Introduction
Although disease-free survival and overall survival
of patients with breast cancer have been improved through intensive
treatment, breast cancer is still an important public health
problem in women worldwide (1,2).
Major breast cancer treatment methods consist, both separately and
in combination of surgery, radiotherapy and chemotherapy. In
particular, tamoxifen, anti-estrogen medicine, is widely used in
the prevention and treatment of estrogen receptor positive breast
cancer (3). Inherent or acquired
tumor drug resistance limits many agents that could be used to
treat this disease and are often associated with severe,
dose-limiting and systemic toxicities. Therefore, new agents acting
on novel targets in breast cancer are currently under investigation
and the need to develop novel non-toxic therapeutic agents active
against breast cancer remains an important goal.
Interest in the pharmacological effects of bioactive
compounds on cancer treatments and prevention has increased
dramatically over the past twenty years. A great number of natural
agents derived from plants can be potentially useful in
complementary therapy for cancer patients. As well, there is a need
to develop a new, more powerfully active drug from natural
resources with lesser side effects that can act as a substitute for
the current chemical therapy. A recent study confirmed that
increasing vegetable and fruit consumption might reduce the risk of
breast cancer (4,5). Also a lower incidence of breast
cancer is associated with a high consumption of phytoestrogens,
which are biologically active plant-derived phenolic compounds that
structurally mimic the mammalian estrogen, estradiol-17b (6,7).
Among many bioactive compounds, basic and preclinical research on
resveratrol (trans-3,4′,5-trihydroxystilbene), a naturally
occurring polyphenol which can be found in grapes and red wine, has
shown pleiotropic, cardioprotective, anti-aging and anticancer
activities (8,9). In addition, resveratrol is not a
potent cytotoxic compound when compared with other chemotherapeutic
drugs. However, exposure to high doses of resveratrol is required
to induce apoptosis in cancer cells and resveratrol’s biological
activity is limited by its photosensitivity and metabolic
instability. Thus, several studies were undertaken to obtain
synthetic analogues of resveratrol with potent anti-cancer activity
(10). We previously investigated,
designed and synthesized analogues of resveratrol that had potent
activity and demonstrated that four synthetic resveratrol analogues
(HS-1784, -1792, -1791 and -1793) displayed stronger antitumor
effects than resveratrol in most cancer cells tested, including
human breast adenocarcinoma cell line MCF-7 (11–13).
Moreover, the resveratrol analogue,
4-(6-hydroxy-2-naphthyl)-1,3-benzenediol (HS-1793), especially
overcomes the resistance conferred by Bcl-2 by inducing apoptosis
(11,13).
Taken together, the evidence from in vitro
studies indicated that the anticancer activity of HS-1793 in
various cancer cells was mediated through apoptosis (11,13).
However, the specific apoptosis mechanisms at work are not yet well
understood. Therefore, the aim of the present study was to
investigate whether HS-1793 induced cytotoxicity via mitochondria
induced apoptosis and explore the potential mechanisms in murine
breast cancer cells.
Materials and methods
Chemical and reagents
RPMI-1640 medium and fetal bovine serum (FBS) were
obtained from Gibco (Gaithersburg, MD, USA). JC-1 was obtained from
Molecular Probes (Eugene, OR, USA). Rabbit monoclonal to cytochrome
c, PARP [poly(ADP-ribose) polymerase], caspase-3 and GAPDH
antibodies were obtained from Abcam (Cambridge, UK). Anti-AIF
(apoptosis-inducing factor) and anti-Endo G (endonuclease G) rabbit
antibodies were obtained from Calbiochem (San Diego, USA).
Propidium iodide (PI) was obtained from Sigma (St. Louis, MO, USA).
The enhanced chemiluminescent western blotting detection reagent
(SuperSignal West Pico chemiluminescent substrate) was obtained
from Pierce (Rockford, IL, USA).
Preparation of HS-1793
To obtain HS-1793, the stilbene double bond present
in resveratrol was substituted with a naphthalene ring as
previously described (11,12). A stock solution was made in
absolute ethanol at 50 mM, and the working dilutions were directly
made in culture media. The control vehicle was culture media
containing amounts of ethanol equivalent to those present in
HS-1793.
Cell culture condition
FM3A (murine breast cancer cells) originated from
the mammary gland of the C3H/He mouse were grown in RPMI-1640
medium (Gibco) containing 10% FBS, 100 U/ml penicillin and 100 U/ml
streptomycin, under the condition of wet air containing 5%
CO2 and 37°C.
HS-1793 treatment and cell viability
assay
HS-1793 dissolved in EtOH (50 mM) was prepared and
stored at −80°C until use. FM3A cells were plated onto 6-well
plates at a density of 2×105 cells/well and grown for
treatment period. For dose-response experiments, different
concentrations of HS-1793 (0, 1.3, 2.5, 5, 10, 15 and 20 μM) were
added to cells and grown at 37°C and 5% CO2 for 24 and
48 h. For time-dependent experiments, the cells were treated with 5
μM of HS-1793 for 0, 12, 24, 36, 48 and 72 h. Cell viability
determined with hemacytometer by trypan blue and MTT assay as
previously described (11).
Sub-G1 phase assay
FM3A cells were precultured onto 6-well plates at a
density of 2×105 cells/well for 24 h and the cells were
treated with HS-1793 (5 μM) according to exposure time (0, 12, 24,
36, 48 and 72 h). The trypsin was added to the cells in plates for
3 min, after which the cells were harvested by centrifugation at
1,500 rpm for 5 min. The pellets were washed twice with cold
phosphate-buffered saline (PBS) and then fixed by using 70% ethanol
at 4°C for 30 min. The cells were then washed twice with cold PBS
and resuspended in PBS containing 40 μg/ml PI soultion, 0.1 mg/ml
RNase and 0.1% Triton X-100 in a dark room for 30 min at 37°C. The
cells were then analyzed by flow cytometer (Beckman Coulter, USA).
The sub-G1 (apoptosis) phase analyzed with software for cell cycle
analysis.
Nuclear morphology analysis of
apoptosis
FM3A cells were pretreated with HS-1793 for each
exposure time (0, 12, 24, 36, 48 and 72 h). Then, cells were
harvested and cell suspension was centrifuged onto a clean,
fat-free glass slide with a cytocentrifuge. The samples were fixed
for 10 min in 4% paraformaldehyde (PFA) and stained with 4 μg/ml of
Hoechst 33342 at 37°C for 30 min. The cells were photographed under
Nikon Eclipse TS100 fluorescence microscopy (Nikon, Tokyo,
Japan).
TUNEL assay
Apoptosis was detected by using TUNEL apoptosis
detection kit (Millipore, USA) and performed according to the
instruction of the supplier. The samples were observed under
fluorescence microscopy.
Assay of mitochondrial membrane potential
(ΔΨm)
Change in ΔΨm were determined by staining
the cells with the potential-sensitive fluorescent probe
5,5′,6,6′-tetrachloro-1,1′3,3′-tetraethylbenzimidazol carbocyanine
iodide JC-1. The JC-1 dye was directly added to 1 μM of final
concentration into the cell culture medium. After incubation for 20
min, the cells were centrifuged at 1,000 rpm for 5 min and removed
supernatant. Then, the cells were washed with medium and added to
400 μl of PBS. ΔΨm was measured by a flow cytometer.
Confocal laser microscopy
FM3A cells were pretreated with HS-1793 for each
exposure time (0, 12, 24, 36, 48 and 72 h). The cells were
incubated for 15 min at 37°C after adding 450 μM of mitotracker
probe (Invitrogen, Carlsbad, CA, USA) and then washed with medium.
The cells were cytospan onto a clean fat-free glass slide.
Cytocentrifuged cells were fixed in 4% PFA for 30 min and incubated
overnight at 4°C with anti-cytochrome c, AIF and Endo G
antibody (1:100 dilution) and then incubated with secondary
antibody (FITC-conjugated goat anti-rabbit IgG at 1:100 dilution)
for 1 h at 37°C. Photomicrographs were obtained using a Zeiss LSM
700 laser-scanning confocal microscope (Zeiss, Goettingen,
Germany).
Immunoblotting
The cell were harvested and washed twice with
ice-cold PBS. Then, cells were resuspended in 200 μl ice-cold
solubilizing buffer (300 mM NaCl, 50 mM Tris-HCl, pH 7.6, 0.5%
Triton X-100, 1 ml protease inhibitor cocktail) and incubated at
4°C for 40 min. The lysates were centrifuged at 14,000 rpm for 20
min. Protein concentrations of cell lysates were determined by
using the Bio-Rad protein assay kit (Bio-Rad Laboratories,
Hercules, CA, USA). Equal amounts of protein were subjected to
7.5–15% SDS-PAGE for caspase-3, PARP, anti-cleaved caspase-3 and
anti-cleaved PARP, respectively, and transferred to a
nitro-cellulose membrane. Western blot analysis was performed using
standard protocols (14).
Immunostaining with antibodies was performed using Super-Signal
West Pico enhanced chemiluminescence substrate and detected with
LAS-3000PLUS (Fuji Photo Film Co., Kanagawa, Japan).
Statistical analysis
All data are expressed as mean standard deviation
(SD). The evaluation of statistical significance was performed
using Student’s t-test or one-way analysis of variance (ANOVA)
using the Statistical Package for the Social Sciences (SPSS)
statistical software for Windows, Version 18.0 (Chicago, IL, USA).
For all analyses, a difference was considered to be significant at
P<0.05.
Results
HS-1793 reduced cell viability in FM3A
cells
To examine the antitumor activity of HS-1793 in
murine breast cancer cells, exponentially dividing FM3A cells were
treated with various concentrations of HS-1793, and cell viability
was measured at each exposure time. HS-1793 significantly decreased
the percentages of viable cells and those effects were
dose-dependent at 24 and 48 h (p<0.05, Fig. 1A). However, HS-1793 induced no
significant inhibition of cell growth in a nonmetastatic human
mammary epithelial cell line (MCF-10A) under tested concentrations
(data not shown). Indeed, HS-1793 caused marked growth inhibition
of 50% (IC50) in FM3A cells at 5 μM for 48 h and we also
showed that HS-1793 treatment significantly inhibited FM3A cell
growth in a time-dependent manner at 5 μM (p<0.05, Fig. 1B). Therefore, our data indicate
that HS-1793 induced effective cell death in FM3A cells in a dose-
and time-dependent manner.
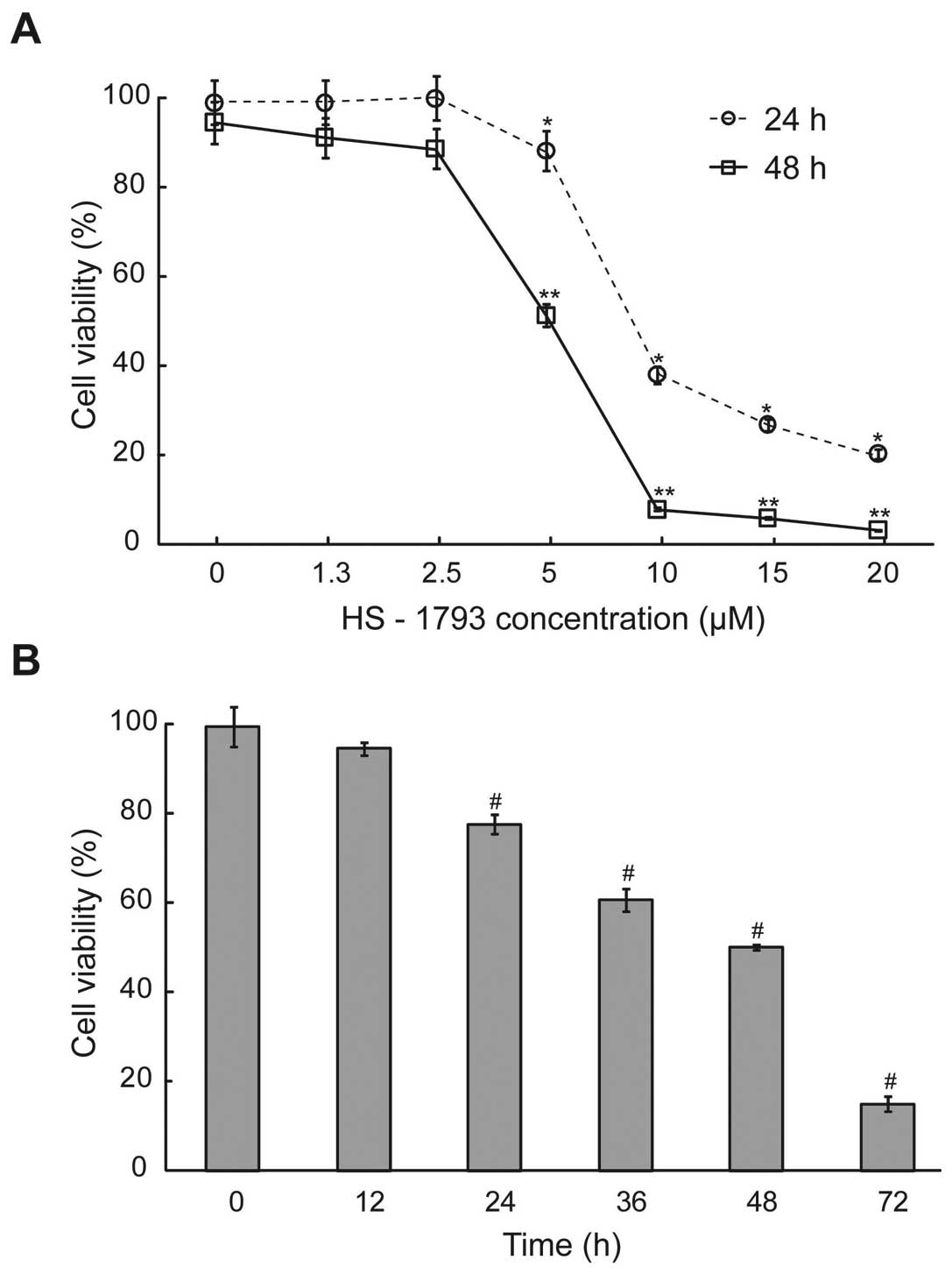 | Figure 1.The effect of cell viability after
HS-1793 treatment in FM3A cells. (A) The cells were treated with or
without different concentrations (0, 1.3, 2.5, 5, 10, 15 and 20 μM)
of HS-1793 for 24 or 48 h. (B) After treatment of HS-1793 5 μM, the
cells were treated for various exposure times (0, 12, 24, 36, 48
and 72 h). Cells were collected by centrifugation and the viable
cells were counted by trypan blue or MTT assay. Data are reported
as the mean ± SD of three experiments. *,**,#P<0.05
as compared with untreated control (0 μM). |
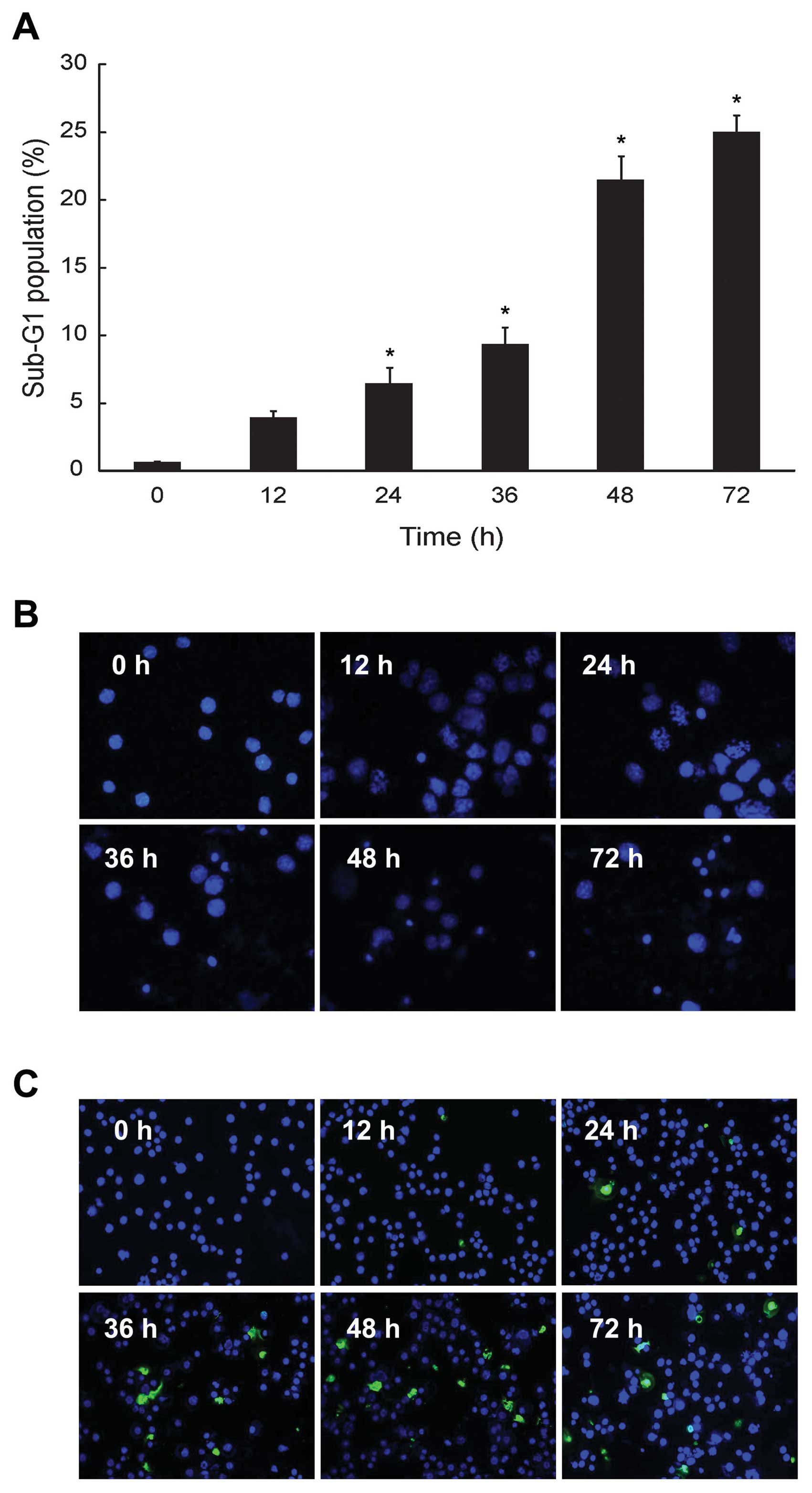
HS-1793 increased sub-G1 phase and
induction of apoptosis in FM3A cells
We investigated whether the HS-1793 induced
cytotoxicity in FM3A cells would be due in part to proapoptotic
effects. Consequently, the effect of HS-1793 on cell cycle
progression was analyzed by flow cytometry in exponentially
dividing cultures of FM3A cells treated with either ethanol
(control) or HS-1793 (5 μM), and the percentages of cells in sub-G1
phases were calculated. HS-1793 induced enhancement of the sub-G1
DNA content in a time-dependent manner at 5 μM (Fig. 2A). In addition, we identified
apoptotic features in the cells using nuclear morphological changes
and DNA fragmentation. FM3A cells exhibited increase of nuclear
fragment (Fig. 2B) and DNA
fragment (Fig. 2C) in a
time-dependent manner by HS-1793 treatment (5 μM). However, HS-1793
did not induced an enhancement of sub-G1 DNA content, nuclear
fragment and DNA fragment at 5 μM in MCF-10A (data not shown).
These results suggest that HS-1793 can have anti-proliferative
activity through induction of apoptosis in FM3A cells while there
is little normal cell toxicity under treatment condition.
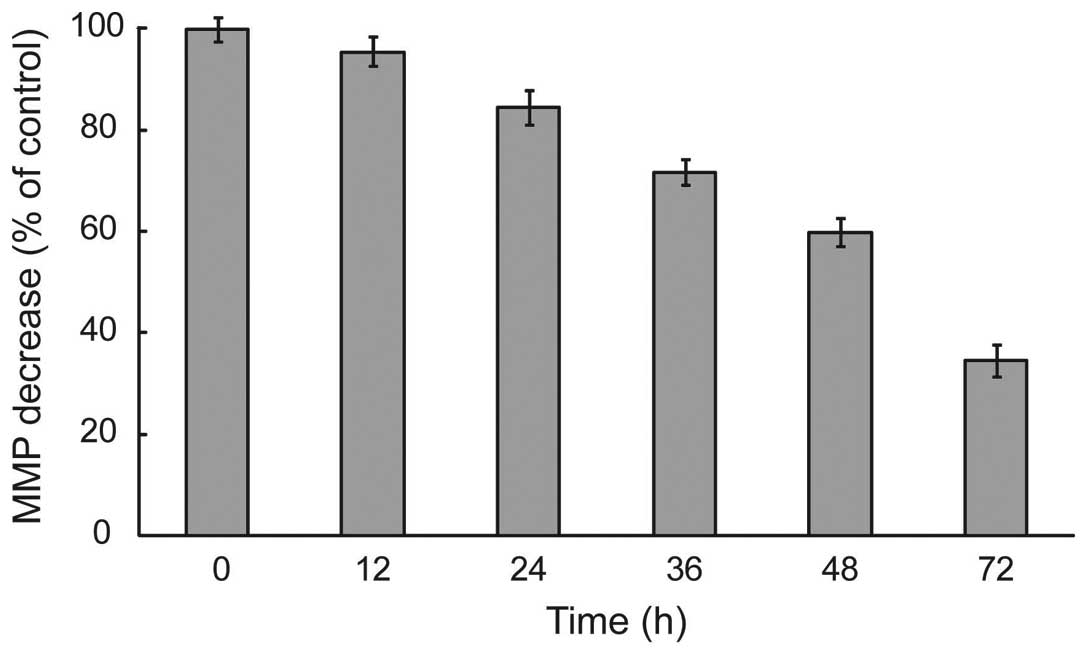
HS-1793 changed ΔΨm levels in
FM3A cells
During apoptosis, early and pivotal events occurred
in the mitochondria that were often, although not always,
associated with the collapse in ΔΨm(14). To delineate this mechanism, we
measured whether HS-1793 induced alterations of ΔΨm by
the use of mitochondrial selective lipophilic cation JC-1 probe.
HS-1793 exerted enhancement of JC-1 green fluorescence intensity
after treatment of HS-1793 (5 μM), representative cells with
depolarized mitochondria in a time-dependent and significant
depolarization with 2.9-fold decrease in ΔΨm were
observed after treatment at 72 h in FM3A cells (p<0.05, Fig. 3). It can be seen in that HS-1793
decreased the levels of ΔΨm in FM3A cells and these
effects were time-dependent.
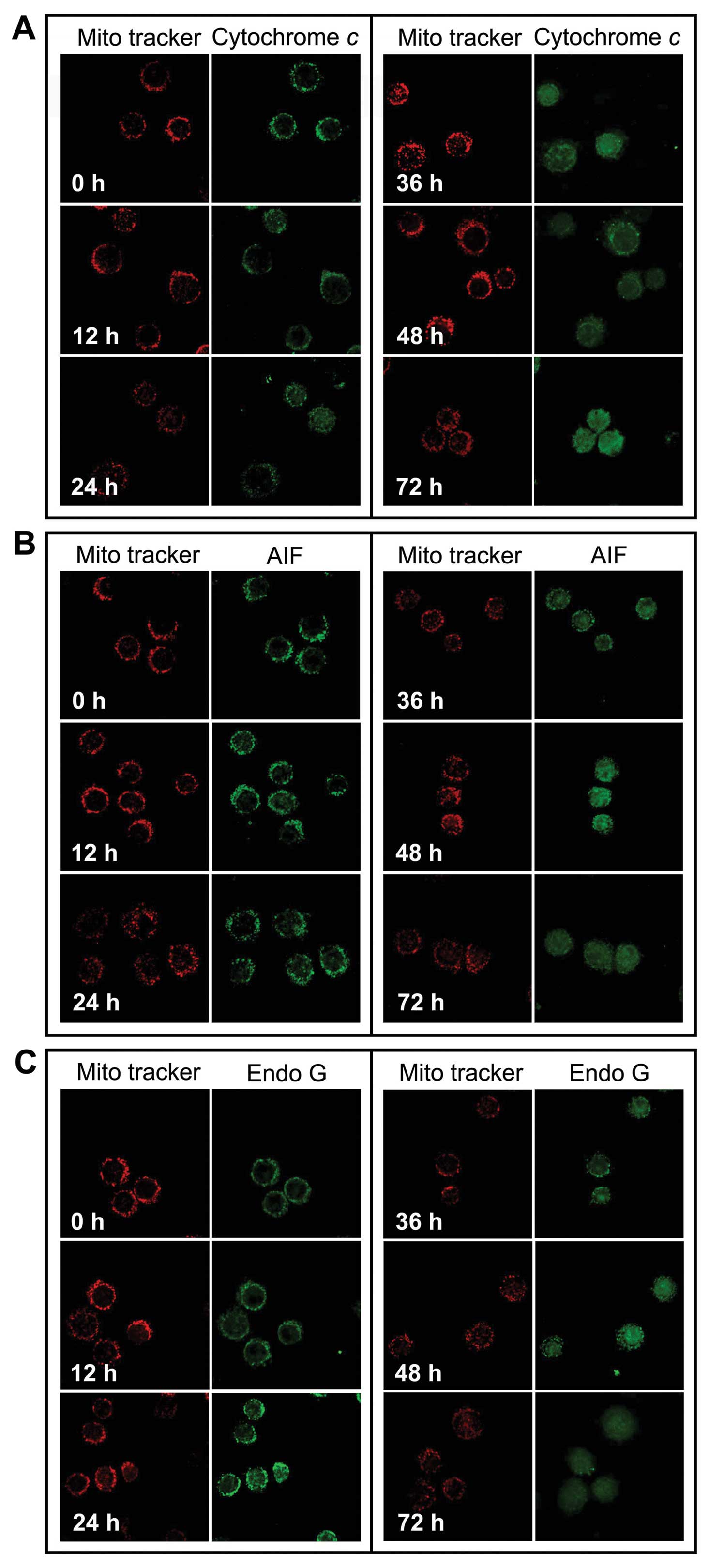
HS-1793 promotes release of cytochrome c,
AIF and Endo G from the mitochondria into the cytosol
We investigated whether cytochrome c, Endo G
and AIF were released from the inner mitochondrial membrane during
HS-1793 treatment. FM3A cells after treatment with 5 μM of HS-1793
for 12, 24, 36, 48 and 72 h, cells were harvested and the
translocation of cytochrome c, AIF and Endo G were
determined using confocal laser microscopy. Fig. 4 shows that HS-1793 promotes the
release of cytochrome c (Fig.
4A), AIF (Fig. 4B) and Endo G
(Fig. 4C) from mitochondria and
that longer treatment time periods increased the release of the
mitochondrial proteins.
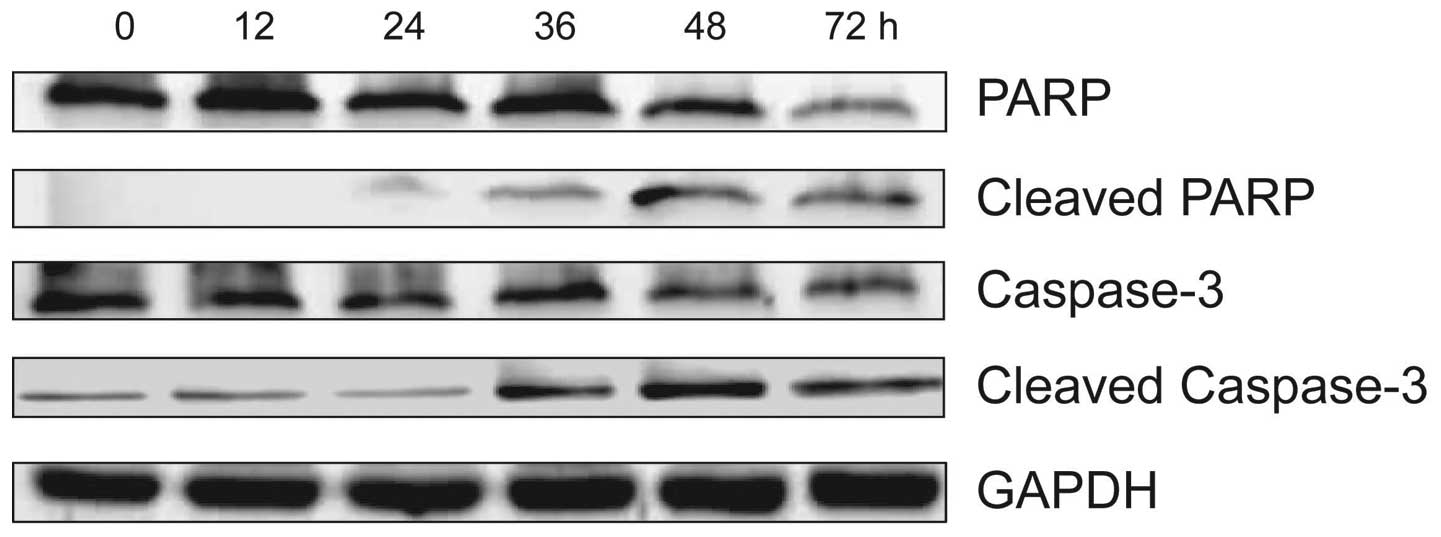
HS-1793 induces apoptosis by caspase-3
and PARP activation in FM3A cells
In order to confirm that HS-1793 induced apoptosis
was also mediated through mitochondria-dependent caspase
activation, we analyzed caspase-3 and PARP activities by using
western blotting and treatment with 5 μM of HS-1793 in FM3A cells
for different time-points (0–72 h). As shown in Fig. 5, HS-1793 decreased caspase-3 and
PARP expression in FM3A cells while cleaved caspase-3 and PARP
expression was increased by HS-1793. These data suggest that
HS-1793 induces activation of caspase-dependent pathway in FM3A
cells, therefore the caspase cascade would be involved in the
apoptosis.
Discussion
The therapeutic goal for cancer is to trigger
tumor-selective cell death and the tumor’s response to therapy
depends mainly on tumor’s ability to undergo cell death. The role
of apoptosis in the cytotoxicity of anticancer drugs has become
clearer (15). Among natural
bioactive compounds, the antiproliferative activity of the
resveratrol against tumor cell lines of different origins has been
extensively characterized (16–23).
From these studies, it was found that resveratrol induced cell
death and that, in certain cell types, it involved an apoptotic
mechanism (18,21–23).
The dose at which an apoptotic effect was seen in resveratrol is
relatively higher (100–200 μM) than the dose used to induce cell
cycle arrest or cancer cell proliferation inhibition (10–30 μM)
(20,22). In vitro studies demonstrated
that resveratrol exerts dose- and time-dependent antiproliferative
and proapoptotic effects in human breast cancer MCF-7 and MDAMB-231
cells, thus decreasing cell viability (22). A key target for identifying methods
of cancer prevention and therapy is the induction of apoptosis or
the debilitation of cancer cells without excessive normal cell
damage by any natural compound (24,25).
In this respect, chemical modification of the stilbene backbone of
resveratrol may need to enhance its biological activity. Previous
studies have reported that several resveratrol analogues
demonstrate stronger anti-tumor effects than resveratrol (10,11).
Among them, HS-1793 does not contain the unstable double bond found
in resveratrol and the position of two of three hydroxyl groups in
HS-1793 at the aromatic ring is different from resveratrol
(12). The term resveratrol
derivative/analogue is used for HS-1784 because HS-1784 is a
derivative of resveratrol and HS-1793 is derived from HS-1784 which
has been reported in previous studies (11,12).
A synthetic analogue having the same structure as HS-1784 was
documented to have a high ceramide-mediated proapoptotic activity
in human breast cancer cells and to block the cell cycle in the
G0–G1 phase in leukemia cells (26). HS-1793 was also noted to display
stronger antitumor effects than resveratrol in most cancer cells,
to overcome the resistance conferred by Bcl-2 in U937 cells via
14-3-3, and to exert its antitumor activity via Bad (11). However, there is still considerable
uncertainty about the cytotoxic effects on HS-1793 induced
apoptosis mechanism in breast cancer cells.
In search for novel strategies for further
management of breast cancer, we have attempted to identify the
molecular mechanisms involved in HS-1793-induced apoptosis, both
caspase-dependent and -independent via mitochondria pathway. In the
present study, we found that HS-1793 was effective in decreasing
cell numbers in the murine FM3A breast cancer cell line through
growth inhibition and/or apoptosis. Furthermore, to understand the
association between HS-1793 and apoptosis, we showed various
apoptotic changes in FM3A cells exposed to 5 μM of HS-1793. In
sub-G1 DNA content, HS-1793 was enhanced in a time-dependent manner
and also increased nuclear fragment and DNA fragment. Our results
showed that HS-1793 induced apoptosis or cell growth inhibition in
lower dose (3–25 μM) than resveratrol (100–300 μM) in breast cancer
cells (11,22). These results suggest that HS-1793,
a novel resveratrol analogue, may be superior to natural
resveratrol as a candidate for chemoprevention agent.
Most of the conventional anticancer treatments are
thought to induce cell death through indirect activation of the
mitochondria-dependent pathway of apoptosis, a pathway often found
altered in drug-resistant cancer cells (27,28).
In most cases, chemotherapeutic drugs first interact with an
intracellular target resulting in stress signals that secondarily
converge to mitochondria and finally result in apoptotic cell
death. Following stress signals generated by conventional
treatments, the permeability of mitochondrial membranes is
increased, leading to the release of proapoptotic proteins which in
turn initiate the caspase cascade and finally result in cell death
(27,29). In particular, a
mitochondria-dependent step, involving outermembrane
permeabilization, is associated with most pro-apoptotic stimuli.
The mitochondria contain several apoptogenic factors (cytochrome
c, Smac/Diablo, HtrA2/Omi, AIF and Endo G) and the release
of these factors regulate apoptosis (30). Endo G and AIF have been reported to
induce caspase-independent nuclear apoptosis and thus it has been
proposed that they are involved in caspase-independent cell death
processes (31–35). AIF is a phylogenetically ancient
mitochondrial intermembrane flavoprotein endowed with the unique
capacity to induce caspase-independent peripheral chromatin
condensation and large-scale DNA fragmentation and provides a
biochemical link between the associated mitochondrial membrane
permeabilization and the nuclear signs of apoptosis (31,35).
Endo G is a mitochondrial nuclease that is likely implicated in
mitochondrial DNA replication and is synthesized as a propeptide
with an amino-terminal presequence that targets the nuclease to
mitochondria (34). Loss of
ΔΨm induced secretion of apoptotic proteins such as AIF
and Endo G proteins from mitochondria to cytosol promoted the
activation of apoptosis. AIF and Endo G cleave DNA in the nucleus
leads to caspase-independent cell death (28,36).
In this study, we investigated whether the mitochondrial AIF and
Endo G release involving ΔΨm were critical for the
HS-1793-mediated apoptosis. Our data clearly indicated that HS-1793
caused loss of ΔΨm the translocations of cytochrome
c, AIF and Endo G protein from the nucleus into cytosol at 5
μM concentration in a time-dependent manner in FM3A cells. It can
be seen that HS-1793 promoted the release of AIF and Endo G from
mitochondria into cytosol and enhanced caspase-independent death in
FM3A cells.
The loss of membrane potential is an early event in
mitochondrial-mediated apoptosis (36,37).
After the reduction of membrane potential and the release of
mitochondrial cytochrome c, a critical step is the formation
of apoptosomes, which ultimately cleave procaspase-3 to form active
caspase-3. Caspases play critical roles in the execution of
apoptosis (38,39). Caspase-3 has also been shown to be
a key component integral for apoptosis, and relies on the actions
of the initiator caspases including caspase-8 and -9 to mediate its
actions (40). In addition,
molecular studies examining the apoptotic process have revealed
that the Bcl-2 protein functions upstream of caspase-3, and that it
prevents the proteolytic activation of caspase-3, thus leading to
cleavage of PARP and apoptosis (40–43).
Our results showed that HS-1793 treatment activated caspase-3 as
well as PARP in FM3A cells. HS-1793 induced cytotoxicity through
increases of DNA fragmentation, nuclear fragmentation and sub-G1
DNA contents as well as the cleavage of PARP, thus confirming that
the apoptosis induced by HS-1793 in FM3A cells might be mediated
through the caspase-3 pathway.
In conclusion, these results demonstrate that
HS-1793 induced cytotoxicity in FM3A cells is due to subsequent
induction of apoptosis via mitochondrial pathway from caspase
activation or cytochrome c, AIF and Endo G release. These
findings suggest that HS-1793 might be a potentially promising
candidate compound that needs to be further explored as an
anticancer agent or mitochondria target drugs for the treatment of
human breast cancer.
Acknowledgements
This study was supported by Nuclear
R&D Program through the Dong Nam Institute of Radiological and
Medical Sciences funded (code: 50493-2012 and 50590-2012) by the
Ministry of Education, Science and Technology.
References
|
1.
|
K ChanGJ MorrisChemoprevention of breast
cancer for women at high riskSemin
Oncol33642646200610.1053/j.seminoncol.2006.08.01717145342
|
|
2.
|
A JemalA ThomasT MurrayM ThunCancer
statistics, 2002CA Cancer J
Clin522347200210.3322/canjclin.52.1.23
|
|
3.
|
P LazarusA Blevins-PrimeauY ZhengD
SunPotential role of UGT pharmacogenetics in cancer treatment and
prevention: focus on tamoxifenAnn NY Acad
Sci115599111200910.1111/j.1749-6632.2009.04114.x19250197
|
|
4.
|
S GandiniH MerzenichC RobertsonP
BoyleMeta-analysis of studies on breast cancer risk and diet: the
role of fruit and vegetable consumption and the intake of
associated micronutrientsEur J
Cancer36636646200010.1016/S0959-8049(00)00022-8
|
|
5.
|
H AdlercreutzPhyto-oestrogens and
cancerLancet Oncol3364373200210.1016/S1470-2045(02)00777-5
|
|
6.
|
JL LimerV SpeirsPhyto-oestrogens and
breast cancer chemopreventionBreast Cancer
Res6119127200410.1186/bcr78115084232
|
|
7.
|
W CraigHealth-promoting properties of
common herbsAm J Clin Nutr70491499199910479221
|
|
8.
|
JF SavouretM QuesneResveratrol and cancer:
a reviewBiomed
Pharmacother568487200210.1016/S0753-3322(01)00158-512000139
|
|
9.
|
BB AggarwalA BhardwajRS AggarwalNP SeeramS
ShishodiaY TakadaRole of resveratrol in prevention and therapy of
cancer: preclinical and clinical studiesAnticancer
Res2427832840200415517885
|
|
10.
|
YJ CallQY WeiJG FangL YangZL LiuJH WhcheZ
HanThe 3,4-Dihydroxyl groups are important for trans-resveratrol
analogs to exhibit enhanced antioxidant and apoptotic
activitiesAnticancer Res249991002200415161055
|
|
11.
|
SH JeongWS JoSH SongA novel resveratrol
analog, HS-1793, overcomes the resistance conferred by Bcl-2 in
human leukemic U937 cellsBiochem
Pharmacol713371347200910.1016/j.bcp.2009.01.002
|
|
12.
|
SH SongH LeeY JinSyntheses of hydroxy
substituted 2-phenyl-naphthalenes as inhibitors of tyrosinaseBioorg
Med Chem Lett17461464200710.1016/j.bmcl.2006.10.02517064896
|
|
13.
|
SH JeongWS JoSH SongA novel resveratrol
analogue HS-1793 treatment overcomes the resistance conferred by
Bcl-2 and is associated with the formation of mature PML nuclear
bodies in renal clear cell carcinoma Caki-1 cellsInt J
Oncol35135313602009
|
|
14.
|
P DeckerD IsenbergS MullerInhibition of
caspase-3-mediated poly(ADP-ribose) polymerase (PARP) apoptotic
cleavage by human PARP autoantibodies and effect on cells
undergoing apoptosisJ Biol
Chem27590439046200010.1074/jbc.275.12.904310722754
|
|
15.
|
JA CallSG EckhardtDR CamidgeTargeted
manipulation of apoptosis in cancer treatmentLancet
Oncol910021011200810.1016/S1470-2045(08)70209-218760670
|
|
16.
|
AK JoeH LiuM SuzuiME VuralD XiaoIB
WeinsteinResveratrol induces growth inhibition, S-phase arrest,
apoptosis, and changes in biomarker expression in several human
cancer cell linesClin Cancer Res88939032002
|
|
17.
|
AW Opipari JrL TanAE BoitanoDR SorensonA
AuroraJR LiuResveratrol-induced autophagocytosis in ovarian cancer
cellsCancer
Res64696703200410.1158/0008-5472.CAN-03-240414744787
|
|
18.
|
J DorrieH GerauerY WachterSJ
ZuninoResveratrol induces extensive apoptosis by depolarizing
mitochondrial membranes and activating caspase-9 in acute
lymphoblastic leukemia cellsCancer Res61473147392001
|
|
19.
|
MV ClementJL HirparaSH ChawdhuryS
PervaizChemo-preventive agent resveratrol, a natural product
derived from grapes, triggers CD95 signaling-dependent apoptosis in
human tumor cellsBlood9299610021998
|
|
20.
|
Y SchneiderF VincentB
DurantonAnti-proliferative effect of resveratrol, a natural
component of grapes and wine, on human colonic cancer cellsCancer
Lett1588591200010.1016/S0304-3835(00)00511-510940513
|
|
21.
|
S BaatoutH DerradjiP JacquetD OomsA
MichauxM MergeayEnhanced radiation-induced apoptosis of cancer cell
lines after treatment with resveratrolInt J Mol
Med13895902200415138632
|
|
22.
|
E Pozo-GuisadoJM MerinoS
Mulero-NavarroResveratrol induced apoptosis in MCF-7 human breast
cancer cells involves caspase independent mechanism with
downregulation of Bcl-2 and NF-κBInt J
Cancer1157484200515688415
|
|
23.
|
SH TsengSM LinJC ChenResveratrol
suppresses the angiogenesis and tumor growth of gliomas in ratsClin
Cancer Res1021902202200410.1158/1078-0432.CCR-03-010515041740
|
|
24.
|
K ViktorssonR LewensohnB
ZhivotovskyApoptotic pathways and therapy resistance in human
malignanciesAdv Cancer
Res94143196200510.1016/S0065-230X(05)94004-916096001
|
|
25.
|
A TaraphdarM RoyR BhattacharyaNatural
products as inducers of apoptosis: Implication for cancer therapy
and preventionCurr Sci80138713962001
|
|
26.
|
RE MewshawRJ Edsall JrC YangER beta
ligands. 3. Exploiting two binding orientations of the
2-phenylnaphthalene scaffold to achieve ER beta selectivityJ Med
Chem4839533979200510.1021/jm058173s15943471
|
|
27.
|
KM DebatinApoptosis pathways in cancer and
cancer therapyCancer Immunol
Immunother53153159200410.1007/s00262-003-0474-814749900
|
|
28.
|
HM KuoHC TsaiYL LinMitochondrial-dependent
caspase activation pathway is involved in baicalein-induced
apoptosis in human hepatoma J5 cellsInt J
Oncol35717724200919724907
|
|
29.
|
WP RoosB KainaDNA damage-induced cell
death by apoptosisTrends Mol
Med12440450200610.1016/j.molmed.2006.07.00716899408
|
|
30.
|
G Van LooX SaelensM Van GurpM MacFarlaneSJ
MartinP Van den AbeeleThe role of mitochondrial factors in
apoptosis: a Russian roulette with more than one bulletCell Death
Differ910311042200212232790
|
|
31.
|
X WangG YangJ ChaiY ShiD XueMechanisms of
AIF-mediated apoptotic DNA degradation in Caenorhabditis
elegansScience29815871592200210.1126/science.107619412446902
|
|
32.
|
N ZamzamiG KroemerThe mitochondrion in
apoptosis: how Pandora’s box opensNat Rev Mol Cell
Biol26771200111413468
|
|
33.
|
SA SusinTwo distinct pathways leading to
nuclear apoptosisJ Exp
Med192571580200010.1084/jem.192.4.57110952727
|
|
34.
|
LY LiX LuoX WangEndonuclease G in an
apoptotic DNase when released from
mitochondriaNature4129599200110.1038/3508362011452314
|
|
35.
|
C CandeI CohenE DaugasApoptosis-inducing
factor (AIF): a novel caspase-independent death effector released
from
mitochondriaBiochimie84215222200210.1016/S0300-9084(02)01374-312022952
|
|
36.
|
JC MartinouS DesagherB AntonssonCytochrome
c release from mitochondria: all or nothingNat Cell
Biol24143200010.1038/3500406910707095
|
|
37.
|
M LiT KondoQL ZhaoApoptosis induced by
cadmium in human lymphoma U937 cells through
Ca2+-calpain and caspase-mitochondria dependent
pathwaysJ Biol
Chem2753970239709200010.1074/jbc.M00736920010970901
|
|
38.
|
GM CohenCaspase: the executioners of
apoptosisBiochem J3261161997
|
|
39.
|
S ShimizuY EguchiW KamiikeH MatsudaY
TsujimotoBcl-2 expression prevents activation of the ICE protease
cascadeOncogene122251225719968649764
|
|
40.
|
RJ YouleA StrasserThe BCL-2 protein
family: opposing activities that mediate cell deathNat Rev Mol Cell
Biol94759200810.1038/nrm230818097445
|
|
41.
|
RT AllenWJ Hunter IIIDK
AgrawalMorphological and biochemical characterization and analysis
of apoptosisJ Pharmacol Toxicol
Meth37215228199710.1016/S1056-8719(97)00033-69279777
|
|
42.
|
TJ PrestonA AbadiL WilsonG
SinghMitochondrial contributions to cancer cell physiology:
potential for drug developmentDrug Deliv
Rev494561200110.1016/S0169-409X(01)00127-211377802
|
|
43.
|
HL XuXF YuAC QuR ZhangXR QuYP ChenXY MaDY
SuiAnti-proliferative effect of Juglone from Juglans mandshurica
Maxim on human leukemia cell HL-60 by inducing apoptosis through
the mitochondria dependent pathwayEur J
Pharmacol6451422201010.1016/j.ejphar.2010.06.07220655907
|