Introduction
Glioblastoma multiforme is the most common primary
brain tumor in humans arising from cells of the glia lineage. The
name denotes a very heterogeneous type of tumor with respect to
cell morphology and chromosome aberrations, which includes extended
deletions, gain of entire chromosomes or chromosome arms and gene
amplification (1). Surgical
resection and radiotherapy are the first and second stage of
treatment of glioblastoma, respectively, while the addition of
chemotherapy to radiation has so far shown limited improved
survival (2,3). In mammals, exposure of cells to
radiation causes DNA double-strand breaks that are generally
repaired via non-homologous end joining following activation of
DNA-dependent protein kinase (DNA-PK) (4). However, when DNA damage is excessive,
cells induce DNA-PK-mediated apoptosis. It has been reported that
exposure to high doses of ionizing radiation leads to autophagy
induction in some types of cancer cells including malignant glioma
cells (5).
Macroautophagy (hereafter referred to as autophagy)
is a biphasic process characterized by the formation of
double-membrane vesicles called autophagosomes (i.e. the formation
phase) often containing subcellular organelles, which fuse with
lysosomes. In the following maturation phase the vesicle content is
degraded generating macromolecules and ATP that are recycled as to
maintain cellular homeostasis (6).
The formation of autophagosomes is mediated by a highly organized
and hierarchical team of autophagy-related gene products (ATG
proteins) of which microtubule-associated protein light chain 3
(LC3), the mammalian homolog of yeast ATG8p, is the most specific
marker as it accumulates on the autophagosomal membrane giving rise
to characteristic punctate patterns (6,7).
Several recent studies have suggested that autophagy also functions
as a pro-death mechanism. In this respect, it has been shown that
different types of cancer cells undergo autophagic cell death in
response to anticancer therapy (8,9). At
present, it remains controversial whether autophagy represents a
survival and cytoprotective process or causes death in cancer cells
as well as the precise molecular mechanisms that regulate this
dynamic process.
Recent studies from our laboratory have revealed
that siRNA-mediated downregulation of protein kinase CK2 leads to
morphological changes resembling activation of autophagic cells
death in human glioblastoma cells (10). Protein kinase CK2 is a
constitutively active and highly conserved serine/threonine kinase
composed of two catalytic α and/or α′ subunits and two regulatory β
subunits. Evidence indicates that the individual subunits do not
exist exclusively within the tetrameric complex but also as free
proteins (11,12). CK2 expression and activity are
deregulated in many human diseases including cancer and while the
overexpression often correlates with enhanced cell survival and
proliferation, cellular depletion generally results in reduced cell
viability and increased cell death, particularly apoptosis
(13). Moreover, although mounting
evidence underlines the importance of targeting CK2 for activation
of apoptosis in cancer cells, the role of this kinase with respect
to induction and/or progression of other types of cell death such
as autophagy is largely unknown.
In this study, we aimed to shed light on the
mechanism by which CK2 silencing induces autophagy in human
glioblastoma cells treated with the radiomimetic drug
neocarzinostatin. We report evidence that downregulation of protein
kinase CK2 results in inhibition of the mammalian target of
rapamycin (mTOR) pathway, downregulation of Raptor expression
levels and activation of the extracellular signaling-regulated
protein kinase 1/2 (ERK1/2) signaling pathway as evidenced by ERK
phosphorylation and enhanced kinase activity. The reported results
support the notion that CK2 may serve as a potential target for
enhancing the cellular response of glioblastoma cells resistant to
multiple drug treatments.
Materials and methods
Cell culture
The human glioblastoma cell lines M059K and T98G
were obtained from the American Type Culture Collection (Rockville,
MD, USA). The cells were cultured in DMEM (Invitrogen, Taastrup,
Denmark) with 10% FBS and maintained at 37°C in a 5% CO2
atmosphere.
Cell culture and treatments
Cells were transfected with a set of 4 small
interfering RNA (siRNA) duplexes directed against CK2α or CK2α′
(On-Target plus SMARTpools, Dharmacon, Lafayette, CO, USA), using
Dharmafect I transfection reagent (Dharmacon) for 72 h according to
the manufacturer’s instructions. Control experiments were performed
transfecting cells with scramble siRNA sequences. Where indicated,
cells were incubated with the radiomimetic drug neocarzinostatin
for 24 h (NCS, a gift from Dr Hiroshi Maeda, Kumamoto University,
Kumamoto, Japan). Endogenous mTOR activity was inhibited by
incubating T98G cells with 100 nM rapamycin (Calbiochem,
Nottingham, UK) and M059K cells with 50 nM rapamycin, respectively.
Maturation of autophagy vacuoles was inhibited by treating cells
with 50 nM bafilomycin A (Calbiochem) for 6 h prior to cell
analysis.
Flow cytometry
Autophagy was analyzed by staining cells with the
vital dye acridine orange (Sigma, Brondby, Denmark). Briefly, cells
were incubated with acridine orange at a final concentration of 1
μg/ml for 15 min prior to trypsinization and flow cytometry
analysis on a FACSCalibur (BD Biosciences, San Diego, CA, USA)
using CellQuest Pro Analysis Software (BD Biosciences). To detect
mitochondrial superoxide production, cells were trypsinized and
incubated with 5 μM MitoSOX™ Red Mitochondrial Superoxide
indicator (Invitrogen) for 15 min prior to FACS analysis. ROS
scavengers treatment was performed by incubating cells with 100
μM BHA or 50 μM BPN (both from Sigma) for 24 h prior
to harvesting and subsequent analysis by flow cytometry.
Western blot analysis
Whole cell extracts and immunoblotting were as
previously described (14). The
primary antibodies employed in this study were: mouse monoclonal
anti-CK2β and -CK2α (both from Calbiochem); rabbit polyclonal
anti-CK2α′ obtained by immunizing rabbits with a specific peptide
(SQPCADNAVLSSGTAAR) deriving from human CK2α′; mouse monoclonal
anti-β-actin (Sigma); mouse monoclonal anti-mTOR and -AKT1 (both
from BD Biosciences), rabbit monoclonal anti-Raptor and -ERK1/2
(p-T202/Y204); rabbit polyclonal anti-mTOR (p-S2481) and -ERK1/2
(all from Cell Signaling Technology, Beverly, MA, USA); mouse
monoclonal anti-AKT (p-S473) and -p70 S6 kinase (p-T389) (both from
Cell Signaling Technology) and rabbit polyclonal anti-p70 S6 kinase
(Santa Cruz Biotechnology, Santa Cruz, CA, USA). Protein bands were
then visualized by a chemiluminescence detection system following
the manufacturer’s guidelines (CDP-Star; Applied Biosystems, Foster
City, CA, USA).
In vitro protein kinase assay
The activity of ERK1/2 was tested using a
non-radioactive p44/42 MAPK kinase assay kit (Cell Signaling
Technology) following the manufacturer’s instructions. Briefly,
whole cells extracts were immunoprecipitated with rabbit monoclonal
anti-phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) antibody
immobilized on sepharose beads. Immunoprecipitates were subjected
to a non-radioactive kinase assay in the presence of 0.25 μg
recombinant GST-fused Elk-1, 1X kinase buffer-containing 200
μM ATP. Incubation time was 30 min at 30°C. Phosphorylation
of Elk-1 was revealed by labeling western blot membranes with mouse
monoclonal anti-phospho-Elk-1 (Ser383) antibody.
EGFP-LC3 translocation assay
Cells were grown on cover-slips and transfected with
CK2-siRNA for 48 h, prior to addition of NCS for 24 h. To visualize
GFP-LC3, cells were infected with Premo Autophagy Sensors (LC3B-FP,
wild-type) BacMam2.0 (Invitrogen) according to the manufacturer’s
instructions. The LC3B (G120A)-FP mutant form was employed in
control experiments, as the mutation of the expressed LC3B prevents
its cleavage and subsequent lipidation following induction of
autophagy. After 24 h, cells were fixed with 4% paraformaldehyde
for 20 min, permeabilized with 0.2% Na-citrate/0.2% Triton X-100
for 5 min and counterstained with 4′,6-diamidino-2-phenylindole
(DAPI) and analyzed on a DMRBE microscope (×400 magnification)
equipped with a Leica DFC420C camera. Pictures were processed using
ImageJ software (NIH, Bethesda, MD, USA). For each condition, at
least 200 GFP-LC3-expressing cells were analyzed and the percentage
of puncta per cell, deriving from the expression of GFP, was
determined by using the ‘analyze particles’ function in the ImageJ
software.
Statistical analysis
The statistical significance of differences between
the means of two groups was evaluated by the two-tailed t-test and
the level of significance was as indicated in the figure
legends.
Results
Downregulation of protein kinase CK2
induces autophagic cell death in human glioblastoma cells
Accumulating evidence indicates that induction of
DNA double-strand breaks in some types of cancer cells, including
malignant glioma cells, induces autophagic cell death (8,9). To
compare the effects of DNA damage on autophagy induction, we
analyzed M059K and T98G cells after exposure to the radiomimetic
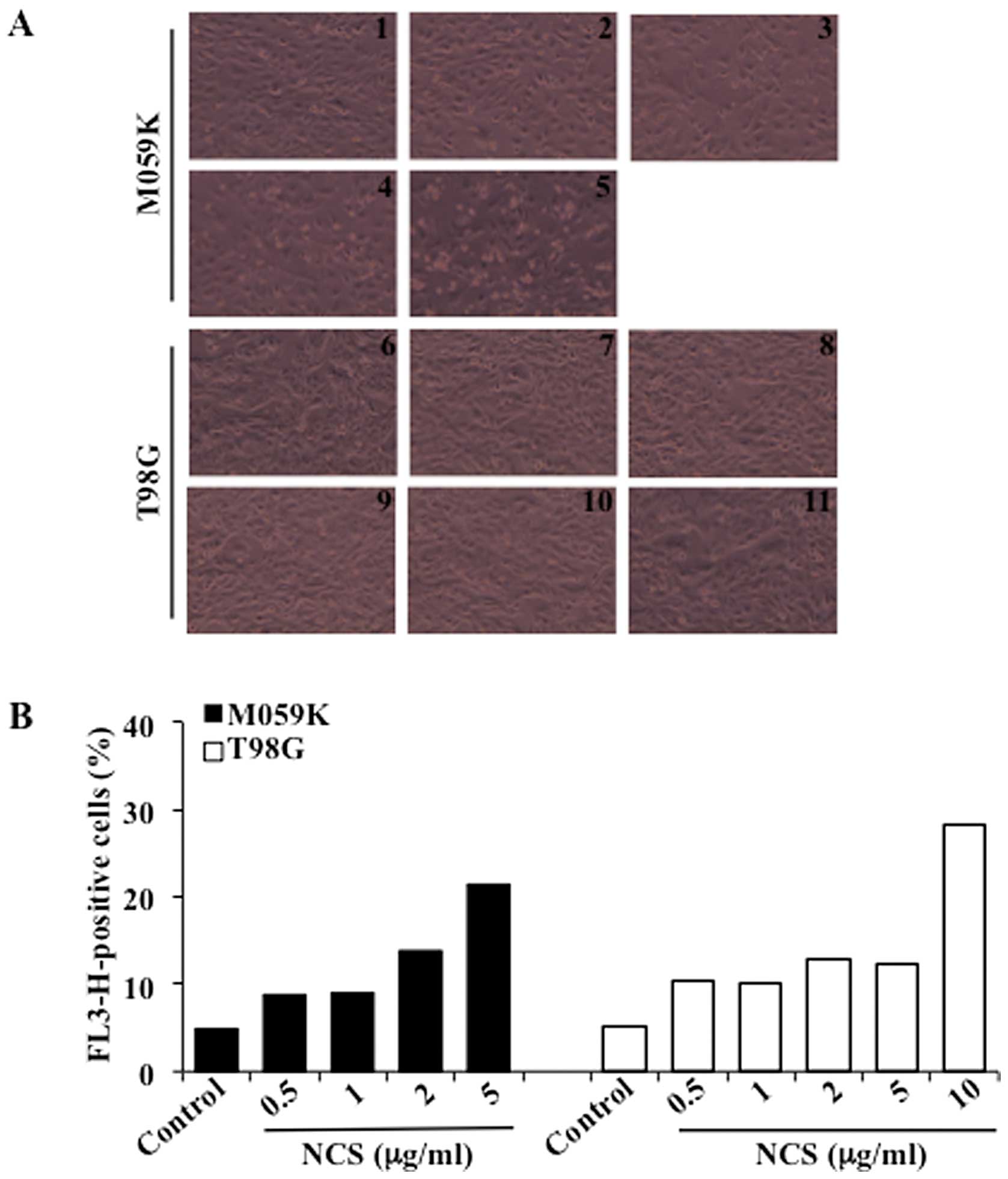
drug neocarzinostatin (NCS) (15).
Treatment with increasing concentrations of NCS for 24 h led to
marked cytotoxicity in M059K cells when incubated with up to 5
μg/ml NCS while a similar effect was observed in T98G cells
exposed to 10 μg/ml NCS (Fig.
1A). Induction of autophagy was determined by flow cytometry
after staining of cells with acridine orange, a vital dye which
accumulates in acidic compartments emitting bright red fluorescence
indicative of autophagic vacuoles formation (AVOs) (16). As shown in Fig. 1B, untreated cells showed negligible
cytoplasmic staining as compared to cells treated with increasing
concentrations of NCS. NCS (5 μg/ml) led to formation of
AVOs in approximately 21% of the M059K cells while a slightly
higher percentage (i.e. 28%) was reached with T98G cells when
incubated with 10 μg/ml NCS. To assess the role of CK2 in
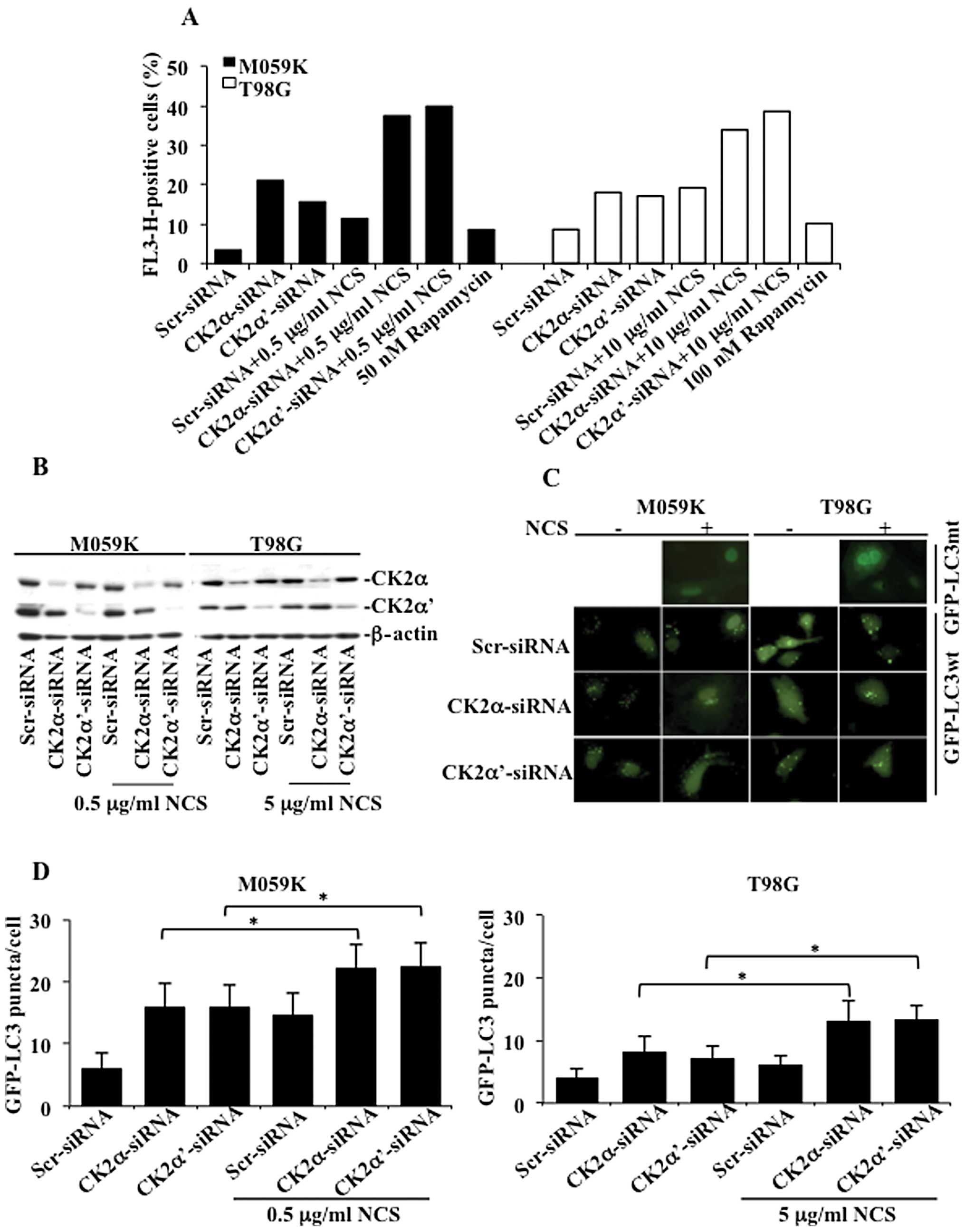
the activation of autophagy, we transfected M059K and T98G cells
with a pool of small interfering RNAs (siRNAs) against the
individual catalytic subunits of CK2 in the presence or absence of
NCS as indicated in Fig. 2A. Cells
were subsequently stained with acridine orange and analyzed by flow
cytometry. In M059K cells, incubation with NCS resulted in AVOs
accumulation in approximately 11% of the total number of cells
while reduction of CK2α and -α′ protein levels led to the formation
of AVOs in 21 and 15% of the cells, respectively. This percentage
increased to approximately 37 and 39% following incubation with
NCS, respectively. In the case of T98G cells, the number of cells
with AVOs following exposure to NCS or treatment with siRNAs
against the CK2 catalytic subunits was approximately 18% while the
combined downregulation of the CK2 subunits and treatment with NCS
led to up to 38% AVOs-positive cells. Exposure of cells to
rapamycin, a known activator of autophagy in malignant glioma cells
(17), resulted in the
accumulation of AVOs in approximately 10% of the total number of
cells.
To confirm the induction of autophagic cell death
induced by the siRNA-mediated CK2 silencing, we assessed the
presence of a punctate pattern of the green-fluorescent protein
(GFP)-tagged LC3 (GFP-LC3) expression. M059K and T98G cells were
transfected with siRNAs against the individual catalytic subunits
of CK2 for 48 h. Subsequently, cells were transduced with GFP-LC3wt
or a mutated form (GFP-LC3mt) to rule out the possibility that
expression of GFP-LC3 would lead to an unspecific aggregation
rather than its translocation to the autophagosomal membranes
(control experiment), and incubated for additional 24 h in the
absence and presence of NCS, respectively. Western blot analysis of
cell lysates confirmed the significant downregulation of the
individual CK2 subunits (Fig. 2B).
As shown in Fig. 2C, cells
transduced with GFP-LC3mt showed diffuse distribution of GFP-LC3
following incubation with NCS while cells transduced with GFP-LC3wt
showed a punctate pattern of GFP-LC3 fluorescence, indicating the
presence of autophagic vacuoles and recruitment of LC3 to their
surface. Quantification of autophagic-positive cells revealed that
downregulation of CK2 increased the incidence of autophagy in both
cell types as compared to control experiments (Fig. 2D). Furthermore, when cells were
additionally incubated with NCS, the percentage of puncta per cell
significantly increased up to 22% in the case of M059K cells and
13% in the case of T98G cells with respect to cells treated with
NCS and transfected with Scr-siRNA. Induction of autophagy was
lower in T98G than in M059K cells. However, these results indicate
that downregulation of CK2 induces autophagic cell death which is
significantly enhanced in the presence of NCS and suggest that
differences in autophagy stimulation might be related to the
intrinsic resistance of different glioblastoma cell types towards
induction of this process.
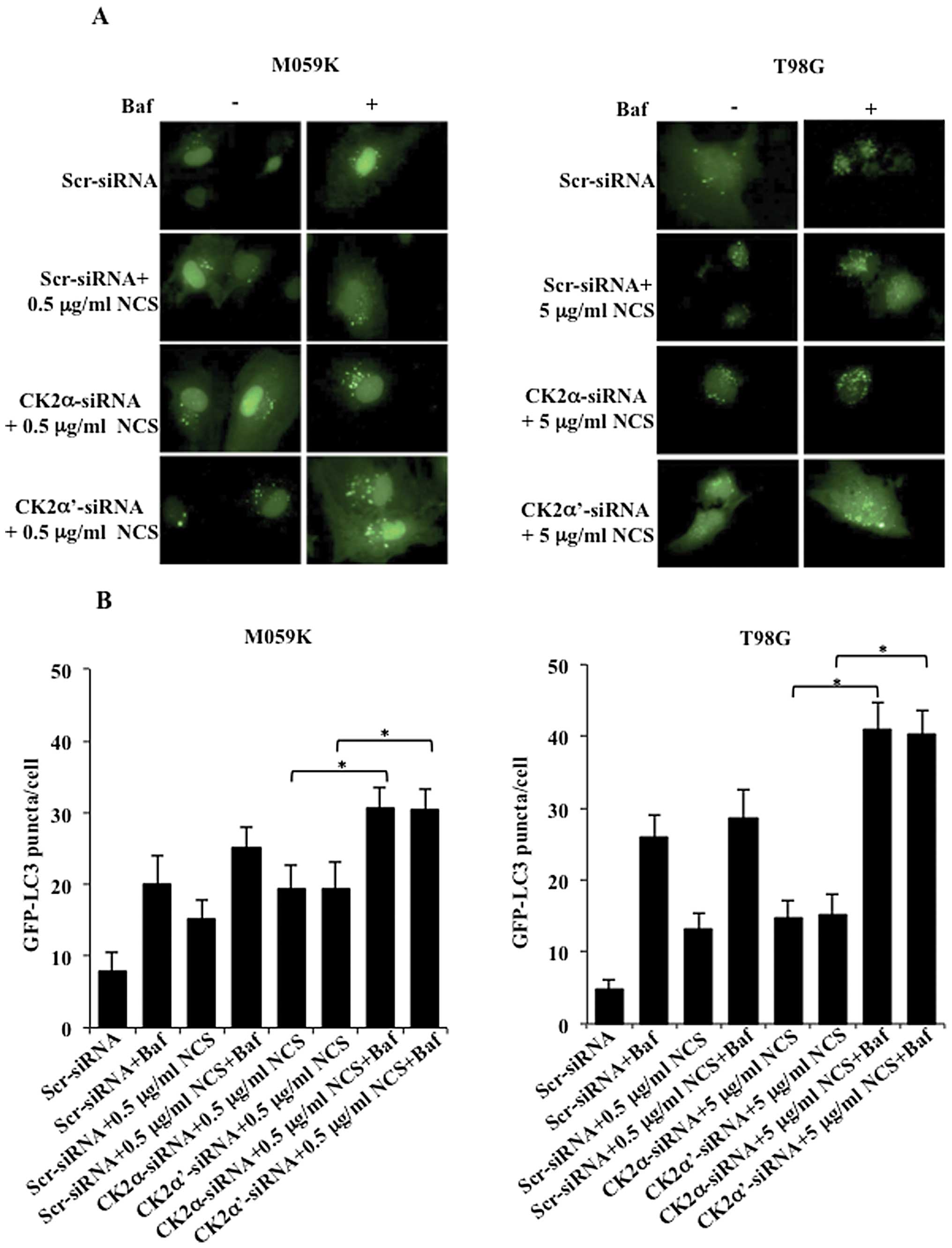
Accumulation of GFP-LC3 puncta may result from
activation of the autophagic process or a defect in the
autophagosome maturation. To distinguish between these two events,
we induced autophagy in the presence or absence of the vacuolar
H+-ATP inhibitor bafilomycin A (Baf) which is expected
to inhibit the fusion between autophagosomes and lysosomes and,
thus, formation of autolysosomes (18). As expected, CK2-depleted cells
revealed a punctate pattern of GFP-LC3 following treatment with NCS
consistent with data presented above, however, the number of puncta
per cell was significantly higher in cells additionally treated
with bafilomycin A (Fig. 3A).
Determination of the number of puncta per cell indicated that both
cell lines depleted of CK2 and treated with NCS and bafilomycin A
accumulated a significantly higher number of GFP-LC3-puncta as
compared to cells subjected to similar treatment but in the absence
of bafilomycin A (Fig. 3B). Flow
cytometry analysis of cells stained with acridine orange confirmed
data reported above (results not shown). Overall, results reported
here suggest that induction of autophagic cell death mediated by
silencing of CK2 by siRNA emerges from stimulation of the formation
phase of AVOs as indicated by the recruitment of GFP-LC3 to
autophagic vacuoles and increased number of puncta per cells upon
incubation with bafilomycin A.
Downregulation of protein kinase CK2
regulates autophagy through the mTOR and ERK1/2 signaling
pathways
The class I phosphatidylinositol 3-phosphate kinase
(PI3K)/AKT/Raptor-mTOR (mTORC1) signaling pathway and the
Ras/Raf/MEK1/2/ERK1/2 pathway are two well-known signaling cascades
involved in the regulation of autophagy (19–21).
Because of their implication in autophagy regulation, we examined
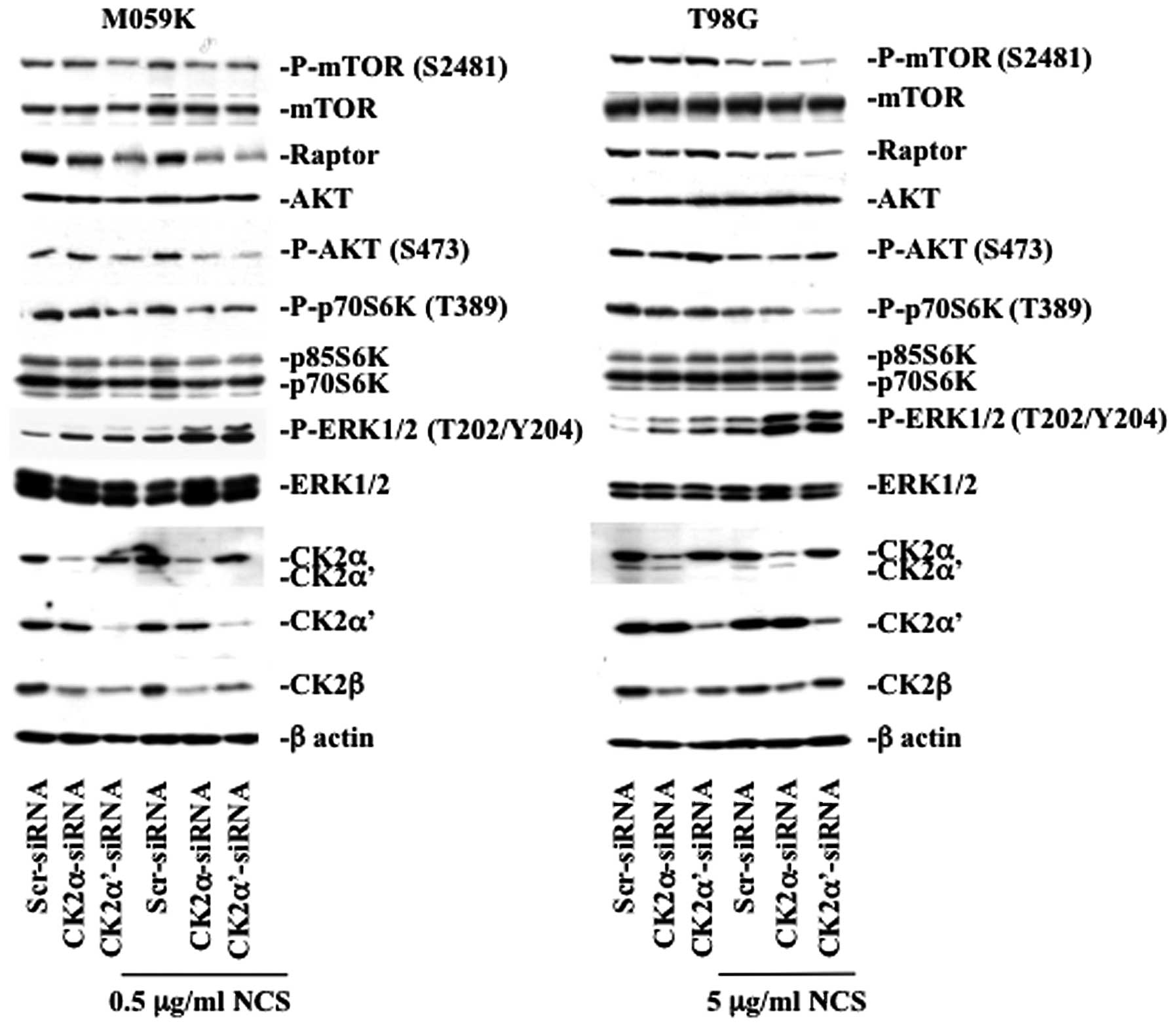
their status upon downregulation of CK2 (Fig. 4). In M059K cells, CK2 depletion led
to decreased kinase activity of mTOR reflected by the diminished
phosphorylation of mTOR at S2481 which is a marker for autokinase
activity in vivo(22),
decreased phosphorylation of the mTOR downstream target, p70 S6
kinase (p70S6K), and lower AKT phosphorylation at S473. The latter
being an important positive regulator of mTOR via pathways
involving the TSC1/TSC2 complex (23). Interestingly, downregulation of CK2
led to marked decreased expression levels of Raptor and this effect
was particularly pronounced in cells depleted of CK2α′ in the
absence or presence of NCS. In T98G cells, the mTOR pathway was
mainly affected in CK2α′-siRNA and NCS-treated cells evidenced by
the significant decreased mTOR and p70S6K phosphorylation levels
and lowered expression of Raptor.
Next, we examined whether cellular depletion of CK2
affected the phosphorylation of ERK1/2, which is considered a
positive regulator of autophagy (24). As predicted, repeated experiments
showed that silencing of CK2α and -α′ in the presence of NCS led to
marked increase in ERK1/2 phosphorylation in both cell lines,
respectively. As CK2 knockdown led to increased phosphorylation of
ERK1/2 in cells treated with NCS, we tested whether this effect was
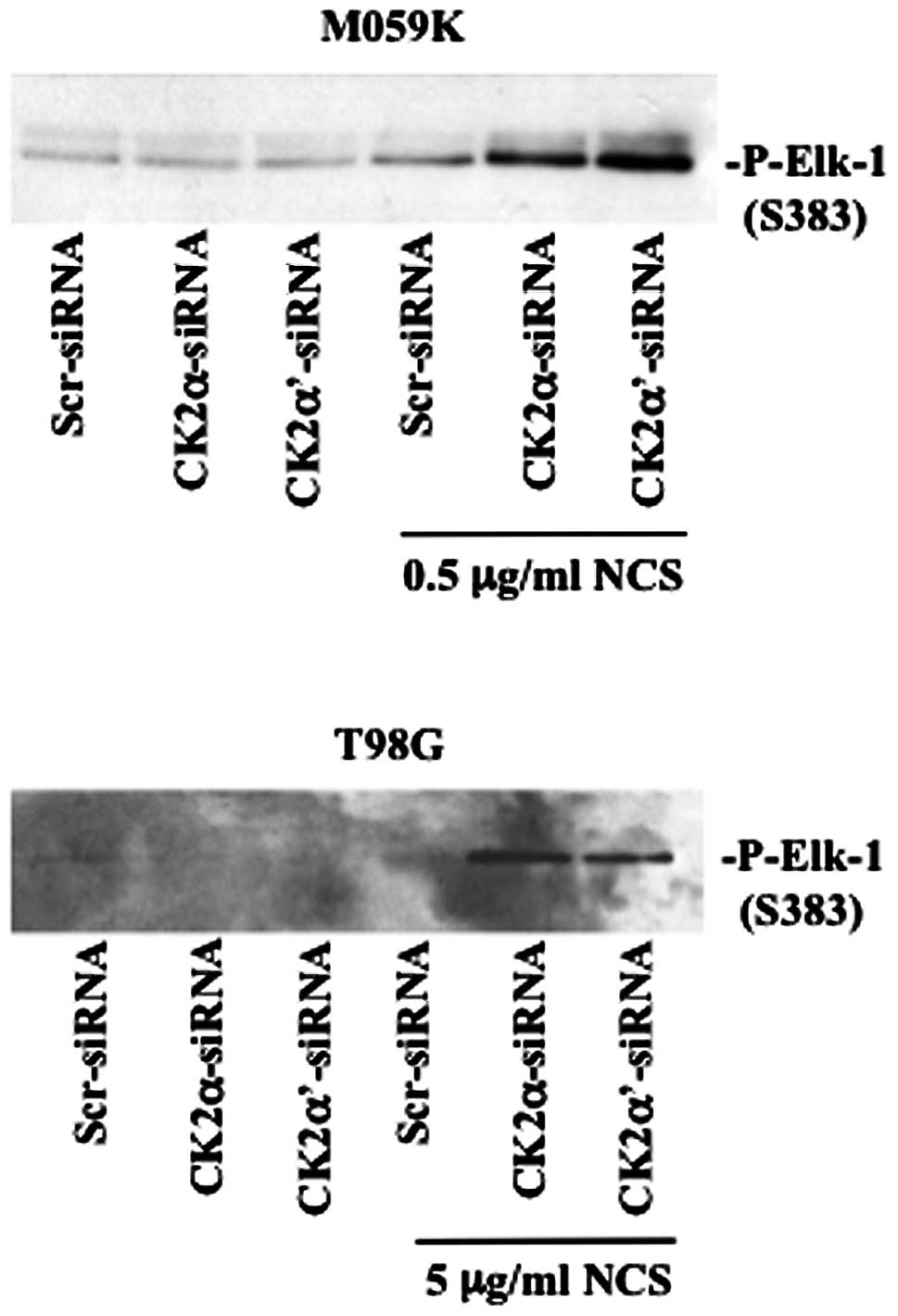
associated with increased ERK1/2 kinase activity (Fig. 5). We performed a non-radioactive
kinase assay where the activity of endogenous ERK1/2, isolated by
immnuoprecipitation, was tested against Elk-1 protein a downstream
target of ERK (25). By employing
a specific antibody directed against phosphorylated Elk-1, we
verified that enhanced phosphorylation of ERK was accompanied by
marked kinase activation in cells depleted of CK2 and treated with
NCS. Overall, western blot analysis of the aforementioned proteins
suggests that induction of autophagy following CK2 knockdown and
NCS treatment occurs through inhibition of the PI3K/AKT/mTOR and
activation of the ERK1/2 signaling pathways. As cellular depletion
of CK2 markedly enhanced the phosphorylation of ERK1/2, we sought
to verify whether autophagy would be blocked by ERK inhibition
using the mitogen-activated protein kinase kinase1/2 (MEK1/2)
inhibitors, U0126 and PD098059 (26) employed in separate experiments.
However, autophagy induction was attenuated but not completely
suppressed by indirect inhibition of ERK in the presence of the
aforementioned MEK1/2 specific inhibitors (results not shown).
Induction of autophagy mediated by CK2
knockdown is accompanied by the generation of ROS in T98G but not
M059K cells
Mitochondria are the main source of reactive oxygen
species (ROS), which includes superoxide, hydrogen peroxide and
hydroxyl radical. It has recently been reported that ROS may
regulate several core autophagic pathways (27). When mitochondrial ROS is elevated,
the mitochondrial membrane is damaged, resulting in ROS leakage
into the cytosol thereby damaging other organelles. Generation of
mitochondrial ROS has been shown to correlate with phosphorylation
and activation of ERK which co-localizes with mitochondria, AVOs
and lysosomes and regulates mitophagy in glioblastoma cells
(28). Hence, to test whether
induction of autophagy mediated by CK2 downregulation was
accompanied by ROS generation, we measured intracellular levels of
superoxide, the predominant ROS in mitochondria (29), by flow cytometry after labeling
cells with MitoSOX Red reagent a specific mitochondrial
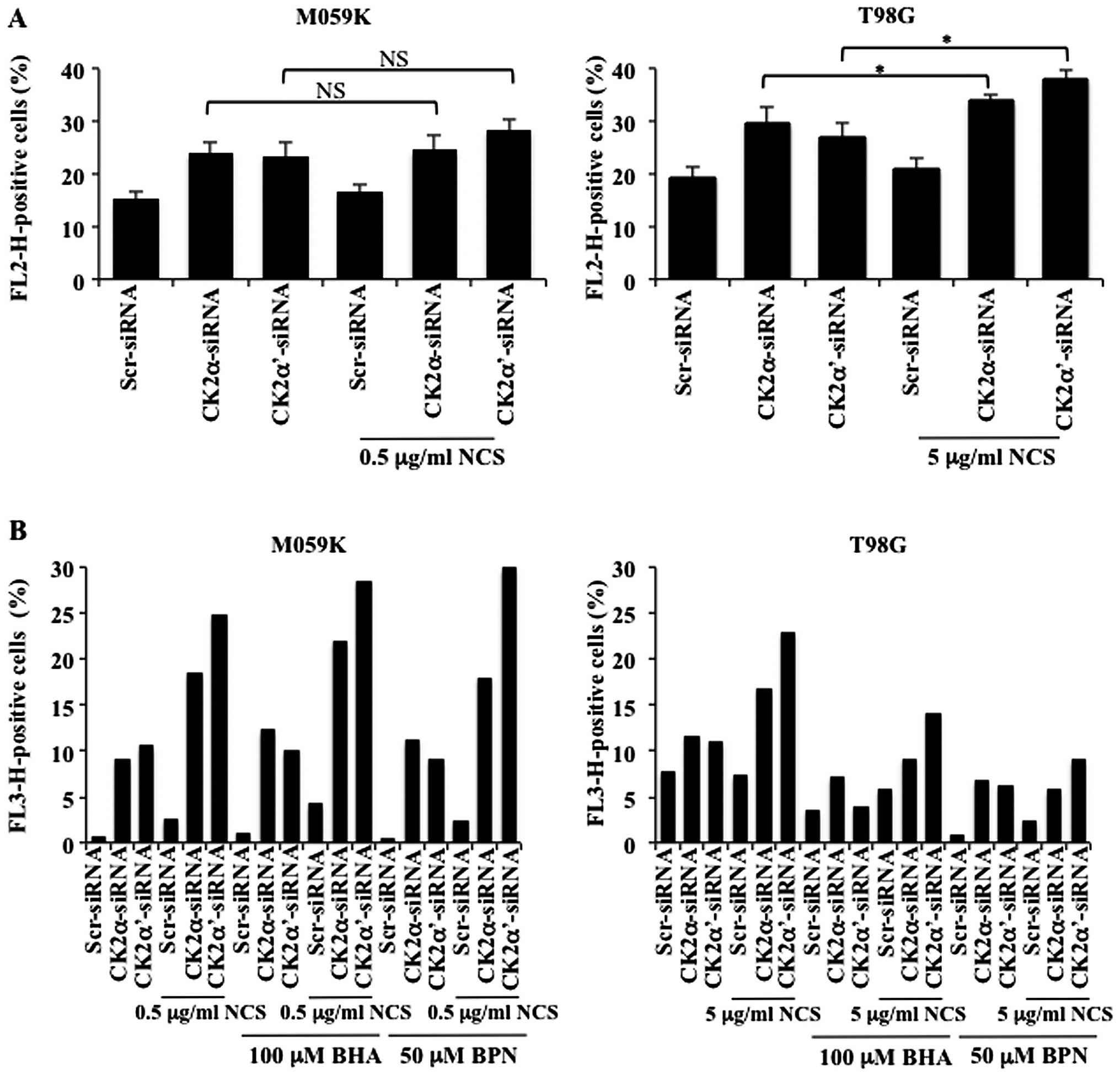
superoxide-detecting fluorescent dye. As shown in Fig. 6A, mitochondrial superoxide
increased in cells depleted of CK2. Additional treatment with NCS
led to slightly higher level of superoxide content which was
significant in T98G but not in M059K cells. Because superoxide has
been reported to be the major regulator of autophagy among
mitochondrial ROS, we investigated whether inhibition of ROS
production would be blocked in the presence of ROS scavengers and
whether this event would have an influence on autophagy induction.
Autophagy was evaluated by flow cytometry of acridine orange
stained cells. As shown in Fig.
6B, treatment with butylated hydroxyanisole (BHA) or
N-tert-Butyl-α-phenylnitrone (BPN) significantly reduced
accumulation of autophagic vacuoles induced by depletion of CK2 in
the absence or presence of NCS in T98G cells however, the same
effect was not observed in M059K cells. Overall, results reported
here indicate that cellular depletion of CK2 is accompanied by ROS
production and that the dependency on ROS generation in the
regulation of autophagy seems to be cell type-dependent. In this
respect, it has been previously reported that terfenadine, a highly
potent H1 histamine receptor antagonist, induces autophagy by
ROS-dependent and -independent mechanisms in human melanoma cell
lines (30).
Discussion
The process of autophagy is tightly controlled by
several signaling cascades and involves various steps including
induction, vesicle formation, autophagosome-lysosome fusion,
digestion and release of macromolecules in the cytosol. Protein
kinases play an important role in the regulation of this process
and while some of them such as mTOR, PI3K and AMPK directly
regulate components of the autophagic machinery, others, less
characterized, may control this process indirectly by influencing
level of expression and/or function of autophagy-related proteins.
In this study, we demonstrated that downregulation of protein
kinase CK2 by RNA interference leads to induction of autophagy in
human glioblastoma cells and that the combined exposure to the
radiomimetic drug NCS, significantly augments this process. The
level of induction of autophagy by CK2-siRNA-mediated silencing was
significantly higher than the one induced by rapamycin treatment as
shown in Fig. 2. Rapamycin and its
derivatives such as CCI-779, RAD001 and AP23573, are
well-established inhibitors of mTOR that have been shown to induce
modest levels of autophagy potentially because these agents rather
than acting as inhibitors of mTOR interfere solely with the
function of the mTOR/raptor complex (i.e. mTORC1) and thus, do not
block all mTOR forms (31).
Accordingly, the fact that CK2 silencing in the absence or presence
of NCS treatment led to marked increase in autophagy-positive cells
as determined by flow cytometry and analysis of the number of
puncta formation in GFP-LC3-positive cells, suggest that CK2 might
affect various components/steps of the autophagic process. In this
respect, we assessed the expression levels and the activity of a
downstream component of the mTORC1 signaling pathway, p70S6K, and
found that its phosphorylation was significantly inhibited in both
cell lines. In M059K cells, downregulation of CK2α’ alone was
sufficient to negatively affect the phosphorylation of p70S6K
kinase while this effect was achieved in T98G cells following
additional treatment with NCS. This difference might reflect the
intrinsic resistance of T98G cells to cell death induction.
Analysis of Raptor protein revealed marked downregulation of its
expression levels. It remains to be determined the mechanism by
which CK2 depletion results in lowered Raptor expression as the
regulation might be at the transcriptional and/or translational
levels. However, one cannot exclude that the observed reduced
phosphorylation of p70S6K, which is a translation regulator, might
suppress overall cellular protein synthesis, hence, Raptor
expression. On the other hand, decreased phosphorylation of p70S6K
seen in CK2 depleted cells, might be a direct consequence of low
Raptor expression as it has been demonstrated that the
siRNA-mediated downregulation of Raptor leads to reduced
phosphorylation of p70S6K and 4E-BP1 (32).
In our study, autophosphorylation of mTOR at S2481
was found repressed. It has recently been demonstrated that, in
contrast to what was previously reported, mTORC-associated mTOR
S2481 autophosphorylation monitors mTORC intrinsic catalytic
activity in vivo(33).
Hence, our results additionally show that CK2 depletion contributes
to the activation of autophagy by blocking the autophosphorylation
of mTOR. The two mTOR complexes are linked through AKT since mTORC2
(i.e. mTOR/Rictor)-activated AKT indirectly stimulates mTORC1. We
reported that CK2 depletion leads to decreased phosphorylation of
AKT at S473. Interestingly, phosphorylation of AKT at S473 has been
shown to be upregulated after mTORC1 inhibition. This effect has
been proposed to confer resistance to drug treatment (34). The reason for this discrepancy is
not entirely clear, however, it suggests that AKT phosphorylation
at S473 is subjected to multiple levels of regulation (35,36).
One of the most striking observations that emerged
from our studies is the significant increased phosphorylation of
ERK1/2 in cells treated with CK2-siRNA and NCS that was accompanied
by upregulation of ERK1/2 kinase activity. ERK activity has been
associated with constitutive autophagy and autophagic cell death in
many cellular models and in response to different stresses
including amino acid depletion and various drug treatments
(24,37–39).
Moreover, direct ERK activation by overexpression of constitutively
active MEK has been shown to promote autophagy without any further
stimulus (40). Hence, our results
support the notion that sustained activation of ERK pathway results
in induction of autophagic cell death in glioblastoma cells. The
observed failure to reverse completely effects induced by CK2
knockdown in cell treated with the specific MEK1/2 inhibitors,
U0126 and PD098059, might depend on the CK2-mediated induction of
autophagy through regulation of multiple pathways, i.e. the mTOR
and ERK1/2 signaling cascades. This is in accordance to previous
data supporting the notion that it is necessary to knockdown more
than one of the members that belong to autophagy-related pathways
in order to induce this process (41).
An additional observation that emerged from this
study is that both cell types showed increased mitochondrial
superoxide production following CK2 silencing, however, autophagy
induction was affected by treatment with ROS scavengers only in
T98G cells. Although it remains to be determined whether
mitochondrial ROS generation is a consequence of autophagy
activation or a catalyst of this process, the intensity of the
death stimulus in the two cell lines might determine the dependency
on ROS generation for the induction of autophagy.
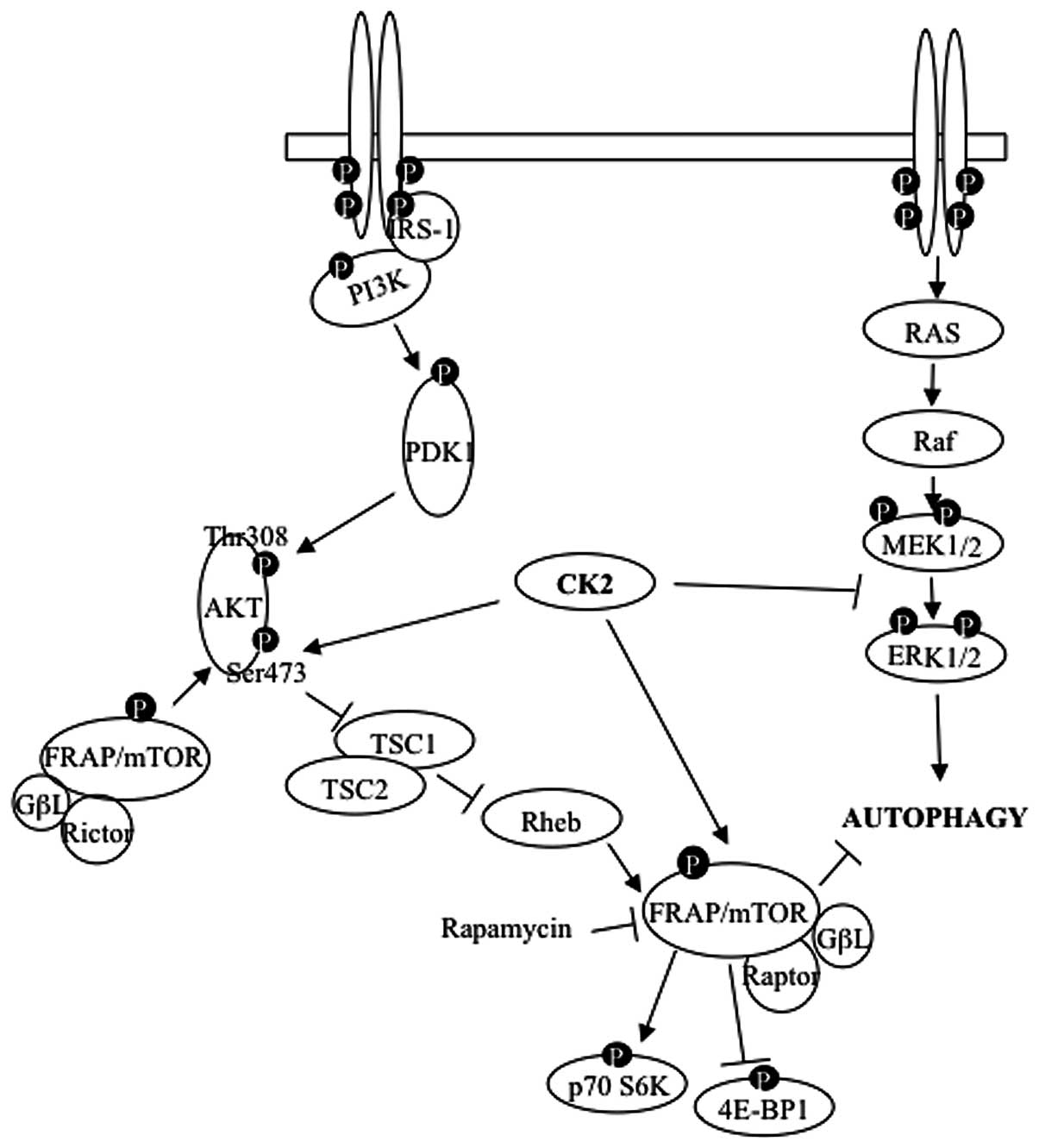
In this study, we have shown that depletion of
protein kinase CK2 leads to marked activation of autophagic
cell-death and this effect is enhanced when cells are additionally
treated with NCS. The effective induction of autophagy is due to
CK2 action at multiple levels and results in the extensive
inhibition of Raptor-mTOR complex, lack of activation of the
AKT-survival pathway and marked activation of the ERK1/2 signaling
cascade (Fig. 7). Further studies
are necessary to shed light on the complex regulation of autophagy
mediated by protein kinases, however, silencing the CK2 signal by
RNA interference represents a potential therapeutic strategy for
sensitizing malignant glioma cells to NCS-mediated autophagic cell
death.
Acknowledgements
This study was supported by a Grant
from the Danish Cancer Society (DP08152) to BG.
References
|
1.
|
DN LouisH OhgakiOD WiestlerWK CaveneePC
BurgerA JouvetBW ScheithauerP KleihuesThe 2007 WHO classification
of tumours of the central nervous systemActa
Neuropathol11497109200710.1007/s00401-007-0243-417618441
|
|
2.
|
M VerheijH BartelinkRadiation-induced
apoptosisCell Tissue Res301133142200010.1007/s004410000188
|
|
3.
|
F LefrancJ BrotchiR KissPossible future
issues in the treatment of glioblastomas: special emphasis on cell
migration and the resistance of migrating glioblastoma cells to
apoptosisJ Clin
Oncol2324112422200510.1200/JCO.2005.03.08915800333
|
|
4.
|
GC SmithSP JacksonThe DNA-dependent
protein kinaseGenes Dev13916934199910.1101/gad.13.8.91610215620
|
|
5.
|
W ZhuangZ QinZ LiangThe role of autophagy
in sensitizing malignant glioma cells to radiation therapyActa
Biochim Biophys Sin
(Shanghai)41341351200910.1093/abbs/gmp02819430698
|
|
6.
|
CW WangDJ KlionskyThe molecular mechanism
of autophagyMol Med965762003
|
|
7.
|
EL EskelinenMaturation of autophagic
vacuoles in Mammalian
cellsAutophagy1110200510.4161/auto.1.1.127016874026
|
|
8.
|
S PaglinT HollisterT DeloheryN HackettM
McMahillE SphicasD DomingoJ YahalomA novel response of cancer cells
to radiation involves autophagy and formation of acidic
vesiclesCancer Res61439444200111212227
|
|
9.
|
KC YaoT KomataY KondoT KanzawaS KondoIM
GermanoMolecular response of human glioblastoma multiforme cells to
ionizing radiation: cell cycle arrest, modulation of the expression
of cyclin-dependent kinase inhibitors, and autophagyJ
Neurosurg98378384200310.3171/jns.2003.98.2.0378
|
|
10.
|
BB OlsenOG IssingerB GuerraRegulation of
DNA-dependent protein kinase by protein kinase CK2 in human
glioblastoma
cellsOncogene2960166026201010.1038/onc.2010.33720711232
|
|
11.
|
B GuerraOG IssingerProtein kinase CK2 in
human diseasesCurr Med
Chem1518701886200810.2174/09298670878513293318691045
|
|
12.
|
NA St-DenisDW LitchfieldProtein kinase CK2
in health and disease: from birth to death: the role of protein
kinase CK2 in the regulation of cell proliferation and survivalCell
Mol Life Sci6618171829200910.1007/s00018-009-9150-219387552
|
|
13.
|
GM UngerAT DavisJW SlatonK AhmedProtein
kinase CK2 as regulator of cell survival: implications for cancer
therapyCurr Cancer Drug
Targets47784200410.2174/156800904348168714965269
|
|
14.
|
BB OlsenB GuerraAbility of CK2beta to
selectively regulate cellular protein kinasesMol Cell
Biochem316115126200810.1007/s11010-008-9817-218560763
|
|
15.
|
LF PovirkDNA damage and mutagenesis by
radiomimetic DNA-cleaving agents: bleomycin, neocarzinostatin and
other enediynesMutat
Res3557189199610.1016/0027-5107(96)00023-18781578
|
|
16.
|
F TraganosZ DarzynkiewiczLysosomal proton
pump activity: supravital cell staining with acridine orange
differentiates leukocyte subpopulationsMethods Cell
Biol41185194199410.1016/S0091-679X(08)61717-37532261
|
|
17.
|
A IwamaruY KondoE IwadoH AokiK FujiwaraT
YokoyamaGB MillsS KondoSilencing mammalian target of rapamycin
signaling by small interfering RNA enhances rapamycin-induced
autophagy in malignant glioma
cellsOncogene2618401851200710.1038/sj.onc.120999217001313
|
|
18.
|
A YamamotoY TagawaT YoshimoriY MoriyamaR
MasakiY TashiroBafilomycin A1 prevents maturation of autophagic
vacuoles by inhibiting fusion between autophagosomes and lysosomes
in rat hepatoma cell line, H-4-II-E cellsCell Struct
Funct233342199810.1247/csf.23.33
|
|
19.
|
D HanahanRA WeinbergThe hallmarks of
cancerCell1005770200010.1016/S0092-8674(00)81683-9
|
|
20.
|
CH JungS-H RoJ CaoNM OttoD-H KimmTOR
regulation of autophagyFEBS
Lett58412871295201010.1016/j.febslet.2010.01.01720083114
|
|
21.
|
EA CorcelleP PuustinenM JäätteläApoptosis
and autophagy: targeting autophagy signalling in cancer cells
-‘trick or treats’FEBS J276608460962009
|
|
22.
|
RT PetersonPA BealMJ CombSL
SchreiberFKBP12-rapamycin-associated protein (FRAP)
autophosphorylates at serine 2481 under translationally repressive
conditionsJ Biol
Chem27574167423200010.1074/jbc.275.10.741610702316
|
|
23.
|
J HuangS WuC-L WuBD ManningSignaling
events downstream of mammalian target of rapamycin complex 2 are
attenuated in cells and tumors deficient for the tuberous sclerosis
complex tumor suppressorsCancer
Res6961076114200910.1158/0008-5472.CAN-09-0975
|
|
24.
|
U SivaprasadA BasuInhibition of ERK
attenuates autophagy and potentiates tumour necrosis
factor-alpha-induced cell death in MCF-7 cellsJ Cell Mol
Med1212651271200810.1111/j.1582-4934.2008.00282.x18266953
|
|
25.
|
CS HillR MaraisS JohnJ WynneS DaltonR
TreismanFunctional analysis of a growth factor-responsive
transcription factor
complexCell73395406199310.1016/0092-8674(93)90238-L8477450
|
|
26.
|
X WangGP StudzinskiPhosphorylation of
raf-1 by kinase suppressor of ras is inhibited by ‘MEK-specific’
inhibitors PD 098059 and U0126 in differentiating HL60 cellsExp
Cell Res2682943002001
|
|
27.
|
B RavikumarS SarkarJE DaviesM FutterM
Garcia-ArencibiaZW Green-ThompsonM Jimenez-SanchezVI KorolchukM
LichtenbergS LuoDC MasseyFM MenziesK MoreauU NarayananM RennaFH
SiddiqiBR UnderwoodAR WinslowDC RubinszteinRegulation of mammalian
autophagy in physiology and pathophysiologyPhysiol
Rev9013831435201010.1152/physrev.00030.200920959619
|
|
28.
|
RK DagdaJ ZhuSM KulichCT
ChuMitochondrially localized ERK2 regulates mitophagy and
autophagic cell stress: implications for Parkinson’s
diseaseAutophagy4770782200818594198
|
|
29.
|
Y ChenMB AzadSB GibsonSuperoxide is the
major reactive oxygen species regulating autophagyCell Death
Differ1610401052200910.1038/cdd.2009.4919407826
|
|
30.
|
F Nicolau-GalmésA AsumendiE
Alonso-TejerinaG Pérez-YarzaS-M JangiJ GardeazabalY
Arroyo-BerdugoJM CareagaJL Díaz-RamónA ApraizMD BoyanoTerfenadine
induces apoptosis and autophagy in melanoma cells through
ROS-dependent and -independent
mechanismsApoptosis1612531267201121861192
|
|
31.
|
S HuangPJ HoughtonTargeting mTOR signaling
for cancer therapyCurr Opin
Pharmacol3371377200310.1016/S1471-4892(03)00071-7
|
|
32.
|
WK WuCW LeeCH ChoFK ChanJ YuJJ SungRNA
interference targeting raptor inhibits proliferation of gastric
cancer cellsExp Cell
Res31713531358201110.1016/j.yexcr.2011.03.00521396933
|
|
33.
|
GA SolimanHA Acosta-JaquezEA DunlopB
EkimNE MajAR TeeDC FingarmTOR Ser-2481 autophosphorylation monitors
mTORC-specific catalytic activity and clarifies rapamycin mechanism
of actionJ Biol
Chem28578667879201010.1074/jbc.M109.09622220022946
|
|
34.
|
M BreuleuxM KlopfensteinC StephanCA
DoughtyL BarysSM MairaD KwiatkowskiHA LaneIncreased AKT S473
phosphorylation after mTORC1 inhibition is rictor dependent and
does not predict tumor cell response to PI3K/mTOR inhibitionMol
Cancer Ther8742753200910.1158/1535-7163.MCT-08-066819372546
|
|
35.
|
G Di MairaM SalviG ArrigoniO MarinS SarnoF
BrustolonLA PinnaM RuzzeneProtein kinase CK2 phosphorylates and
upregulates Akt/PKBCell Death Differ12668677200515818404
|
|
36.
|
JN KreutzerM RuzzeneB GuerraEnhancing
chemosensitivity to gemcitabine via RNA interference targeting the
catalytic subunits of protein kinase CK2 in human pancreatic cancer
cellsBMC Cancer10440452201010.1186/1471-2407-10-44020718998
|
|
37.
|
E Ogier-DenisS PattingreJ Benna ElP
CodognoErk1/2-dependent phosphorylation of Galpha-interacting
protein stimulates its GTPase accelerating activity and autophagy
in human colon cancer cellsJ Biol
Chem2753909039095200010.1074/jbc.M006198200
|
|
38.
|
AA EllingtonMA BerhowKW
SingletaryInhibition of Akt signaling and enhanced ERK1/2 activity
are involved in induction of macroautophagy by triterpenoid B-group
soyasaponins in colon cancer
cellsCarcinogenesis27298306200610.1093/carcin/bgi21416113053
|
|
39.
|
Y ChengF QiuSI TashiroS OnoderaT
IkejimaERK and JNK mediate TNFalpha-induced p53 activation in
apoptotic and autophagic L929 cell deathBiochem Biophys Res
Commun376483488200810.1016/j.bbrc.2008.09.01818796294
|
|
40.
|
S CagnolJ-C ChambardERK and cell death:
mechanisms of ERK-induced cell death - apoptosis, autophagy and
senescenceFEBS
J277221200810.1111/j.1742-4658.2009.07366.x19843174
|
|
41.
|
P SzyniarowskiE Corcelle-TermeauT FarkasM
Høyer-HansenJ NylandstedT KallunkiM JaattelaA comprehensive siRNA
screen for kinases that suppress macro-autophagy in optimal growth
conditionsAutophagy7112201110.4161/auto.7.8.1577021508686
|