Introduction
The insulin like growth factor (IGF) system is
composed of two related growth factors (IGF-I and IGF-II) and a
group of IGF binding proteins (IGFBPs). This system has important
roles in human development and maintenance of normal functions of
many cells of the mammalian organism. In human, both IGF-I and
IGF-II are produced in multiple tissues throughout life and have
potentially divergent roles in pathophysiology. IGF-II has various
roles in pathological conditions and a particularly prominent role
in tumor development. This association between cancer and IGFs has
long been a subject for investigation (1). Dysregulation of IGF2, its
receptors (the type 1 and 2 IGFR), and IGFBPs provide part of the
underlying mechanism for uncontrolled increase in cellular
proliferation, as is evident in cancer. The IGF2 locus shows
loss of imprinting (LOI) in several types of cancer (reviewed in
ref. 2) and it was shown to be a
marker for colorectal carcinoma risk (3).
Genome wide epigenetic marks are maintained with
high fidelity in normal cells but frequently become destabilized in
human cancer (4,5). This is exemplified by the
observations that the normally silenced state of the maternally
inherited IGF2 allele is lost in several cancer forms, such
as Wilms’ tumor (6) and colon
cancer (7), resulting in LOI and
an activation of biallelic expression patterns. The finding that
IGF2 LOI in peripheral blood cells is the earliest
predictive marker for colon cancer reinforces the notion that
constitutive epigenetic lesions predispose for cancer (3). The normally repressed states of the
paternal H19 and the maternal IGF2 alleles are
coordinated by the differentially methylated imprinting control
region (ICR or DMR) in the 5′-flank of the H19 gene
(8). This feature involves a
methylation-sensitive, long-range chromatin insulator (9), that via target sites for the 11 zinc
finger protein CTCF represses the maternal IGF2 allele in
mammals (10,11).
It has further been shown that in mouse, this
insulator region is in physical contact with the promoter region of
the Igf2 gene (10,12) and that cohesin is maintaining the
insulation properties in the H19/IGF2 locus (13). In essence, the binding of CTCF to
the ICR, prevents the Igf2/IGF2 gene from being in close
proximity to the enhancers located in the 3′-flank of the
H19 gene, while they can interact with the H19
promoter. The absence of CTCF binding lets the same enhancers
interact with the Igf2/IGF2 gene. The fact that the binding
of CTCF to the ICR is methylation sensitive further strengthens the
notion that the methylation status of the ICR is the most crucial
key for proper maintenance of the mono-allelic expression patterns
of these genes.
Reports have shown that the CTCF target sites within
the H19 ICR are de novo methylated in a wide range of
human cancers (14,15), which would lead to prevention of
CTCF binding and loss of insulator function at the maternal allele,
followed by IGF2 allelic reactivation. This phenomenon can,
however, not explain the appearance of LOI of IGF2 in
correlation with hypomethylation at the maternal ICR which has been
reported in colon cancer (16) and
bladder cancer (17). These
findings suggest alternative, ICR independent, mechanisms that can
reactivate the silenced IGF2 gene on the maternal
allele.
PLAG1 is a proto-oncogene discovered in
pleomorphic adenoma of the salivary glands (18). It has been described as a seven
zinc finger transcription factor which is developmentally regulated
and expressed during fetal development in several tissues, but is
downregulated after birth. One reported mechanism for deregulation
of PLAG1 is promoter swapping with the β-catenin gene,
resulting in ectopic expression of PLAG1 in pleomorphic
adenoma of the salivary glands (18). PLAG1 has been shown to have
oncogenic capacity and to potently trigger IGF2 expression
by its binding to the P3 promoter as shown by electromobility shift
assay (EMSA) (19). In addition
PLAG1 has been shown to be overexpressed in several other
tumor types, such as hepatoblastoma (20), lipoblastoma (21) and acute myeloid leukemia (22).
Here, we set out to investigate the role of PLAG1 in
the regulation of IGF2 in an insulator-reporter system as
well as at the endogenous level in cell lines. The findings provide
evidence that PLAG1 can activate IGF2 P3 promoter-dependent
expression, despite the presence of the ICR in a reporter system.
The endogenous sensitivity of IGF2 to PLAG1 displayed a cell
type-specific response, and the IGF2 P3 promoter revealed
clear differential capacity to bind the PLAG1 transcription factor
which was however, not reflected by the IGF2
transcription.
Materials and methods
Cell lines and stably transfected cell
clones
JEG-3 (human choriocarcinoma) and Hep3B (human
hepatocellular carcinoma) cell lines were maintained as previously
described (23). For stable
transfection, 2 μg of the pSAR-PLAG1 (19) construct and 0.1 μg of the
pBK-CMV plasmid (neo +) was co-transfected into JEG-3 cells using
Fugene 6 (Roche) according to the protocol recommended by the
manufacturer, followed by neomycin selection (500 μM) for 4
weeks. Resistant cell clones were picked, expanded and analysed for
zinc-inducible (100 μM ZnCl2) PLAG1
expression using quantitative real-time PCR (qRT-PCR). One zinc
responsive cell clone which had undetectable PLAG1 mRNA in the
absence of zinc was selected for further experimental
procedures.
Quantitative real-time PCR analysis of
IGF2 and PLAG1
qRT-PCR was performed to evaluate the IGF2
and PLAG1 mRNA levels in wild-type Hep3B and JEG-3 cells.
Total-RNA was extracted using Trizol and phenol-chloroform,
quantified by Nanodrop, and 6–9 μg of RNA was treated with
DNase I (6–7 units, at 37°C for 30 min). DNase I treated RNA (1
μg) was used for cDNA synthesis with iScript cDNA synthesis
kit (Bio-Rad) in 20 μl reactions. Reverse transcription was
performed in 20 μl reactions following the protocol from the
manufacturer. A total of 2 μl of product was used for PCR
amplification of individual transcripts from IGF2 and
PLAG1 in an Applied Biosystem 7500 Fast Real-Time PCR
instrument using the iScrip SYBR-Green kit (Bio-Rad). The primers
for IGF2 are F: 5′-AAC TGG CCA TCC GAA AAT AGC and R: 5′-TTT
GCA TGG ATT TTG GTT TTC AT. For PLAG1 primers sequences are
F: 5′-CAC ACA GGA GAG AGG CCC TAC and R: 5′-CAC AAT AAT TAC ACT
TGT. Thermo cycling conditions were 50°C for 2 min and 95°C for 5
min, followed by 40 cycles of 95°C for 20 sec and 57° or 67°C for
30 sec for IGF2 and PLAG1, respectively. The GAPDH
mRNA was used for normalization to correct for the amount of RNA
loaded in each sample. Primer sequences are F: 5′-GAA GGT GAA GGT
CGG AGT C and R: 5′-GAA GAT GGT GAT GGG ATT TC. Thermo cycling
conditions were 50°C for 2 min and 95°C for 5 min, followed by 40
cycles of 95°C for 20 sec and 60°C for 30 sec.
Semi-quantitative RT-PCR
Semi-quantitative RT-PCR of PLAG1 transcripts was
performed in JEG-3 stable clones before and after Zn-induction
using the primers mentioned above. Thermo cycling conditions were
94°C for 5 min, followed by 30 cycles of 94°C for 30 sec, 59°C for
50 sec and 70°C for 50 sec and final extension at 72°C for 10
min.
Transient transfection and expression of
IGF2 P3 driven GFP reporter constructs
The IGF2 P3 promoter was cloned as a
BamHI-SalI fragment into the BglII-SalI
site of the pd2EGFP-1 vector (Clontech) to create the
pd2EGFP1-IGF2-P3-GFP intermediate vector. The IGF2-P3-GFP cassette
was excised from the intermediate vector by digesting with
Eco47III-SspI and cloned into a Bstz17I
digested pREP4 H19A vector (23)
to create the pA-GFP plasmid. The pB-GFP was obtained by cloning a
3.3 kb KpnI-XhoI mouse H19 ICR fragment into
pA-GFP. The pO-GFP plasmid was generated by deleting the SV40
enhancer from the pA-GFP plasmid by ClaI digestion followed
by re-ligation. pRep9RFP was created by cloning a
KpnI-XhoI fragment containing the RFP-gene (derived
from pDsRed1, Clontech) into the pRep9 vector (Invitrogen).
Equimolar amounts (3 μg) of the GFP
constructs were co-transfected with 0.25 μg pDsRed2-C1
(Clonetech) or 1 μg pRep9RFP into JEG-3 and Hep3B cells
using Fugene 6 (Roche) according to the protocol recommended by the
manufacturer. GFP expression was analysed using confocal microscopy
(Leica DM Irbe) and flow cytometry (BD FACSCalibur), whereby the
expression levels of GFP were estimated as a ratio between GFP and
RFP expression (constant).
Chromatin conformation analysis
Nuclei from Hep3B and JEG-3 cells, transfected with
the pB-GFP construct, were isolated and DNase I treated as
previously described (23). DNase
I treated DNA (20 μg) was digested with StuI and
analysed in 1.7% agarose gels followed by Southern blotting
according to standard procedures. The chromatin conformations were
analysed by indirect end-labelling as has been accounted for before
(23). The chromatin conformation
was analysed by using a 170 bp StuI-EcoRI fragment
spanning the upstream region outside of the ICR as previously
described (9).
Transient transfection of PLAG1
Hep3B and JEG-3 cells were transiently transfected
with 3 μg of the PLAG1 expression vector pCAGGS-PLAG1
(20) on 60-mm plates for 72 h. As
negative control, mock transfection with a β-galactosidase
containing plasmid was done in parallel. Transfection was performed
using the Lipofectamine and Plus reagent transfection kit
(Invitrogen) according to the manufacturer’s protocol.
Chromatin immunoprecipitation (ChIP)
analysis
Binding of PLAG1 to the IGF2 P3 promoter was
investigated in Hep3B and JEG-3 cells using Chromatin
Immunoprecipitation (ChIP). ChIP was performed from total cellular
extracts according to the Quick and Quantitative Chromatin
Immunoprecipitation (Q2 ChIP) method (described in ref. 24). After trypsinization cells were
cross-linked with 1% formaldehyde and chromatin was sheared by
Bioruptor 10 times (10 sec each) to fragments of 200 to 1,000 base
pairs and cleared for cellular debris by sedimentation. The
supernatant was used for immunoprecipitation of specific
protein-DNA complex using a PLAG1 antibody (Sigma, cat no. AV
38177) and normal rabbit IgG coupled to Dyna beads protein-G
(Invitrogen) at 4°C overnight. IgG was used as a control for
non-specific binding due to Fc receptor binding or other
protein-protein interactions. ChIP complexes were washed with RIPA
buffer and subsequently cross-links were reversed, proteins
digested and DNA eluted in elution buffer. DNA was purified with
PCR Purification Kit (Qiagen). The immunoprecipitated DNA was then
quantified by SYBR-Green qPCR using primers flanking the
PLAG1 consensus binding site in the IGF2 P3 promoter
region (20). Primer sequences are
F: 5′-CTGCCTGCCCGGAGAC and R: 5′-CATGCTGAATGCCCGTTCT. Thermo
cycling conditions were 50°C for 2 min and 95°C for 5 min, followed
by 40 cycles of 95°C for 20 sec and 65°C for 30 sec.
Results
Expression of IGF2 and PLAG1 in cell
lines
Since Hep3B and JEG-3 cells were used as model
systems, we assessed the endogenous mRNA levels of PLAG1 and
IGF2 by qRT-PCR using the housekeeping gene GAPDH as
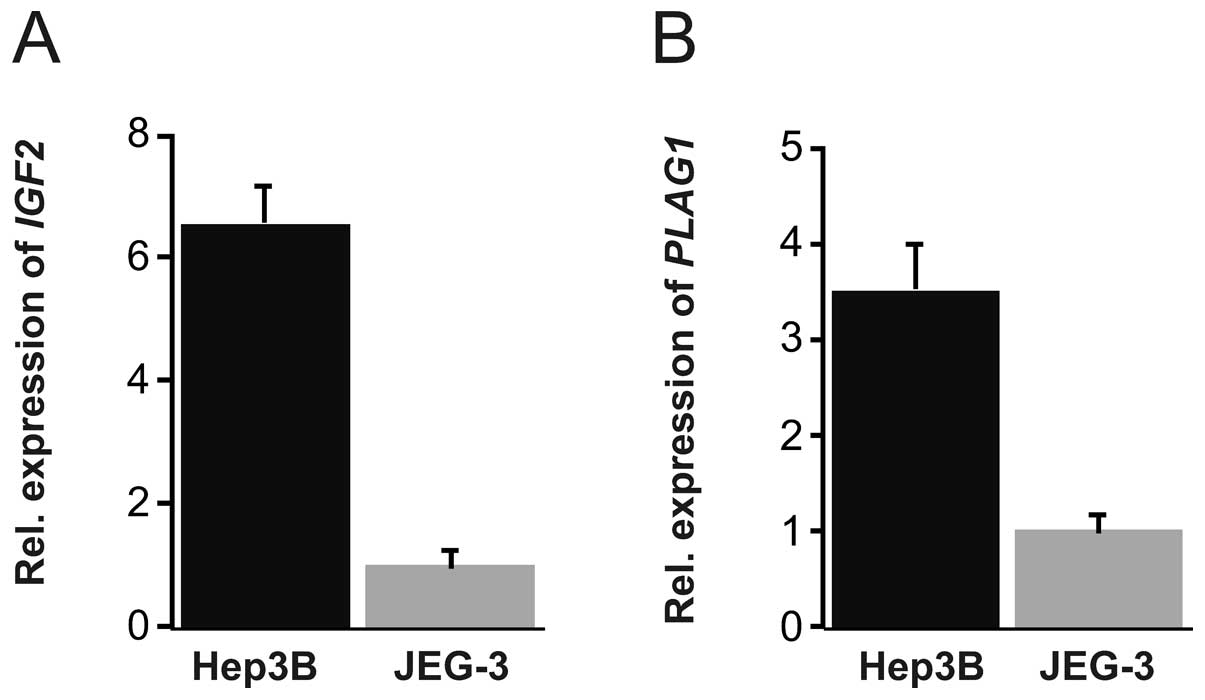
normalization factor. Substantially higher level of IGF2
(around 6-fold) was found in Hep3B as compared to JEG-3 cells
(Fig. 1A). PLAG1 expression
level was approximately 3 times higher in Hep3B compared to JEG-3
cells (Fig. 1B). We therefore
investigated whether PLAG1 expression may be an important
player for higher expression of IGF2 in Hep3B cells, and if
transient over-expression of PLAG1 could elevate IGF2
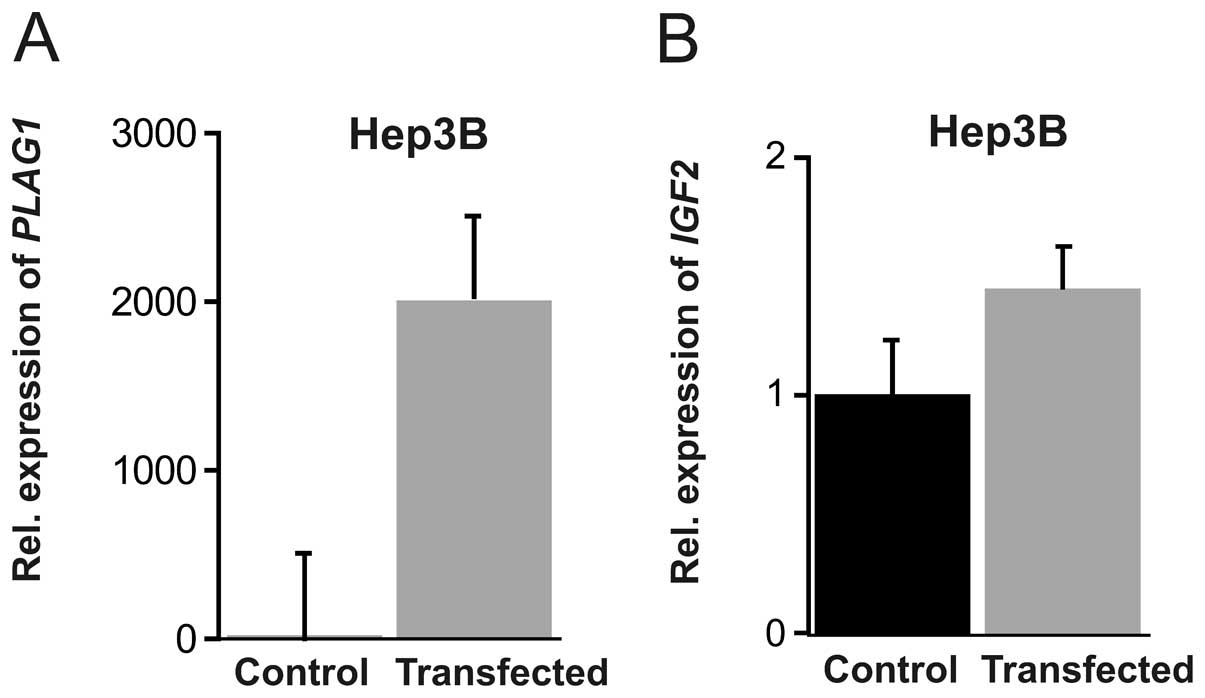
transcription. For this purpose we transfected Hep3B and JEG-3
cells with a PLAG1 expression plasmid. In Hep3B cells, a
monumental 2,000-fold induction of PLAG1 mRNA was achieved
(P=0.0026), together with a modest increase of IGF2
expression (Fig. 2). In JEG-3
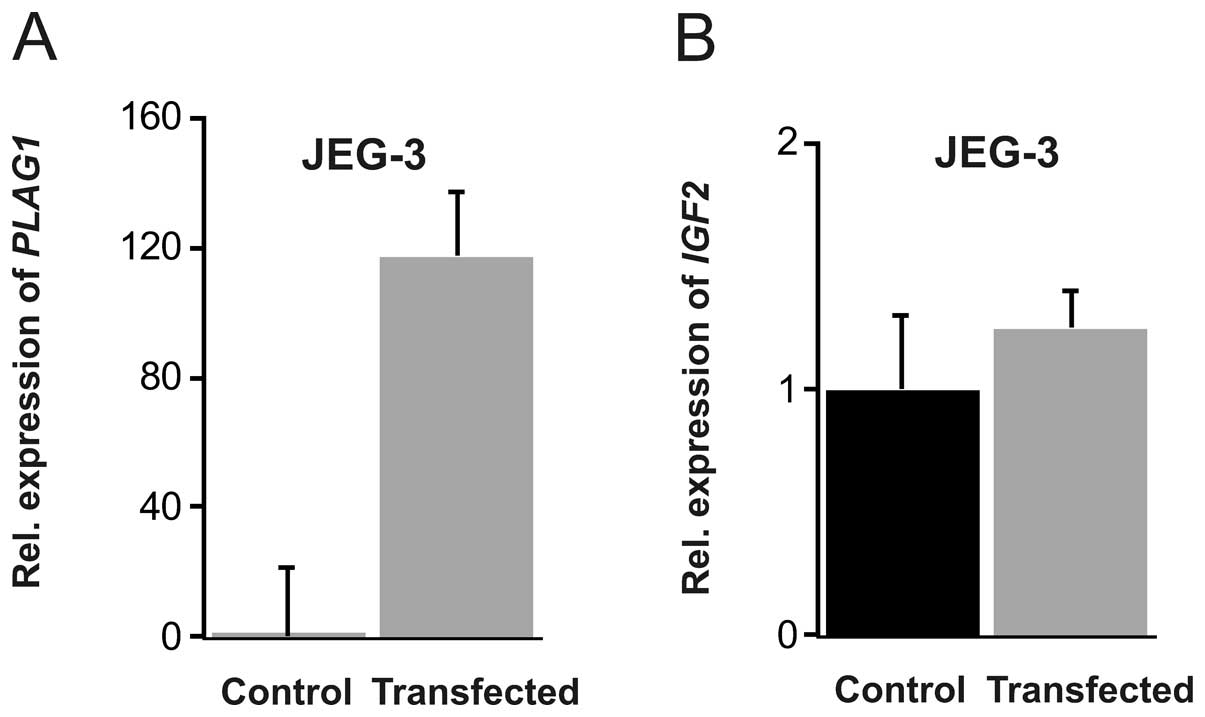
cells, the transfection was less efficient in comparison to Hep3B,
but a robust 130-fold increase in PLAG1 mRNA was achieved
(P=0.017). This did not, however, result in any enhanced
IGF2 expression (Fig.
3).
ChIP analysis
The finding that ectopic PLAG1 expression slightly
induced endogenous IGF2 expression in already high
IGF2 expressing Hep3B cells, but not in JEG-3 cells,
prompted us to analyze binding of PLAG1 to the IGF2 P3
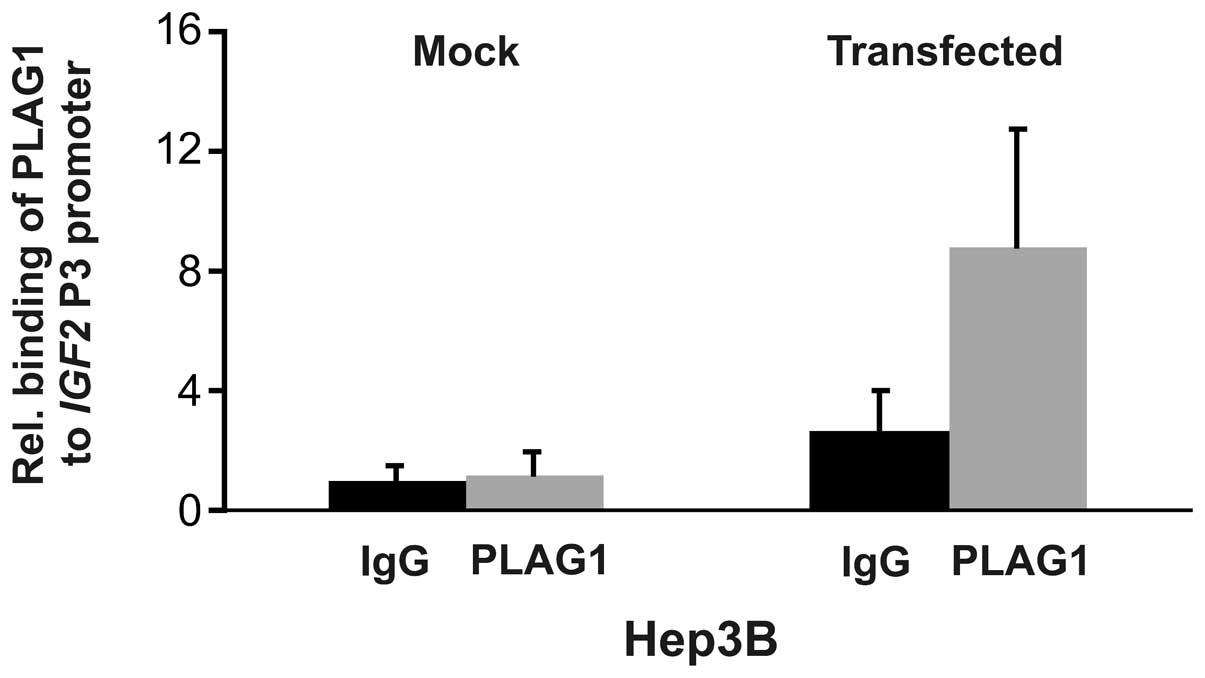
promoter using chromatin immunoprecipitation. Mock transfected
Hep3B cells did not display increased binding of PLAG1 to the P3
promoter. However, transient transfection with PLAG1 resulted in
increased binding (Fig. 4). The
situation for JEG-3 cells was dramatically different. No binding of
PLAG1 to the P3 promoter could be detected in non-transfected, or
PLAG1 transfected cells (data not shown), consistent with the low
endogenous IGF2 expression and the inability to boost this
by additional expression of PLAG1 (Fig. 3).
IGF2 P3 promoter reporter expression in
the presence of a functional insulator
Our results on the relatively weak and cell
type-specific endogenous relationship between IGF2
expression and PLAG1 binding to the P3 promoter, prompted us to
turn to a simpler system that allowed us to separate the P3
promoter from the important H19 3′-enhancers. We hypothesized that
Hep3B and JEG-3 cells have different sensitivity to the H19
upstream insulator in the regulation of IGF2 P3 promoter and
that PLAG1 might be involved in this.
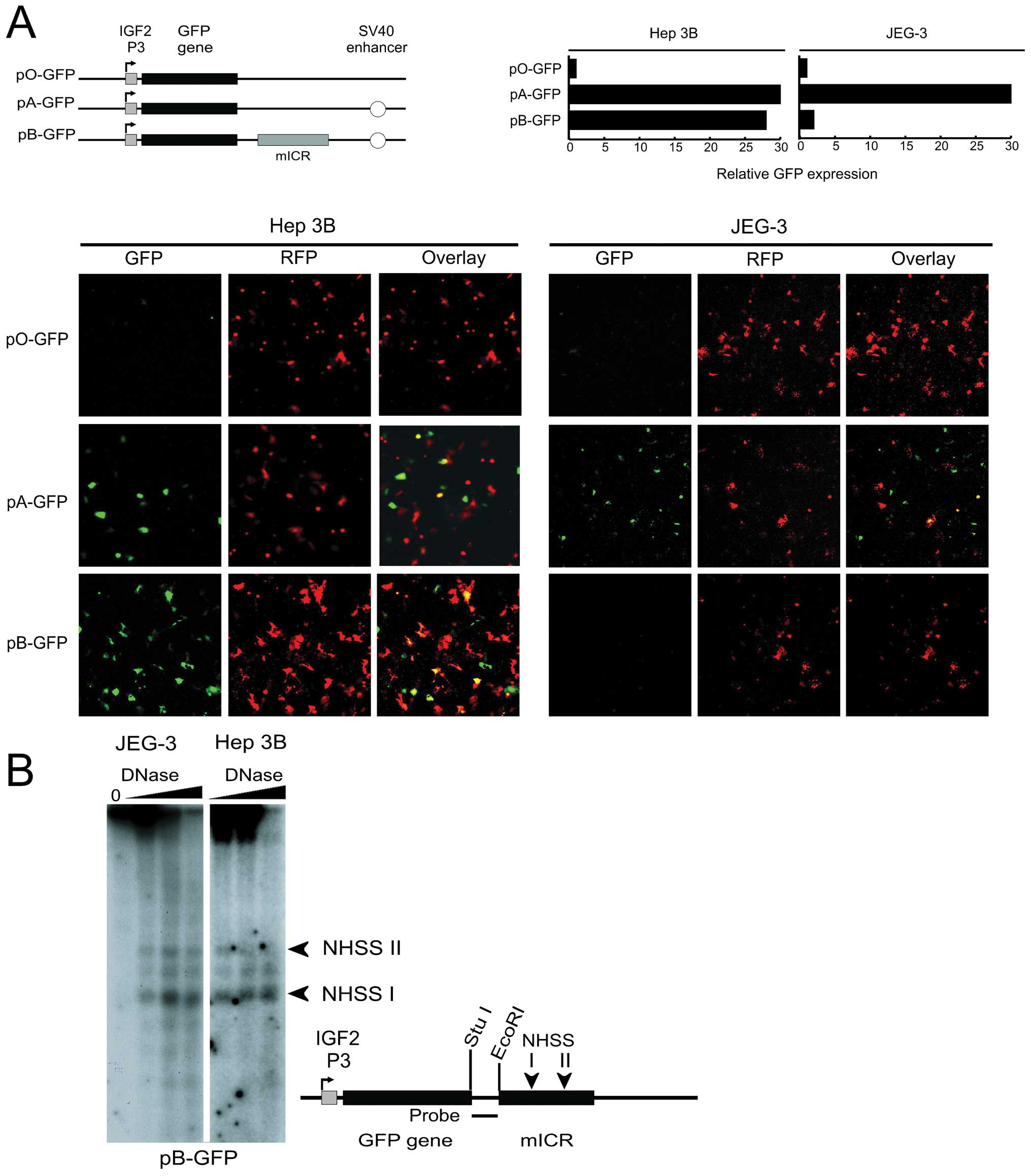
In order to investigate the insulator effect of the
ICR, we developed a GFP based assay system (Fig. 5A) where we could analyze the
effects of insulator function in real-time using confocal
microscopy as well as expression analysis using FACS. JEG-3 and
Hep3B cells were simultaneously transfected with the P3/GFP
episomal constructs (Fig. 5A) and
analyzed at 48h post-transfection. In JEG-3 cells the GFP
expression was low in the enhancer-free promoter construct, pO-GFP,
and substantially higher in the SV40 enhancer containing construct,
pA-GFP (Fig. 5A). The construct
which also contains an insulator, pB-GFP, showed an almost total
enhancer insulation of GFP expression, as expected (Fig. 5A). The Hep 3B cell line, however,
displayed a different expression pattern. As in JEG-3 cells, GFP
expression was low in the enhancer-less promoter construct pO-GFP
and robust in the enhancer-containing pA-GFP construct. However, in
contrast to JEG-3 cells, this high expression was not attenuated in
Hep3B for the insulator containing construct pB-GFP (Fig. 5A). The possible explanation that
the insulator was non-functional by the absence of binding by CTCF
at the ICR, was ruled out, since the chromatin conformations at the
CTCF target-sites of the transfected H19 ICR were virtually
identical, in JEG-3 cells and Hep3B cells (Fig. 5B).
To analyse if this aberrant expression of GFP was
promoter-dependent, the IGF2 P3 promoter was replaced with
the H19 promoter in the otherwise identical constructs. FACS
analyses verified that the insulator function was restored in Hep3B
with these constructs, suggesting that the loss of insulator
function is IGF2 P3 promoter-dependent (data not shown).
This finding is further supported by previous observations where
the ICR has been shown to be a potent insulator of the H19
promoter in Hep3B (23). Since it
was previously shown that PLAG1 can upregulate IGF2
expression and that binding to the P3 promoter could occur in an
EMSA (19), together with our
expression and ChIP results above, we hypothesized that PLAG1 might
be involved in the cell type-specific expression of IGF2
that involves the H19 ICR.
PLAG1 expression attenuates the insulator
function of the H19 ICR
Since the expression of PLAG1 was
significantly lower in JEG-3 than in Hep3B (Fig. 1B), and the GFP-reporter construct
displayed insulator activity in JEG-3 but not Hep3B cells, we
examined whether increased PLAG1 expression in JEG-3 would
result in a Hep3B-like situation with the GFP reporter construct.
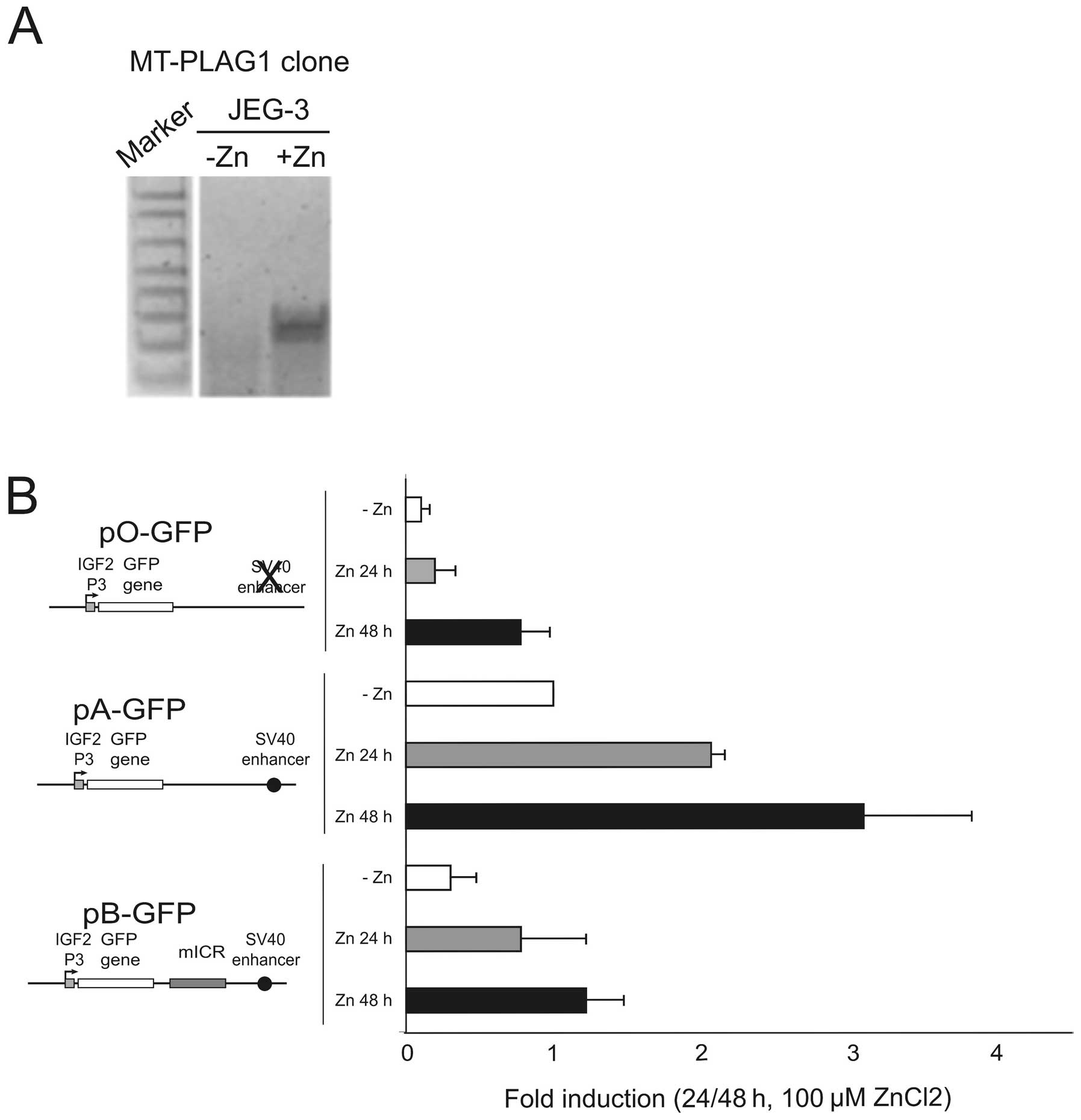
We created a zinc-inducible PLAG1 cell clone derived from
JEG-3 by the stable incorporation of the pSAR-PLAG1
construct (19). Several cell
clones were analysed and one zinc-inducible clone, but with
undetectable PLAG1 expression in the absence of
ZnCl2, was selected for the subsequent analyses. Upon
induction with ZnCl2 the level of PLAG1 expression
increased substantially in this clone (Fig. 6A). The episomal GFP constructs
(Fig. 5A) were co-transfected with
RFP control constructs into the PLAG1 inducible JEG-3 cell
line, followed by qRT-PCR analysis of GFP expression. Upon
Zn-induction of PLAG1, the expression of the GFP reporter
gene was considerably elevated in all constructs (Fig. 6B). By employing the enhancer- and
insulator-free construct (pO-GFP) it was shown that after 48 h of
induction, PLAG1 can elevate the levels of transcription from the
P3 promoter to levels similar to the enhancer driven construct
(pA-GFP) in low-PLAG1 expressing cells. In the presence of
the enhancer, PLAG1-induction dramatically increased the P3 driven
GFP expression. Interestingly, PLAG1 could also induce high
expression in the insulator-containing pB-GFP construct. Although
the insulator still appears to maintain an ability to attenuate
enhancer-promoter interaction in the presence of PLAG1
overexpression (Fig. 6B, pB-GFP
construct, Zn 48 h), the level of this expression is higher than
both the induced pO-GFP construct (Fig. 6B, +Zn 48 h), and more importantly,
even from the non-induced enhancer-containing construct lacking an
insulator, pA-GFP (Fig. 6B, −Zn).
Thus, after 48 h of PLAG1 induction, the levels of P3-driven
GFP in the presence of insulator slightly supersede
non-induced cells containing the insulator-free construct. This
finding suggests that the stringency of the system normally
regulated by insulator-action is partially lost in JEG-3 cells when
PLAG1 is overexpressed.
Discussion
In the current study we report on a novel aspect of
cell line specific IGF2 regulation involving the PLAG1
transcription factor, its binding to the P3 promoter region, and
the possible consequence of PLAG1 expression in the context
of promoter/enhancer interaction. Although, it has been shown
previously by EMSA that the IGF2 P3 promoter contains PLAG1
consensus-binding sites (19), the
binding of PLAG1 to the IGF2 P3 promoter in live cells has
not been reported before. We therefore investigated this by ChIP
analysis. The JEG-3 and Hep3B cells were analyzed in order to
verify the notion that PLAG1 is important for activity of the
IGF2 P3 promoter. We show here that PLAG1
overexpression only increase binding of PLAG1 to the P3 promoter in
Hep3B cells, leading to moderate increase of the endogenous
IGF2 expression in Hep3B, while neither promoter binding nor
expression were affected in JEG-3 cells. On the other hand, PLAG1
stimulated P3 promoter driven GFP transcription in JEG-3 cells as
well as overcame, at least in part, the H19 insulator in a
GFP-plasmid reporter system.
Transcriptional insulators are specialized
cis-acting elements that isolate promoters from positive and
negative influences by distal enhancers. The H19 upstream
insulator is also an ICR, which regulates the allele-specific
transcription of the IGF2 gene in a parent of
origin-dependent manner. This is achieved by methylation of the ICR
on the paternal allele leading to hindrance of the insulator
protein CTCF and loss of communication between the shared
H19/IGF2 enhancer downstream of H19 and the
IGF2 promoter(s) (12).
Methylation of the ICR also spread into the H19 promoter
leading to transcriptional silencing. Conversely, the unmethylated
ICR on the maternal allele binds CTCF and then functions as an
enhancer-blocking insulator that prevents expression of the
maternal IGF2 allele.
Our findings suggest a new role for PLAG1 in
IGF2 overexpression in cancer. Not only does PLAG1
expression affect the activity of the promoter, it also appears to
increase the effect of enhancer activated transcription. The
function of PLAG1 as a regular transcription factor and
proto-oncogene has been reported previously, and there is previous
evidence to show that it binds to a consensus binding site in the
IGF2 P3 promoter and activates its transcription in
vitro. We propose, however, that PLAG1 has additional, more
complex functions, one of which may be to act as a
promoter/enhancer facilitator. The reporter constructs used in this
study were designed to interrogate the influence of insulator
function of the H19 ICR on IGF2 P3 promoter activity
in cells with different levels of PLAG1 expression. We
observed that JEG-3 cells are relying on an exogenous PLAG1
expression to overcome the silencing effect of the insulator in the
construct, although they do transcribe low levels of PLAG1
mRNA. Hep3B cells on the other hand, which have a considerably
higher endogenous PLAG1 expression, are already insensitive
to the insulator. In this connection, it is important to note that
the chromatin structure of the ICR in the episomal construct, was
found to be similar in both JEG-3 and Hep3B cells (Fig. 5B), indicating presence of binding
by CTCF, and thus promoting insulation, in both cases. In this
scenario the induction of PLAG1 was apparently able to partially
overcome an active insulation since the ICR containing construct
(pB-GFP) showed a substantially higher GFP expression than
the enhancer-free and ICR-free construct (pO-GFP), after PLAG1
induction (Fig. 6B). The inability
to completely overcome insulation was evident, however, since
PLAG1 expression in the ICR containing construct did not
reach the GFP expression level of the ICR-less/enhancer
containing construct (pA-GFP).
It is unknown why the low endogenous PLAG1
expression in JEG-3 cells does not affect the expression of the GFP
reporter. It is possible that a threshold concentration of PLAG1 is
required to push a transcription factor complex towards
assembly.
Considering that in the GFP reporter construct,
PLAG1 is able to influence the IGF2 P3 promoter activity in
JEG-3 cells, it is challenging to understand why the functional
response to PLAG1 is different from that in the endogenous P3
promoter. Maybe PLAG1 binding in the endogenous IGF2 P3
promoter requires additional factors, absent in JEG-3 cells but
present in Hep3B. Although we did demonstrate that highly
overexpressed PLAG1 actually increased binding to endogenous P3 in
Hep3B cells, only moderate activation of IGF2 transcription
was achieved and non-transfected cells did not demonstrate
substantial endogenous PLAG1 binding to the P3 promoter suggesting
that other factors, not excluding chromatin structure, are
important for the endogenous IGF2 transcription. This was in
contrast to the JEG-3 cells where neither binding nor activation
could be demonstrated despite substantial PLAG1 expression. These
cells are apparently not geared at all for PLAG1 directed
IGF2 expression, and since the GFP-reporter construct was
PLAG1 inducible, chromatin structure in the endogenous gene may be
an important factor. The lack of additional IGF2 activation
in Hep3B cells after PLAG1 overexpression may also be due to
an already saturated promoter by other permissive transcription
factors, or lack of required additional factors. The PLAG1 protein
may also be sumoylated and thus inactivated, as was shown by others
(25). To our knowledge, whether
or not sumoylation of PLAG1 influences its binding to the
IGF2 P3 promoter has not been demonstrated. It is also
possible that overexpression of PLAG1 does not stimulate the
promoter P3-dependent transcription of IGF2 in JEG-3 cells,
due to methylation mediated inactivation of the P3 promoter or the
H19 DMR. These questions are currently being analyzed.
Although it would be a possibility that PLAG1
interferes with allele-specific expression of IGF2 by
interfering with the ICR, we did not study this issue here. In
addition, Declercq et al concluded that only the paternal
Igf2 allele is involved in the PLAG1 induced formation of
pleomorphic adenomas of the salivary glands, in a transgenic mouse
model (26). This suggests that
although PLAG1 in certain contexts may interefere with the H19 ICR,
it is not involved in the imprinting mechanism.
In conclusion, we show here that the role of PLAG1
in activation of IGF2 is context-specific in that it readily
activates an episomal reporter driven by the IGF2 P3
promoter, it is cell type-specific in its sensitivity to promoter
enhancer insulation and in the endogenous context, and that both
binding to the IGF2 P3 promoter and endogenous transcription
activation is cell type-specific. We also suggest that PLAG1 may
act as a facilitator since it partially overcomes the insulator
function of the H19 DMR, at least in vitro.
Acknowledgements
This study was financially supported
by the Swedish Cancer Society, the Swedish Research Council, the
Swedish Society for Medical Research, the Stockholm County Council,
and funds from Karolinska Institutet. We would like to greatly
acknowledge Marianne Voz for the Zn-inducible PLAG1 plasmid
construct, and Katharina Wimmer for the PLAG1 expression
vector.
References
|
1.
|
D LeRoithCT Roberts JrThe insulin-like
growth factor system and cancerCancer
Lett195127137200310.1016/S0304-3835(03)00159-912767520
|
|
2.
|
H CuiLoss of imprinting of IGF2 as an
epigenetic marker for the risk of human cancerDis
Markers23105112200710.1155/2007/36346417325430
|
|
3.
|
H CuiM Cruz-CorreaFM GiardielloLoss of
IGF2 imprinting: a potential marker of colorectal cancer
riskScience29917531755200310.1126/science.108090212637750
|
|
4.
|
AP FeinbergH CuiR OhlssonDNA methylation
and genomic imprinting: insights from cancer into epigenetic
mechanismsSemin Cancer
Biol12389398200210.1016/S1044-579X(02)00059-712191638
|
|
5.
|
R OhlssonC KanduriJ WhiteheadS PfeiferV
LobanenkovAP FeinbergEpigenetic variability and the evolution of
human cancerAdv Cancer
Res88145168200310.1016/S0065-230X(03)88306-912665055
|
|
6.
|
MJ SteenmanS RainierCJ DobryP GrundyIL
HoronAP FeinbergLoss of imprinting of IGF2 is linked to reduced
expression and abnormal methylation of H19 in Wilms’ tumourNat
Genet743343919947920665
|
|
7.
|
H CuiIL HoronR OhlssonSR HamiltonAP
FeinbergLoss of imprinting in normal tissue of colorectal cancer
patients with microsatellite instabilityNat
Med412761280199810.1038/32609809551
|
|
8.
|
CR KafferM SrivastavaKY ParkA
transcriptional insulator at the imprinted H19/Igf2 locusGenes
Dev1419081919200010921905
|
|
9.
|
C HolmgrenC KanduriG DellCpG methylation
regulates the Igf2/H19 insulatorCurr
Biol1111281130200110.1016/S0960-9822(01)00314-111509237
|
|
10.
|
S KurukutiVK TiwariG TavoosidanaCTCF
binding at the H19 imprinting control region mediates maternally
inherited higher-order chromatin conformation to restrict enhancer
access to Igf2Proc Natl Acad Sci
USA1031068410689200610.1073/pnas.0600326103
|
|
11.
|
V PantP MarianoC KanduriThe nucleotides
responsible for the direct physical contact between the chromatin
insulator protein CTCF and the H19 imprinting control region
manifest parent of origin-specific long-distance insulation and
methylation-free domainsGenes Dev17586590200310.1101/gad.254903
|
|
12.
|
YS YoonS JeongQ RongKY ParkJH ChungK
PfeiferAnalysis of the H19ICR insulatorMol Cell
Biol2734993510200710.1128/MCB.02170-0617339341
|
|
13.
|
R NativioKS WendtY ItoCohesin is required
for higher-order chromatin conformation at the imprinted IGF2-H19
locusPLoS
Genet5e1000739200910.1371/journal.pgen.100073919956766
|
|
14.
|
H CuiEL NiemitzJD RavenelLoss of
imprinting of insulin-like growth factor-II in Wilms’ tumor
commonly involves altered methylation but not mutations of CTCF or
its binding siteCancer Res61494749502001
|
|
15.
|
GA UlanerTH VuT LiLoss of imprinting of
IGF2 and H19 in osteosarcoma is accompanied by reciprocal
methylation changes of a CTCF-binding siteHum Mol
Genet12535549200310.1093/hmg/ddg03412588801
|
|
16.
|
H CuiP OnyangoS BrandenburgY WuCL HsiehAP
FeinbergLoss of imprinting in colorectal cancer linked to
hypomethylation of H19 and IGF2Cancer Res6264426446200212438232
|
|
17.
|
D TakaiFA GonzalesYC TsaiMJ ThayerPA
JonesLarge scale mapping of methylcytosines in CTCF-binding sites
in the human H19 promoter and aberrant hypomethylation in human
bladder cancerHum Mol
Genet1026192626200110.1093/hmg/10.23.261911726548
|
|
18.
|
K KasML VozE RoijerPromoter swapping
between the genes for a novel zinc finger protein and beta-catenin
in pleiomorphic adenomas with t(3;8)(p21;q12) translocationsNat
Genet15170174199710.1038/ng0297-1709020842
|
|
19.
|
ML VozNS AgtenWJ Van de VenK KasPLAG1, the
main translocation target in pleomorphic adenoma of the salivary
glands, is a positive regulator of IGF-IICancer
Res60106113200010646861
|
|
20.
|
A ZatkovaJM RouillardW
HartmannAmplification and overexpression of the IGF2 regulator
PLAG1 in hepatoblastomaGenes Chromosomes
Cancer39126137200410.1002/gcc.1030714695992
|
|
21.
|
A AstromES D’AmoreL SainatiEvidence of
involvement of the PLAG1 gene in lipoblastomasInt J
Oncol1611071110200010811981
|
|
22.
|
SF LandretteYH KuoK HensenPlag1 and Plagl2
are oncogenes that induce acute myeloid leukemia in cooperation
with
Cbfb-MYH11Blood10529002907200510.1182/blood-2004-09-363015585652
|
|
23.
|
C KanduriC HolmgrenM PilartzThe 5′ flank
of mouse H19 in an unusual chromatin conformation unidirectionally
blocks enhancer-promoter communicationCurr Biol104494572000
|
|
24.
|
JA DahlP CollasQ2ChIP, a quick and
quantitative chromatin immunoprecipitation assay, unravels
epigenetic dynamics of developmentally regulated genes in human
carcinoma cellsStem
Cells2510371046200710.1634/stemcells.2006-0430
|
|
25.
|
F Van DyckEL DelvauxWJ van de VenMV
ChavezRepression of the transactivating capacity of the oncoprotein
PLAG1 by SUMOylationJ Biol Chem2793612136131200415208321
|
|
26.
|
J DeclercqF van DyckB van DammeWJ van de
VenUpregulation of Igf and Wnt signalling associated genes in
pleomorphic adenomas of the salivary glands in PLAG1 transgenic
miceInt J Oncol3210411047200818425330
|