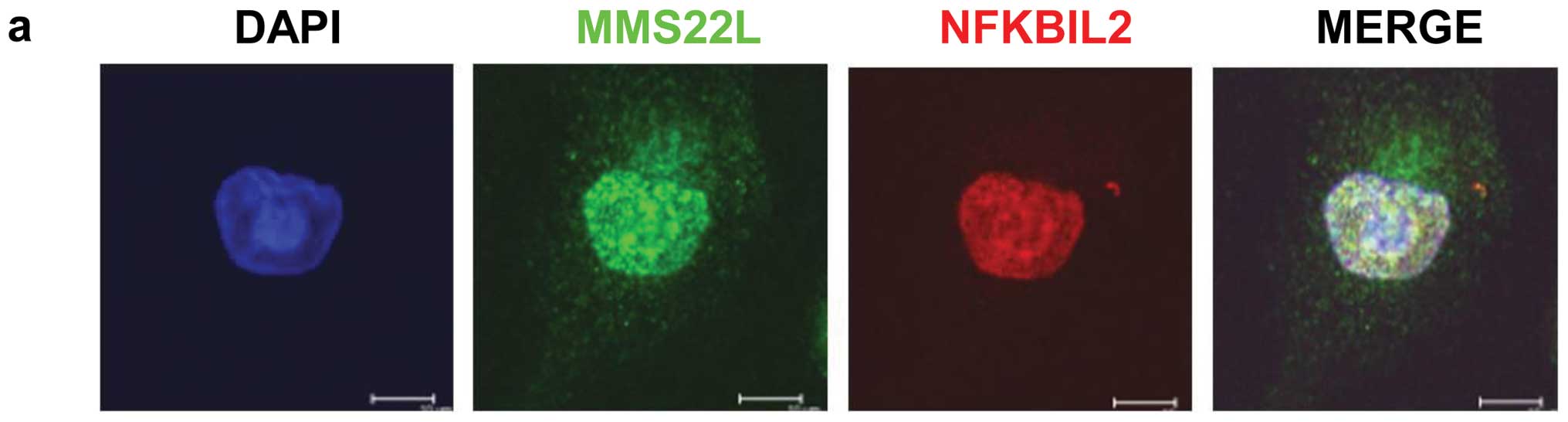
immunofluorescence analysis to determine the subcellular
localization of endogenous MMS22L and NFKBIL2 in various cancer
cell lines including A549, LC319 and HeLa cells, and found that
endogenous MMS22L and NFKBIL2 proteins were mainly co-localized in
the nucleus (representative data of HeLa cells was shown in
Fig. 3a). To examine the
importance of MMS22L-NFKBIL2 interaction in cellar localization of
these proteins, we transiently co-expressed exogenous MMS22L and
NFKBIL2 proteins using mammalian COS-7 or NIH3T3 cells that
expressed these two proteins at very low levels. We found that
exogenous MMS22L was mainly located in the cytoplasm and weakly in
the nucleus of the cells in which exogenous NFKBIL2 protein was not
introduced. However, the nuclear staining of MMS22L was
significantly enhanced when both exogenous MMS22L and NFKBIL2
proteins were introduced in the cells (Fig. 3b). On the other hand, exogenous
NFKBIL2 was mainly present in the nucleus of cells regardless to
the presence or absence of exogenous MMS22L. In addition, we
performed western blot analysis using fractionated cytoplasmic and
nuclear lysates from COS-7 cells that were introduced exogenous
MMS22L and NFKBIL2 proteins. When we transfected both MMS22L-Flag
and NFKBIL2-HA expressing vectors, the amounts of nuclear MMS22L
was significantly increased, compared with the cells transfected
with MMS22L alone (Fig. 3c).
Furthermore, we found that knockdown of endogenous MMS22L with
siRNA for MMS22L (si-MMS22L) reduced NFKBIL2 protein
level in lung cancer LC319 cells and that reduction of NFKBIL2 with
si-NFKBIL2 reduced MMS22L levels and significantly suppressed
cancer cell growth (Fig. 3d; data
not shown). These data suggest that the expression of NFKBIL2 is
likely to promote nuclear localization and stability of MMS22L
protein, and a complex including these two proteins could
coordinately play pivotal roles in cell growth and/or survival.
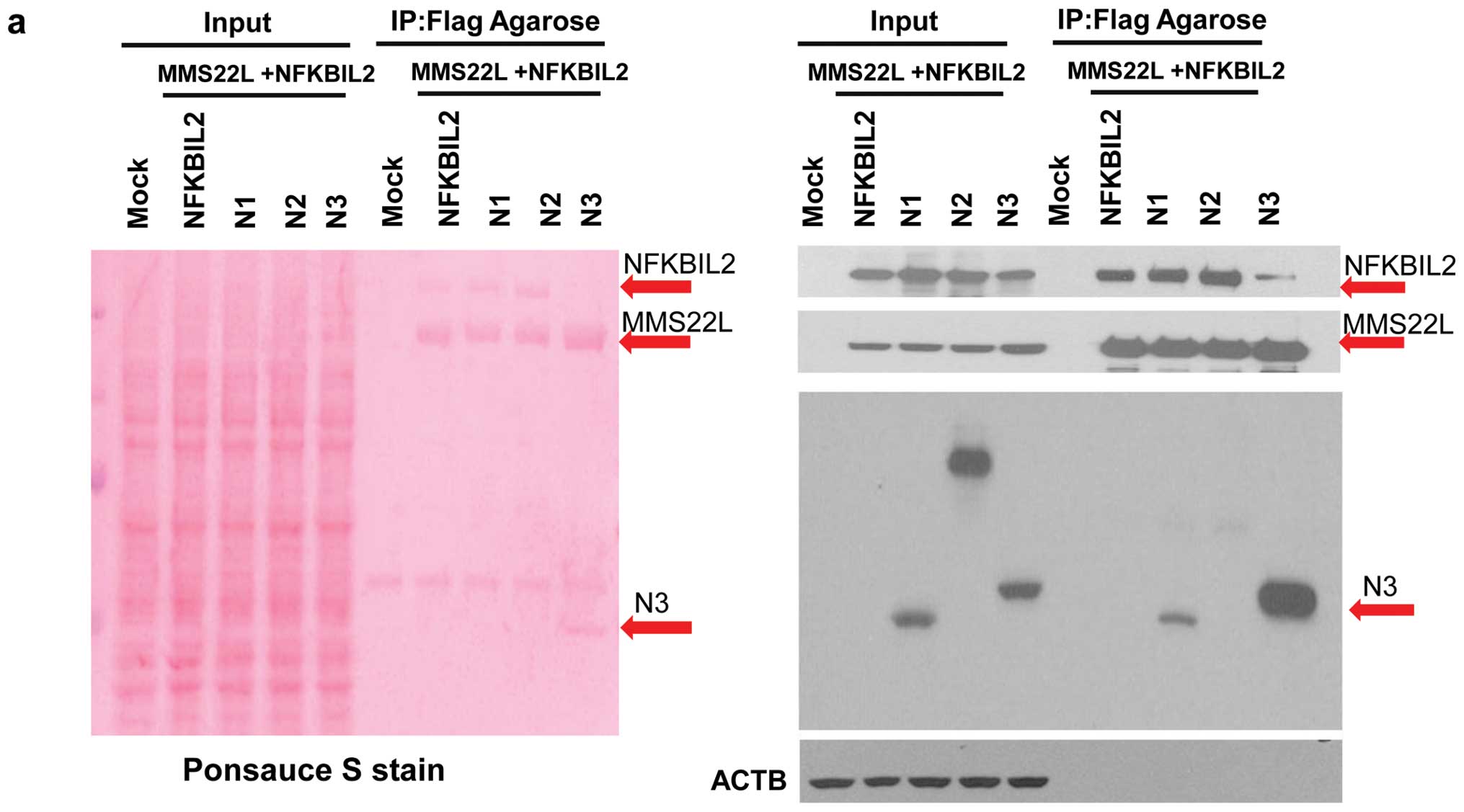
C-terminal portion of NFKIL2 protein is
crucial for binding to MMS22L protein
To examine whether the MMS22L-NFKBIL2 protein
complex may play important roles in carcinogenesis, we subsequently
constructed various plasmids expressing partial MMS22L proteins
with Flag tag or partial NFKBIL2 proteins with HA tag, and
transfected them into COS-7 cells (data not shown).
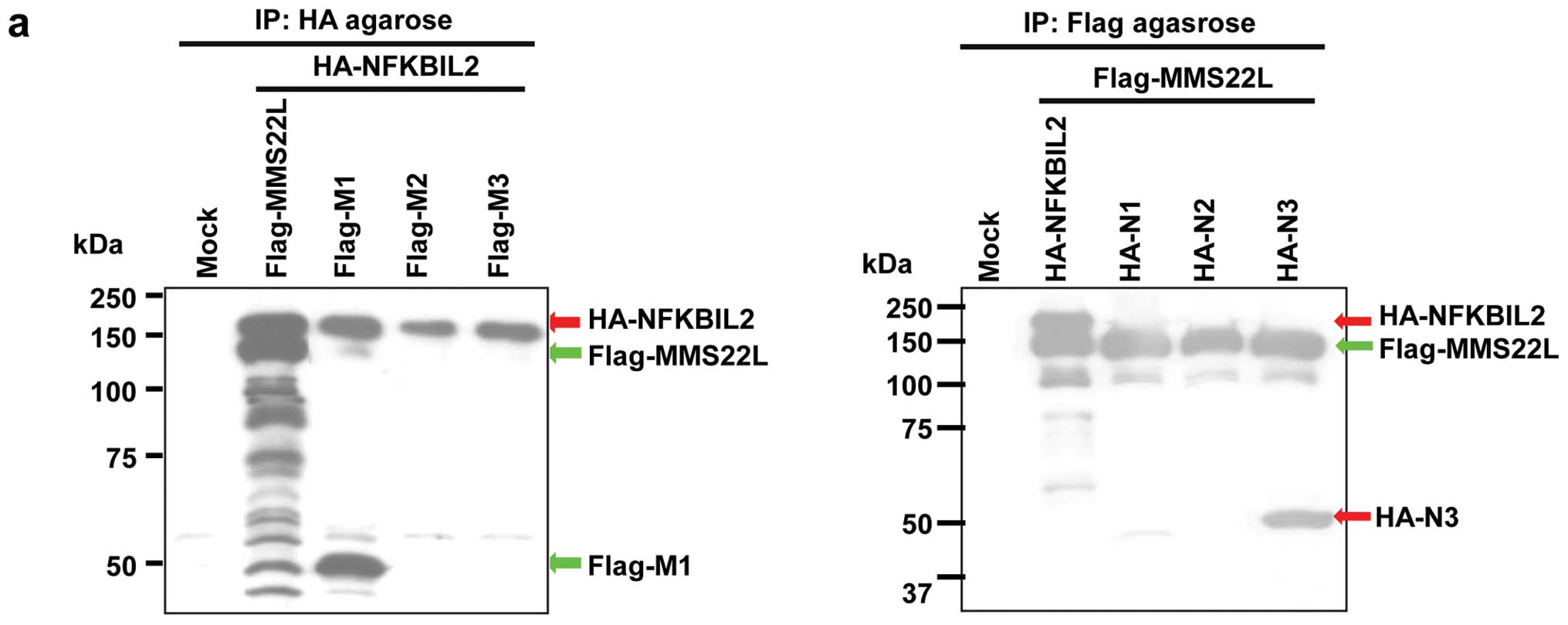
Immunoprecipitation and western blotting assays using antibodies to
Flag- or HA-tags revealed that an N-terminal portion of MMS22L
protein (M1; codon 1–414) could bind to a C-terminal region of
NFKBIL2 (N3; codon 823–1244) (Fig.
4a). Because immunocytochemical analysis revealed that nuclear
localization of MMS22L protein appeared to require the presence of
NFKIL2 protein in the nucleus (Fig.
3b), we subsequently investigated which part of NFKBIL2 protein
is essential for subcellular localization of MMS22L protein in
cultured cells. Plasmids expressing partial proteins of NFKBIL2
were co-transfected with full-length MMS22L expression vector into
COS-7 cells. Interestingly, N-terminal (N1; codon 1–450) and
central part (N2; codon 403–836) of NFKBIL2 proteins could be
localized in the nucleus, while aggregated MMS22L protein was
mainly located in the cytoplasm of the same cells (Fig. 4b). It is concordant with the data
that these two partial proteins (N1 and N2) could not bind to
MMS22L protein as indicated by immunoprecipitation analyses. In
contrast, MMS22L protein and C-terminal part of NFKBIL2 protein
(N3; codon 823–1244) that could bind to MMS22L protein were mainly
localized in the cytoplasm of the cells (Fig. 4b). The data indicate that
N-terminal (N1; codon 1–450) and central (N2; codon 403–836) parts
of NFKBIL2 are more important for nuclear localization of NFKBIL2,
while its C-terminal part (N3; codon 823–1244) is essential for
binding to MMS22L.
Dominant negative growth suppressive
effect of partial NFKBIL2 protein including MMS22L-binding
site
According to the data above, we hypothesized that if
nuclear localization of MMS22L protein is important for cancer
cells growth, reduction of MMS22L protein in the nucleus by
inhibiting the interaction between MMS22L and NFKBIL2 could
suppress the cancer cell growth. To examine whether exogenous
expression of partial N3 protein can inhibit the MMS22L-NFKBIL2
interaction and cell growth, we co-transfected full-length MMS22L
and either of full-/partial-length NFKBIL2 expressing vectors (N1,
N2 or N3) into HEK293 cells, and found that the amount of exogenous
full-length NFKBIL2 protein that binds to exogenous MMS22L was
significantly decreased after introduction of the partial N3
protein, as demonstrated by immunoprecipitation assays, while it
was not changed in the cells transfected with N1 or N2 vectors
(Fig. 5a). To investigate the
functional significance of the interaction between MMS22L and
NFKBIL2 for growth of cancer cells, we transfected either of
vectors expressing partial NFKBIL2 proteins or mock vectors into
two cancer cell lines, HeLa and LC319, which highly expressed both
endogenous MMS22L and NFKBIL2 proteins and lung fibroblast CCDlu-19
cells in which MMS22L expression was hardly detectable. Expectedly,
exogenous expression of the C-terminal portion of NFKIL2 protein
(N3) reduced the levels of MMS22L protein in the nucleus and
inhibited the growth of HeLa and LC319 cells as measured by MTT
assay, while it did not affect the growth of MMS22L-negative
CCDLu-19 cells (Figs. 5b–d). Our
findings imply that inhibition of the interaction between the
MMS22L and NFKBIL2 protein can suppress the nuclear localization of
MMS22L protein, and resulted in the reduction of cancer growth, and
that inhibition of the interaction in cancer cells by small
molecules might be a potential therapeutic strategy for new cancer
treatment.
MMS22L protein acts as an upstream
molecule of NFKB pathway
Since NFKIL2 protein was indicated to be involved in
the NFKB pathway that plays an essential role in the promotion of
cell proliferation and anti-apoptosis (42–45),
we examined the expression of NFKB p65/RelA protein in HeLa cells
in which both exogenous MMS22L and NFKIL2 were introduced, and
found that the level of endogenous p65/RelA protein was elevated
compared with those of cells introduced NFKIL2 alone (data not
shown). The result suggests that the expression of MMS22L-NFKBIL2
complex may positively regulate the NFKB pathway. Subsequently, we
attempted to examine the effect of endogenous MMS22L expression on
the NFKB pathway molecules using cytoplasmic and nuclear fraction
of HeLa and LC319 cells that were treated with TNF-α. We first
confirmed that the level of RelA/p65 was increased in the nucleus
of the cells by TNF-α stimulation (data not shown), but that of
endogenous RelA/p65 protein was decreased in these cells
transfected with siRNA for MMS22L (si-MMS22L) after
TNF-α treatment, compared to cells with control siRNA (si-LUC)
(Fig. 6a). We then examined the
relationship between MMS22L protein and downstream molecules of
RelA/p65 such as Bcl-XL and TRAF1/2 that were the anti-apoptosis
factors. When we treated the si-LUC-transfected HeLa cells with
TNF-α, Bcl-XL and TRAF1 were increased in accordance with the
elevation of RelA/p65 (Fig. 6b).
However, the elevation of p65, Bcl-XL and TRAF1 were not detected
in the TNF-α-stimulated cells transfected with si-MMS22L.
The expression level of MMS22L protein showed good correlation with
those of p65, Bcl-XL and TRAF1 proteins in lung cancer cell lines
(data not shown).
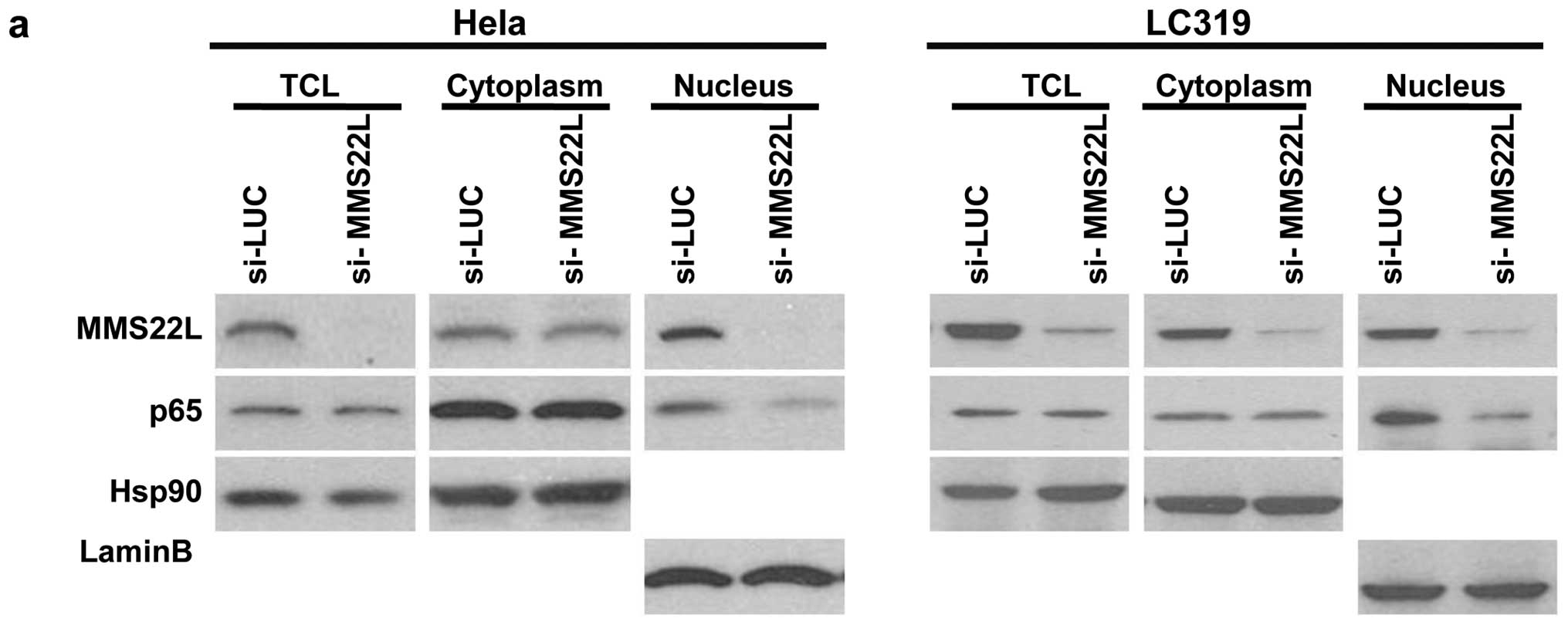 | Figure 6Involvement of MMS22L as an upstream
molecule of NFKB pathway. (a) Western blot analysis using
antibodies to endogenous MMS22L and RelA/p65, and HeLa and LC319
cells transfected with siRNA oligonucleotides for MMS22L
(si-MMS22L) or control siRNA (si-LUC). These cell lines were
stimulated with 50 ng/ml TNF-α for 15 min. The nuclear and
cytoplasmic fraction was isolated using NE-PER™ Nuclear and
Cytoplasmic Extraction Reagents kit (Thermo). (b) Western blot
analysis using antibodies to endogenous MMS22L, RelA/p65, Bcl-XL
and TRAF1 and HeLa cells transfected with si-MMS22L or
si-LUC. These cell lines were treated with 50 ng/ml TNF-α for 15,
30, 60 or 120 min. N.T. indicates no treatment with TNF-α. (c)
Western blot analysis using antibodies to endogenous MMS22L,
RelA/p65, Bcl-XL, ATM, CSB, p53 and HeLa cells which were
transfected with si-MMS22L or si-LUC oligonucleotides, and
were subsequently treated with cisplatin (CDDP; 50 or 100
μg/ml). (d) Schematic summary of MMS22L pathway. |
To further examine the effect of MMS22L expression
on apoptosis pathway in cancer cells, we cultured cancer cells that
were transfected with si-MMS22L under DNA damage condition
using DNA-damaging agents (cisplatin/CDDP or 5-fluorouracil/5-FU).
After knockdown of MMS22L expression with si-MMS22L
in HeLa cells, we treated the cells with CDDP (50 μg/ml) or
5-FU (50 μg/ml) for 48 h and harvested the cells for flow
cytometric analysis. The sub G1 population of the cells which were
transfected with si-MMS22L was significantly increased
compared with those with control siRNA (si-LUC) under DNA damage
condition (data not shown). When we exposed the cells that were
transfected with si-MMS22L or si-LUC with 20 J of
ultraviolet for 48 h, the similar results were observed (data not
shown). Western blot analysis using the HeLa cells which were
transfected with si-MMS22L or si-LUC, and subsequently
treated with CDDP as mentioned above revealed that induction of DNA
repair molecules such as ATM, CSB and p53 as well as RelA/p65 and
its downstream anti-apoptosis factor Bcl-XL were significantly
suppressed in the cells transfected with si-MMS22L compared
with those transfected with si-LUC (Fig. 6c). The data suggest that MMS22L can
function as an upstream molecule of these anti-apoptosis factors
and also affect the induction of some DNA repair pathway molecules
(Fig. 6d).
Discussion
Despite the recent development of surgical
techniques combined with various treatment modalities such as
radiotherapy and chemotherapy, clinical outcome of lung and
esophageal cancer patients still remains poor. Therefore,
development of new types of anticancer drugs is eagerly awaited. To
identify novel target molecules for drug development, we combined
genome-wide expression profile analysis of genes that were
overexpressed in lung and esophageal cancer cells with
high-throughput screening of loss-of-function effects by means of
the RNAi technique and tumor tissue microarray analysis (5–41).
Through this systematic approach we found MMS22L to be
upregulated frequently in clinical lung and esophageal cancer
samples, and showed that this gene product plays an indispensable
role in the growth and/or survival of cancer cells.
We demonstrated that MMS22L is a putative oncogene
and that its nuclear localization and stabilization was enhanced by
binding to NFKBIL2. In addition, we revealed that introduction of
the C-terminal portion of NFKBIL2 protein into cancer cells could
dominant-negatively inhibit the nuclear localization of MMS22L
possibly by blocking the MMS22L-NFKBIL2 interaction, and resulted
in the suppression of cancer cell growth/ survival. Furthermore,
transfection of siRNAs against MMS22L or NFKBIL2 into cancer cells
suppressed their expression and the cell growth. Therefore,
inhibition of the MMS22L-NFKBIL2 interaction or suppressing MMS22L
protein function can be an effective approach for development of
novel cancer therapy.
To date, NFKB transcription factors are known to be
the key regulators of immune, inflammatory and acute phase
responses, and to be involved in the control of cell proliferation
and apoptosis (42–45). Activation of NFKB activity and
consequent induction of its downstream genes lead to the
oncogenesis in mammalian cells. MMS22L protein appeared to act as
an upstream molecule of RelA/p65 and be indispensable for induction
of anti-apoptosis factors, Bcl-XL or TRAF1. Further studies on the
regulation and function of MMS22L protein will contribute to the
understanding of molecular mechanism of carcinogenesis through the
activation of MMS22L and NFKB pathway.
In cancer chemotherapy, many kinds of DNA damaging
agents are being used. The most common approach for targeting the
cell cycle is to exploit the effect of DNA-damaging
chemotherapeutic agents like 5-FU or CDDP, whose effects are
mediated through diverse intracellular targets inducing apoptosis
in various cancer cells (50).
However, the toxicity of DNA-damaging drugs can be diminished by
the activities of several DNA repair pathways as well as
anti-apoptotic factors. Therefore, inhibitors of specific DNA
repair and/or anti-apoptotic pathways might be promising
therapeutic strategy for novel cancer treatments which can improve
the efficacy of DNA damage-based cancer therapy (50). Our data suggested the involvement
of MMS22L in cellular response to DNA damaging agents. In fact,
knockdown of MMS22L expression also enhanced the apoptosis of
cancer cells that were exposed to DNA-damaging agents including
5-FU and CDDP probably due to inhibition of induction of DNA repair
molecules such as ATM, CSB and p53 as well as RelA/p65 and its
downstream anti-apoptosis factor Bcl-XL. The combined data of our
experiments suggest that MMS22L might function as an upstream
molecule of these anti-apoptosis factors and DNA-repair molecules
and that targeting MMS22L could have a significant advantage in
avoiding the resistance of cancer cells to anticancer treatments,
although the detailed function of MMS22L in drug response of the
cells and in carcinogenesis remains to be elucidated.
In summary, our data indicate that MMS22L is
involved in NFKB pathway in cancer cells through its interaction
with NFKBIL2 and that it might be a promising candidate target for
developing highly specific anticancer drugs with minimal risk of
adverse effects.
Acknowledgements
This study was supported in part by
Grant-in-Aid for Scientific Research (B) and Grant-in-Aid for
Scientific Research on Innovative Areas from The Japan Society for
the Promotion of Science to Y.D. Y.D. is a member of Shiga Cancer
Treatment Project supported by Shiga Prefecture (Japan).
References
|
1
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J and
Thun MJ: Cancer statistics, 2009. CA Cancer J Clin. 59:225–249.
2009. View Article : Google Scholar
|
|
2
|
Shimada H, Nabeya Y, Okazumi S, et al:
Prediction of survival with squamous cell carcinoma antigen in
patients with resectable esophageal squamous cell carcinoma.
Surgery. 133:486–494. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Berwick M and Schantz S: Chemoprevention
of aerodigestive cancer. Cancer Metastasis Rev. 16:329–347. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Daigo Y and Nakamura Y: From cancer
genomics to thoracic oncology: discovery of new biomarkers and
therapeutic targets for lung and esophageal carcinoma. Gen Thorac
Cardiovasc Surg. 56:43–53. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kikuchi T, Daigo Y, Katagiri T, et al:
Expression profiles of non-small cell lung cancers on cDNA
microarrays: identification of genes for prediction of lymph-node
metastasis and sensitivity to anti-cancer drugs. Oncogene.
22:2192–205. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kakiuchi S, Daigo Y, Tsunoda T, Yano S,
Sone S and Nakamura Y: Genome-wide analysis of organ-preferential
metastasis of human small cell lung cancer in mice. Mol Cancer Res.
1:485–499. 2003.PubMed/NCBI
|
|
7
|
Kakiuchi S, Daigo Y, Ishikawa N, et al:
Prediction of sensitivity of advanced non-small cell lung cancers
to gefitinib (Iressa, ZD1839). Hum Mol Genet. 13:3029–43. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kikuchi T, Daigo Y, Ishikawa N, et al:
Expression profiles of metastatic brain tumor from lung
adenocarcinomas on cDNA microarray. Int J Oncol. 28:799–805.
2006.PubMed/NCBI
|
|
9
|
Taniwaki M, Daigo Y, Ishikawa N, et al:
Gene expression profiles of small-cell lung cancers: molecular
signatures of lung cancer. Int J Oncol. 29:567–575. 2006.PubMed/NCBI
|
|
10
|
Yamabuki T, Daigo Y, Kato T, et al:
Genome-wide gene expression profile analysis of esophageal squamous
cell carcinomas. Int J Oncol. 28:1375–1384. 2006.PubMed/NCBI
|
|
11
|
Suzuki C, Daigo Y, Kikuchi T, Katagiri T
and Nakamura Y: Identification of COX17 as a therapeutic target for
non-small cell lung cancer. Cancer Res. 63:7038–7041.
2003.PubMed/NCBI
|
|
12
|
Kato T, Daigo Y, Hayama S, et al: A novel
human tRNA-dihydrouridine synthase involved in pulmonary
carcinogenesis. Cancer Res. 65:5638–5646. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Furukawa C, Daigo Y, Ishikawa N, et al:
Plakophilin 3 oncogene as prognostic marker and therapeutic target
for lung cancer. Cancer Res. 65:7102–7110. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Suzuki C, Daigo Y, Ishikawa N, et al: ANLN
plays a critical role in human lung carcinogenesis through the
activation of RHOA and by involvement in the phosphoinositide
3-kinase/AKT pathway. Cancer Res. 65:11314–11325. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ishikawa N, Daigo Y, Takano A, et al:
Characterization of SEZ6L2 cell-surface protein as a novel
prognostic marker for lung cancer. Cancer Sci. 97:737–745. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Takahashi K, Furukawa C, Takano A, et al:
The neuromedin u-growth hormone secretagogue receptor
1b/neurotensin receptor 1 oncogenic signaling pathway as a
therapeutic target for lung cancer. Cancer Res. 66:9408–9419. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hayama S, Daigo Y, Kato T, et al:
Activation of CDCA1-KNTC2, members of centromere protein complex,
involved in pulmonary carcinogenesis. Cancer Res. 66:10339–10348.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kato T, Hayama S, Yamabuki T, et al:
Increased expression of IGF-II mRNA-binding protein 1 is associated
with the tumor progression in patients with lung cancer. Clin
Cancer Res. 13:434–442. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Suzuki C, Takahashi K, Hayama S, et al:
Identification of Myc-associated protein with JmjC domain as a
novel therapeutic target oncogene for lung cancer. Mol Cancer Ther.
6:542–551. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hayama S, Daigo Y, Yamabuki T, et al:
Phosphorylation and activation of cell division cycle associated 8
by aurora kinase B plays a significant role in human lung
carcinogenesis. Cancer Res. 67:4113–4122. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Taniwaki M, Takano A, Ishikawa N, et al:
Activation of KIF4A as a prognostic biomarker and therapeutic
target for lung cancer. Clin Cancer Res. 13:6624–6631. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mano Y, Takahashi K, Ishikawa N, et al:
Fibroblast growth factor receptor 1 oncogene partner as a novel
prognostic biomarker and therapeutic target for lung cancer. Cancer
Sci. 98:1902–1913. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kato T, Sato N, Hayama S, et al:
Activation of holliday junction recognizing protein involved in the
chromosomal stability and immortality of cancer cells. Cancer Res.
67:8544–8553. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kato T, Sato N, Takano A, et al:
Activation of placenta specific transcription factor distal-less
homeobox 5 predicts clinical outcome in primary lung cancer
patients. Clin Cancer Res. 14:2363–2370. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dunleavy EM, Roche D, Tagami H, et al:
HJURP is a cell-cycle-dependent maintenance and deposition factor
of CENP-A at centromeres. Cell. 137:485–497. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hirata D, Yamabuki T, Miki D, et al:
Involvement of epithelial cell transforming sequence-2 oncoantigen
in lung and esophageal cancer progression. Clin Cancer Res.
15:256–266. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sato N, Koinuma J, Fujita M, et al:
Activation of WD repeat and high-mobility group box DNA binding
protein 1 in pulmonary and esophageal carcinogenesis. Clin Cancer
Res. 16:226–239. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sato N, Koinuma J, Ito T, et al:
Activation of an oncogenic TBC1D7 (TBC1 domain family, member 7)
protein in pulmonary carcinogenesis. Genes Chromosomes Cancer.
49:353–367. 2010.PubMed/NCBI
|
|
29
|
Nguyen MH, Koinuma J, Ueda K, et al:
Phosphorylation and activation of cell division cycle associated 5
by mitogen-activated protein kinase play a crucial role in human
lung carcinogenesis. Cancer Res. 70:5337–5347. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ishikawa N, Daigo Y, Yasui W, et al: ADAM8
as a novel serological and histochemical marker for lung cancer.
Clin Cancer Res. 10:8363–8370. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ishikawa N, Daigo Y, Takano A, et al:
Increases of amphiregulin and transforming growth factor-alpha in
serum as predictors of poor response to gefitinib among patients
with advanced non-small cell lung cancers. Cancer Res.
65:9176–9184. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yamabuki T, Takano A, Hayama S, et al:
Dickkopf-1 as a novel serologic and prognostic biomarker for lung
and esophageal carcinomas. Cancer Res. 67:2517–2525. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ishikawa N, Takano A, Yasui W, et al:
Cancer-testis antigen lymphocyte antigen 6 complex locus K is a
serologic biomarker and a therapeutic target for lung and
esophageal carcinomas. Cancer Res. 67:11601–11611. 2007. View Article : Google Scholar
|
|
34
|
Takano A, Ishikawa N, Nishino R, et al:
Identification of nectin-4 oncoprotein as a diagnostic and
therapeutic target for lung cancer. Cancer Res. 69:6694–6703. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sato N, Yamabuki T, Takano A, et al: Wnt
inhibitor Dickkopf-1 as a target for passive cancer immunotherapy.
Cancer Res. 70:5326–5336. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Suda T, Tsunoda T, Daigo Y, Nakamura Y and
Tahara H: Identification of human leukocyte antigen-A24-restricted
epitope peptides derived from gene products upregulated in lung and
esophageal cancers as novel targets for immunotherapy. Cancer Sci.
98:1803–1808. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mizukami Y, Kono K, Daigo Y, et al:
Detection of novel cancer-testis antigen-specific T-cell responses
in TIL, regional lymph nodes, and PBL in patients with esophageal
squamous cell carcinoma. Cancer Sci. 99:1448–1454. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Harao M, Hirata S, Irie A, et al:
HLA-A2-restricted CTL epitopes of a novel lung cancer-associated
cancer testis antigen, cell division cycle associated 1, can induce
tumor-reactive CTL. Int J Cancer. 123:2616–2625. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kono K, Mizukami Y, Daigo Y, et al:
Vaccination with multiple peptides derived from novel cancer-testis
antigens can induce specific T-cell responses and clinical
responses in advanced esophageal cancer. Cancer Sci. 100:1502–1509.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yokomine K, Senju S, Nakatsura T, et al:
The forkhead box M1 transcription factor, as a candidate of target
for anti-cancer immunotherapy. Int J Cancer. 126:2153–2163.
2010.PubMed/NCBI
|
|
41
|
Tomita Y, Imai K, Senju S, et al: A novel
tumor-associated antigen, cell division cycle 45-like can induce
cytotoxic T-lymphocytes reactive to tumor cells. Cancer Sci.
102:697–705. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rayet B and Gelinas C: Aberrant rel/nfkb
genes and activity in human cancer. Oncogene. 18:6938–6947. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tergaonkar V: NFκB pathway: A good
signaling paradigm and therapeutic target. Int J Biochem Cell Biol.
38:1647–1653. 2006.
|
|
44
|
Yamamoto Y and Gaynor RB: Therapeutic
potential of inhibition of the NF-kappaB pathway in the treatment
of inflammation and cancer. J Clin Invest. 107:135–142. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kim HJ, Hawke N and Baldwin AS: NF-κB and
IKK as therapeutic targets in cancer. Cell Death Differ.
13:738–747. 2006.
|
|
46
|
O’Donnell L, Panier S, Wildenhain J, et
al: The MMS22L-TONSL complex mediates recovery from replication
stress and homologous recombination. Mol Cell. 40:619–631.
2010.PubMed/NCBI
|
|
47
|
Duro E, Lundin C, Ask K, et al:
Identification of the MMS22L-TONSL complex that promotes homologous
recombination. Mol Cell. 40:632–644. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Brenda C, O’Connell L, Adamson B, et al: A
genome-wide camptothecin sensitivity screen identifies a mammalian
MMS22L-NFKBIL2 complex required for genomic stability. Mol Cell.
40:645–657. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Piwko W, Olma MH, Held M, et al:
RNAi-based screening identifies the Mms22L-Nfkbil2 complex as a
novel regulator of DNA replication in human cells. EMBO J.
29:4210–4222. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lee BJ, Chon KM, Kim YS, et al: Effects of
cisplatin, 5-fluorouracil, and radiation on cell cycle regulation
and apoptosis. Chemotherapy. 51:103–110. 2005. View Article : Google Scholar : PubMed/NCBI
|