Introduction
Prostate cancer is the most common non-skin cancer
in men, and the second leading cause of death in male cancer
patients after lung cancer (1).
Epidemiological studies show that in Asia the incidence of prostate
cancer is lower compared to that in the West (2). There is increasing evidence showing
that habitual consumption of tea, soybean and its products, which
are rich in catechins and flavonoids, by Asian men is correlated
with a reduction in a number of cancers or their prevention,
including cancer of the prostate gland. Treatments for prostate
cancer also include surgery, orchiectomy, hormonal therapy,
radiation and chemotherapy (3).
Recent studies have been carried out to find
chemo-preventive and/or chemo-therapeutic agents targeting on
cancer from edible and natural resources such as fruits,
vegetables, and terrestrial plants (4). As one of the well-known natural
resources, seaweeds are well-balanced natural sources with a high
degree of trace elements, and strongly advised for fast growing
children and pregnant women (5).
Edible seaweeds have high nutritional values as sources of
vitamins, minerals, and non-caloric dietary fibers and as potential
sources of biological active ingredients (6). Marine organisms have emerged as one
of the important sources for dietary supplements and a number of
them are potentially active and useful source of medicine. Their
metabolites such as flavonoid and other phenolic compounds are
widely used in life science research and provide chances for
discovery-phase of new drug development (7,8).
The brown alga Saccharina japonica (S.
japonica) is frequently consumed in Korea, Japan and China, and
has been used for more than a thousand years as a nutraceutical in
traditional Chinese medicine. Dietary application of Saccharina
sp. has been reported to reduce the risk of intestinal or
mammary cancer in animal studies (9). Furthermore, research has been focused
on the isolation and function of Saccharina sp., which
revealed multiple biological activities such as immunopotentiating
substances, anti-inflammatory, anti-tumor, anti-oxidant,
anti-coagulant, and anti-viral activities (10).
Apoptosis (programmed cell death) is characterized
by specific morphological and biochemical changes, including cell
shrinkage, chromatin condensation, and inter-nucleosomal cleavage
of genomic DNA. Apoptotic cells ultimately break up into plasma
membrane-bound vesicles called apoptotic bodies that are rapidly
phagocytosed by macrophages and neighboring epithelial cells. At
the molecular level, apoptosis is tightly regulated and mainly
harmonized by the activation of the caspases (11,12).
Caspases are aspartate-specific cysteine proteases
and are expressed as pro-enzymes. Caspase activation involves the
proteolytic processing and a substantial body of evidence supports
a cascade model. Initiator caspases such as caspase-2, -8, -9 and
-10 instigate the apoptotic cascade and lead to the activation of
effector caspases, which include caspase-3, -6 and -7. Caspases
cause cell death by cleaving a number of cellular proteins
including nuclear lamins, DNA repair enzymes such as
poly(ADP-ribose) polymerase (PARP), and cytoskeletal proteins such
as actin, fodrin and gelsolin (13–17).
Fragmentation of DNA during apoptosis is caused by an enzyme known
as DNA fragmentation factor (DFF) (18).
Cell proliferation is governed primarily by
regulation of the cell cycle, which consists of four distinct
sequential phases (G0/G1, S, G2, and M). Transition through the
G1-phase of the cell cycle and entry into the S-phase requires
binding and activation of cyclin/cyclin-dependent kinase (CDK)
complexes, predominantly cyclin D/CDK4 or CDK6 and cyclin E/CDK2
(19). Cyclin-dependent kinase
inhibitors (CKIs) are naturally occurring gene products that
inhibit cyclin/CDK activity, leading to G1-phase arrest.
p21waf1 (p21) is a primary negative regulator and
negatively regulates the kinase activity of cyclin/CDK complexes.
These inhibitors block cell cycle progression by binding and
inactivating the cyclin/CDK complex in the G1-phase, leading to
cell cycle arrest (20).
In this study, we examined anticancer effects which
are mediated by multiple pathways of endocellular reticulum (ER)
stress, death receptor and mammalian target of rapamycin (mTOR)-Fox
leading apoptosis and cell cycle arrest in 267B1/K-ras, 267B1 human
prostate cancer cells transformed by K-ras, by treatment
with the extract (n-hexane sub-fraction) of S. japonica,
marine macro-algae.
Materials and methods
Sample preparation
Preparation of seaweeds. S. japonica was
harvested from Kijang aquaculture farm, Korea. The samples were
dried in cold-air drier (60°C) for 40 h, ground with a hammer mill,
and then the dried powder was stored at −20°C until used.
Extraction and fractionation. The dried
powder (2 kg) of S. japonica was refluxed with ethyl alcohol
(95%, v/v, 5 l × 3 times) for 3 h. The extract (446.0 g) was
suspended in H2O:ethyl alcohol (9:1, v/v) and
partitioned with n-hexane, dichloromethane, ethyl acetate (EtOAc),
n-butanol (n-BuOH), and water in sequence, yielding the n-hexane
(135.5 g), dichloromethane (18.1 g), EtOAc (39.6 g), n-BuOH (55 g),
and water (162.8 g) fractions. The n-hexane fraction was subjected
to preparative size exclusion column of Shim-pack PREP-ODS (500 mm
× 21.2 mm, Shimadzu Co., Tokyo, Japan). An exclusion HPLC apparatus
consisted of a pump (Shimadzu LC-6AD), a photodiode array detector
(Shimadzu SPDM20A), an online degasser (Shimadzu DUG-20A3), an
autosampler (SIL-20A), a fraction collector (Shimadzu FRC-10A), a
system controller (CBM-20A), and a Shimadzu LC solution (ver.
1.22sp). The n-hexane fraction was chromatographed on a Shim-pack
PREP-ODS column eluting with methanol at a flow rate of 5.0 ml/min
and monitored at 240 nm. The fraction was separated into four
fractions (GS1-GS4). The GS3 fraction was chromatographed over
Phenomenex C18-ODS (5 μm, 100 Å, 300 mm × 5 mm, Phenomenex Co.,
Tokyo, Japan). A preparative ODS HPLC system was similar to the
exclusion HPLC system except for a binary pump (Phenomenex LC-6AD)
and a column oven (35°C, Phenomenex CTO-20A). The separation of GS3
fraction was conducted using mobile phase water (solvent A) and
methanol (solvent B). The elution profile consisted of a linear
gradient from 20 to 70% B solvent for 90 min. The flow rate was 7.0
ml/min, and detection was performed at 216 nm. Fifteen
sub-fractions (GS3-ODS1-GS3-ODS15) were tested for cell viability
against prostate cancer cells and GS3-ODS4 sub-fraction is selected
for the studies.
Cell culture and treatment of the
extract
Human epithelial cell line (267B1) established from
fetal prostate tissue can be malignantly transformed by a
biological carcinoma, and can serve as a useful model for research
of the progression steps of carcinogenesis. Activated K-ras was
introduced into 267B1 cells by infection with the Kirsten murine
sarcoma virus (1,21). The established 267B1/K-ras cells
are immortalized and contained the essential characteristics of
primary human prostate epithelial cells such as morphology,
expression of cytokeratins. The transformed 267B1/K-ras human
prostate cancer cells and HEK293 human embryonic kidney cells were
used for the experiments. The 267B1/K-ras cells were cultured in
RPMI-1640 with L-glutamine (Lonza Group Ltd., Basel, Switzerland)
containing 10% heat-inactivated fetal bovine serum (Lonza Group
Ltd.) and 1% 100 U/ml penicillin and 100 μg/ml streptomycin (PAA
Laboratories GmbH, PA, Austria). HEK293 cells were cultured in
Dulbecco's modified Eagle's medium with high glucose (DMEM/high
glucose, Thermo Scientific Hyclone, Logan, UT, USA), 10%
heat-inactivated fetal bovine serum (Thermo Scientific Hyclone) and
1% 100 U/ml penicillin and 100 μg/ml streptomycin (PAA Laboratories
GmbH) at 37°C in a humidified atmosphere of 5% CO2. The
fraction, dissolved in dimethylsulfoxide (DMSO) (ATCC, Manassas,
VA, USA), was added to the culture media to the final concentration
specified in the text. The final concentration of DMSO in the
culture medium was not exceeded 0.04% (v/v), and the same
concentration of DMSO was added to the control dishes.
Cell viability assay
For the cell viability assay, 267B1/K-ras cells and
HEK293 cells in exponential growth phase were suspended at a final
concentration of 1×105 cells/ml in each medium
containing 10% FBS in 96-well cell culture plate (SPL Lifesciences,
Gyeonggi, Korea) in triplicate. The cells were then treated with
various concentrations (10, 20, 30, 40 and 50 μg/ml) of extracted
fraction and incubated for 24 h. After the treatment, 10 μl of
EZ-Cytox Cell Viability Assay Solution WST-1® (Daeil Lab
Service, Jong-No, Korea) was added onto each well and the cells
were further incubated at 37°C for 3 h and then read at 460 nm with
ELISA reader (Molecular Devices, Sunnyvale, CA, USA).
Western blot analysis
The cultured 267B1/K-ras cells in 150 mm dish
(Corning Incorporated Life Science, Lowell, MA, USA) were treated
with the extract for 24 h and then washed with ice-cold PBS
(phosphate-buffered saline) and harvested by scrapping. The
harvested cells were collected by centrifugation, lysed in ice-cold
lysis buffer [50 mM Tris-Cl (pH 7.5), 150 mM NaCl, 1 mM DTT, 0.5%
NP-40, 1% Triton X-100, 1% deoxycholate, 0.1% SDS and proteinase
inhibitors (PMSF, EDTA, aprotinin, leupeptin, prostatin A)] (Intron
Biotechnology, Gyeonggi, Korea). After incubation on ice for 30
min, the insoluble materials were removed by centrifugation at
14,000 rpm for 20 min at 4°C. The protein contents of the cell
lysates were determined by a Protein Quantification kit (CBB
solution®) (Dojindo Molecular Technologies, Rockville,
MD, USA) with bovine serum albumin (BSA) as standard. An aliquot
from each sample was boiled for 4 min, and then resolved by 12%
SDS-polyacrylamide gel electrophoresis (SDS-PAGE). The proteins
were electro-transferred to a nitrocellulose membrane (PALL Life
Sciences, MI, USA) and then blocked in PBST buffer (135 mM NaCl,
2.7 mM KCl, 4.3 mM NaPO4, 1.4 mM
KH2PO4, 0.5% Tween-20) containing 5% skim
milk for overnight at 4°C. The blots were probed with the primary
antibody (Cell Signaling Technology Inc., Beverly, MA, USA) and
then washed three times in PBST, followed by incubation for 1 h
with horseradish peroxidase-coupled anti-rabbit IgG or anti-mouse
IgG as second antibodies (Cell Signaling Technology Inc.). The
blots were then washed in PBST and visualized by an enhanced
chemiluminescent (ECL) detection solution (Pierce, Rockford, IL,
USA).
Treatment of zVAD-FMK
To investigate the involvement of caspases in
apoptosis, 267B1/K-ras cells were seeded at a final concentration
of 1×105 cells/ml in RPMI-1640 media containing 10% FBS
and 1% penicillin-streptomycin in 96-well cell culture plate in
triplicate. The cells were then pre-incubated with 20 μM of a
caspase inhibitor, zVAD-FMK (dissolved in DMSO) for 1 h prior to
treating with the concentration of 30 μg/ml of the extract.
zVAD-FMK
(carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone,
Tocris Bioscience, Ellisville, MO, USA) is a cell-permeant
pan-caspase inhibitor that irreversibly binds to the catalytic site
of caspase proteases and can inhibit induction of apoptosis. After
treatment of extract for 24 h, 10 μl of EZ-Cytox Cell Viability
Assay Solution WST-1 (Daeil Lab Service) was added onto each well
and the cells were further incubated at 37°C for 3 h and then read
at 460 nm with ELISA reader (Molecular Devices).
DAPI staining
267B1/K-ras cells were cultured at 37°C with 5%
CO2 on coverglass-bottom dishes (SPL Life Sciences) in
RPMI-1640 media containing 10% FBS and 1% penicillin-streptomycin
and then incubated for 24 h with the concentration of 20 and 30
μg/ml of the extract. To see the formation of apoptosome, cells
were rinsed twice with 1 μg/ml of DAPI
(4′,6-diamidine-2′-phenylindole dihydrochloride, Roche Applied
Science, Indianapolis, IN, USA) in methanol (Sigma-Aldrich, St.
Louis, MO, USA) and stained by the addition of DAPI solution. After
incubation in dark at 37°C for 20 min, cells were rinsed once with
methanol (Sigma-Aldrich). Stained cells on the slides were mounted
in Prolong Gold Antifade Reagent (Invitrogen, Eugene, OR, USA)
followed by observation under a Nikon ECLIPS 50i microscope
equipped with charged-coupled device (CDD) camera. Images were
captured and processed with High-Content Analysis Software
(Cambridge Healthtech Ins., Needham, MA, USA).
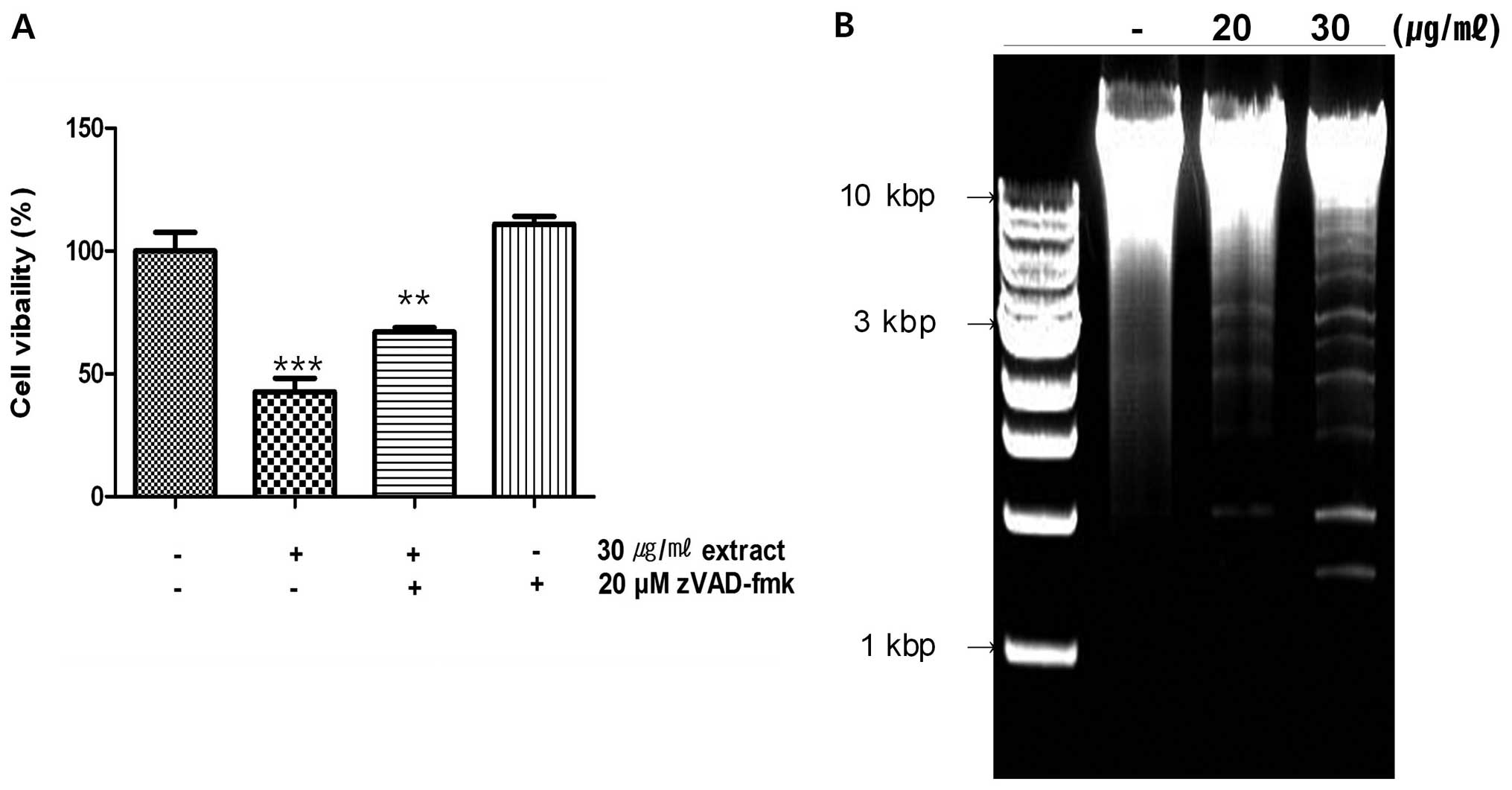
DNA fragmentation
For detecting genomic DNA fragmentation, the treated
267B1/K-ras cells were washed with ice-cold PBS buffer and
harvested. The collected cells were handled by following protocol
of DNeasy® blood & tissue kit (Qiagen GmbH, Hilden,
Germany). The isolated DNA was analyzed on a 1.5% agarose gel (Life
Technologies Inc., Grand Island, NY, USA) with UV transilluminator
(Vilber Lourmat, Marne-la-Vallee, France) after ethidium bromide
(Sigma-Aldrich) staining. Isolated DNA was mixed with 6× loading
dye (Promega, Madison, WI, USA) and loaded on the agarose gel with
1 kb DNA Ladder (Promega) for identify the size of DNA.
Immunofluorescence stain
267B1/K-ras cells were cultured at 37°C with 5%
CO2 on coverglass-bottom dishes in RPMI-1640 media
containing 10% FBS and 1% penicillin-streptomycin, were incubated
for 24 h with the concentration of 20 and 30 μg/ml of the extract.
Cells were fixed with 4% formaldehyde (Junsei Chemical Co., Ltd.,
Japan) for 15 min at room temperature and blocked for 1 h in 5%
mouse and rabbit normal serum (Santa Cruz Biotechnology, Inc., CA,
USA) of host of primary antibodies and 0.3% Triton X-100. Fixed and
blocked cells were incubated with 0.1 μg/ml of primary antibodies
(cleaved caspase-3 and β-actin) (Cell Signaling Technology Inc.)
for 3 h and then treated with 0.1 µg/ml of anti-mouse IgG (H+L),
F(ab')2 Fragment (Alexa Fluor® 555 Conjugate) and/or
anti-rabbit IgG (H+L), F(ab')2 Fragment (Alexa Fluor®
488 Conjugate) (Cell Signaling Technology Inc.) for 1 h. Stained
cells on the slides were mounted in Prolong Gold Antifade Reagent
followed by observation under a Nikon ECLIPS 50i microscope
equipped with CDD camera. Images were captured and processed with
High-Content Analysis software.
Fluo-3/AM assay
267B1/K-ras cells cultured at 37°C with 5%
CO2 on cover glass-bottom dish in RPMI-1640 media
containing 10% FBS and 1% penicillin-streptomycin, were incubated
for 24 h with the concentration of 20 and 30 μg/ml of the extract.
In order to detect the change of intracellular Ca2+
concentration, the treated cells were incubated for 30 min at room
temperature with 1.5 μM of Fluo-3/AM (Invitrogen). The cells on the
slides were mounted in Prolong Gold Antifade Reagent followed by
observation under a Nikon ECLIPS 50i microscope equipped with CDD
camera and images were captured and processed with High-Content
Analysis software.
FACS analysis
To investigate the mechanisms of growth inhibition,
267B1/K-ras cells in 100-mm cell culture dish (SPL Life Sciences)
were incubated with various dose of the extract for 24 h.
Distribution of cell cycle was examined using flow cytometry.
Briefly, cells were harvested by trypsinization, fixed in 99%
ethanol at 4°C for overnight. And then resuspended in PBS
containing RNase A (10 μg/ml) and incubated for 1 h at 37°C. DNA
was stained with propidium iodide (40 μg/ml) (Sigma-Aldrich) for 10
min and cells were then examined by FACSCalibur (Becton-Dickinson,
Mountain View, CA, USA).
Statistical analysis
The GraphPad Prism 5.0 for window was used to
determine the statistical significance of differences between
values for various experimental and control group. Determinations
were performed in triplicate and the results are presented as mean
± SEM. In cases where no error bar is seen in the graph, the
variation is small and thus, the bar is hidden behind the bar.
ANOVA post hoc and subsequently Dunnett's multiple comparison tests
were used for statistical analysis.
Results and Discussion
Effects of the extract on cell
proliferation
In order to understand the effects of the extract on
cell proliferation, 267B1/K-ras cells were treated with the extract
(n-hexane sub-fraction) of S. japonica followed by cell
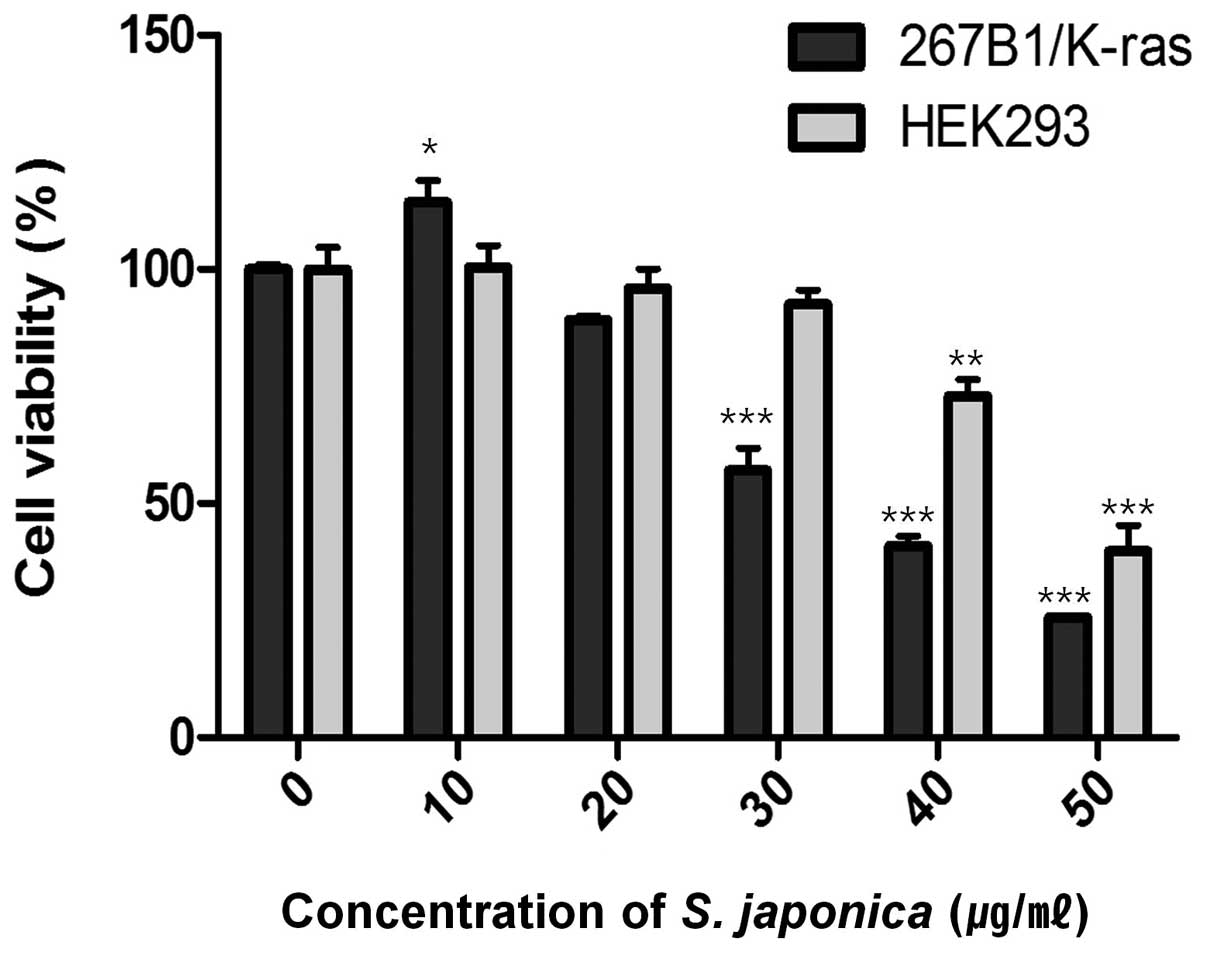
viability assay. The assay showed decreased cell viability at
concentration of 35 μg/ml (IC50) (Fig. 1). Treatment with 10 and 20 μg/ml of
the extract had lower effect on the viability of the 267B1/K-ras
cells which suggested that extensive death of the cells was only
occurring at the higher concentration. However, the effect of 20
and 30 μg/ml of the extract on HEK293 human embryonic kidney cells
are relatively non-toxic. To examine the effects of the extract on
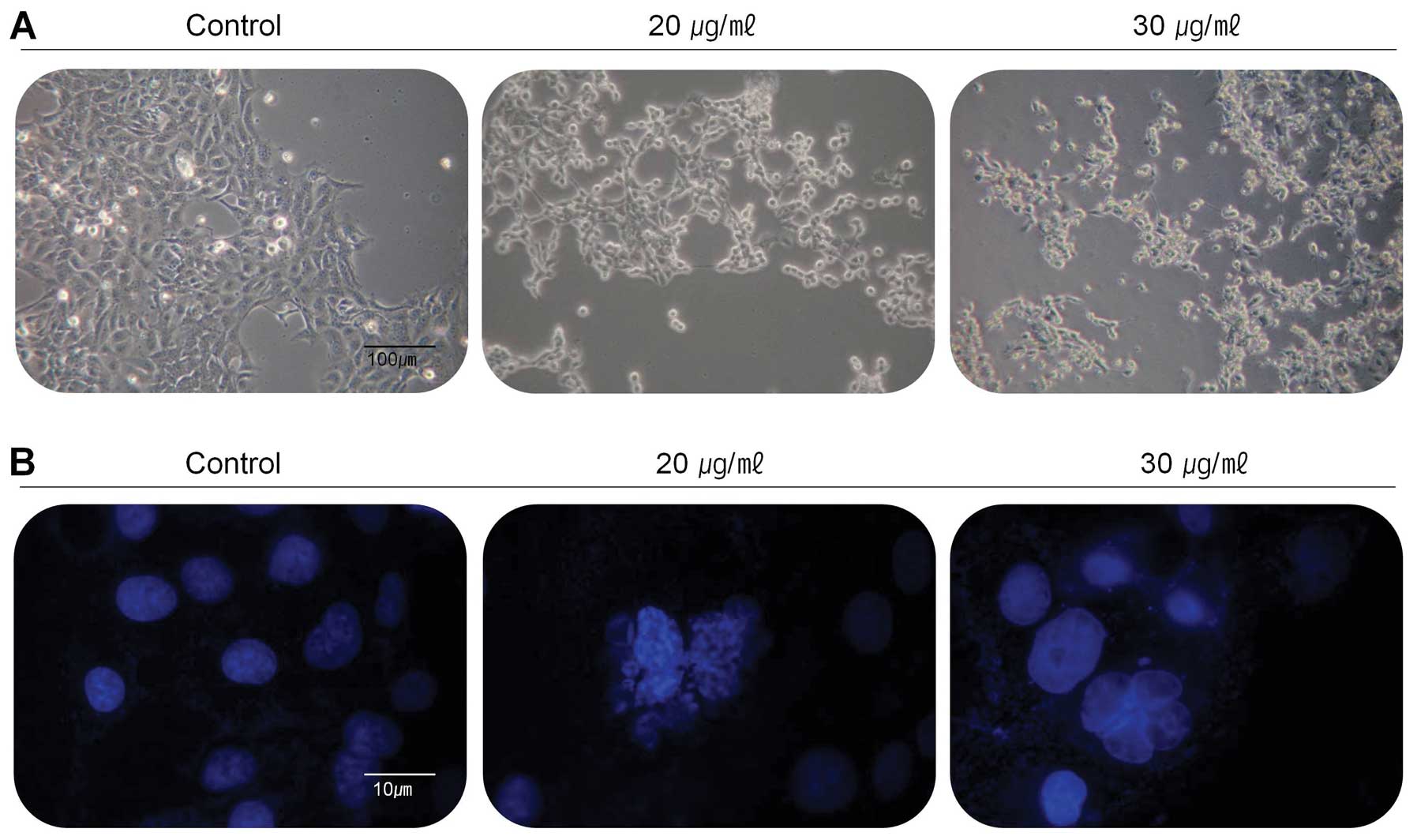
the changes of cell population and morphology, the cells were
treated with the extract at the concentration of 20 and 30 μg/ml
for 24 h and then observed under the inverted microscope (Fig. 2A). Compared with control, most of
the cells were shrunken and some of cells floated on the medium.
Cell population was also dramatically reduced in the medium
containing the extract with the concentration of 30 μg/ml. Further
experiments were carried out to determine whether this
anti-proliferative effect of the extract on the viability of
267B1/K-ras cells was closely associated with apoptotic cell death.
Morphological analysis following DAPI stain was performed to
analyze the cells with nuclear chromatin condensation and apoptotic
bodies (Fig. 2B). The results of
DAPI stain showed overall different forms in the treated cells
compared to untreated control cells. The cells treated with 20 and
30 μg/ml of the extract had reduced cell density, increased
chromatin condensation and incomplete nuclear membrane, so the
extract may be considered as an inducer of apoptosis on 267B1/K-ras
cells.
The extract of S. japonica induces
apoptosis
There are two apoptotic pathways; the extrinsic
death receptor-mediated pathway and the intrinsic
mitochondria-mediated pathway. The truncated Bid protein provides
cross-talk between the two. Both of these pathways are regulated by
caspases, which are responsible, either directly or indirectly, for
the cleavages of cellular proteins, a characteristic of
apoptosis.
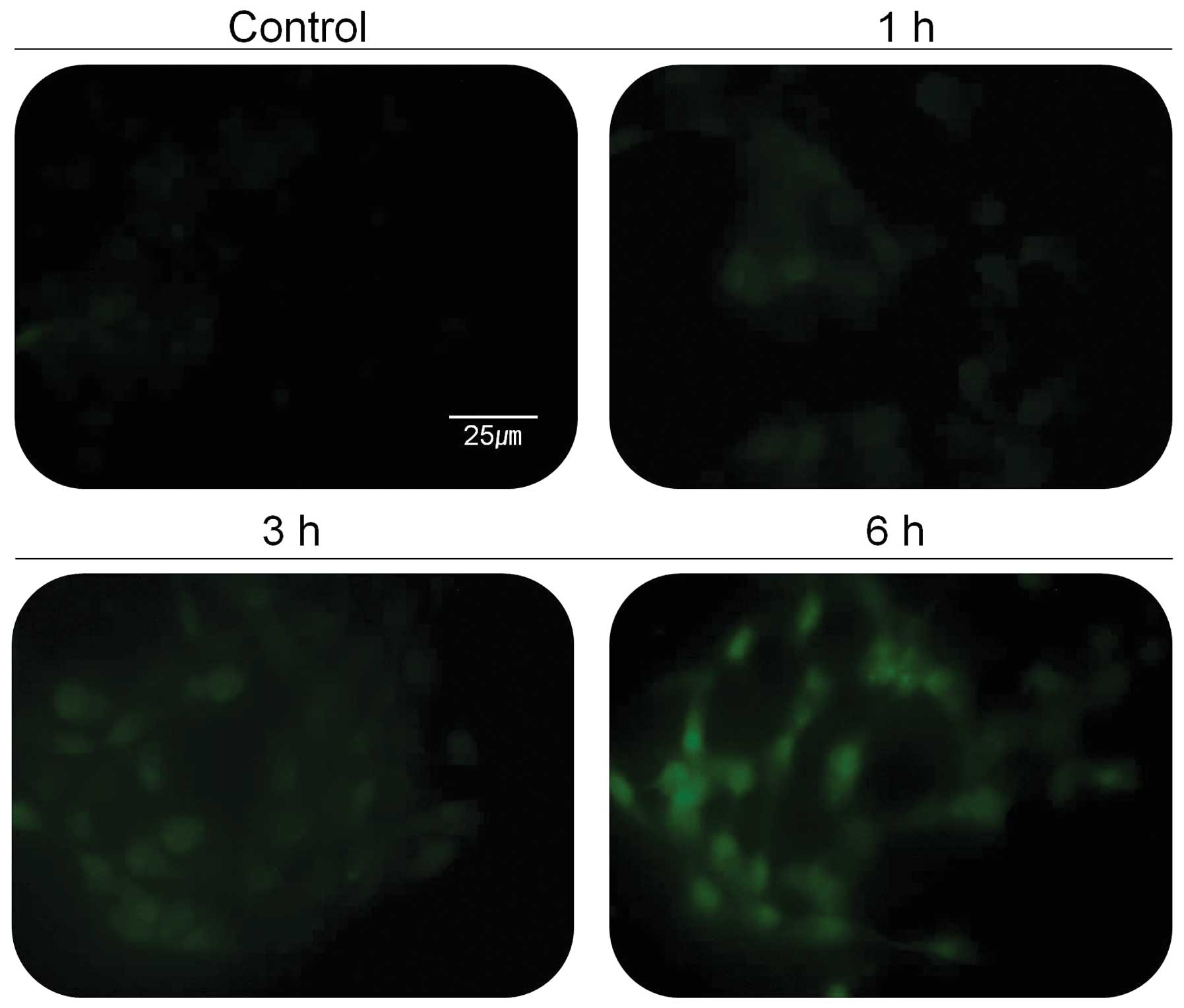
Stressed endocellular reticulum (ER) induces
increased level of cellular calcium ion that activates specific
proteins in cells. Activation of calcium-dependent proteases such
as calpain is thought to play an important role in apoptotic cell
death. In addition, several studies have shown that calpain can
contribute to tumor death, and calpain inhibitors have been used to
block apoptosis. Since calpain is activated by elevated
intracellular Ca2+ concentration, 267B1/K-ras cells were
tested for the change of intracellular Ca2+
concentration (Fig. 4). The green
fluorescent light, indicating the concentration of cellular calcium
ion, was increased based on the treatment time with 30 μg/ml of the
extract (Fig. 4). The activity of
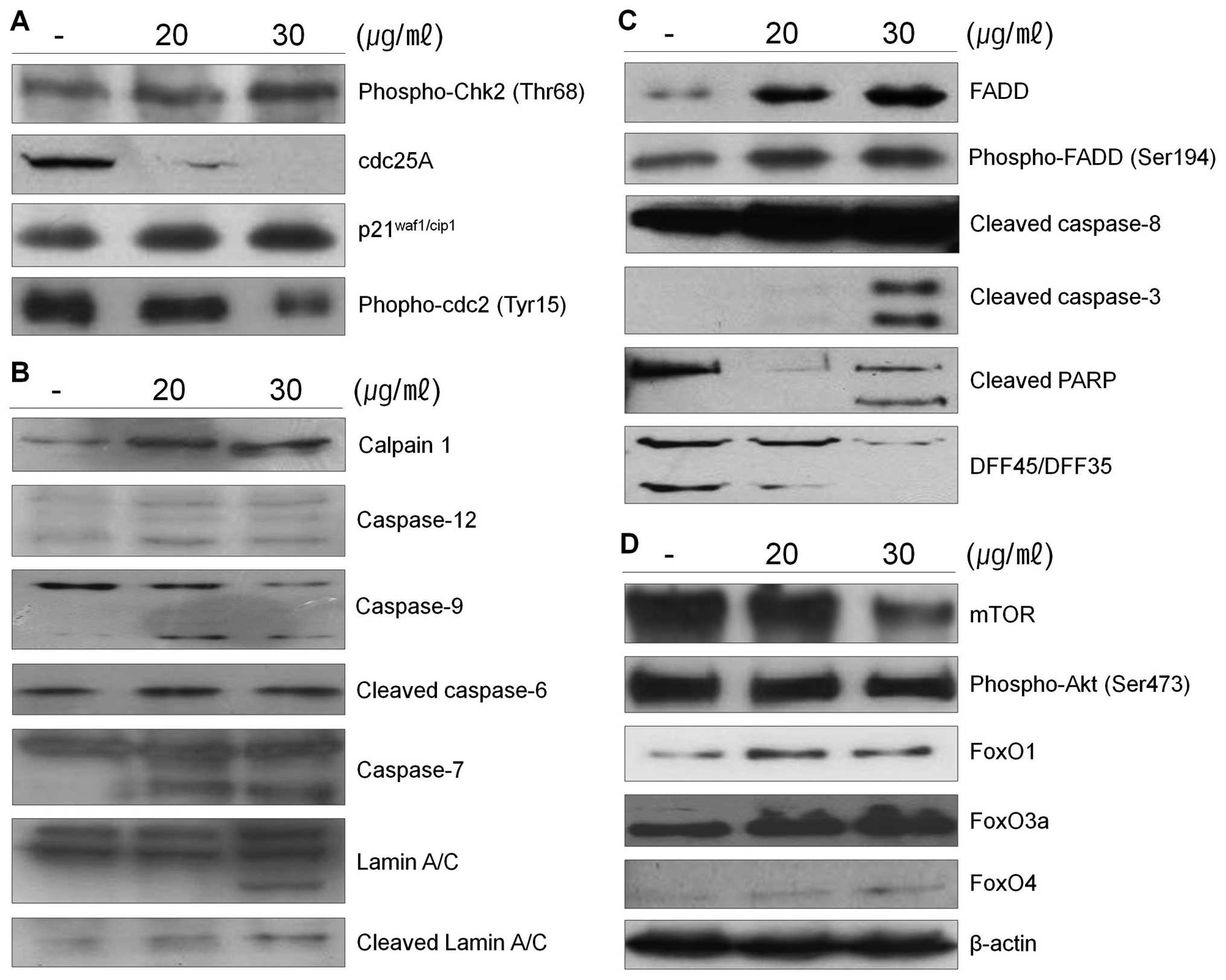
calpain in 267B1/K-ras cells treated with the extract was assessed
in cell lysates using western blot analysis (Fig. 3B). The results shown that
267B1/K-ras cells treated with the extract contained the lysed form
of calpain. Lysis of the calpain is associated with calpain
activation during apoptosis (22).
To examine whether calpain is involved in the extract-induced
apoptosis, ER-specific effectors are examined (Fig. 3B). Caspase-12 locates on the
cytoplasmic side of ER and is proteolytically activated following
ER stress and calpain activation. Activation of caspase-12 was
observed after treatment of the extract (Fig. 3B). After its activation at the ER,
caspase-12 cleaves procaspase-9, leading to activation of
caspase-3. ER stress induced processing of procaspase-9 can occur
in the absence of cytochrome c release, which argues that
caspase-12 triggers directly caspase-9 activation and apoptosis,
independently of the mitochondrial cytochrome c/Apaf-1 pathway
(23).
The death receptor mediated apoptotic pathway has
been proposed as a therapeutic target of cancer. Since the Fas/FasL
system is also a key signal transduction pathway of apoptosis, we
examined the involvement of the Fas/FasL system in 267B1/K-ras
cells after treated with the extract. The cell surface receptor Fas
has a fas-associated protein with death domain (FADD) in the
cytolasmic domain. Once the death signal factors bind to Fas, the
FADD is phosphorylated. To determine the pathways of the
extract-induced apoptosis, the levels of FADD and phospho-FADD were
examined using western blot analysis (Fig. 3C). The levels of FADD and
phospho-FADD were increased after the treatment. These data suggest
that the extract induced apoptosis following the death receptor
mediated extrinsic pathway. Increased phospho-FADD stimulates
binding of caspase-8 to FADD, and this leads the activation of
caspase-8, which can then cleave and activate downstream effector
caspases such as caspase-3 or -7 leading the death of 267B1/K-ras
cells (Fig. 3C) (24).
The mTOR pathway is a central regulator of cell
growth that couples the control of protein synthesis to the
availability of growth factors, nutrients, and energy. This is
accomplished via the regulation of mTOR by multiple signals,
including the PI3-kinase/Akt-FoxO pathway (25). Recently, the FoxO family of
Forkhead transcription factors has been reported to regulate cell
proliferation via the PI3K/Akt pathway (26). Therefore, to determine the status
of PI3K/Akt-FoxO signaling pathway in 267B1/K-ras cells, we treated
267B1/K-ras cells with the extract followed by analyzing the
changes of phosphoryation of Akt and FoxO proteins using western
blot analysis (Fig. 3D). Activated
mTOR up-regulates Akt by its phosphorylation. However, exposure of
the extract resulted in reduced phosphorylation of Akt and mTOR,
but increased FoxO proteins, FoxO1, FoxO3a and FoxO4 in a
dose-dependent manner. Decreased expression level of mTOR induces
dephosphorylation of Akt which cannot repress the transcription
factor, FoxO. Then expression level of FoxO is elevated (Fig. 3D). From these results, we suggest
that PI3K/Akt-FoxO signaling pathway may be activated by the
treatment of S. japonica extract in 267B1/K-ras cells.
Involvement of caspase-3 activation in
apoptosis
The family of caspases can be divided into major
subgroups based on substrate-specificity, sequence homology and
biochemical function; in particular, caspase-3 plays a pivotal role
in the terminal phase of apoptosis degrading several proteins.
Before checking the expression levels of proteins affected by
caspase-3, 267B1/K-ras cells were investigated for their caspase
activity (Fig. 5A) to evaluate
whether the extract affects caspases, the viability of cells were
increased in case of treatment with zVAD-fmk, inhibitor of caspase.
As shown in the results, more cells were alive when treated with
both the extract and zVAD-fmk than treated with the extract alone
(Fig. 5A). It suggested that the
extract is able to affect the activity of caspases. The PARP
protein is one of the cleavage targets of caspase-3 and is also
involved in DNA repair in response to environmental stress. Its
molecular weight is changed from 116 to 86 kDa by apoptosis.
Another target protein of caspase-3 is DFF45 which functions in
normal cells as chaperone for DFF40 during its synthesis. The
association of DFF45 (or its isoform DFF35) with DFF40 inhibits the
DNase activity of the latter (16). Caspase-3 cleaves DFF45, inactivates
its inhibitory function on DFF40 and causes nuclear DNA degradation
by DFF40, leading to cell death (Figs.
3C and 5B) (17). Caspase-3 also plays a role as a
major protein to cleave lamins maintaining nuclear membrane
structure. Lamins function in cell cycle control, DNA replication
and chromatin organization, are are cleaved by caspase-6 by
activating caspase-3. During apoptosis, lamin is cleaved into a
large (41–50 kDa) and small (28 kDa) fragment, and the cleavage of
lamins results in nuclear dysregulation and cell death (Fig. 3B) (27). Results indicate that the extract
activates caspase-3 and it cleaves specific target proteins leading
to apoptosis (Fig. 3).
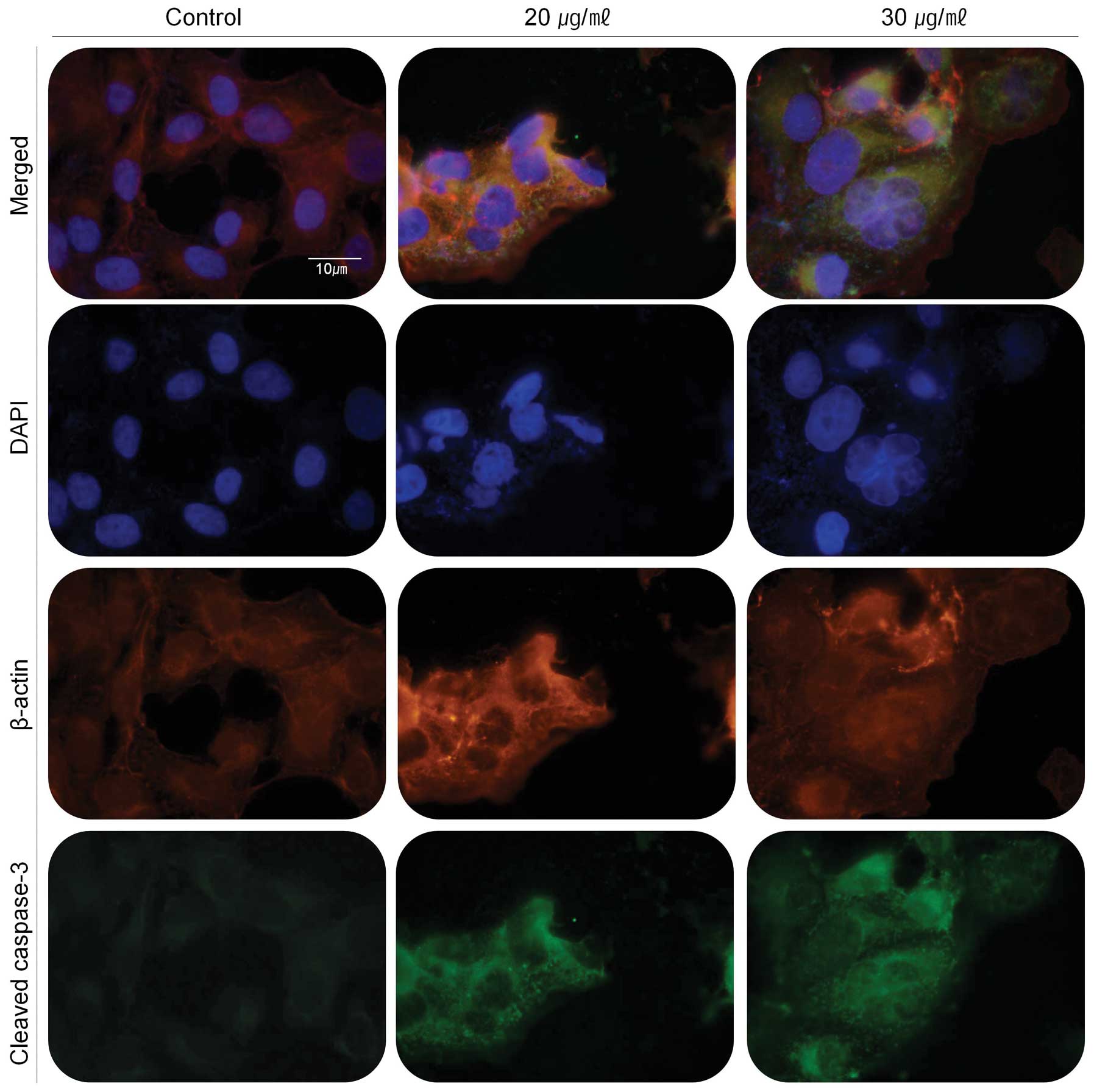
Immunofluorescence assay was also performed to prove
the involvement of caspase-3 in the apoptosis event (Fig. 6). The cleaved caspase-3 was
expressed in green fluorescent light as immunofluorescence makes
use of fluorophores to visualize the location of the antibodies
against cleaved caspase-3. In the cells treated with the extract,
cleaved caspase-3 (green) exists around the cell with stronger
fluorescence in comparison with untreated cells (control).
The extract induced cell cycle arrest
at G2 phase
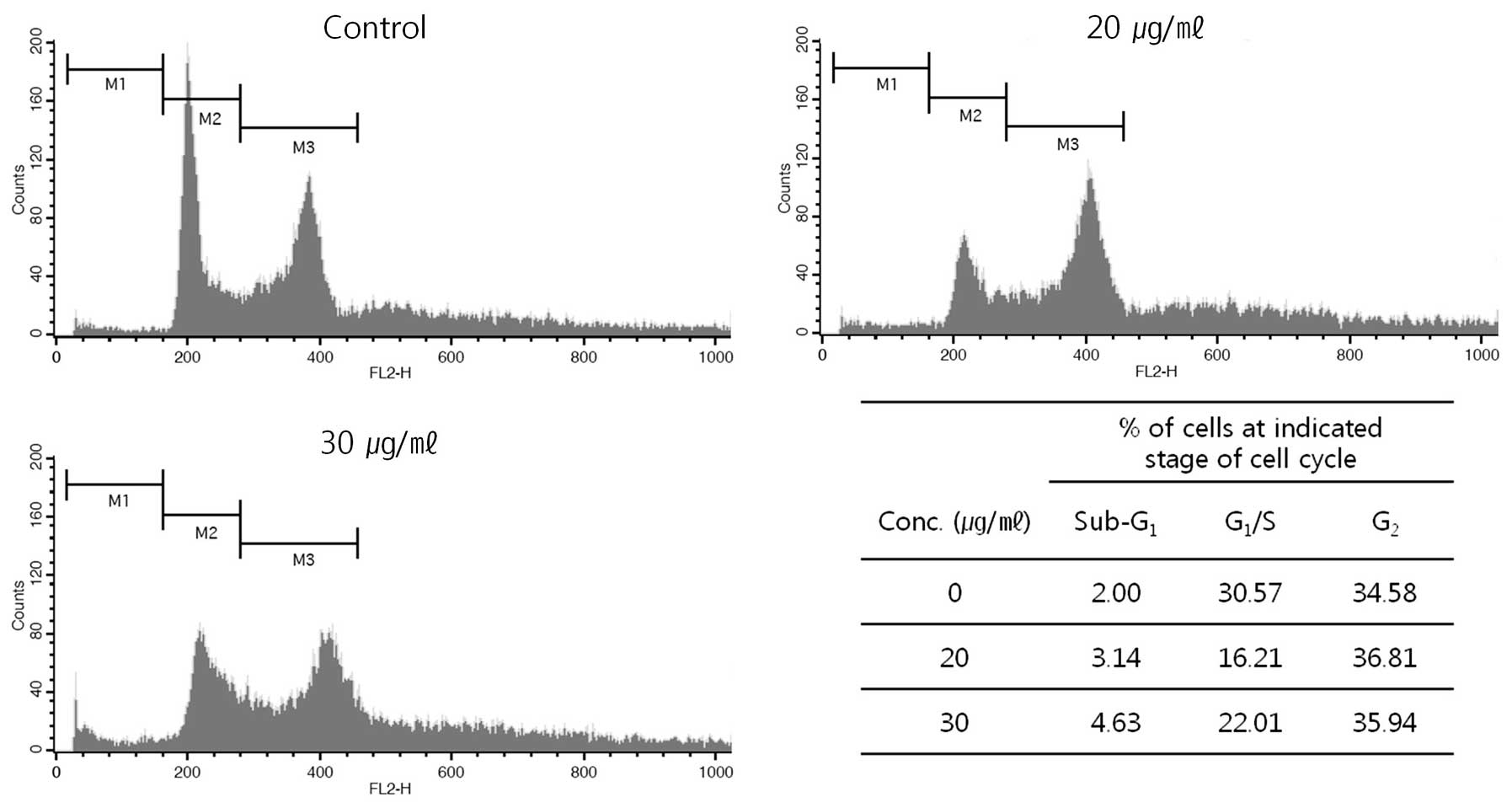
With the apoptosis induced by the extract, we also
analyze distribution of cell cycle by FACS (Fig. 7). After the treatment, the cells
were harvested and then distribution of cell cycle was analyzed.
The harvested cells, which had fragmented DNA, were increased in G2
phase in DNA histogram from 34.58% at control (without the
treatment) to 36.81 and 35.94% of the total population at 20 and 30
μg/ml of the extract, respectively. Western blot analysis was
performed to determine whether cyclin-dependent kinase (Cdk)
inhibitor p21waf1/cip1, phospho-Chk2, phospho-cdc2 and
cdc25A are involved in the extract-mediated anti-proliferative
effect on 267B1/K-ras cells (Fig.
3A). Increased expression level of phospho-Chk2 stimulates
up-regulation of p21waf1/cip1 proteins and
down-regulation of phospho-cdc2 and cdc25A in 267B1/K-ras cells
treated with the extract in a concentration-dependent manner. Taken
together, these results demonstrated that the cytotoxic effects
observed in response to the extract are associated with the
induction of apoptotic cell death.
Phosphorylation in Chk2, the checkpoint kinase, by
DNA damage leads to phosphorylation of cdc25A. The cdc25
phosphatase may be responsible for activation of cdc2 regulating
the entry of eukaryotic cells into mitosis by phosphorylation
(28). The tumor suppressor
protein p21waf1/cip1 acts as an inhibitor of cell cycle
progression. Besides being an inhibitor of cell cycle progression,
p21 acts as a mediator of the apoptotic pathway. Its enforced
expression induces apoptosis or enhances the apoptotic response to
chemotherapeutic agents (29).
The results shown in this study suggested that the
extract (n-hexane sub-fraction) of S. japonica induced
inhibition of cell growth resulted from multiple signaling pathways
mediated by apoptosis and cell cycle arrest at G2 phase.
In conclusion, the anticancer effects of the S.
japonica extract (n-hexane sub-fraction) on 267B1/K-ras human
prostate cancer cells were assessed by detecting apoptosis and
cycle arrest. The exposure of the cells to both 20 and 30 μg/ml of
the extract for 24 h resulted in the inhibition of cell growth and
morphology changes. The activation of caspases by calpain, FADD and
FoxO makes the cells undergoing apoptosis. Elevated intracellular
calcium concentration is assumed to activate calpain and to induce
activation of other proteins involved in ER stress. FADD as
intracellular domain of Fas, indicates that the extract has similar
role to FasL. Thus, the extract can induce apoptosis by the
activation of caspase-8 and the caspase cascade. The cells can also
undergo apoptosis when the FoxO transcription factor is
inactivated. For the cell cycle arrest, the expression level of
cell cycle controlling proteins such as phospho-Chk2, phospho-cdc2,
p21waf1/cip1 and cdc25A were examined. The results
suggested that the anticancer effects of the extract originated
from a brown macro-alga, S. japonica act on apoptosis, cell
cycle arrest and growth inhibition. The extract might be a useful
candidate to develop compounds against human prostate cancer, and
more studies will be required to find mitochondrial apoptosis and
anti-metastasis effect using a further fractionized and selected
single compound from this extract.
Acknowledgements
This study was financially supported
by the Ministry for Food, Agriculture, Forestry and Fisheries,
Republic of Korea.
References
|
1.
|
Parda DS, Thraves PJ, Kuettel MR, et al:
Neoplastic transformation of a human prostate epithelial cell line
by the v-Ki-ras oncogene. Prostate. 23:91–98. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Wynder EL, Rose DP and Cohen LA: Nutrition
and prostate cancer: a proposal for dietary intervention. Nutr
Cancer. 22:1–10. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Giovannucci E: Epidemiologic
characteristics of prostate cancer. Cancer. 75:1766–1777. 1995.
View Article : Google Scholar
|
|
4.
|
Stan SD, Kar S, Stoner GD and Singh SV:
Bioactive food components and cancer risk reduction. J Cell
Biochem. 104:339–356. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Booth E: Trace elements and seaweeds.
Proceeding of the 4th International seaweed symposium. De Virville
AD and Feldmann J: MacMillan; London: pp. 385–393. 1964
|
|
6.
|
Yan X, Chuda Y, Suzuki M and Nagata T:
Fucoxanthin as the major antioxidant in Hijikia fusiformis, a
common edible seaweed. Biosci Biotechnol Biochem. 63:605–607. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Munro MHG, Blunt JW, Dumdei EJ, et al: The
discovery and development of marine compounds with pharmaceutical
potential. J Biotechnol. 70:15–25. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Rice-Evans C, Miller N and Paganga G:
Antioxidant properties of phenolic compounds. Trends Plant Sci.
2:152–159. 1997. View Article : Google Scholar
|
|
9.
|
Yuan YV and Walsh NA: Antioxidant and
antiproliferative activities of extracts from a variety of edible
seaweeds. Food Chem Toxicol. 44:1144–1150. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Lee NY, Ermakova SP, Zvyagintseva TN, Kang
KW, Dong Z and Choi HS: Inhibitory effects of fucoidan on
activation of epidermal growth factor receptor and cell
transformation in JB6 Cl41 cells. Food Chem Toxicol. 46:1793–1800.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Lieberthal W, Koh JS and Levine JS:
Necrosis and apoptosis in acute renal failure. Semin Nephrol.
18:505–518. 1998.PubMed/NCBI
|
|
12.
|
Zimmermann KC, Bonzon C and Green DR: The
machinery of programmed cell death. Pharmacol Ther. 92:57–70. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Cryns VL, Bergeron L, Zhu H, Li H and Yuan
J: Specific cleavage of α-fodrin during Fas-and tumor necrosis
factor-induced apoptosis is mediated by an
interleukin-1β-converting enzyme/Ced-3 protease distinct from the
poly(ADP-ribose) polymerase protease. J Biol Chem. 271:31277–31282.
1996.
|
|
14.
|
Kothakota S, Azuma T, Reinhard C, et al:
Caspase-3-generated fragment of gelsolin: effector of morphological
change in apoptosis. Science. 278:294–298. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Lazebnik Y, Kaufmann S, Desnoyers S,
Poirier G and Earnshaw W: Cleavage of poly(ADP-ribose) polymerase
by a proteinase with properties like ICE. Nature. 371:346–347.
1994. View
Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Lazebnik YA, Takahashi A, Moir RD, et al:
Studies of the lamin proteinase reveal multiple parallel
biochemical pathways during apoptotic execution. Proc Natl Acad Sci
USA. 92:9042–9046. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Mashima T, Naito M, Fujita N, Noguchi K
and Tsuruo T: Identification of actin as a substrate of ICE and an
ICE-like protease and involvement of an ICE-like protease but not
ICE in VP-16-induced U937 apoptosis. Biochem Biophys Res Commun.
217:1185–1192. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Sakahira H, Enari M and Nagata S: Cleavage
of CAD inhibitor in CAD activation and DNA degradation during
apoptosis. Nature. 391:96–99. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Sherr CJ: Gl phase progression: cycling on
cue. Cell. 79:551–555. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Xiong Y, Hannon G, Zhang H, Casso D,
Kobayashi R and Beach D: p21 is a universal inhibitor of cyclin
kinases. Nature. 366:701–704. 1993. View
Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Kwon O, Kim KA, Kim SO, et al: NF-κB
inhibition increases chemosensitivity to trichostatin A-induced
cell death of Ki-Ras-transformed human prostate epithelial cells.
Carcinogenesis. 27:2258–2268. 2006.
|
|
22.
|
Kuo PL, Hsu YL, Cho CY, Ng LT, Kuo YH and
Lin CC: Apoptotic effects of Antrodia cinnamomea1 fruiting bodies
extract are mediated through calcium and calpain-dependent pathways
in Hep 3B cells. Food Chem Toxicol. 44:1316–1326. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Rasheva VI and Domingos PM: Cellular
responses to endoplasmic reticulum stress and apoptosis. Apoptosis.
14:996–1007. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Muzio M, Chinnaiyan AM, Kischkel FC, et
al: FLICE, a novel FADD-homologous ICE/CED-3-like protease, is
recruited to the CD95 (Fas/APO-1) death-inducing signaling complex.
Cell. 85:817–827. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Sabatini DM: mTOR and cancer: insights
into a complex relationship. Nat Rev Cancer. 6:729–734. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Park SJ, Sohn HY, Yoon J and Park SI:
Down-regulation of FoxO-dependent c-FLIP expression mediates
TRAIL-induced apoptosis in activated hepatic stellate cells. Cell
Signal. 21:1495–1503. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Oberhammer FA, Hochegger K, Fröschl G,
Tiefenbacher R and Pavelka M: Chromatin condensation during
apoptosis is accompanied by degradation of lamin A+B, without
enhanced activation of cdc2 kinase. J Cell Biol. 126:827–837.
1994.
|
|
28.
|
Sørensen CS, Syljuåsen RG, Falck J, et al:
Chk1 regulates the S phase checkpoint by coupling the physiological
turnover and ionizing radiation-induced accelerated proteolysis of
Cdc25A. Cancer Cell. 3:247–258. 2003.PubMed/NCBI
|
|
29.
|
Gartel AL and Tyner AL: The role of the
cyclin-dependent kinase inhibitor p21 in apoptosis 1 supported in
part by NIH grant R01 DK56283 (to ALT) for the p21 research and
Campus Research Board and Illinois Department of Public Health
Penny Severns Breast and Cervical Cancer grants (to ALG). 1. Mol
Cancer Ther. 1:6392002.
|