Introduction
Metastatic spread of tumor cells is the most lethal
aspect of cancer and often occurs via the lymphatic vessels
(1). Tumor-associated lymphatic
vessels, also referred to as tumor lymphangiogenesis (2), i.e., new lymphatic vessel formation
in cancer (3), act as a conduit by
which disseminating tumor cells access regional lymph nodes and
form metastases (4,5). Lymphangiogenic growth factors, which
include the secreted glycoproteins vascular endothelial growth
factor-C (VEGF-C), vascular endothelial growth factor-D (VEGF-D)
and their cognate receptor tyrosine kinase vascular endothelial
growth factor receptor-3 (VEGFR-3) located on lymphatic endothelial
cells can advance or regulate proliferation, migration, metastasis
and survival of lymphatic endothelial cells (LECs), and lymphatic
tube formation in physiological and pathological conditions such as
wound repair, tissue regeneration and tumorigenesis (6–8).
These molecules play an important role on lymphangiogenesis and
metastatic spread of tumor cells to lymph nodes (9,10).
So, a targeted approach to block lymphangiogenesis pathways, such
as VEGF-C, or/and -D/VEGFR-3 axis, seems to be an attractive
anticancer treatment strategy.
Norcantharidin (NCTD), a demethylated form of
cantharidin with anti-tumor properties and an active ingredient of
traditional Chinese medicine-Mylabris, has been reported to possess
potent antiangiogenesis and antitumor properties in several cell
lines and tumor xenograft models (11–16).
However, its role in tumor-associated lymphangiogenesis and
lymphatic metastasis remains unclear. Here, we investigated for the
first time the effect of NCTD on proliferation, migration,
tube-formation, i.e., lymphangiogenesis of human lymphatic
endothelial cells (HLECs), and their VEGF-C or/and -D/VEGFR-3
pathways in vitro, so as to explore wether it served as a
target inhibitor for the HLEC lymphangiogenesis and whether it is a
potential anti-lymphangiogenic agent of cancer therapeutic
strategy.
Materials and methods
Cell lines and cell culture
HLECs used in this study were human dermal lymphatic
endothelial cells (HDLECs). Primary HDLECs were purchased from
ScienCell Research Laboratories, USA, and were identified by using
immunofluorescent cyto-chemical technique via CD31,
podoplanin and Lyve1. Tissue culture flasks were peridiumed with
fibronectin (1 mg/ml; Chemicon, USA). Cells were cultured in
endothelial cell growth medium (ECGM) with endothelial cell growth
factor (ScienCell Research Laboratories) in an incubator (Forma
Series II HEPA Class 100, Thermo Co., USA) with 5% CO2
at 37°C. The medium was changed every 2 days. When the cells became
confluent, at 80% plating efficiency, they were treated with 5 ml
phosphate-buffered saline (PBS) solution, digested with 1 ml 0.025%
trypsin and 1 ml 0.02% ethylenediamine tetraacetic acid (EDTA)
solution (ScienCell Research Laboratories), and observed by
inversion microscope (Nikon TS100, Japan). Then the cells were
retuned to culture at 37°C in 5% CO2, and continued
transfer of culture. The cells, which were at fifth generation,
were used in the experiment.
Proliferation assay
The cultured HDLEC suspensions (5×105
cells/ml) were used in this assay. The cultures were divided into
NCTD groups and control groups. The tetrazolium-based colorimetric
assay (MTT) was used to evaluate the inhibitory effect of NCTD on
proliferation of HDLECs in vitro (tumor cytotoxicity test).
After HDLECs were cultured in a 96-well plate peridiumed with
fibronectin (5×104 cells/ml × 100 μl/well) in culture
medium (ECGM, 100 μl/well) overnight, they were without (negative
control, equal ECGM solution) or treated with various
concentrations (1.25–80 μg/ml, experimental groups; 6 wells per
concentration) of NCTD (10 mg/2 ml, Jiangsu Kangxi Pharmaceutical
Works, China) in fresh culture medium at 37°C in 5% CO2
for 24 h. The tumor cell cytotoxicity was determined by MTT
(methyltiazolyl tetrazolium, Sigma, MO, USA). The optical densities
(A value) at 490 nm were measured with an ELISA reader (Elx800UV,
Bio-Tek Co., USA). The A490 value of the experimental groups was
divided by the A490 value of untreated controls and presented as a
percentage of the cells. The inhibitory percent of NCTD on HDLECs
(%) = (1-A490 value in the experimental group / A490 value of
control group) × 100%. Three separate experiments were performed.
The concentration of drug giving 50% growth inhibition
(IC50) was calculated from the formula IC50=
lg−1{Xm-I [P-(3-Pm-Pn)/4]}. In equal experimental
conditions, HDLECs were treated with 5 μg/ml concentration of NCTD
at 6-h interval for 48 h, the optical densities (A value) and the
inhibitory percent of NCTD on HDLECs were measured and
calculated.
Apoptosis assay
HDLECs (1×105 cells/ml) were cultured on
the fibronectin-peridium slides in a 96-well plate overnight. When
slides were filled with 50–80% cells, they were treated without
(negative control, equal ECGM solution) or treated with various
concentrations (1.25–10 μg/ml, experimental groups) of NCTD,
respectively. Then, the slides were fixed with 0.5 ml 4% formalin,
washed with PBS solution, and stained with 0.5 ml fluorescence
agent Hoechst 33258 (Sigma) and CY3 NHS ester (Lumiprobe, USA).
Apoptosis of HDLECs was observed under a fluorescence microscope
(Nikon Eclipse TE2000-U, Japan). Ten sample slides in each group
were chosen by analysis. Visual fields (>10) were observed or
>500 cells were counted per slide. The apoptotic percent of each
group = apoptotic cells/cells in all × 100%.
Flow cytometry
HDLECs (1×105 cells/ml) cultured in a
96-well plate peridiumed with fibronectin were without (negative
control, equal ECGM solution) or treated with various
concentrations (1.25–15.00 μg/ml, experimental groups; 6 wells per
concentration) of NCTD at 37°C in 5% CO2 for 24 h, then
were made up into the cell suspension (5×105 cells/ml),
and suspended in 500 μl binding buffer. Tumor DNA was then stained
for 15 min with 5 μl Annexin V-FITL and propidium iodine (PI,
Sigma). DNA value and apoptotic rate of HDLECs in each group were
determined by Cell Apoptotic Detection Kit (BioDev Co., China) and
Fluorescent Activated Cell Sorter (420 type FACS flow cytometry,
Becton-Dickinson, CA).
Morphological observation
HDLECs (1×105 cells/ml) cultured in a
96-well plate peridiumed with fibronectin overnight were without
(negative control, equal ECGM solution) or treated with various
concentrations (1.25–5.00 μg/ml, experimental groups; 6 wells per
concentration) of NCTD for 24 h, and then observed under an
inverted microscope (I×50/I×70, Olympus, Japan). Simultaneously in
the control group and 2.50 μg/ml NCTD group, they were digested
with 0.025% trypsin and 0.002% EDTA solution, centrifuged (200 g
for 10 min), washed with PBS solution, and fixed in 2.5%
glutaraldehyde-1% osmium tetroxide buffered with PBS (pH 7.2). The
specimens were dehydrated in a graded ethanol series, embedded in
Spury Resin, cut into 50–70 nm sections, double-stained with uranyl
acetate-lead citrate, and analyzed by standard procedures under a
transmission electron microscope (JEM-1230, Jeol Co., Japan).
Migration assay
The cultured HDLECs were inoculated in 4 culture
dishes peridiumed with fibronectin (1×105 cells/ml × 100
μl/dish) in fresh culture medium (ECGM, 100 μl/dish). When HDLECs
were fused into a single layer in dishes, a straight line on the
surface of the growthing cells was scraped with sterile blades by
scraping line method. Then, the cells at side of the line were
scraped off completely using a plastic scraper, washed with
D-Hanks. The cultures were treated without (negative control, equal
ECGM solution) or treated with various concentrations (1.25–5
μg/ml, experimental groups, 4 dishes per concentration) of NCTD in
fresh culture medium at 37°C in 5% CO2 for 24 h. Lastly,
cell migration distance onto the scraped areas and migrating cells
were measured and counted in 4 independent microscopic visual
fields (×100) under inversion microscope (Nikon TS100, Japan), and
expressed as mean number per 1 field, so as to determine the
inhibitory effect of NCTD on proliferation and migration of
HDLECs.
Invasion assay
Living HDLECs were trypsinized with 0.25% trypsin
and washed with fresh culture medium, suspended in the culture
medium with 10% bovine calf serum (1×105 cells/ml). The
cell suspension was transferred to the above layer of the Matrigel
invasion chamber (0.3 ml/every chamber), while 0.8 ml of RPMI-1640
medium with 10% bovine calf serum was only added to the bottom
layer of the Matrigel invasion chamber. Then the cells were
cultured in 50 ml/l CO2 at 37°C for 24 h. The cells were
treated without (untreated control group) or with various
concentrations of NCTD (the six-concentration groups, every
concentration ×6) in fresh culture medium (0.3 ml/every chamber),
were cultured in an incubator with 50 ml/l at 37°C for 72 h. The
passing-membrane cells were collected from the above layer of the
Matrigel invasion chamber, centrifuged (200 r/min, 10 min), dyed by
trypan blue dye, and counted in a hemocytometer. Each experiment
was performed thrice.
Lymphatic tube formation assay
First, the mixed fibrinogen gel was prepared with
fibrin gel [which was from human fibrinogen (3 mg/ml; Bite Bio Co.,
Beijing, China) and human thrombin (50 U/ml; Sigma) dissolved with
PBS], solidified by adding in order 500 μl fibrinogen, 100 μl ECGM
and 10 μl thrombin solution, and incubated at 37°C in 5%
CO2 for 30 min to promote gelling, according to the
literature. Then, 6-well plates were coated with the fibrinogen gel
(0.05 ml/well) and incubated at 37°C for 1 h to promote gelling.
HDLECs (5th-generation cells) suspension (1×105/ml, 300
μl/well) were seeded on the gel surface, cultured in ECGM (2 ml) in
an incubator with 5% CO2 at 37°C. The medium was changed
every 2 days. After 1 week, the cells were treated without (equal
ECGM solution; the control group) or with 2.5 μg/ml NCTD, 100 μl
mF4-31C1 (a soluble VEGFR-3 antibody) and NCTD+mF4-31C1
(experimental groups, 6/each group), respectively, for 2–4 days.
Inverted phase-contrast light microscope (Olympus) was used to
observe and record the morphological changes between the groups and
the tube formation by HUVECs, i.e., lymphangiogenesis in
vitro. Tube formation ability was quantified by counting the
total number of cell clusters and branching in 5 randomly chosen
microscopic fields per well under magnification ×100. Results were
expressed as the mean percentage of branching over total cell
clusters and expressed as a ratio to the control.
Immunocytochemistry
HDLEC suspension (3×105 cells/ml, 400
μl/well) was seeded on the fibronectin-peridium slides in a 96-well
plate overnight, cultured continually for 48 h. When cells were
fused into a single layer, they were treated without (equal ECGM
solution; the control group) or treated for 24 h with 2.5 μg/ml
NCTD, 100 μl mF4-31C1 (a soluble VEGFR-3 antibody), and
NCTD+mF4-31C1, respectively. Then, the slides were fixed with the
solution of methanol and acetone (v/v=1: 1), were used in the
experiment.
VEGF-C, VEGF-D, VEGFR-3 protein products from HDLECs
of each group were determined by streparidinperoxidase (S-P)
staining method, according to the kit brochure (Jinmei
Biotechnology Co., Ltd., Shanghai). The slides from each group were
washed with PBS solution, treated with 3%
H2O2 and 1% TritonX-100 solution, were added
in order with A reagents, i.e., primary antibody [rabbit anti-human
monoclonal antibody VEGF-C (invitrogen, USA), VEGF-D (Abcam, USA),
VEGFR-3 (Cell Signaling, USA)], B reagent (biotinylated anti-rabbit
secondary), C reagent (HRP logo Streptavidin) and DAB solution,
respectively. Then, slides were rinsed in distilled water,
dehydrated through alcohol and xylene and mounted coverslip using a
permanent mount medium and observed by optic microscope (Olympus).
For negative control, the slides were treated with PBS in place of
primary antibody. Six sample slides in each group were chosen by
analysis. Visual fields (>10) were observed or >500 cells
were counted per slide.
Western blot analysis
The drug experiment for HDLECs in vitro was
the same as for immunocytochemistry. Expression of VEGF-C, VEGF-D,
VEGFR3 proteins from HDLECs of each group were determined by
western blot analysis (Lowry method; Lowry method protein kit, Puli
Lai Co., Shanghai, China). Cells were lysed with 200 ml of cell
lysis buffer (Promega) containing a cocktail of protease inhibitors
(Nacalai Tesque) and the supernatant of the lysed cells was
recovered. An aliquot of 20 mg of proteins was subjected to sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) under
reducing condition, and were then transferred to a PVDF membrane.
One hour after being blocked with PBS containing 5% non-fat milk,
the membrane was incubated overnight, was then added in order with
each primary antibody [(rabbit anti-human VEGF-C, VEGF-D, VEGFR-3
antibodys (1:500; Abcam), and rabbit anti-human β-actin antibody
(1:5000; Cell Signaling, USA)] diluted with PBST containing 5%
non-fat milk at 4°C, an appropriate anti-rabbit HRP-labeled
secondary antibody (1:5000; Abcam), HistoFine (Dako, Glostrup,
Denmark) at room temperature for 2 h. The target proteins were
visualized by an enhanced chemiluminescent (ECL) reagent (GE
Healthcare, USA), imaged on the Bio-Rad chemiluminescence imager.
The gray value and gray coefficient ratio of each protein was
analyzed and calculated.
RT-PCR analysis
The drug experiment for HDLECs in vitro was
the same as immunocytochemistry. Expression of VEGF-C, VEGF-D,
VEGFR3 mRNAs from HDLECs of each group were determined by
quantitative real-time polymerase chain reaction (QRT-PCR) assay.
QRT-PCR was performed as described by the manufacturer. Total RNA
was extracted from HDLECs using the TRIzol reagent. Concentration
of RNA was determined by the absorption at 260. The primers for
amplification were designed and synthesized by Shanghai Sangon Co.
The primers for VEGF-C, VEGF-D, VEGFR3 and GAPDH were as follows:
VEGF-C (106 bp) 5′AGT CGC GAC AAA CAC CTT CT3′ (sense), 5′GTA GCT
CGT GCT GGT GTT CA3′ (anti-sense); VEGF-D (194 bp) 5′CAG TGG GTT
GAT CTG GGA CT3′ (sense), 5′CAT GCA GGG TAG GAT GGT CT3′
(anti-sense); VEGFR-3 (117 bp) 5′CGG CTT CAG CTG TAA AGG AC3′
(sense), 5′ TTG TAA AAC ACC TGG CCT CC3′ (anti-sense); GAPDH (128
bp) 5′GCC TCC AAG GAG TAA GAC CC3′ (sense), 5′TGT GAG GAG GGG AGA
TTC AG3′ (anti-sense). Polymerse chain reactions were performed in
a 20-μl reaction volume. RT-PCR reaction was run in the following
conditions: at 94°C for 5 min; at 94°C for 30 sec, at 55°C for 30
sec, at 72°C for 30 sec, 35 circles; at 72°C for 10 min. The
amplifying conditions were control group: at 50°C for 2 min, NCTD
group: at 95°C for 5 min, mF4-31C1 group: at 95°C for 45 sec, and
NCTD+mF4-31C1 group: at 60°C, 45 sec; 40 circles. PCR products (10
μl) were placed onto 15 g/l agarose gel and observed by EB
(ethidium bromide) staining using the ABI PRISM 7300 SDS
software.
Statistical analysis
Statistical analyses were performed using SPSS 13.0.
The data are presented as mean ± SD. Statistical differences were
evaluated using Student’s t-test or the χ2 test.
P<0.05 was considered statistically significant.
Results
NCTD inhibits HLEC proliferation
As shown in Fig. 1,
the cultured HDLECs began to grow at 6 h, maturated at 24 h, which
were predominantly of shuttle-shape, or accumulation, with abundant
cytoplasm, clear nuclei (control group, Fig. 1Aa1); after NCTD treatment, the
morphology of HDLECs showed visible cell aggregation, floating,
nuclear condensation or fragmentation, cataclysm, apoptotic bodies,
or even death (Fig. 1Aa2–a4).
Furthermore, NCTD inhibited markedly the HDLEC proliferation in a
dose- and time-dependent manner with the IC50 value 6.8
μg/ml (Fig. 1B). Thus, NCTD
inhibits significantly the proliferation and growth of HLECs.
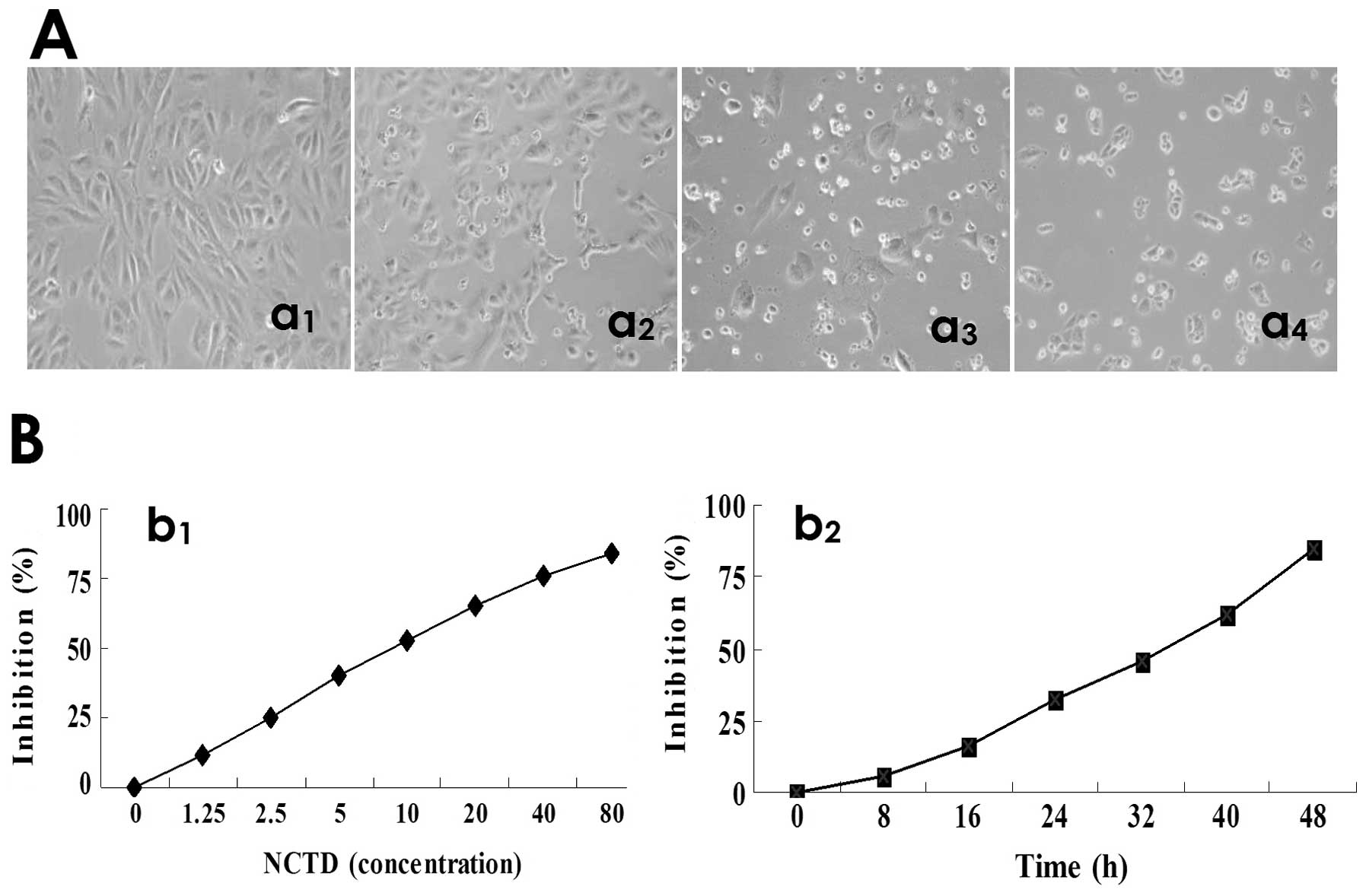 | Figure 1.Inhibitory effects of NCTD on HDLEC
proliferation in vitro. (A) Histomorphology of HDLECs under
inversion optic microscope (magnification ×200) at 24 h, such as
predominantly shuttle-shape, with abundant cytoplasm, clear nuclei
in the control group (a1); visible cell aggregation, floating,
nuclear condensation, chromatin redistributing, nuclear
fragmentation, cataclysm, apoptotic bodies, or even death in
different concentration groups of NCTD (a2, NCTD 1.25 μg/ml; a3,
NCTD 2.5 μg/ml; a4, NCTD 7.5 μg/ml). (B) The dose (b1)-, and time
(b2)-response curves of NCTD effect on HDLECs with the
IC50 value of 6.8 μg/ml. Cell number was counted by the
MTT method. |
NCTD induces HLEC apoptosis
In this study, apoptosis of HLECs was evaluated by
flow cytometry (FCM), Hoechst immunofluorescence dye and
transmission electron microscopy. As shown in Fig. 2, after treatment with NCTD for 24
h, NCTD induced significantly HDLEC apoptosis in vitro when
compared with control group (1.280±0.010 vs. 16.380±1.400%,
P<0.05), with a dose- and time-dependent manner, i.e., apoptosis
percent of HDLECs (total cells under right quadrant of cells)
increased (FCM analysis, Fig. 2A);
viable HDLECs decreased, apoptotic cells which presented bright
blue or orange dyes, even with fine or fragmented nuclei increased
(fluorescence microscopy observation, Fig. 2B), along with increase of NCTD
concentration. In addition, Fig.
2C shows under electron microscope irregular cells with
abundant microvilli, clear cell organelles, larger
nucleus/cytoplast ratio, irregular nuclei and chromatin enrichment
compared to the control group (Fig.
2Cc1); and decreased microvilli, golgiosome atrophy,
mitochondria swelling, cytoplast vacuoles, nuclear shrinkage,
chromosome condensation, chromatin aggregation and typical
apoptosis bodies in NCTD (2.5 μg/ml) group (Fig. 2Cc2–c4). It is thus clear that NCTD
induces significantly apoptosis of HLECs.
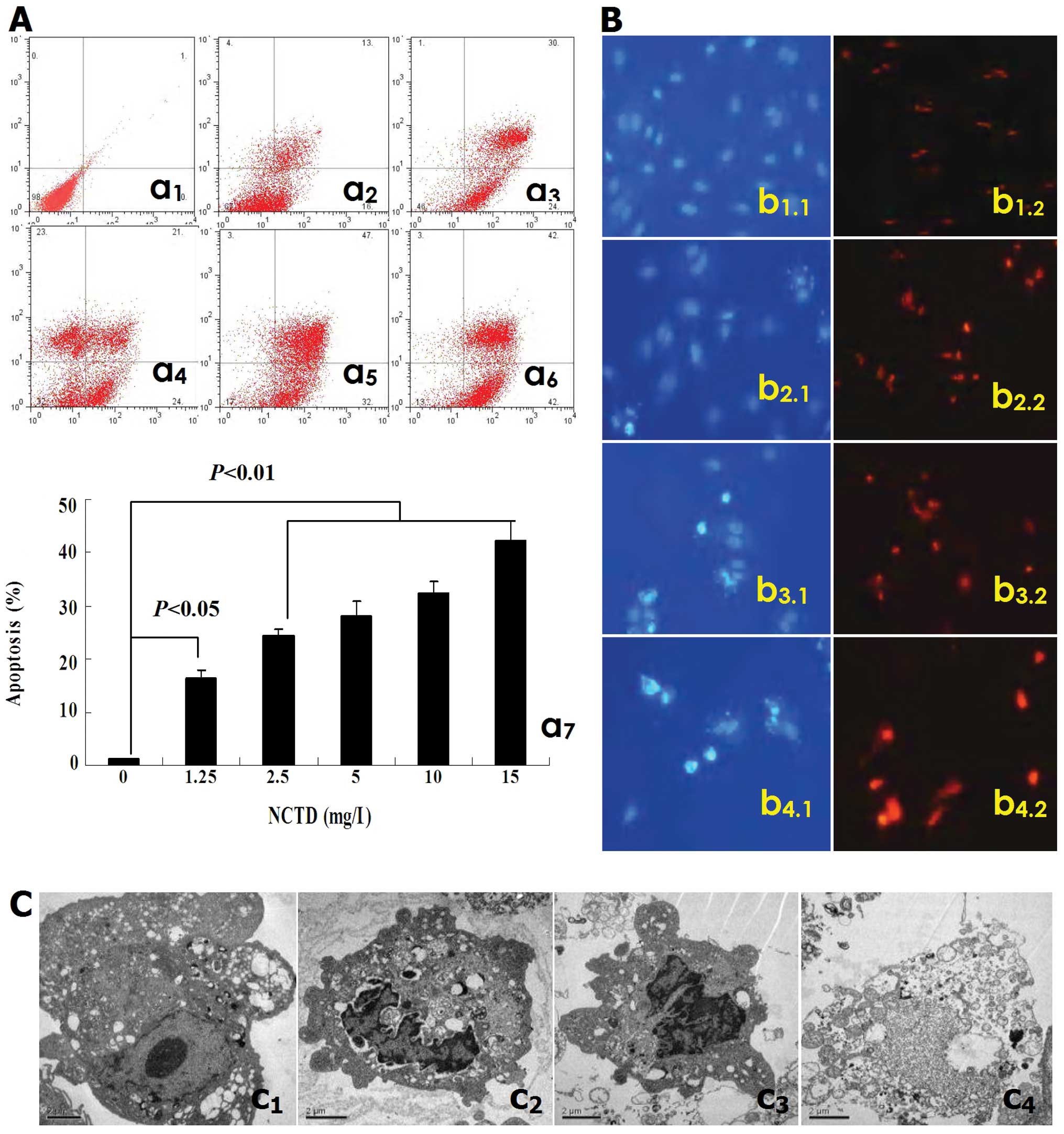 | Figure 2.Inductive effect of NCTD on HDLECs
apoptosis in vitro. (A) a1, control group; a2–a6, NCTD
groups, i.e., a2, 1.25 μg/ml; a3, 2.5 μg/ml; a4, 5.0 μg/ml; a5, 10
μg/ml; a6, 15 μg/ml. a7, the effect of NCTD on HDLECs’ apoptosis;
FCM analysis. NCTD induced significantly HDLEC apoptosis when
compared with control group. (B) b1, control group; b2, NCTD 1.25
μg/ml; b3, NCTD 5.0 μg/ml; b4, NCTD 10 μg/ml; Hoechst and cy3 dye,
fluorescent microscope ×200. Many cells with normal blue- (b1.1) or
brown-dye (b1.2) nuclei, without apoptosis in control group; but
with increase of NCTD concentration, viable HDLECs decreased,
apoptotic cells which presented bright blue-dye (b2.1–b4.1) or
orange-dye (b2.2–b4.2), even with fine or fragmented nuclei
increased (b2.1–b4.1). (C) NCTD 2.5 μg/ml; transmission electron
microscope ×8000. Irregular cells with abundant microvilli, clear
organelles, larger nucleus/cytoplast ratio, irregular nuclei and
chromatin enrichment in control group (c1); and microvilli
decreasing, golgiosome atrophy, mitochondria swelling, cytoplast
vacuole, nuclear shrinkage, chromosome condensation, chromatin
aggregation and typical apoptosis bodies in NCTD group (c2–c4). |
NCTD inhibits HLEC migration
The effects of NCTD on migration of the cultured
HDLECs, i.e., cell migration distance onto the scraped areas and
migrated cells were measured by scraping line method under an
inversion microscope. As shown in Fig.
3, after treatment with NCTD for 24 h and along with increase
of NCTD concentration, migrated cells (11.5±3.4, 17.7±2.8 or
32.5±4.3 vs. 56.3±5.2 n, P=0.000; Fig.
3Bb1) onto the scraped areas were decreased, cell migration
distance (75.6±9.7, 163.4±15.3 or 258.7±28.7 vs. 405.7±20.5 μm,
P=0.000; Fig. 3Bb2) was shortened
significantly, when compared with control group. It was shown that
NCTD inhibited significantly migration of HLECs in vitro, in
a dose-dependent manner.
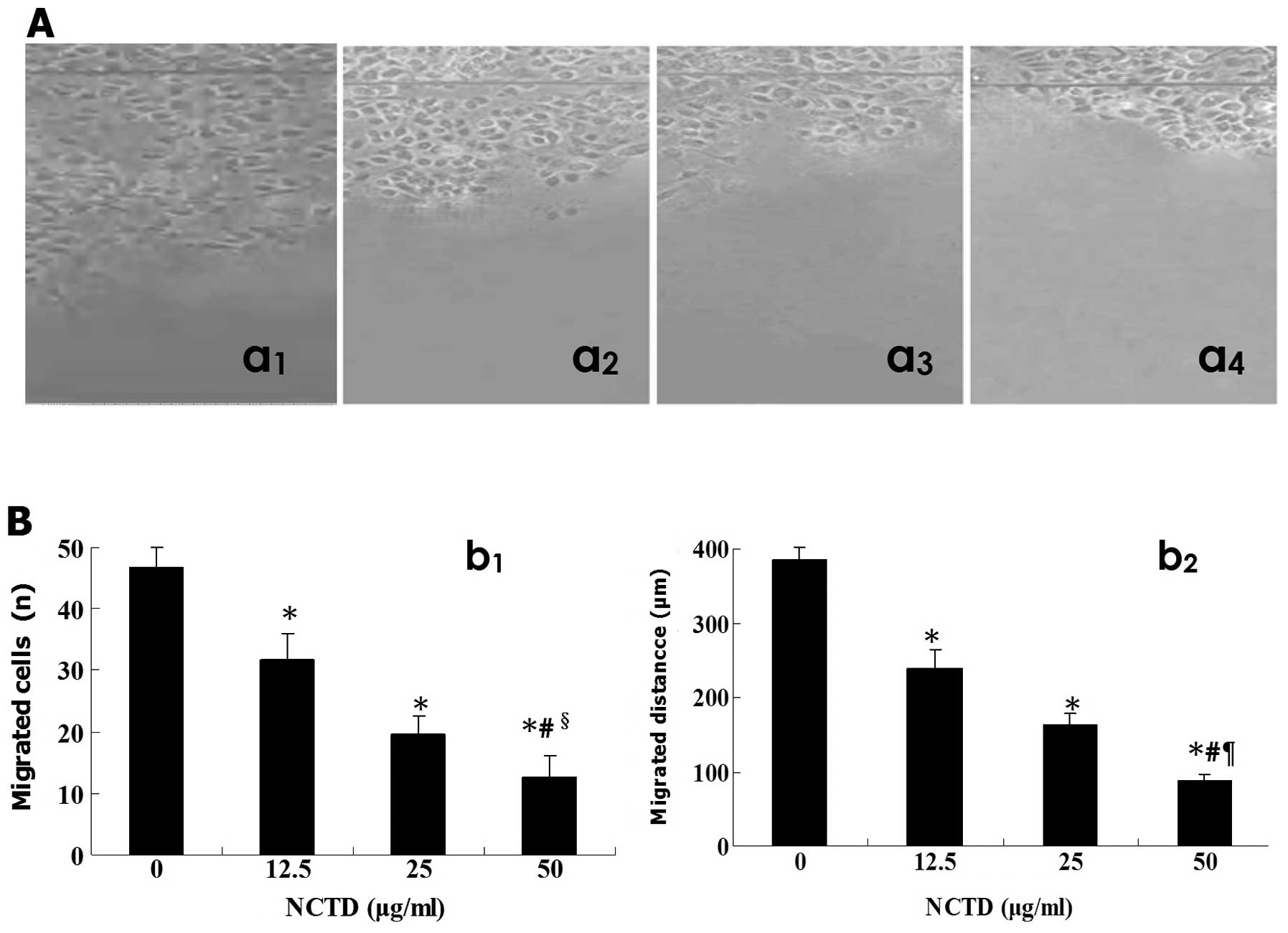 | Figure 3.Inhibitory effect of NCTD on HDLEC
migration in vitro. (A) Migration assay of HDLECs by
scraping line method under an inversion optic microscope,
magnification ×200 (a1, control group; a2, 1.25 NCTD μg/ml; a3, 2.5
NCTD μg/ml; a4, 5.0 NCTD μg/ml). (B) The effect of NCTD on HDLEC
migration (b1, migrated cells onto the scraped areas; b2, migration
distance of cells onto the scraped areas in different groups). NCTD
inhibited significantly HDLEC migration when compared with control
group. *P=0.000, vs. control group; #P=0.000,
vs. 1.25 μg/ml NCTD group; §P=0.015;
¶P=0.034, vs. 2.5 μg/ml NCTD group. |
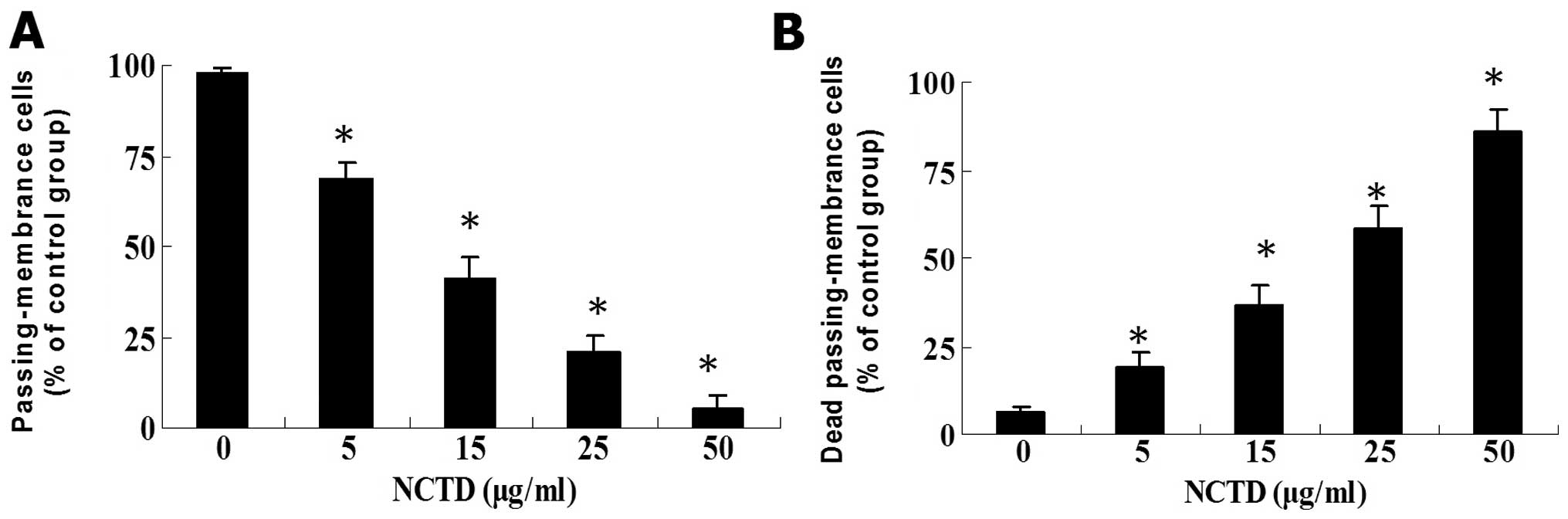
NCTD inhibits HLEC invasion
The effect of NCTD on invasion of the cultured
HDLECs was measured by Matrigel invasion experiment. As shown in
Fig. 4, HDLECs in control group
passed more of the membrane and had more invasive capability in
vitro; NCTD began to inhibit the invasion of HDLECs at the
concentration of 5 μg/ml and as its concentration increased, their
passing membrane cells markedly decreased, the trypan blue dyed
cells, namely the dead passing-membrane cells obviously increased
(P<0.01). At 50 μg/ml of NCTD, the invasive action of HDLECs was
inhibited almost completely. Thus, NCTD inhibited significantly
invasion of HLECs in a dose-dependent manner.
NCTD inhibits HLEC lymphatic tube
formation
The lymphatic capillary-like structures (i.e.,
lymphangiogenesis) formed from the three-dimensional culture of
HDLECs in vitro was observed by inverted phase-contrast
light microscopy. As shown in Fig.
5, when seeded on the mixed fibrinogen gel for 24 h, HDLECs
started to paste the well walls, grew, spread out, formed cell
groups composed of multi-angular or pseudopod cells (Fig. 5Aa1); formed tubular rudiment
structure at the third day (Fig.
5Aa2); had some typical lymphatic capillary-like tubes, with
obvious pipe walls, the lumen and progressive branches after 1 week
(Fig. 5Aa3). At the second day in
the process of lymphatic tube formation, HDLECs started to grow in
the form of pasting the wall of the well, to spread out and form
tubular rudimental structures in control group (Fig. 5Bb1); HDLECs lost the capacity of
the above lymphatic tube formation, with visible cell aggregation,
floating, nuclear fragmentation, apoptosis and necrosis, after
using NCTD (Fig. 5Bb2), mF4-31C1
(Fig. 5Bb3) or NCTD+mF4-31C1
(Fig. 5Bb4) for 2 days. By
counting the total number of cell clusters and branching of
lymphatic tube formation, it was found that the lymphatic
capillary-tube number in NCTD, mF4-31C1 or NCTD+mF4-31C1 group was
less than that in the control group (10.9±2.3, 32.8±5.4 or 9.8±1.5,
vs. 68.4±5.2, all P<0.000), while the lymphatic number in NCTD
or NCTD+mF4-31C1 group was less than that in mF4-31C1 group
(10.9±2.3 or 9.8±1.5, vs. 32.8±5.4, all P<0.01). One week after
lymphatic tube formation, there are some typical lymphatic tube
structures in the control group (Fig.
5Cc1); the formed structure of lymphatic tubes was destroyed,
with visible cell aggregation, floating, nuclear fragmentation,
apoptosis and necrosis, after using NCTD (Fig. 5Cc2), mF4-31C1 (Fig. 5Cc3) or NCTD+mF4-31C1 (Fig. 5C44) for 4 days. It was shown that
NCTD or mF4-31C1 obviously inhibited and destroyed the forming of
lymphangiogenesis and formed lymphangiogenesis from the 3-D culture
of HDLECs in vitro, while this effect of NCTD or
NCTD+mF4-31C1 was stronger. Thus, NCTD or mF4-31C1 effectively
inhibits lymphatic tube formation of HLECs.
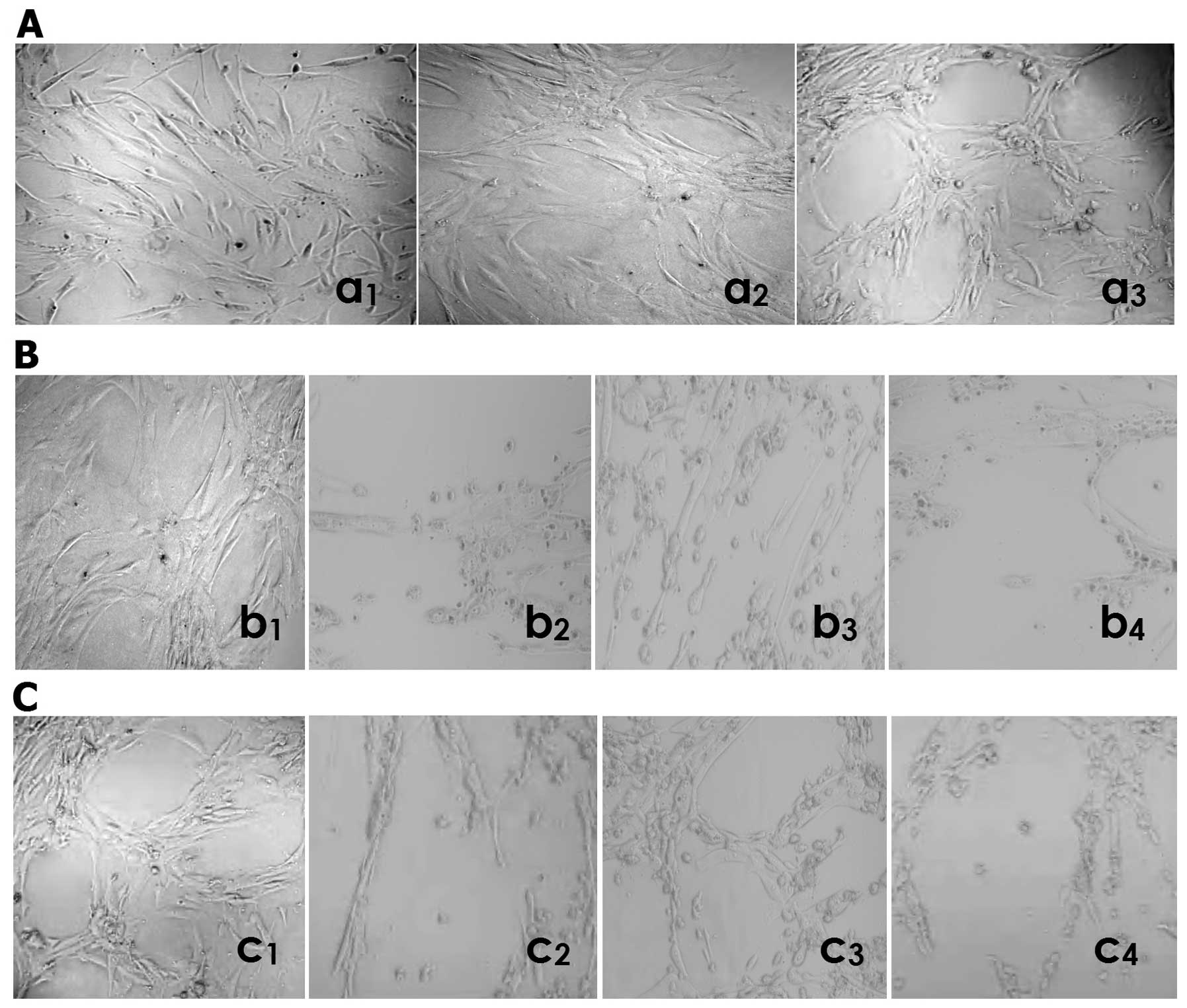 | Figure 5.Lymphatic tube formation i.e.,
lymphangiogenesis of HDLECs in different groupsby inverted
phase-contrast light microscopy (magnification ×200) in
vitro. (A) (in control group): when seeded on the mixed
fibrinogen gel, HDLECs started to paste the wall of the well, grew,
spread out, formed cell groups composed of multi-angular or
pseudopod cells at 24 h (a1); formed tubular rudiment structure at
the third day (a2); had some typical lymphatic tube formation, with
obvious pipe walls, lumen and progressive branches (a3) after 1
week. (B) (at the second day in the process of lymphatic tube
formation): in control group, HDLECs started to grow in the form of
pasting the wall of well, to spread out and form tubular rudiment
structure (b1); using NCTD (b2), mF4-31C1 (b3) or NCTD+mF4-31C1
(b4) for 2 days, HDLECs lost the capacity of the above lymphatic
tube formation, with visible cell aggregation, floating, nuclear
fragmentation, apoptosis and necrosis. (C) One week after lymphatic
tube formation, there are some typical lymphatic tube structures in
the control group (c1); the formed structure of lymphatic tubes was
destroyed, with visible cell aggregation, floating, nuclear
fragmentation, apoptosis and necrosis, after using NCTD (c2),
mF4-31C1 (c3) or NCTD+mF4-31C1 (44) for 4 days. |
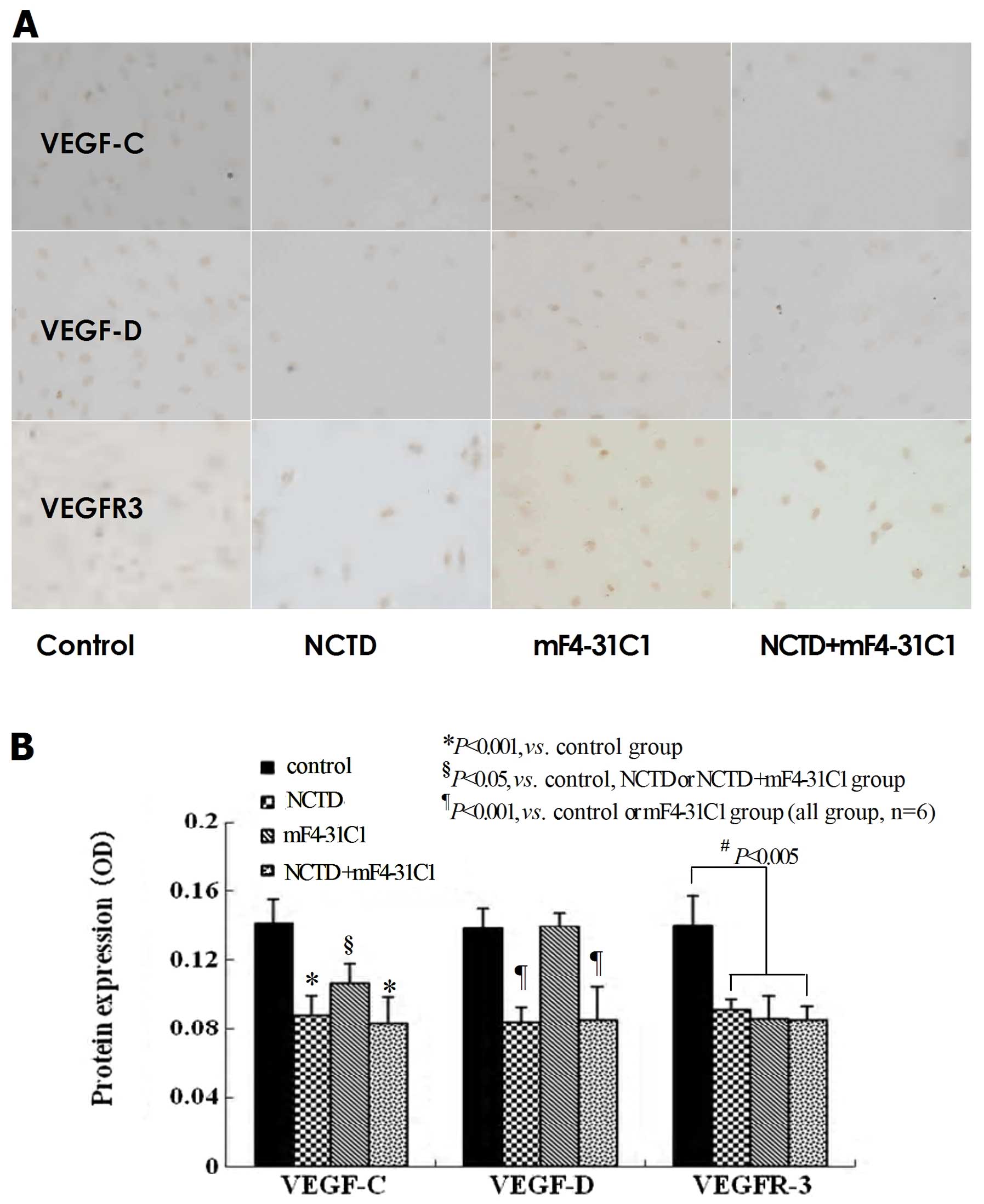
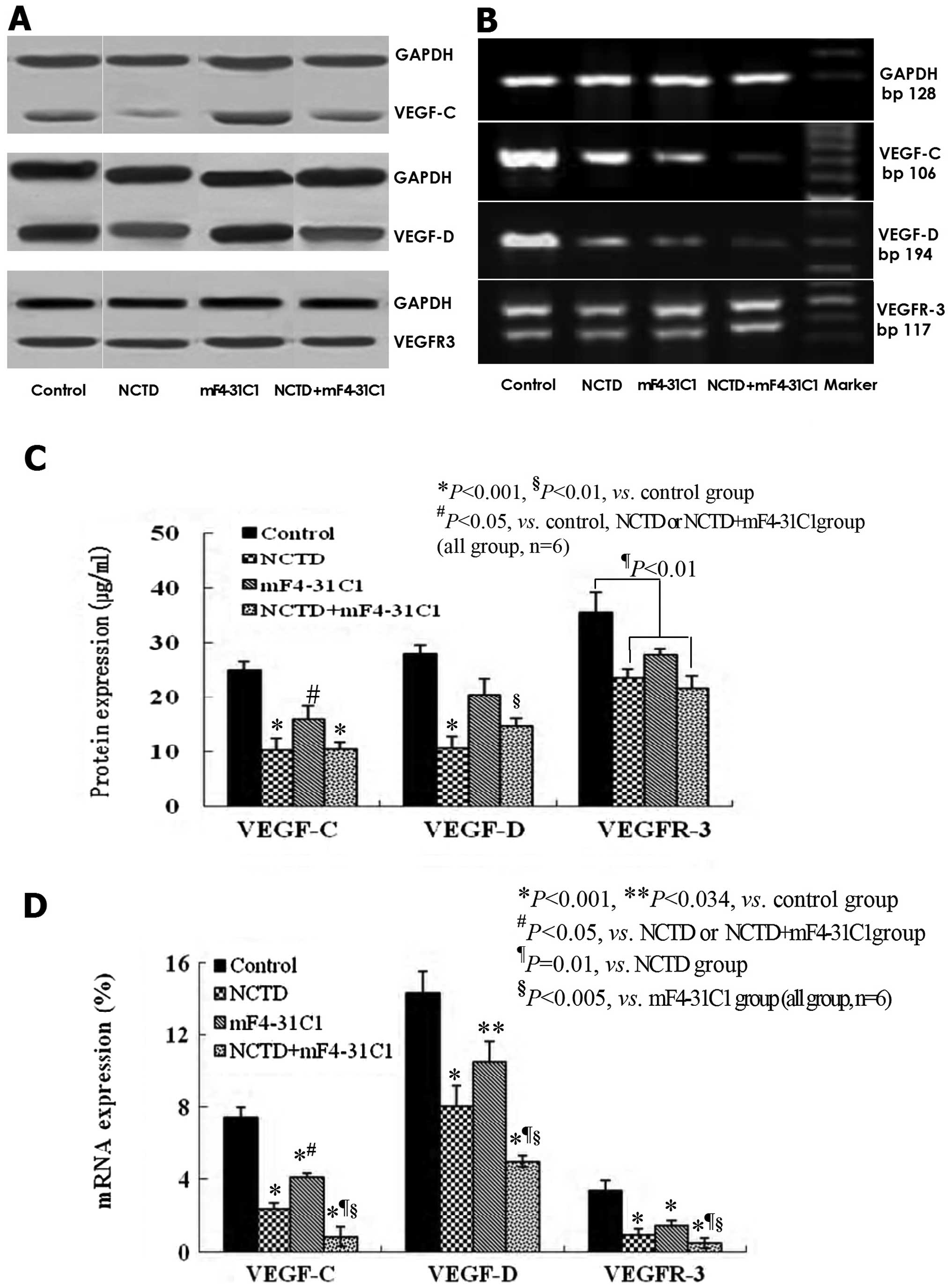
NCTD downregulates the expression of
VEGF-C, VEGF-D and VEGFR-3 in the process of lymphatic
tube-formation of HLECs
VEGF-C, VEGF-D, VEGFR-3 at protein and mRNA levels
in lymphangiogenesis on 3-D culture of HDLECs were measured by
immunohistochemistry (streparidin-peroxidase staining method),
western blotting and quantitative real-time polymerase chain
reaction (QRT-PCR). As shown in Figs.
6 and 7, at protein level, the
expression of VEGF-C, VEGF-D and VEGFR-3 in NCTD group or
NCTD+mF4-31C1 group was decreased significantly when compared with
control group (P<0.001); but the expression of VEGF-C in NCTD or
NCTD+mF4-31C1 group was lower than that of mF4-31C1 group
(P<0.05); and there was no statistical difference in VEGF-D
expression between control group and mF4-31C1 group (Figs. 6 and 7A–C). At the mRNA level, the expression
of VEGF-C, VEGF-D and VEGFR3 mRNAs was decreased significantly in
all experimental groups when compared with control group; but the
expression of VEGF-C mRNA in NCTD or NCTD+mF4-31C1 group was also
lower than that of mF4-31C1 group (P<0.05); and the expression
of VEGF-C, VEGF-D or VEGFR3 mRNAs in NCTD+mF4-31C1 group was
significantly lower than those of mF4-31C1 (P<0.005) or NCTD
(P=0.01) group (Fig. 7C and D). It
was shown that NCTD downregulated the expression of VEGF-C, VEGF-D
and VEGFR-3 at protein and mRNA levels in the process of lymphatic
tube-formation of HLECs; mF4-31C1 did not effect VEGF-D expression;
the downregulation of VEGF-C, VEGF-D/VEGFR3 by the combined
application of NCTD and mF4-31C1 can be enhanced. Thus, we believe
that NCTD inhibit lymphangiogenesis in HLECs by simultaneously
blocking VEGF-C, VEGF-D and VEGFR-3.
Discussion
The metastatic spread of tumor cells via the
lymphatic vessels is a lethal aspect of cancer. Tumor-associated
lymphatic vessels act as a conduit by which disseminating tumor
cells access regional lymph nodes and form metastases (17). The presence of meta-static cells in
the sentinel lymph node is a prognostic indicator and the degree of
dissemination determines the therapeutic course of action (18). A growing body of evidence indicates
that tumor lymphangiogenesis is a predictive indicator of
metastasis to lymph nodes. According to the centrifugal theory of
lymphangiogenesis, in the early stage of the development of
lymphatics, lymph sacs develop independently by the further
sprouting of venous endothelium and extend by the sprouting of
endothelial cells into the surrounding tissues and organs to form
local lymphatic capillary networks (19). In other words, lymphangiogenesis is
the growth of newly formed lymphatic vessels; this process with
multiple steps is similar to the well-known mechanism of
angiogenesis: endothelial cell proliferation, migration,
rearrangement and tube formation, along with degradation,
reconstruction and production of extracellular matrix. Lymph node
lymphangiogenesis and increased lymph flow through tumor-draining
lymph nodes are speculated to actively promote metastasis via the
lymphatics (20). Tumor- or
stromal-secreted cytokines, such as VEGF-C and VEGF-D, bind to VEGF
receptors on LECs and induce proliferation and growth of new
lymphatic capillaries (21).
Therefore, inhibition of tumor-associated lymphangiogenesis or its
VEGF-C, -D/VEGF receptors may be a potential therapy for primary
tumors and metastasis via the lymphatics.
In recent years, some drugs are used to treat tumors
with the anti-lymphangiogenic mechanism. Researchers have focused
on hot-spots of treating carcinoma by inhibition or destroying of
tumor lymphatic tube formation early, i.e., lymphangiogenesis
(9,10,18).
These lymphangiogenic inhibitors such as deguelin, endostar,
silencing Id-1, liposomal honokiol, mF4-31C1, Ki23057, sVEGFR3-Ig,
VEGFR31-Ig, artemisinin, etodolac, MMI270, CSDA and AZM475271 have
been reported as adjuvant anti-lymphangiogenic and anti-tumor drugs
against some metastatic cancers in experimental and in clinical
setting (22–37). NCTD has been reported to possess
potent anti-angiogenesis and antitumor properties in several cell
lines and tumor models (11–16,38–41).
However, it is still unclear whether NCTD effectively inhibits
tumor-associated lymphangiogenesis and lymphatic metastasis. Here,
we investigated the antilymphangiogenic effect of NCTD on HLECs. It
was shown that NCTD significantly inhibited proliferation,
migration, invasion, lymphatic tube formation of HLECs and induced
apoptosis of HLECs (all P<0.01), dose- and time-dependently
(IC50 6.8 μg/ml). Over the past few years, understanding
of the cellular and molecular aspects of physiologic
lymphangiogenesis and tumor-induced lymphangiogenesis has advanced.
Proliferation of the lymphatics is an active biological behavior of
tumor cells, with heterogeneity of interactions of tumor cells with
lymphatic vessels in tumors and regional lymph system. Although
lymphatic vessels constitute the most important channel of
lymphatic spread, lymphatic endothelium is an interactive surface
for cancer cells, and the ability of cancer cells to interact with
the lymphatic endothelial cells is a key step in allowing them to
invade the lymphatic system (20).
In this study, we identified for the first time that NCTD inhibited
lymphangiogenesis in HLECs in vitro. Thus, we believed that
NCTD might be a potential target agent for lymphangiogenesis of
HLECs, and the key of the antitumor properties or mechanisms.
Tumors promote lymphangiogenesis by secreting
molecules (lymphangiogenic growth factors) from tumor cells and
stromal cells that stimulate lymphatic endothelial cell growth
(42). These lymphangiogenic
growth factors include VEGF-A, VEGF-C, VEGF-D, PDGF, HGF, Ang-1,
Ang-2, IGF-1/2 and FGF-2 (bFGF) (18). Of them, most important
lymphangiogenic growth factors are VEGF-C, VEGF-D and their cognate
receptor VEGFR-3 located on lymphatic endothelial cells (6–10,18–21).
These molecules play an important role on lymphangiogenesis and
distinguish subtypes in tumor metastasis. In xenotrans-planted
tumors, cancer cells transfected by VEGF-C gene induce the growth
of functional lymphatics and result in hyperplastic vessels
(43). VEGF-C not only induces the
hyperplasia of peritumoral vessels, but also increases the
volumetric flow rate of fluid in lymphatics that results in the
increased delivery of tumor cells to the lymph node (44). Indeed, many other growth factors
(FGF-2, Ang-1, VEGF-A, IGF-1, HGF) stimulate lymphangio-genesis
indirectly through VEGF-C. VEGF-D also plays a role in the
regulation of tumor lymphangiogenesis (45,46).
Functional autocrine stimulation of VEGF-D in cancer not only
stimulates the proliferation of cancer cells and LECs, but also
plays a role in the maintenance of anti-apoptotic characteristics
of tumor-derived endothelial cells (20). Activation of VEGF-C/VEGF-D/VEGFR-3
axis increases motility and invasiveness of LECs, promote formation
of tumor lymphatics (lymphangiogenesis) (47). Most anti-lymphangiogenic
pre-clinical studies to date have targeted the
VEGF-C/VEGF-D/VEGFR-3 signaling pathway. A targeted approach to
block pathways of lymphangiogenesis seems to be an attractive
anticancer treatment strategy. These strategies have included
targeting and knock down of lymphangiogenic ligands, targeting
receptors on lymphatic endothelium and inhibiting tumor
lymphangiogenesis with soluble receptors. The downmodulation of
VEGF-C with stable RNAi transfection in murine breast cancer cells
results in reduced tumor lymphangiogenesis and lymph node
metastasis (48). Deguelin
suppresses tumor-associated lymphangiogenesis and lymphatic
metastasis in lung tumor model by downregulation of VEGF-D both
in vitro and in vivo (22). Blocking the expression of VEGFR-3
using interference vector-based RNA interference inhibits tumor
growth of colorectal cancer (29).
Adeno-associated virus-mediated gene transfer of sVEGFR3-Fc
potently blocks tumor-associated lymphangiogenesis and metastasis
to the lymph nodes in human prostate and melanoma tumor models
(49). mF4-31C1, a novel VEGFR-3
neutralizing antibody, potently antagonizes the binding of VEGF-C
to VEGFR-3. Because blocking of VEGFR-3 using mF4-31C1 completely
and specifically prevented both physiologically normal and tumor
VEGF-C-enhanced lymphangiogenesis in the adult mouse tail skin
model of lymphatic regeneration but had no effect on either blood
angiogenesis or the survival or function of existing lymphatic
vessels, targeting VEGFR-3 with the specific inhibitor blocked new
lymphatic growth exclusively (26). In the present study, NCTD
downregulated the expression of VEGF-C, VEGF-D and VEGFR-3 at
protein and mRNA levels in the process of lymphatic tube-formation,
i.e., lymphangiogenesis of HLECs. Because VEGF-C and VEGF-D are
primarily lymphangiogenic factors, VEGFC,-D/VEGFR axis or signal
pathway seem probably the main target of the anti-lymphangiogenesis
in physiological and pathological conditions, we may deduce that
NCTD target and inhibit lymphangiogenesis in HLECs by simultaneous
blocking and suppression of VEGF-C and -D/VEGFR-3 axis or signal
pathway. The present findings may be of importance to explore the
therapeutic strategy of NCTD as an anti-lymphangiogenic agent for
tumor lymphangiogenesis and lymphatic metastasis. However, further
experiments are needed to explore whether NCTD inhibits tumor
lymphangio-genesis and lymphatic metastasis in tumor models, and
whether it works through the same mechanism in vivo.
Acknowledgements
This study was supported by a grant
from the National Nature Science Foundation of China (no.
81072004).
References
|
1.
|
Achen MG and Stacker SA: Tumor
lymphangiogenesis and metastatic spread - new players begin to
emerge. Int J Cancer. 119:1755–1760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Nagahashi M, Ramachandran S, Rashid OM and
Takabe K: Lymphangiogenesis: a new player in cancer progression.
World J Gastroenterol. 16:4003–4012. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Tobler NE and Detmar M: Tumor and lymph
node lymphangio-genesis-impact on cancer metastasis. J Leukoc Biol.
80:691–696. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Liersch R, Biermann C, Mesters RM and
Berdel WE: Lymph-angiogenesis in cancer: current perspectives.
Recent Results Cancer Res. 180:115–135. 2010. View Article : Google Scholar
|
|
5.
|
Sleeman JP and Thiele W: Tumor metastasis
and the lymphatic vasculature. Int J Cancer. 125:2747–2756. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Veikkola T, Jussila L, Makinen T, Karpanen
T, Jeltsch M, Petrova TV, Kubo H, Thurston G, McDonald DM, Achen
MG, Stacker SA and Alitalo K: Signaling via vascular endothelial
growth factor receptor-3 is sufficient for lymphangiogenesis in
transgenic mice. EMBO J. 20:1223–1231. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Joukov V, Sorsa T, Kumar V, Jeltsch M,
Claesson-Welsh L, Cao Y, Saksela O, Kalkkinen N and Alitalo K:
Proteolytic processing regulates receptor specificity and activity
of VEGF-C. EMBO J. 16:3898–3911. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Siegfried G, Basak A, Cromlish JA,
Benjannet S, Marcinkiewicz J, Chrétien M, Seidah NG and Khatib AM:
The secretory proprotein convertases furin, PC5, and PC7 activate
VEGF-C to induce tumorigenesis. J Clin Invest. 111:1723–1732. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Kowanetz M and Ferrara N: Vascular
endothelial growth factor signaling pathways: therapeutic
perspective. Clin Cancer Res. 12:5018–5022. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Wissmann C and Detmar M: Pathways
targeting tumor lymphangiogenesis. Clin Cancer Res. 12:6865–6868.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Ho YP, To KK, Au-Yeung SC, Wang X, Lin G
and Han X: Potential new antitumor agents from an innovative
combination of demethylcantharidin, a modified traditional Chinese
medicine, with a platinum moiety. J Med Chem. 44:2065–2068. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Deng L and Tang S: Norcantharidin
analogues: a patent review (2006–2010). Expert Opin Ther Pat.
21:1743–1753. 2011.PubMed/NCBI
|
|
13.
|
Fan YZ, Fu JY, Zhao ZM and Chen CQ:
Inhibitory effect of norcantharidin on the growth of human
gallbladder carcinoma GBC-SD cells in vitro. Hepatobiliary Pancreat
Dis Int. 6:72–80. 2007.PubMed/NCBI
|
|
14.
|
Fan YZ, Zhao ZM, Fu JY, Chen CQ and Sun W:
Norcantharidin inhibits growth of human gallbladder carcinoma
xenografted tumors in nude mice by inducing apoptosis and blocking
the cell cycle in vivo. Hepatobiliary Pancreat Dis Int. 9:414–422.
2010.PubMed/NCBI
|
|
15.
|
Chen YJ, Tsai YM, Kuo CD, Ku KL, Shie HS
and Liao HF: Norcantharidin is a small-molecule synthetic compound
with anti-angiogenesis effect. Life Sci. 85:642–651. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Zhang JT, Fan YZ, Chen CQ, Zhao ZM and Sun
W: Norcantharidin: a potential antiangiogenic agent for gallbladder
cancers in vitro and in vivo. Int J Oncol.
40:1501–1514. 2012.PubMed/NCBI
|
|
17.
|
Achen MG and Stacker SA: Molecular control
of lymphatic metastasis. Ann NY Acad Sci. 1131:225–234. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Zwaans BM and Bielenberg DR: Potential
therapeutic strategies for lymphatic metastasis. Microvasc Res.
74:145–158. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Kotani M: The lymphatics and
lymphoreticular tissues in relation to the action of sex hormones.
Arch Histol Cytol. 53(Suppl): 1–76. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Zhang Z, Helman JI and Li LJ:
Lymphangiogenesis, lymphatic endothelial cells and lymphatic
metastasis in head and neck cancer - a review of mechanisms. Int J
Oral Sci. 2:5–14. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Nathanson SD: Insights into the mechanisms
of lymph node metastasis. Cancer. 98:413–423. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Hu J, Ye H, Fu A, Chen X, Wang Y, Chen X,
Ye X, Xiao W, Duan X, Wei Y and Chen L: Deguelin - an inhibitor to
tumor lymphangiogenesis and lymphatic metastasis by downregulation
of vascular endothelial cell growth factor-D in lung tumor model.
Int J Cancer. 127:2455–2466. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Dong X, Zhao X, Xiao T, Tian H and Yun C:
Endostar, a recombined humanized endostatin, inhibits
lymphangiogenesis and lymphatic metastasis of lewis lung carcinoma
xenograft in mice. Thorac Cardiovasc Surg. 59:133–136. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Dong Z, Wei F, Zhou C, Sumida T, Hamakawa
H, Hu Y and Liu S: Silencing Id-1 inhibits lymphangiogenesis
through down-regulation of VEGF-C in oral squamous cell carcinoma.
Oral Oncol. 47:27–32. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Wen J, Fu AF, Chen LJ, Xie XJ, Yang GL,
Chen XC, Wang YS, Li J, Chen P, Tang MH, Shao XM, Lu Y, Zhao X and
Wei YQ: Liposomal honokiol inhibits VEGF-D-induced
lymphangio-genesis and metastasis in xenograft tumor model. Int J
Cancer. 124:2709–2718. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Pytowski B, Goldman J, Persaud K, Wu Y,
Witte L, Hicklin DJ, Skobe M, Boardman KC and Swartz MA: Complete
and specific inhibition of adult lymphatic regeneration by a novel
VEGFR-3 neutralizing antibody. J Natl Cancer Inst. 97:14–21. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Rinderknecht M, Villa A, Ballmer-Hofer K,
Neri D and Detmar M: Phage-derived fully human monoclonal antibody
fragments to human vascular endothelial growth factor-C block its
interaction with VEGF receptor-2 and 3. PLoS One. 5:e119412010.
View Article : Google Scholar
|
|
28.
|
Zhang D, Li B, Shi J, Zhao L, Zhang X,
Wang C, Hou S, Qian W, Kou G, Wang H and Guo Y: Suppression of
tumor growth and metastasis by simultaneously blocking vascular
endothelial growth factor (VEGF)-A and VEGF-C with a
receptor-immunoglobulin fusion protein. Cancer Res. 70:2495–2503.
2010. View Article : Google Scholar
|
|
29.
|
Lui Z, Ma Q, Wang X and Zhang Y:
Inhibiting tumor growth of colorectal cancer by blocking the
expression of vascular endothelial growth factor receptor 3 using
interference vector-based RNA interference. Int J Mol Med.
25:59–64. 2010.PubMed/NCBI
|
|
30.
|
Yashiro M, Shinto O, Nakamura K, Tendo M,
Matsuoka T, Matsuzaki T, Kaizaki R, Ohira M, Miwa A and Hirakawa K:
Effects of VEGFR-3 phosphorylation inhibitor on lymph node
metastasis in an orthotopic diffuse-type gastric carcinoma model.
Br J Cancer. 101:1100–1106. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Guo B, Zhang Y, Luo G, Li L and Zhang J:
Lentivirus-mediated small interfering RNA targeting VEGF-C
inhibited tumor lymphangiogenesis and growth in breast carcinoma.
Anat Rec. 292:633–639. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Wang J, Zhang B, Guo Y, Li G, Xie Q, Zhu
B, Gao J and Chen Z: Artemisinin inhibits tumor lymphangiogenesis
by suppression of vascular endothelial growth factor C.
Pharmacology. 82:148–155. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Iwata C, Kano MR, Komuro A, Oka M, Kiyono
K, Johansson E, Morishita Y, Yashiro M, Hirakawa K, Kaminishi M and
Miyazono K: Inhibition of cyclooxygenase-2 suppresses lymph node
metastasis via reduction of lymphangiogenesis. Cancer Res.
67:10181–10189. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Cao G, Wu JX and Wu QH: Low molecular
weight heparin suppresses lymphatic endothelial cell proliferation
induced by vascular endothelial growth factor C in vitro. Chin Med
J. 122:1570–1574. 2009.PubMed/NCBI
|
|
35.
|
Nakamura ES, Koizumi K, Kobayashi M and
Saiki I: Inhibition of lymphangiogenesis-related properties of
murine lymphatic endothelial cells and lymph node metastasis of
lung cancer by the matrix metalloproteinase inhibitor MMI270.
Cancer Sci. 95:25–31. 2004. View Article : Google Scholar
|
|
36.
|
Matsumoto G, Yajima N, Saito H, Nakagami
H, Omi Y, Lee U and Kaneda Y: Cold shock domain protein A (CSDA)
overexpression inhibits tumor growth and lymph node metastasis in a
mouse model of squamous cell carcinoma. Clin Exp Metastasis.
27:539–547. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Ischenko I, Seeliger H, Camaj P, Kleespies
A, Guba M, Eichhorn ME, Jauch KW and Bruns CJ: Src tyrosine kinase
inhibition suppresses lymphangiogenesis in vitro and in vivo. Curr
Cancer Drug Targets. 10:546–553. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
An WW, Wang MW, Tashiro S, Onodera S and
Ikejima T: Norcantharidin induces human melanoma A375-S2 cell
apoptosis through mitochondrial and caspase pathways. J Korean Med
Sci. 19:560–566. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Yi SN, Wass J, Vincent P and Iland H:
Inhibitory effect of norcantharidin on K562 human myeloid leukemia
cells in vitro. Leuk Res. 15:883–886. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Yang PY, Chen MF, Tsai CH, Hu DN, Chang FR
and Wu YC: Involvement of caspase and MAPK activities in
norcantharidin-induced colorectal cancer cell apoptosis. Toxicol In
Vitro. 24:766–775. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Chen YJ, Kuo CD, Tsai YM, Yu CC, Wang GS
and Liao HF: Norcantharidin induces anoikis through Jun-N-terminal
kinase activation in CT26 colorectal cancer cells. Anticancer
Drugs. 19:55–64. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
McColl BK, Loughran SJ, Davydova N,
Stacker SA and Achen MG: Mechanisms of lymphangiogenesis: targets
for blocking the metastatic spread of cancer. Curr Cancer Drug
Targets. 5:561–571. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Cohen-Kaplan V, Naroditsky I, Zetser A,
Ilan N, Vlodavsky I and Doweck I: Heparanase induces VEGF C and
facilitates tumor lymphangiogenesis. Int J Cancer. 123:2566–2573.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Hoshida T, Isaka N, Hagendoorn J, di
Tomaso E, Chen YL, Pytowski B, Fukumura D, Padera TP and Jain RK:
Imaging steps of lymphatic metastasis reveals that vascular
endothelial growth factor-C increases metastasis by increasing
delivery of cancer cells to lymph nodes: therapeutic implications.
Cancer Res. 66:8065–8075. 2006. View Article : Google Scholar
|
|
45.
|
Bierer S, Herrmann E, Kopke T, Neumann J,
Eltze E, Hertle L and Wülfing C: Lymphangiogenesis in kidney
cancer: expression of VEGF-C, VEGF-D and VEGFR-3 in clear cell and
papillary renal cell carcinoma. Oncol Rep. 20:721–725.
2008.PubMed/NCBI
|
|
46.
|
Choi JH, Oh YH, Park YW, Baik HK, Lee YY
and Kim IS: Correlation of vascular endothelial growth factor-D
expression and VEGFR-3-positive vessel density with lymph node
metastasis in gastric carcinoma. J Korean Med Sci. 23:592–597.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Su JL, Yen CJ, Chen PS, Chuang SE, Hong
CC, Kuo IH, Chen HY, Hung MC and Kuo ML: The role of the
VEGF-C/VEGFR-3 axis in cancer progression. Br J Cancer. 96:541–545.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Chen Z, Varney ML, Backora MW, Cowan K,
Solheim JC, Talmadge JE and Singh RK: Down-regulation of vascular
endothelial cell growth factor-C expression using small interfering
RNA vectors in mammary tumors inhibits tumor lymphangiogenesis and
spontaneous metastasis and enhances survival. Cancer Res.
65:9004–9011. 2005. View Article : Google Scholar
|
|
49.
|
Lin J, Lalani AS, Harding TC, Gonzalez M,
Wu WW, Luan B, Tu GH, Koprivnikar K, VanRoey MJ, He Y, Alitalo K
and Jooss K: Inhibition of lymphogenous metastasis using
adeno-associated virus-mediated gene transfer of a soluble VEGFR-3
decoy receptor. Cancer Res. 65:6901–6909. 2005. View Article : Google Scholar : PubMed/NCBI
|