Introduction
Breast cancer is the most prevalent form of cancer
diagnosed in women, and there continues to be limited drug
treatment options for the ∼30% of patients with estrogen receptor
(ER)-negative breast cancer (1,2). In
the search for effective drugs for ER-negative breast cancer,
several lead compounds from natural products have emerged including
curcumin (diferuloylmethane), the primary bioactive compound
isolated from the rhizome of turmeric (Curcuma longa Linn.).
Curcumin has numerous pharmacological, chemopreventative and
chemotherapeutic actions, and in vitro studies have also
demonstrated that curcumin exhibits potent cytoxicity toward
numerous cell lines including ER-negative human breast cancer cells
(3–9). Furthermore, in vivo studies
have demonstrated decreased tumorigenesis of many organs, including
the mammary gland (10–15). However, curcumin has shown limited
clinical efficacy, due to its low bioavailability and low stability
(11). Therefore, numerous groups
have concentrated on improving drug efficacy, bioavailability and
stability by synthesizing analogs of curcumin. Specifically,
cyclohexanone analogs of curcumin have shown enhanced activity and
stability compared to curcumin (16). Specifically, the
cyclohexanone-containing curcumin derivative
2,6-bis((3-methoxy-4-hydroxyphenyl)methylene)-cyclohexanone (BMHPC)
was cytotoxic towards ER-negative breast cancer cells
(IC50 of 5.0 μM) (17),
although bioavailability and in vivo efficacy were still
problematic. More recently the fluorinated cyclohexanone derivative
EF24 has shown potent cytotoxicity toward MDA-MB-231 cells with an
IC50 value of 0.8 μM (18,19).
Our laboratory has been involved in the search for
new drug treatments for ER-negative breast cancer and we have shown
that 2nd generation heterocyclic cyclohexanone curcumin analogs
exhibit potent cytotoxicity toward ER-negative breast cancer cells.
This work demonstrated that
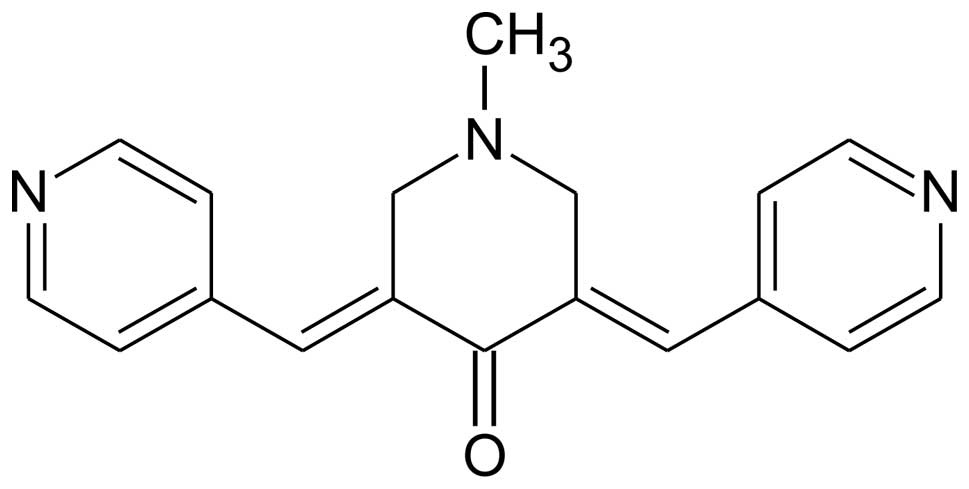
1-methyl-3,5-bis[(E)-4-pyri-dyl)methylidene]-4-piperidone (RL66)
(Fig. 1) exhibited IC50
values of 0.8, 0.5 and 0.6 μM for MDA-MB-231, MDA-MB-468 and SKBr3
breast cancer cells, respectively (20). It also induced apoptosis, as ∼18%
of MDA-MB-231 cells underwent apoptosis after 12 h of RL66
treatment (2 μM) (20). Only one
other compound synthesized (RL71) showed a more potent effect in
vitro (21), but this compound
did not suppress tumor growth in vivo. Therefore, this study
was designed to comprehensively investigate the potency and
mechanisms of action of RL66 in vitro and in vivo in
order to determine its potential to be developed into a drug for
ER-negative breast cancer.
Materials and methods
Materials
HUVEC, MDA-MB-231, MDA-MB-468 and SKBr3 cells were
purchased from American Type Culture Collection (Manassas, VA,
USA). Primary antibodies to NF-κB, p38, pp38, NF-κB, JNK, pJNK,
cleaved caspase-3, 4EBP1, p4EBP1, p27, mTOR, pmTOR, HER2, pHER2 and
β-actin were purchased from Cell Signaling Technology (Danvers, MA,
USA). Akt and pAkt primary antibodies were purchased from BD
Biosciences (Auckland, New Zealand). Dulbecco’s modified Eagle’s
medium (DMEM) nutrient mixture Ham’s F-12, sulforhodamine B salt,
propidium iodide (PI), ammonium persulfate, horseradish peroxidase
were purchased from Sigma-Aldrich (Auckland, New Zealand).
Acrylamide, bisacrylamide, sodium dodecylsulfate and PVDF membrane
were purchased from Bio-Rad Laboratories (Hercules, CA, USA).
Complete mini EDTA-free protease inhibitor cocktail and Annexin
V-FLUOS were purchased from Roche Diagnostics Corporation
(Mannheim, Germany). RL66 was prepared as described previously
(18). All other chemicals were of
the highest purity commercially available.
Cell maintenance
MDA-MB-231, MDA-MB-468 and SKBr3 cells were
maintained in complete growth media composed of DMEM/Ham’s F12
supplemented with 5% fetal bovine serum (FBS), 2 mM L-glutamine,
100 U/ml streptomycin, 250 ng/ml amphotericin B, and 100 U/ml
penicillin and 2.2 g/l NaHCO3.
Cytotoxicity
MDA-MB-231, MDA-MB-468 and SKBr3 cells
(95×104 cells/well) were seeded in 12-well plates in 1
ml DMEM/HamF12 supplemented with 5% FBS, 100 U/ml penicillin, 100
μg/ml streptomycin, 25 ng/ml amphotericin B and 2.2 g/l
NaHCO3 and incubated for 24 h at 37°C. To determine
cytotoxicity over a time-course, cells were treated with RL66 (1.5
or 2 μM) for 6, 12, 24, 36, 48 and 72 h. Vehicle control cells were
treated with DMSO (0.1%). Cell number in each well was determined
using the sulforhodamine B (SRB) assay (22).
Cell cycle analysis
Flow cytometry was used to analyze DNA content in
order to determine cell cycle distribution. MDA-MB-231, MDA-MB-468
and SKBr3 cells were plated and treated with RL66 (1.5 or 2 μM) or
0.1% DMSO as control for 6–48 h in 6-well plates. The cells were
harvested, washed with PBS and then fixed in 70% ethanol. Following
rehydration with PBS, the cells were stained with PI in the dark at
4°C as described (23). The
samples were analyzed via flow cytometry using a FACSCalibur flow
cytometer (BD Biosciences). The percentage of cells in each phase
of cell cycle was determined using Cell Quest Pro software. Results
are expressed as percent of cells in each phase of the cell
cycle.
Induction of apoptosis
MDA-MB-468, and SKBr3 cells were seeded in 6-well
culture plate in 2 ml of DMEM/HamF12 supplemented with 5% FBS, 100
U/ml penicillin, 100 μl/ml streptomycin, 25 ng/ml amphotericin B
and 2.2 g/l NaHCO3. The cells were treated with RL66
(1.5 or 2 μM) or vehicle control for 12–36 h. Apoptosis was
assessed using Annexin V-FLUCOS/PI staining, as described (24). The samples were analyzed using a
FACSCalibur flow cytometer (BD Biosciences) and the proportion of
apoptotic cells was determined using CellQuest Pro software.
Preparation of cell lysates
MDA-MB-231, MDA-MB-468 and SKBr3 cells were seeded
in 10-cm culture dishes at 2.5×106 cells per well in 10
ml of DMEM/HamF12 supplemented with 5% FBS, 100 U/ml penicillin,
100 μg/ml streptomycin, 25 ng/ml amphotericin B and 2.2 g/l
NaHCO3. Cells were treated with RL66 (2 or 3 μM) or
vehicle control for 0–36 h. At the end of treatment, whole cell
lysates were prepared and protein concentration of the lysates was
determined using the bicinchoninic acid (BCA) method (25).
Western blot analysis
Cell lysates were resolved by SDS-PAGE (40 μg
protein per well) and then the proteins were transferred to a PVDF
membrane. Protein levels were analyzed with the desired primary
antibodies, followed by horseradish peroxidase-conjugated secondary
antibodies (Bio-Rad Laboratories). The digital chemiluminescence
images were taken by a Versadoc densitometer (Bio-Rad
Laboratories).
Transwell migration
Transwell migration was performed using 24-well
plates containing BioCoat™ Matrigel™ Invasion Chamber inserts (BD
Biosciences, Bedford, MA, USA). HUVEC cells (50,000/well) were
plated on rehydrated Matrigel coated culture inserts. The bottom
chamber contained 500 μl of EGM serum free media. The cells were
treated with 0.1% DMSO or RL66 (1 μM) and incubated for 18 h at
37°C in a humidified 5% CO2 incubator. After incubation,
all contents from well inserts were aspirated and non-migrated
cells were removed with a cotton swab. Migrated cells on the bottom
of the filters were stained with DiffQuick solution for 1 min and
excess stain was washed with water and dried. Cells on the filters
were counted using a Zeiss Axioplan camera and compared to the
control well insert that contained no matrigel. Results are
expressed as migrated cells as a percent of total cell
population.
Endothelial tube formation
Geltrex matrigel (125 μl) was added onto 24-well
plates which was then incubated at 37°C, 5% CO2 for 30
min. HUVEC cells (5×104/well) were loaded into each
well, followed by addition of DMSO (0.1%) or RL66 (1 μM). The plate
was incubated at 37°C, 5% CO2 for 18 h and photos (×200)
were taken by an individual blinded to the treatment groups.
Animals and housing
Female CD-1 mice (6-week-old) were purchased from
the Hercus Taieri Resource Unit (Dunedin, New Zealand). All
procedures were approved by the University of Otago (AEC no.
91/07). Mice were housed in pathogen-free conditions with woodchip
bedding with access to food (Reliance rodent diet, Dunedin, NZ) and
water ad libitum. Mice were housed in a 21–24°C environment
on a scheduled 12 h light/dark cycle and acclimatized for 3 days
prior to experimentation.
MDA-MB-468 xenografts
Female CD1 athymic nude mice (5–6-week-old) were
purchased from Hercus Taieri Resource Unit. They were maintained at
21–24°C with a 12-h light/dark cycle in a specifically designed
pathogen-free isolation facility and allowed to acclimatize for 1
week before experimentation. Mice were inoculated into the right
flank with MDA-MB-468 cells (2×106/50 μl matrigel),
which were left to form palpable tumors. Tumor volume was measured
weekly with electronic calipers (L × W × H). When the tumor volume
reached approximately 100 mm3, mice (5/group) were
randomly assigned to the various treatment groups. Mice were then
orally gavaged with RL66 (0.85 or 8.5 mg/kg/day), or water as the
vehicle control (5 ml/kg/day) for 10 weeks. Dosing solutions were
prepared fresh each day.
Assessment of animal health
Food consumption and body weight were monitored
daily throughout the treatment period. Mice were euthanized by
CO2 inhalation 24 h following the last dose and
necropsies were then performed. Blood was collected via the
inferior vena cava and placed on ice, while major organs, as well
as tumors were excised and weighed. Organ weights are expressed as
a percentage of body weight. Plasma was separated and used to
determine hepatotoxicity via the plasma marker alanine
aminotransferase (ALT) activity using a commercially available kit
(Medica Pacifica, Auckland, New Zealand). Results are expressed as
IU/l.
Immunohistochemistry of tumor
sections
Tumors were embedded in OCT compound and then
sectioned (12 mm), fixed in acetone, and air-dried overnight.
Sections were then washed twice in Tween-20 PBS, incubated with
normal serum for 30 min at room temperature, and then incubated
overnight with primary CD105 antibody. Slides were then rinsed and
peroxidase blocked using hydrogen peroxide (3%) before incubation
with the appropriate secondary antibody for 30 min at room
temperature in a humidified chamber. The sections were then
incubated with ExtrAvidin (Bio-Rad Laboratories, Auckland, New
Zealand) (1:20) for 30 min at room temperature in a humidified
chamber before development with 3,30-diaminobenzidine
tetrahydrochloride as the chromogen and counterstaining with
Mayer’s haematoxylin. Once slides were dehydrated, DPX mounting
medium and coverslips were applied. The sections were analyzed from
tumors obtained from each mouse and a representative slide is shown
in the results section.
Statistical analysis
All time course data were analyzed using a two-way
ANOVA coupled with a Bonferroni post hoc test. Tumor volume was
analyzed using a repeated measures 2-way ANOVA coupled with a
Bonferroni post hoc test. For all data in which time was not a
factor, the data were analyzed using a one-way ANOVA coupled with a
Bonferroni post hoc test. p<0.05 was the minimum requirement for
a statistically significant difference.
Results
Previously we have shown that RL66 elicited sub
micromolar IC50 values in three different ER-negative
breast cancer cell lines (20).
Therefore, the first aim of this study was to examine the
time-course of RL66-mediated cytotoxicity toward MDA-MB-231,
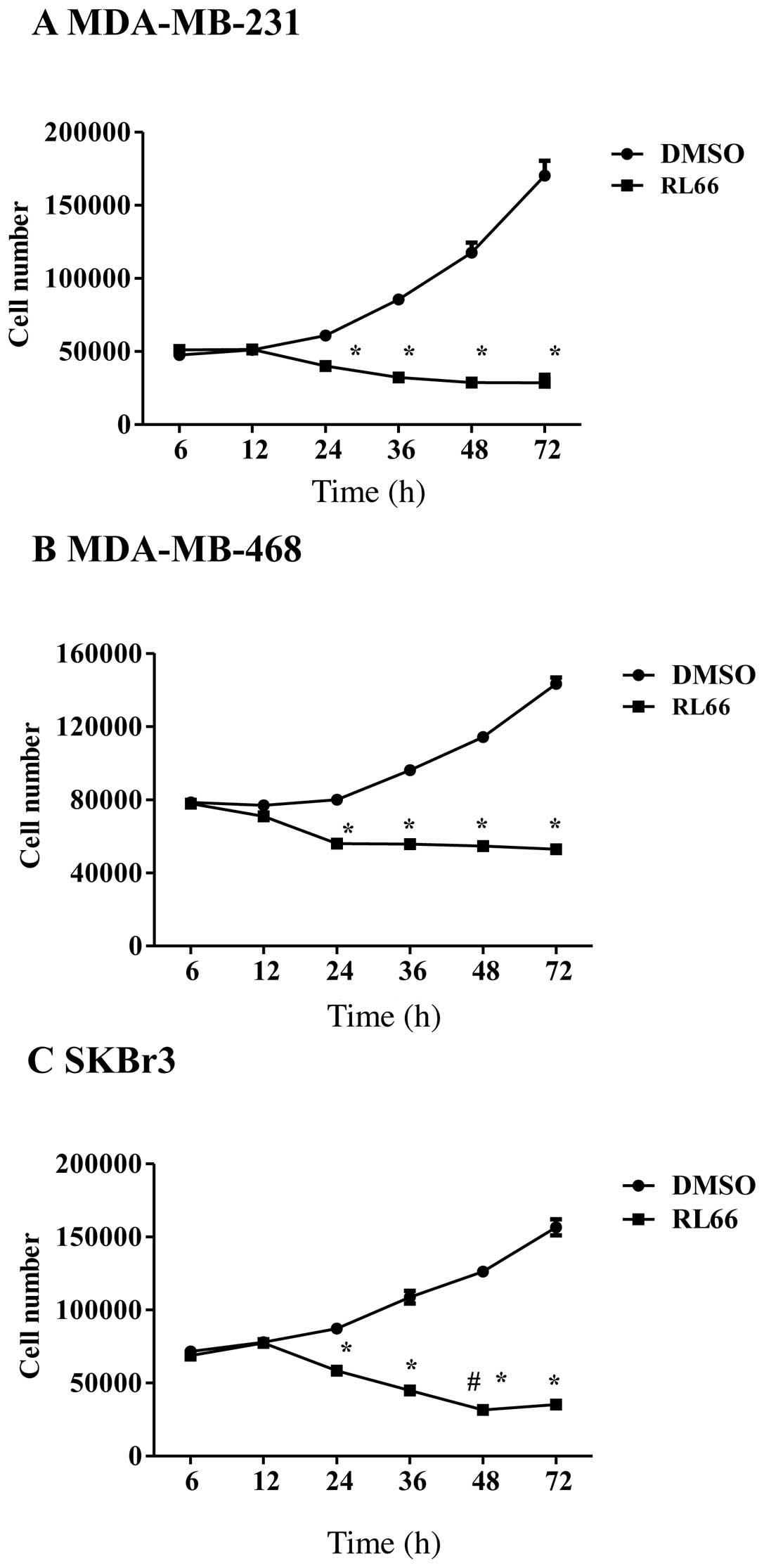
MDA-MB-468 and SKBr3 cells. The results showed that RL66 elicited
time-dependent and cell line-dependent cytotoxicity (Fig. 2A–C). Specifically, time-dependent
cytotoxicity was elicited in SKBr3 cells where 2 μM signficantly
increased cytotoxicity at 48 h compared with all previous time
points (Fig. 2C). However, in the
two TNBC cell lines no further cytotoxicity was elicited after 24
h. Thus, RL66 showed potent cytotoxicity toward SKBr3 cells
compared to a cytostatic effect in TNBC cells.
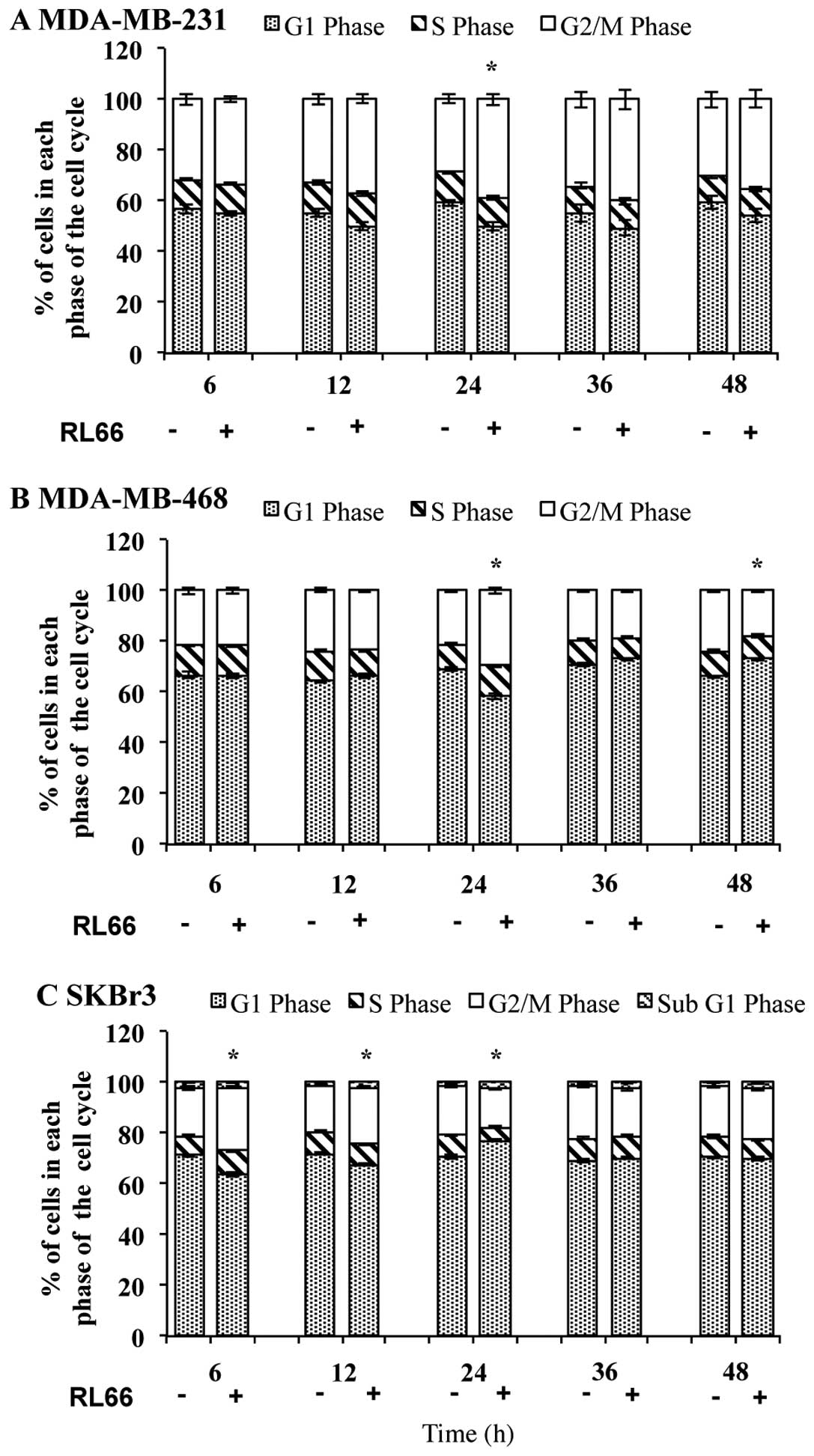
We next examined whether the cytotoxicity of RL66
was due to cell cycle arrest. Treatment of MDA-MB-231, MDA-MB-468,
and SKBr3 cells with RL66 produced G2/M phase arrest in all three
cell lines. Specifically, at 24 h, RL66 caused an 150% increase in
the proportion of G2/M phase cells over control in both MDA-MB-231
(Fig. 3A) and MDA-MB-468 cells
(Fig. 3B). In SKBr3 cells, after 6
and 12 h, the proportion of cells undergoing G2/M phase was
increased by 130 and 120%, respectively, compared to control
(Fig. 3C). Moreover, there was a
significant reduction in the proportion of cells in S phase at 24
h. SKBr3 cells were also the only cell type to show an increase in
subG1 cells.
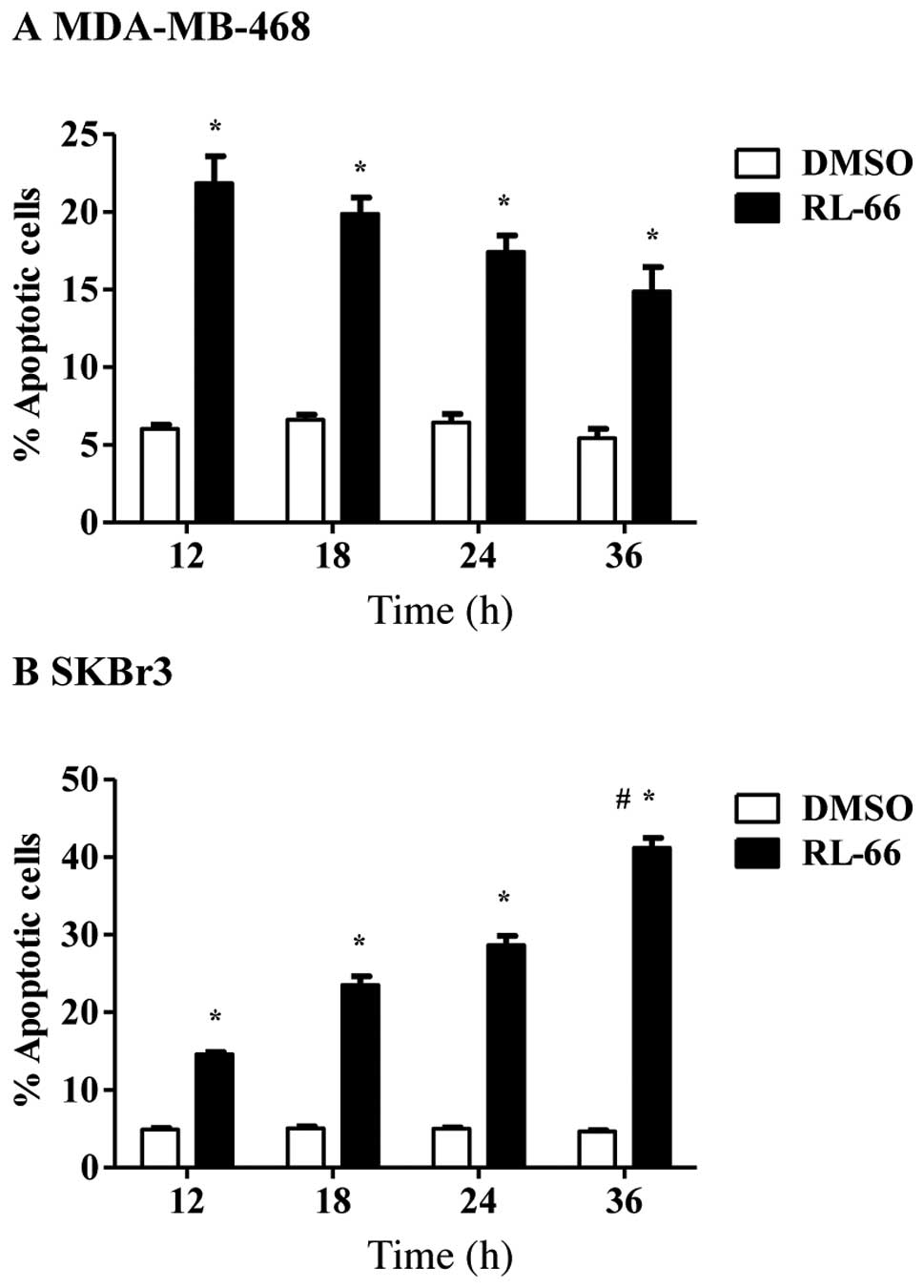
To determine if RL66-mediated cell cycle arrest
resulted in apoptosis, time-dependent changes in apoptosis were
examined. The induction of apoptosis was time-dependent in SKBr3
cells, as 42% of cells were apoptotic after 36 h and this was
significantly elevated compared to all other time points (Fig. 4B). In contrast 15–22% of MDA-MB-468
cells underwent apoptosis and this effect was maintained from 12–36
h (Fig. 4A) indicating the lack of
a time-dependent effect. G2/M arrest did not drive apoptosis in
MDA-MB-468 cells, as apoptosis was increased at 12 h, which was
prior to the increase in G2/M phase arrest. However, the early
appearance of G2/M phase arrest at 6 h in SKBr3 cells is a likely
reason why these cells show a strong apoptotic response over time.
Our previous work with RL66 in MDA-MB-231 cells indicated that the
induction of apoptosis was weakest in this cell line, as 18% of
cells underwent apoptosis and this decreased to 8% after 18–36 h
(20). Overall RL66 displayed a
more potent cytotoxic effect in SKBr3 cells. To determine if this
was due to the inhibition of HER2/neu expression, drug-mediated
changes in HER2/neu and other downstream cell signaling proteins
were determined via western blot analysis.
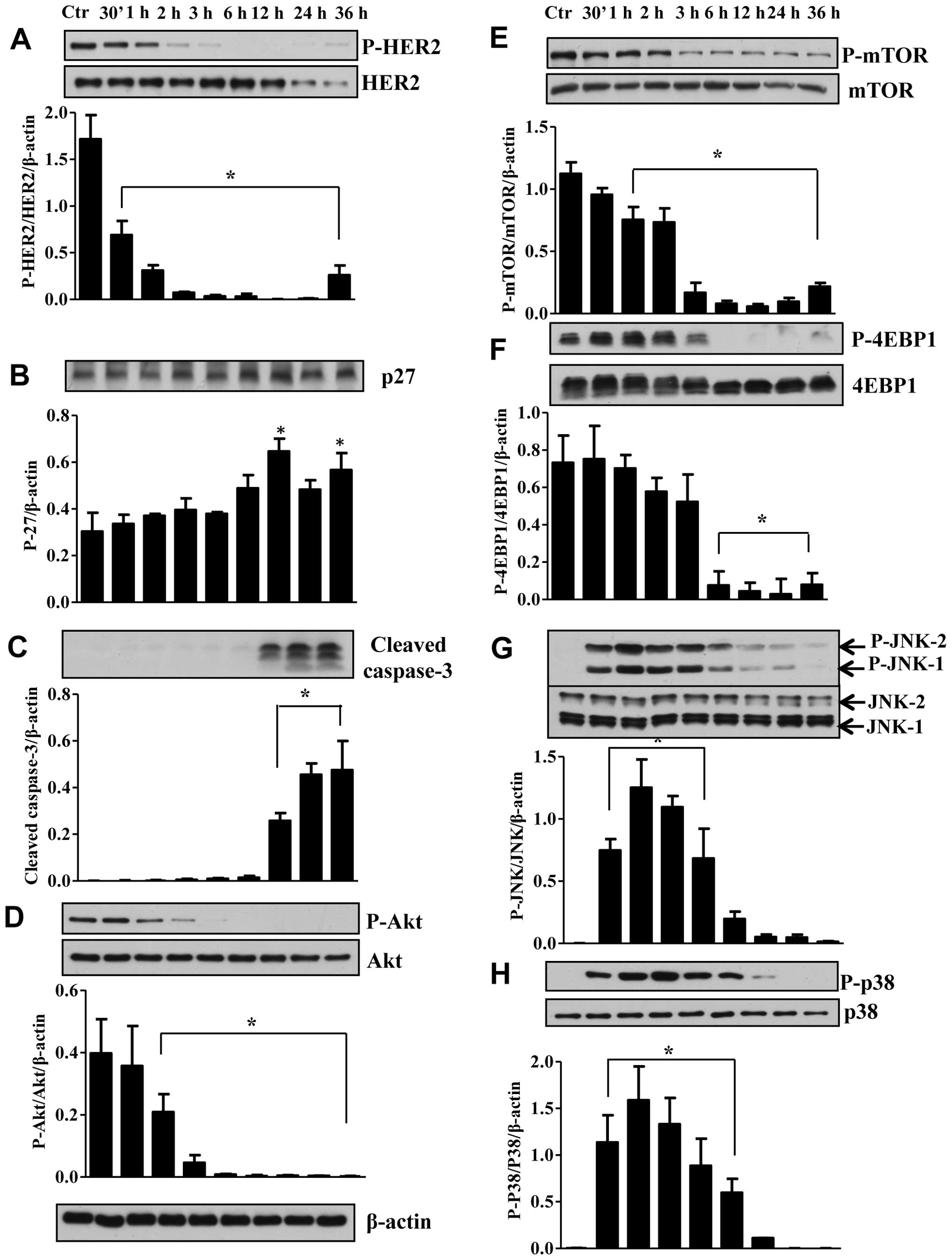
Treatment of SKBr3 cells with RL66 (2 μM) decreased
the ratio of pHER2/HER2, with an almost complete inhibition for
2−24 h (Fig. 5A). To link the
changes in HER2/neu with cell cycle progression protein changes in
the cyclin dependent kinase inhibitor, p27 were determined. The
results showed that decrease in HER2/neu correlated with a
significant increase in the expression of p27 at 12 and 36 h
(Fig. 5B). Thus the decrease in
HER2/neu leads to an increase in p27 leading to the observed G2/M
arrest and apoptosis. The presence of apoptosis was also confirmed
in SKBr3 cells by the signficant increase in cleaved caspase-3
(Fig. 5C). Proteins downstream of
EGFR were also examined and RL66 treatment resulted in a 90%
decrease in the ratio of pAkt/Akt (Fig. 5D). This correlated with a decrease
in mTOR and its downstream effector 4EBP1 (Fig. 5E and F). Conversely, the stress
kinases JNK1/2 and p38 were transiently incrased from 30 min-6 h
(Fig. 5G and H). Thus, RL66
produces a consistent time-dependent disruption of cell signaling
proteins in SKBr3 cells that begins with potent inhibition of
HER2/neu.
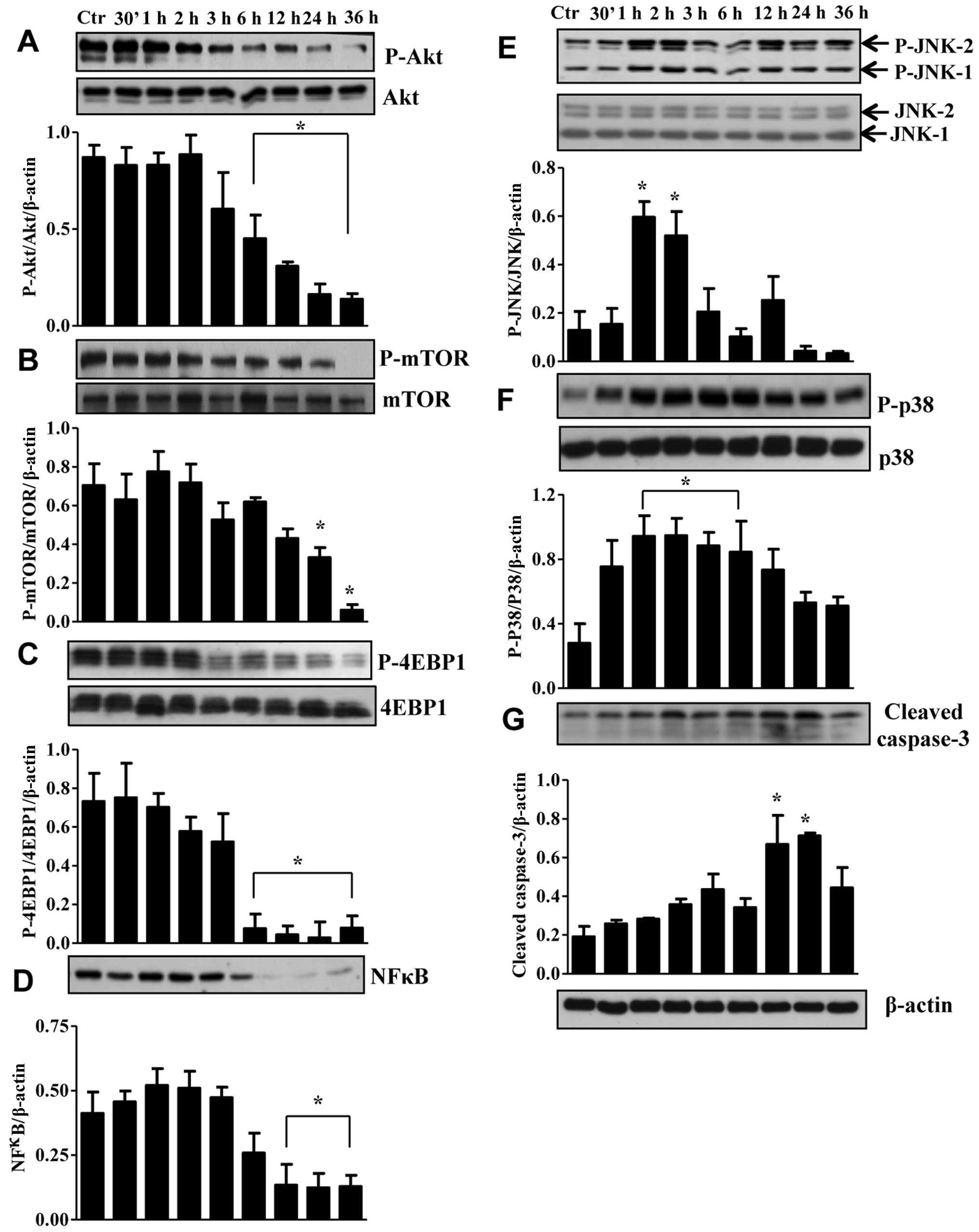
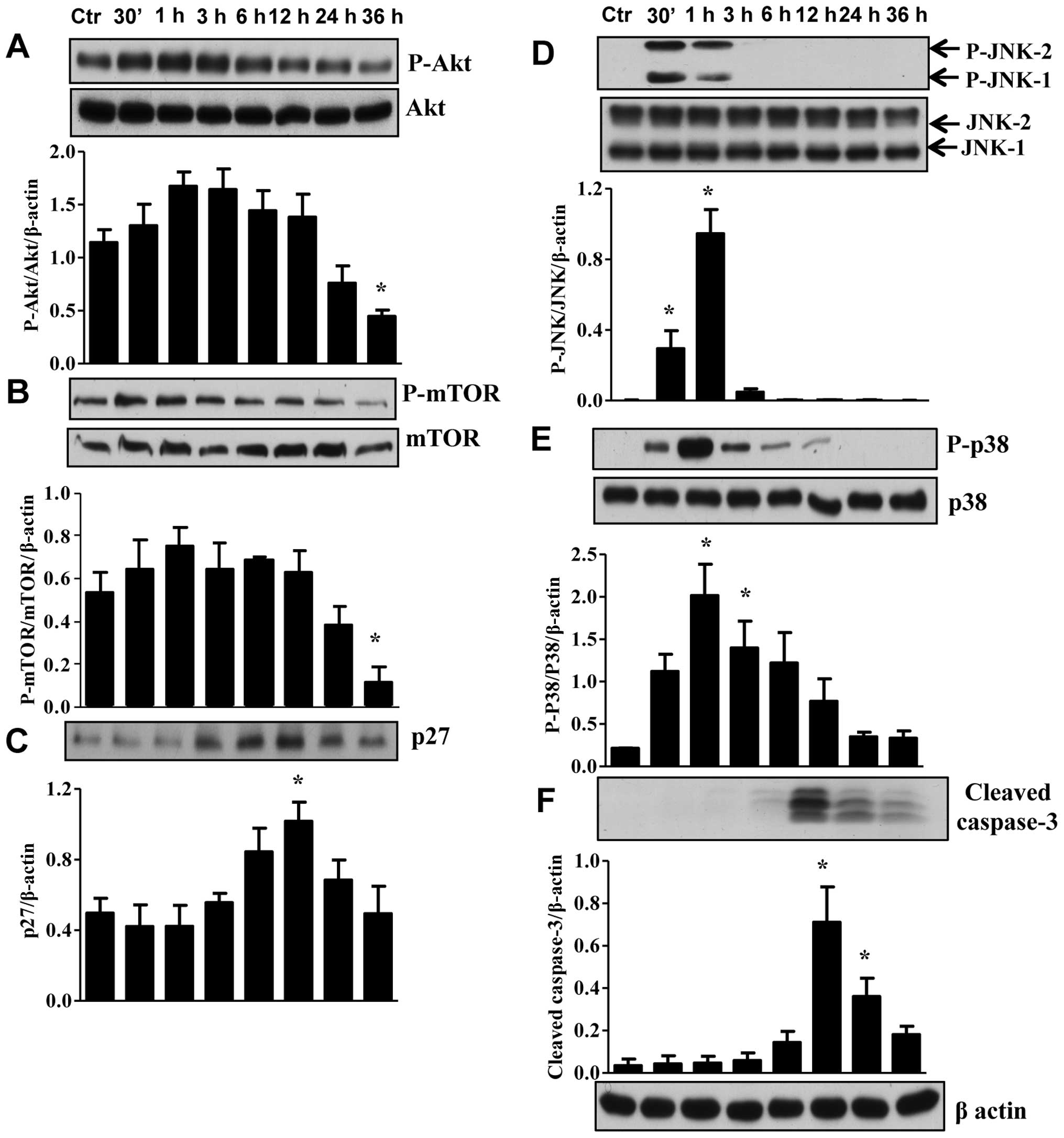
To determine the molecular mechanisms responsible
for apoptosis and cell cycle arrest in TNBC cells we first examined
for other isoforms of the EGFR. RL66 failed to alter the ratio of
pEGFR/EGFR protein levels (data not shown). However, RL66 did
modulate the expression of Akt, mTOR, 4EBP1, NF-κB, JNK1/2, p38 and
caspase-3 in MDA-MB-231 (Fig. 6)
and Akt, mTOR, p27, JNK1/2, p38 and caspase-3 in MDA-MB-468 cells
(Fig. 7). Specifically, RL66
significantly decreased the ratio of pAkt/Akt from 6–36 h in
MDA-MB-231 cells and at 36 h in MDA-MB-468 cells (Figs. 6A and 7A). The stress initiated by the treatment
of MDA-MB-231 and MDA-MB-468 cells resulted in a transient increase
in both JNK1/2 and p38 MAPK phosphorylation (Figs. 6E, F and 7D, E). Furthermore, RL66 increased levels
of cleaved caspase 3 at 12 and 24 h (Figs. 6G and 7F). MDA-MB-231 cells were the only cell
line to show a signficant decrease in NF-κB which occurred at 12–36
h (Fig. 6D).
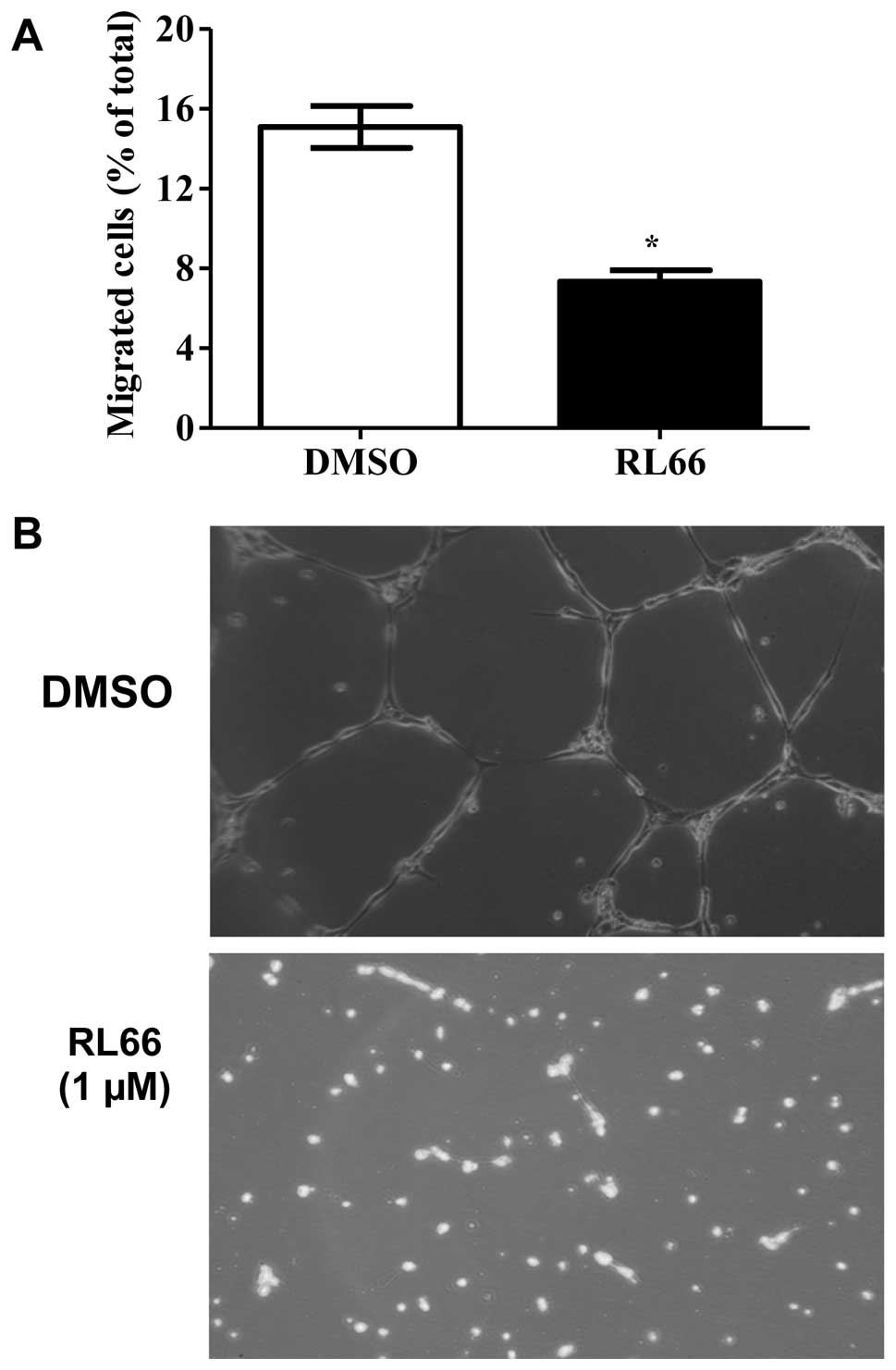
To determine if RL66 could modulate angiogenesis,
in vitro assays using HUVEC cells were performed, as the
ability of these cells to migrate through matrigel and their
ability to form tube-like networks are hallmarks of angiogenesis.
Quantifiable and visual assys were used to form a more complete
in vitro picture. The results showed that RL66 (1 μM)
significantly reduced HUVEC cell migration by 46% compared to
vehicle control (Fig. 8A) and
completely inhibited endothelial tube formation (Fig. 8B).
To determine if these potent in vitro effects
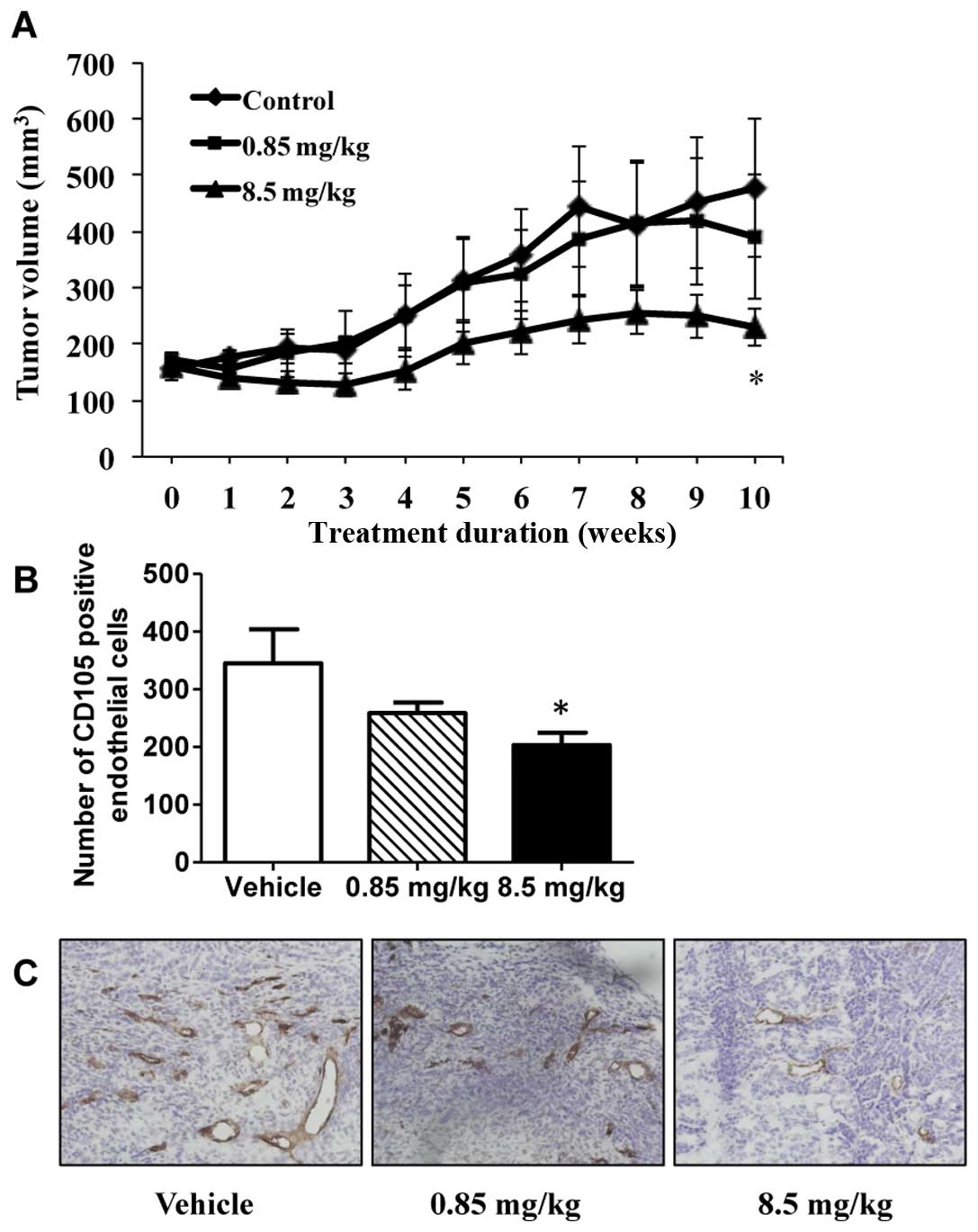
would translate to in vivo tumor suppression, RL66 was
examined for its ability to modulate tumor growth in a xenograft
model of TNBC. The results showed that tumors in mice treated with
RL66 (8.5 mg/kg) were significantly smaller in volume compared to
tumors from vehicle treated mice (Fig.
9A). This effect was apparent after 4 weeks of treatment and
continued throughout the 10 weeks of treatment. At the conclusion
of the study tumor volume and weight was 48% smaller in the RL66
treated mice compared to vehicle control. Importantly, RL66
treatment was non-toxic to the mice as body weight, gross organ
weight of the liver, kidney, spleen and uterus was not different
between treatment and control (data not shown). Additionally,
plasma alanine aminotransferase activity, a marker of
hepatotoxicity, remained in the normal range (23–85 IU/l). To
determine if the tumor suppression elicited by RL66 was accompanied
with a decrease in microvessel density, tumors were sectioned and
stained for the blood vessel marker CD105. The results demonstrated
that RL66 (8.5 mg/kg) significantly decreased CD105 staining 57%
compared to control tumors (Fig. 9B
and C).
Discussion
We have previously shown that RL66 elicited high
cytotoxic potency towards ER-negative breast cancer cells (20). Therefore, this study was designed
to further characterize this cytotoxic effect in vitro and
in vivo. The results show that RL66 promoted G2/M cell cycle
arrest, induced apoptosis and modulated the Akt-dependent signaling
pathway and stress response MAPK pathway. RL66 also downregulated
the expression of HER2/neu in SKBr3 cells. Importantly, RL66
suppressed tumor growth in vivo, while it remained non-toxic
to major organs. In addition, RL66 exhibited anti-angiogenic
effects in vitro by inhibiting the invasion of HUVEC cells
and their ability to form endothelial tube-like network and in
vivo by decreasing microvessel density in tumor slices.
RL66 displays potent cytotoxicity in ER-negative
breast cancer cells compared to other cyclohexanone curcumin
analogs (18,20). Moreover, it had superior
cytotoxicity compared with other curcumin analogs such as
3,5-bis(flurobenzylidene) piperidin-4-one (EF24) (26), 5-bis
(4-hydroxy-3-methoxy-benzylidnen)-N-methyl-4-piperidone (PAC)
(27) and GO-Y030 (28) in MDA-MB-231 cells. Specifically
IC50 values of 1.2, 1 and 0.3 μM were reported for EF24,
GO-Y030 and RL66, respectively (20,26,28).
While EF24 induced G2/M phase arrest and apoptosis in MDA-MB-231
cells (19) and inhibited the
NF-κB pathway in a TNFα-dependent manner (26), it has not been examined in other
breast cancer cells. Additionally, RL66 has a stronger ability to
induce apoptosis compared to the analog 4-hydroxy-3-methoxybenzoic
acid methyl ester (HM-BME), where 25 μM was required to cause 37%
of LNCaP prostate cancer cells to undergo apoptosis after 24 h
(29). The curcumin analogs FLLL11
and FLLL12 were equally potent as RL66 in MDA-MB-468 cells with
similar IC50 values (0.3 μM). However, this did not
translate to other breast cancer cell types as these analogs had
IC50s of 2-5 μM in MDA-MB-231 and SkBr3 cells (27). These analogs also downregulated Akt
phosphorylation and HER2/neu expression in SKBr3 breast cancer
cells but at concentrations of 10 μM, 5-fold greater than RL66
(30). Overall RL66 is more potent
as all of its anticancer actions were elicited at concentrations of
3 μM or less.
Breast cancer patients whose tumors overexpress
HER2/neu have a poor prognosis, shorter relapse time and lower
survival time (31). In this study
we showed that RL66 decreased HER2/neu expression in SKBr3 cells,
which led to a decrease in Akt and mTOR as well as an increase in
p27 and cleaved caspase-3. Since p27 is a key regulator of G2/M
phase arrest and apoptosis (32,33),
inhibition of HER2/neu is a key initial mechanism for the
apoptotoic effect elicited by RL66 in SKBr3 cells. RL66 was more
potent than some curcumin analogs at down-regulating the expression
of HER2/neu, as 4 μM concentrations of RL90 and RL91 and 10 μM
concentrations of and FLLL11 and FLLL12, were required to elicit a
similar effect (30,34). However, it was not more effective
than RL71, which inhibited HER2/neu at 1 μM (21). However, RL66 remains a strong drug
candidate for ER-negative/HER2-positive breast cancer, especially
since it has shown efficacy in vivo, which to date RL71 has
not displayed (21).
MAPK signaling which includes activation of JNK and
p38 is involved in the regulation of the cell cycle and induction
of apoptosis in breast cancer cells (35). Various cytotoxic agents induce
apoptotic cell death via activation of MAPK signaling and induction
of caspase-3 (36–38). Our studies showed that RL66
treatment induced JNK1/2 and p38 MAPK in MDA-MB-231 and MDA-MB-468
cells. Anticancer agents such as curcumin, which causes activation
of p38, JNK1/2 and caspase-3, also induce similar apoptotic events
(39,40). The MAPK pathway may also upregulate
the cell cycle regulatory protein, p27 in breast cancer cells
(41). Our results demonstrated
that in MDA-MB-468 RL66 enhanced the expression of p27 which would
contribute to the observed G2/M cell cycle arrest.
We further studied the effect of RL66 on the
PI3K/Akt/mTOR pathway. Akt is an important oncoprotein which is
constitutively active in breast cancer cells and has been
implicated in numerous regulatory mechanisms involving protein
synthesis, cell cycle progression and inhibition of apoptosis
(42,43). Our results showed that RL66
decreased the phosphorylation of Akt on Ser-473 in a cell line and
time-dependent manner. Specifically, in MDA-MB-231 cells, RL66
decreased Akt phosphorylation after 6 h whereas in MDA-MB-468 cells
the phosphorylation of Akt was only decreased after 36 h. However,
RL66 was more potent than the analogs RL90 and RL91, which did not
decrease the ratio of pAkt/Akt at concentrations of 4 μM (34). The decreased activity of Akt led to
decreased activation of its substrate mTOR in both cell lines.
Akt contributes to the activity of NF-κB by
controling its translocation to the nucleus (44) and a decrease in Akt activity may
affect the stability and level of NF-κB (45). NF-κB belongs to a family of
transcription factors which has been associated with inhibition of
apoptosis by promoting the expression of anti-apoptotic proteins
such as Bcl-xL, c-Myb and caspase inhibitors (46,47).
RL66 downregulated the expression of NF-κB in MDA-MB-231 cells.
However, higher concentrations were required to downregulate NF-κB
in MDA-MB-468 cells (data not shown) and this is consistent with
other curcumin analogs (21,23).
Curcumin has also been shown to interfere with the functions of Akt
and MAPKs and to inhibit its downstream target NF-κB (48,49)
and thus RL66 retains many of the same actions as curcumin.
Importantly, the potent in vitro actions of
RL66 translated to tumor suppression in a xenograft model of TNBC.
Tumor suppression was accompanied by a 57% decrease in tumor
microvessel density. While other 2nd generation heterocyclic
cyclohexanone curcumin derivatives have shown potent in
vitro actions, RL66 is the first drug in this class to elicit
tumor suppression in vivo. This provides evidence for the
need to continue examining this class of curcumin analogs.
In summary, we showed that RL66 causes cell cycle
arrest and induces apoptosis in ER-negative breast cancer cells and
also modulates a variety of signaling pathways that culminate in
potent cytotoxicity. Specifically, inhibition of Akt pathway and
the activation of p38/JNK pathway may contribute to the anticancer
activity of RL66 in TNBC cells, while inhibition of HER2/neu and
induction of p27 are key mechanisms in SKBr3 cells. RL66 suppresses
tumor growth in an in vivo model of TNBC and this correlates
with a decrease in tumor microvessel density. This anti-angiogenic
effect was also shown in vitro. Thus, RL66 shows potential
as a new drug therapy for ER-negative breast cancer that warrants
further investigation.
Abbreviations:
|
RL66
|
1-methyl-3,5-bis[(E)-4-pyridyl)methylidene]-4-piperidone;
|
|
ER
|
estrogen receptor;
|
|
TNBC
|
triple negative breast cancer
|
Acknowledgements
This study was supported by a grant
from the Breast Cancer Cure Research Trust (RJR) and a University
of Otago postgraduate scholarship (BY).
References
|
1.
|
Parl FF, Schmidt BP, Dupont WD and Wagner
RK: Prognostic significance of estrogen receptor status in breast
cancer in relation to tumor stage, axillary node metastasis, and
histopathologic grading. Cancer. 54:2237–2242. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Doane AS, Danso M, Lal P, Donaton M, Zhang
L, Hudis C and Gerald WL: An estrogen receptor-negative breast
cancer subset characterized by a hormonally regulated
transcriptional program and response to androgen. Oncogene.
25:3994–4008. 2006. View Article : Google Scholar
|
|
3.
|
Chiu TL and Su CC: Curcumin inhibits
proliferation and migration by increasing the Bax to Bcl-2 ratio
and decreasing NF-κBp65 expression in breast cancer MDA-MB-231
cells. Int J Mol Med. 23:469–475. 2009.PubMed/NCBI
|
|
4.
|
Kang HJ, Lee SH, Price JE and Kim LS:
Curcumin suppresses the paclitaxel-induced nuclear factor-kappaB in
breast cancer cells and potentiates the growth inhibitory effect of
paclitaxel in a breast cancer nude mice model. Breast J.
15:223–229. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Liu Q, Loo WT, Sze SC and Tong Y: Curcumin
inhibits cell proliferation of MDA-MB-231 and BT-483 breast cancer
cells mediated by down-regulation of NFkappaB, cyclinD and MMP-1
transcription. Phytomed. 16:916–922. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Prasad CP, Rath G, Mathur S, Bhatnagar D
and Ralhan R: Potent growth suppressive activity of curcumin in
human breast cancer cells: modulation of Wnt/beta-catenin
signaling. Chem Biol Interact. 181:263–271. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Rowe DL, Ozbay T, O’Regan RM and Nahta R:
Modulation of the BRCA1 protein and induction of apoptosis in
triple negative breast cancer cell lines by the polyphenolic
compound curcumin. Breast Cancer (Auckl). 3:61–75. 2009.PubMed/NCBI
|
|
8.
|
Somers-Edgar TJ, Scandlyn MJ, Stuart EC,
Le Nedelec MJ, Valentine SP and Rosengren RJ: The combination of
epigallocatechin gallate and curcumin suppresses ER alpha-breast
cancer cell growth in vitro and in vivo. Int J Cancer.
122:1966–1971. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Wu X and Wu K: Antiproliferative effect of
curcumin on human breast cancer of MCF-7 cells. Di-San Junyi Daxue
Xuebao. 28:1870–1872. 2006.
|
|
10.
|
Anand P, Thomas Sherin G, Kunnumakkara
Ajaikumar B, et al: Biological activities of curcumin and its
analogues (Congeners) made by man and Mother Nature. Biochem
Pharmacol. 76:1590–1611. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Cheng AL, Hsu CH, Lin JK, et al: Phase I
clinical trial of curcumin, a chemopreventive agent, in patients
with high-risk or pre-malignant lesions. Anticancer Res.
21:2895–2900. 2001.PubMed/NCBI
|
|
12.
|
Inano H, Onoda M, Inafuku N, et al:
Chemoprevention by curcumin during the promotion stage of
tumorigenesis of mammary gland in rats irradiated with gamma-rays.
Carcinogenesis. 20:1011–1018. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Pereira MA, Grubbs CJ, Barnes LH, et al:
Effects of the phytochemicals, curcumin and quercetin, upon
azoxy-methane-induced colon cancer and 7,12-dimethylbenz[a]
anthracene-induced mammary cancer in rats. Carcinogenesis.
17:1305–1311. 1996.PubMed/NCBI
|
|
14.
|
Schaaf C, Shan B, Buchfelder M, et al:
Curcumin acts as anti-tumorigenic and hormone-suppressive agent in
murine and human pituitary tumour cells in vitro and in vivo.
Endocr Relat Cancer. 16:1339–1350. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Singletary K, MacDonald C, Wallig M and
Fisher C: Inhibition of 7,12-dimethylbenz[a]anthracene
(DMBA)-induced mammary tumorigenesis and DMBA-DNA adduct formation
by curcumin. Cancer Lett. 103:137–141. 1996.
|
|
16.
|
Liang G, Shao L, Wang Y, et al:
Exploration and synthesis of curcumin analogues with improved
structural stability both in vitro and in vivo as cytotoxic agents.
Bioorg Med Chem. 17:2623–2631. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Markaverich BM, Schauweker TH, Gregory RR,
Varma M, Kittrell FS, Medina D and Varma RS: Nuclear type II sites
and malignant cell proliferation: inhibition by
2,6-bis-benzylidenecyclohexanones. Cancer Res. 52:2482–2488.
1992.PubMed/NCBI
|
|
18.
|
Adams BK, Ferstl EM, Davis MC, et al:
Synthesis and biological evaluation of novel curcumin analogs as
anti-cancer and anti-angiogenesis agents. Bioorg Med Chem.
12:3871–3883. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Adams BK, Cai J, Armstrong J, et al: EF24,
a novel synthetic curcumin analog, induces apoptosis in cancer
cells via a redox-dependent mechanism. Anticancer Drugs.
16:263–275. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Yadav B, Taurin S, Rosengren RJ,
Schumacher M, Diederich M, Somers-Edgar TJ and Larsen L: Synthesis
and cytotoxic potential of heterocyclic cyclohexanone analogues of
curcumin. Bioorg Med Chem. 18:6701–6707. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Yadav B, Taurin S, Larsen L and Rosengren
RJ: RL71, a second-genration curcumin analog, induces apoptosis and
downregulates Akt in ER-negative breast cancer cells. Int J Oncol.
41:1119–1127. 2012.PubMed/NCBI
|
|
22.
|
Skehan P, Storeng R, Scudiero D, et al:
New colorimetric cytotoxicity assay for anti-cancer drug screening.
J Natl Cancer Inst. 82:1107–1112. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Somers-Edgar TJ, Taurin S, Larsen L,
Chandramouli A, Nelson MA and Rosengren RJ: Mechanisms for the
activity of heterocyclic cyclohexanone curcumin derivatives in
estrogen receptor negative human breast cancer cell lines. Invest
New Drugs. 29:87–97. 2011. View Article : Google Scholar
|
|
24.
|
Stuart EC and Rosengren RJ: The
combination of raloxifene and epigallocatechin gallate suppresses
growth and induces apoptosis in MDA-MB-231 cells. Life Sci.
82:943–948. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Smith PK, Krohn RI, Hermanson GT, et al:
Measurement of protein using bicinchoninic acid. Anal Biochem.
150:76–85. 1985. View Article : Google Scholar
|
|
26.
|
Kasinski AL, Du Y, Thomas SL, et al:
Inhibition of IkappaB kinase-nuclear factor-kappaB signaling
pathway by 3,5-bis(2-flurobenzylidene)piperidin-4-one (EF24), a
novel monoketone analog of curcumin. Mol Pharmacol. 74:654–661.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Al-Hujaily EM, Mohamed AG, Al-Sharif I, et
al: PAC, a novel curcumin analogue, has anti-breast cancer
properties with higher efficiency on Er-negative cells. Breast
Cancer Res Treat. 128:97–107. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Hutzen B, Friedman L, Sobo M, et al:
Curcumin analogue GO-Y030 inhibits STAT3 activity and cell growth
in breast and pancreatic carcinomas. Inter J Oncol. 35:867–872.
2009.PubMed/NCBI
|
|
29.
|
Kumar AP, Garcia GE, Ghosh R, Rajnarayanan
RV, Alworth WL and Slaga TJ: 4-Hydroxy-3-methoxybenzoic acid methyl
ester: a curcumin derivative targets Akt/NF kappa B cell survival
signaling pathway: potential for prostate cancer management.
Neoplasia. 5:255–266. 2003. View Article : Google Scholar
|
|
30.
|
Lin L, Hutzen B, Ball S, et al: New
curcumin analogues exhibit enhanced growth-suppressive activity and
inhibit AKT and signal transducer and activator of transcription 3
phosphorylation in breast and prostate cancer cells. Cancer Sci.
100:1719–1727. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Wang SC and Hung MC: HER2 overexpression
and cancer targeting. Semin Oncol. 28:115–124. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Hsieh WT, Huang KY, Lin HY and Chung JG:
Physalis angulata induced G2/M phase arrest in human breast
cancer cells. Food Chem Toxicol. 44:974–983. 2006. View Article : Google Scholar
|
|
33.
|
Hsu JD, Kao SH, Ou TT, Chen YJ, Li YJ and
Wang CJ: Gallic acid induces G2/M phase arrest of breast cancer
cell MCF-7 through stabilization of p27(Kip1) attributed to
disruption of p27(Kip1)/Skp2 complex. J Agric Food Chem.
59:1996–2003. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Somers-Edgar TJ, Taurin S, Larsen L,
Chandramouli A, Nelson MA and Rosengren RJ: Mechanisms for the
activity of heterocyclic cyclohexanone curcumin derivatives in
estrogen receptor negative human breast cancer cell lines. Invest
New Drugs. 29:87–97. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Santen RJ, Song RX, McPherson R, Kumar R,
Adam L, Jeng MH and Yue W: The role of mitogen-activated protein
(MAP) kinase in breast cancer. J Steroid Biochem Mol Biol.
80:239–256. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Wada T and Penninger JM: Mitogen-activated
protein kinases in apoptosis regulation. Oncogene. 23:2838–2849.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Liu B, Han M, Sun RH, Wang JJ, Zhang YP,
Zhang DQ and Wen JK: ABL-N-induced apoptosis in human breast cancer
cells is partially mediated by c-Jun NH2-terminal kinase
activation. Breast Cancer Res. 12:R92010. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Kuo PL, Chen CY and Hsu YL:
Isoobtusilactone A induces cell cycle arrest and apoptosis through
reactive oxygen species/apoptosis signal-regulating kinase 1
signaling pathway in human breast cancer cells. Cancer Res.
67:7406–7420. 2007. View Article : Google Scholar
|
|
39.
|
Collett GP and Campbell FC: Curcumin
induces c-jun N-terminal kinase-dependent apoptosis in HCT116 human
colon cancer cells. Carcinogenesis. 25:2183–2189. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Weir NM, Selvendiran K, Kutala VK, et al:
Curcumin induces G2/M arrest and apoptosis in cisplatin-resistant
human ovarian cancer cells by modulating Akt and p38 MAPK. Cancer
Biol Ther. 6:178–184. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Eto I: Nutritional and chemopreventive
anti-cancer agents up-regulate expression of p27Kip1, a
cyclin-dependent kinase inhibitor, in mouse JB6 epidermal and human
MCF7, MDA-MB-321 and AU565 breast cancer cells. Cancer Cell Int.
6:202006. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Dillon RL, White DE and Muller WJ: The
phosphatidyl inositol 3-kinase signaling network: implications for
human breast cancer. Oncogene. 26:1338–1345. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Burow ME, Weldon CB, Melnik LI, Duong BN,
Collins-Burow BM, Beckman BS and McLachlan JA: PI3-K/AKT regulation
of NF-kappaB signaling events in suppression of TNF-induced
apoptosis. Biochem Biophys Res Commun. 271:342–345. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Gong L, Li Y, Nedeljkovic-Kurepa A and
Sarkar FH: Inactivation of NF-kappaB by genistein is mediated via
Akt signaling pathway in breast cancer cells. Oncogene.
22:4702–4709. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Barkett M and Gilmore TD: Control of
apoptosis by Rel/NF-kappaB transcription factors. Oncogene.
18:6910–6924. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Lauder A, Castellanos A and Weston K:
c-Myb transcription is activated by protein kinase B (PKB)
following interleukin 2 stimulation of T cells and is required for
PKB-mediated protection from apoptosis. Mol Cell Biol.
21:5797–5805. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Shehzad A, Wahid F and Lee YS: Curcumin in
cancer chemoprevention: molecular targets, pharmacokinetics,
bioavailability, and clinical trials. Arch Pharm (Weinheim).
343:489–499. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Dhandapani KM, Mahesh VB and Brann DW:
Curcumin suppresses growth and chemoresistance of human
glioblastoma cells via AP-1 and NFkappaB transcription factors. J
Neurochem. 102:522–538. 2007. View Article : Google Scholar : PubMed/NCBI
|