Introduction
Pancreatic adenocarcinoma is one of the most lethal
and poorly understood human malignancies. Because of the lack of
effective systemic therapies the 5-year survival rate for patients
with pancreatic adenocarcinoma has remained at 1–3% without a
change over the past 25 years (1,2). To
date, the only potential curative means is surgical resection, of
which only 20% of patients are eligible. Alternative therapies,
such as radiotherapy and chemotherapy remain largely ineffective.
Therefore, the development and evaluation of novel targeted
therapeutic agents that reduce the intrinsic drug resistance of
this disease poses one of the greatest challenges in pancreatic
cancer research and other intractable cancers.
AMP-activated protein kinase (AMPK), a
serine/threonine kinase, is a highly conserved sensor of cellular
energy status in eukaryotes and is widely known as a regulator of
cell metabolism (3). AMPK is a
heterotrimeric protein consisting of a catalytic α-subunit and
regulatory β-/γ-subunits (4,5). It
is phosphorylated at Thr172 in response to an increase in the ratio
of AMP-to-ATP within its activation domain of α-subunit by upstream
kinases LKB1 (6–8) and calmodulin-dependent protein kinase
kinase β (CaMKKβ) (9–11). Several previous studies show that
excessive AMPK activation by treatment of AMPK activator (such as
Metformin, 5-aminoimidazole-4-carboxamide riboside (AICAR) or
A769662) inhibits the growth and/or survival of various cancer cell
lines (12–19). Moreover, BML-275 (compound C), a
potent, selective, and reversible ATP-competitive inhibitor of AMPK
induces cell death in various types of cancers including myeloma,
glioma, prostate and breast carcinoma cells (20–23).
In addition, inhibition of AMPK pathway by compound C sensitizes
apoptosis by co-treatment with tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL), doxorubicin or cisplatin in
human renal, leukemia, gastric carcinoma, colon carcinoma, and
cervix adenocarcinoma cell lines (24–26).
Therefore, pharmacological inhibition of AMPK activity might be
potentially useful in therapy of human solid tumors. However, the
effect of AMPK inhibition of pancreatic cancer cell proliferation
or survival has not been investigated.
Cell cycle deregulation resulting in uncontrolled
cell proliferation is the one of the most frequent alterations that
occurs during tumor development (27) and targeting of cell cycle
progression and/or machinery is effective strategy to control
aberrant proliferation of cancer cell (28,29).
There are two major checkpoints, G1/S and G2/M checkpoints, are
known to regulate the cell cycle. The G2/M checkpoint plays a key
role in the maintenance of chromosomal integrity by allowing cells
to repair DNA damage before entering mitosis. A key regulator of
the cell cycle at G2/M checkpoint is cyclin dependent kinase 1
(CDK1), especially cell division cycle 2 (Cdc2). Cdc2 activation
depends on the dephosphorylation of Tyr15 by Cdc25C (30). In addition, Cdc2 can be further
regulated by GADD45 and 14-3-3 by p53 pathway (31). Reactive oxygen species (ROS)
generation causes oxidative stress and has been shown to
significantly function to controlling cancer cell survival
(32). Oxidation of DNA bases and
breakage of DNA strand may occurs as results of oxidative DNA
damage and parts of these lesions are converted to DNA
double-strand breaks (33–35). BML-275 was reported to induce cell
cycle arrest at G2/M-phase and ROS generation in U251 glioma cells
(22). Therefore, understanding
the molecular mechanisms of BML-275 to sensitize these cells to
undergo BML-275-mediated G2/M arrest and apoptosis is an important
issue for effective cancer therapy.
In this study, we performed experiments to determine
anti-tumor effect(s) by BML-275 in human pancreatic cancer cell
lines. Our results suggest that BML-275 regulates cell survival via
targeting AMPK and generating ROS in multiple human pancreatic
cancer cells.
Materials and methods
Cell culture and reagents
MIA PaCa-2, Panc-1, CFPAC-1 and BxPC-3 cells were
purchased from American Type Culture Collection (ATCC, Manassas,
VA, USA) and AsPC-1, Capan-1 and Colo-357 cells were obtained from
Tissue Culture Shared Resource of Georgetown University Lombardi
Comprehensive Cancer Center (Washington, DC, USA). Immortal human
pancreatic ductal epithelial cells, HPDE6-C7 were acquired from Dr
M.S. Tsao (36). AsPC-1, BxPC-3,
Capan-1 and Colo-357 cells were cultured in RPMI-1640 media
supplemented with fetal bovine serum (FBS; 20% for AsPC-1, 10% for
Colo-357, Capan-1 and BxPC-3 cells), 100 U/ml penicillin, 100
μg/ml streptomycin and 1% sodium pyruvate. MIA PaCa-2 cells
were cultured in Dulbecco’s modified Eagle’s medium (DMEM)
containing 10% FBS, 2.5% horse serum (HS), 100 U/ml penicillin and
100 μg/ml streptomycin. Panc-1 and CFPAC-1 cells were
cultured in DMEM containing 10% FBS, 10 U/ml penicillin and 10
μg/ml streptomycin. HPDE6-C7 cells were cultured in
keratinocyte serum-free (KSF) medium supplemented by an epidermal
growth factor and bovine pituitary extract and 1X
antibiotic-antimycotic. Cell culture reagents were purchased from
BioWhittaker (Walkersville, MD, USA) and Invitrogen (Carlsbad, CA,
USA). BML-275 was purchased from Tocris Bioscience (Ellisville, MO,
USA), and A769662 was obtained from LC Laboratories (Woburn, MA,
USA).
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
A total of 2,000 human pancreatic cancer cells,
counted by the Luna Cell Counter (Logos Biosystems, Gyeonggi-Do,
Korea) were plated in 96-well flat-bottom plates and then exposed
to test the effects of BML-275 in various concentrations. At the
indicated times, 10 μl of 1 mg/ml MTT (Sigma, St. Louis, MO,
USA) in PBS was added to each well for 4 h. After centrifugation
and removal of the medium, 150 μl of DMSO (Sigma) was added
to each well to dissolve the formazan crystals. The absorbance was
measured at 560 nm using an ELx808 Absorbance Microplate Reader
(BioTek Instruments Inc., Winooski, VT, USA). Absorbance of
untreated cells was designated as 100% and cell survival was
expressed as a percentage of this value. Triplicate wells were
assayed for each condition and standard deviation (SD) was
determined.
Western blot (WB) analysis
Cells were grown to ∼70% confluence and reagents
were added at the indicated concentrations. After exposure to
BML-275 alone or in combination with NAC, cells were lysed in cell
lysis buffer containing 20 mM Tris-HCl, 0.5 M NaCl, 0.25% Triton
X-100, 1 mM EDTA, 1 mM EGTA, 10 mM β-glycophosphate, 10 mM NaF, 300
μM Na3VO4, 1 mM benzamidine, 2
μM PMSF and 1 mM DTT. Protein concentrations were determined
by a BCA protein assay kit (Thermo Scientific, Rockford, IL, USA).
Proteins were separated by SDS-PAGE, transferred on to PVDF
membranes, blocked in 1X blocking buffer (Sigma) and probed with
the following primary antibodies: phospho-ACC (S79), ACC,
phospho-AMPKα (T172), AMPK, phospho-ATM (S1981), phospho-CHK2
(T68), phospho-Histone H2A.X (S139), XIAP and Survivin (Cell
Signaling Technology, Boston, MA), Bcl2 and
Poly-ADP-Ribose-Polymerase (PARP; BD Biosciences, Franklin, NJ,
USA) and α-tubulin (Sigma). Then, the membranes were incubated with
horseradish peroxidase (HRP)-conjugated secondary antibodies
(Sigma) and visualized with a chemiluminescence kit (Santa Cruz
Biotechnology, Santa Cruz, CA, USA) according to the manufacturer’s
recommended protocol and exposed with X-ray film (American X-ray
and Medical Supply, Jackson, CA, USA).
Clonogenic assay
Human pancreatic cancer cells (4x105
cells) were seeded in 60-mm dishes. Twenty-four hours after
plating, varying concentrations of the drugs, either as a single
agent or in combination, were added to the dishes. After treatment,
cells (2,000 cells) were re-seeded in 60-mm dishes (triplicate).
Each culture dish was incubated for 14 days and photographed after
staining with 0.5% crystal violet in 1X PBS including 25% methanol.
Colonies were examined under a light microscope and counted after
capturing images by scanner. Colony numbers were calculated
according to the percentage of the untreated cells (37).
Flow cytometry
Human pancreatic cancer cell lines were collected
after treatment of BML-275 by trypsinization, washed with PBS and
fixed overnight in 70% ethanol at −20°C. Cells were incubated with
20 μg/ml propidium iodide and 40 μg/ml RNase A in 1X
PBS. Cells were analyzed on a FACSCalibur flow cytometer (Becton
Dickinson, San Jose, CA, USA) at the Flow Cytometry and Cell
Sorting Shared Resource, Georgetown University Lombardi
Comprehensive Cancer Center. The acquired data were analyzed by
Cell Quest Pro Analysis software (Becton Dickinson).
Small interfering RNA (siRNA)
For the RNA interfering experiment, AMPKα-siRNA,
5′-CUGAGUUGCAUAUACUGUA-3′ and control-siRNA,
5′-GACGAGCGGCACGUGCACA-3′ were purchased from Bioneer (Daejeon,
Korea). AMPKα-siRNA or control-siRNA were transfected into MIA
PaCa-2 and Panc-1 cells using Lipofectamine 2000 (Invitrogen)
according to the manufacturer’s procedure. After 48 h transfected
cells were processed for cell cycle analysis, WB analysis and
measurement of ROS generation.
ROS generation
For measurement of ROS generation, human pancreatic
cancer cell lines were treated with BML-275 with or without
N-acetyl cysteine (NAC) for the indicated times and then loaded
with 50 μM 2′, 7′-dichlorofluorescin diacetate (DCFDA;
Molecular Probes, Eugene, OR, USA) and 0.5 μg/ml Hoechst
33342 (HO; Sigma) for 30 min. After rinsing, fluorescent images
were taken with fluorescence intensities were obtained with a
Fluorocount at excitation/emission wavelengths of 490/530 nm
(DCFDA) and 340/425 (HO), and values of ROS generation were
obtained by determining the ratio of DCFDA/HO signals per well.
Statistical methods
Statistical comparisons were made using the
two-tailed Student’s t-test where appropriate. Results were
considered significant in at means *P<0.05,
**P<0.01 and ***P<0.005. Data were
expressed as the mean ± SD.
Results
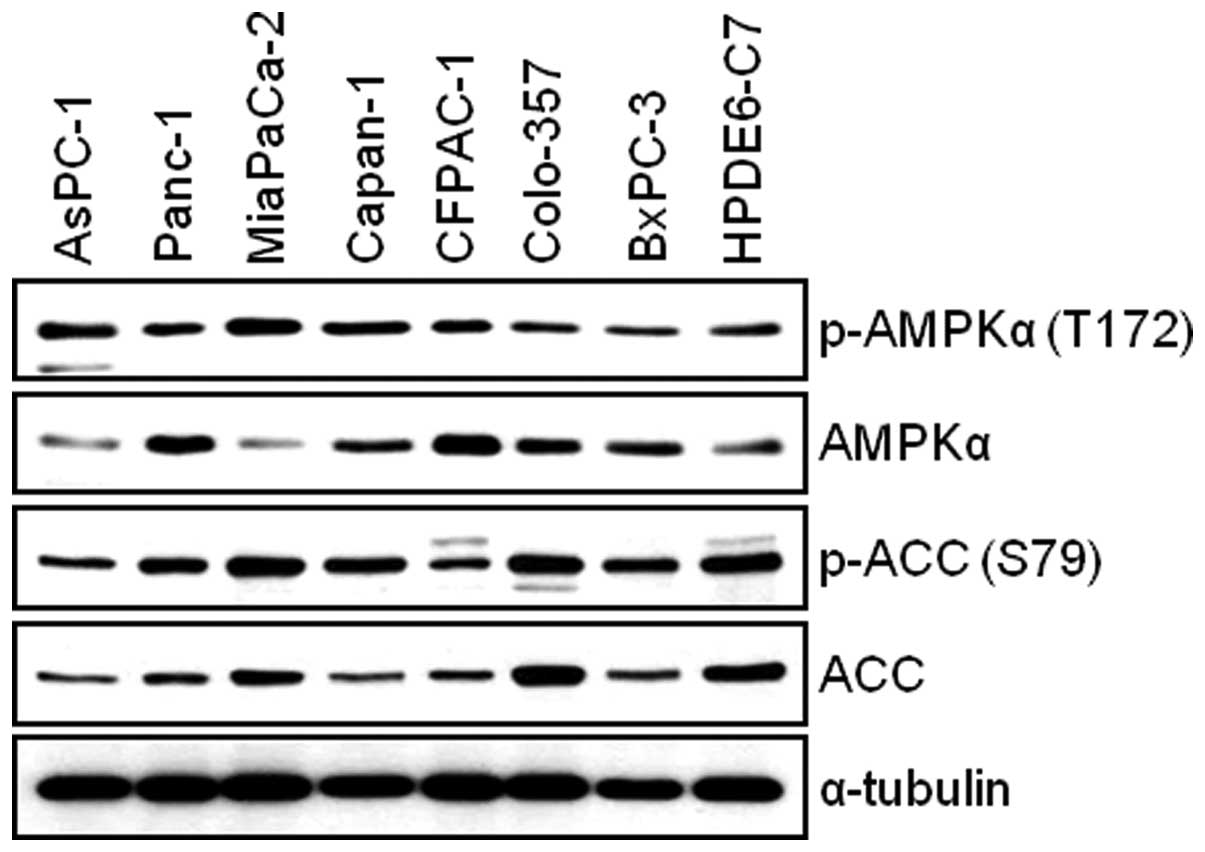
Human pancreatic cancer cells and
immortal human pancreatic duct epithelial cells express AMPKα
We first examined the total and phosphorylated form
of AMPKα in AsPC-1, Panc-1, MIA PaCa-2, Capan-1, CFPAC-1, Colo-357,
BxPC-3 and HPDE6-C7 cells. The WB result reveals that all
pancreatic cell lines used for this study expressed the levels of
both phosphorylated-AMPK and total-AMPK (Fig. 1). Next, we investigated the
expression level of AMPK target protein, Acetyl-CoA Carboxylase
(ACC). We found that there is relatively good correlation between
the levels of phosphorylated-AMPK and phosphorylated-ACC among
pancreatic cell lines (Fig. 1). To
study the antitumor effect(s) by BML-275, AMPK inhibitor, in
pancreatic cancer cells, we chose four pancreatic cancer cell
lines: MIA PaCa-2, Panc-1, Colo-357 and AsPC-1 for further
studies.
BML-275 induces apoptotic cell death
BML-275 is a potent ATP-mimetic competitive
inhibitor of AMPK. In order to explore the antitumor effect(s) by
BML-275, MIA PaCa-2, Panc-1, Colon-357 and AsPC-1 cells were
treated with different concentrations of BML-275 (0, 1, 3, 5 and/or
10 μM) for 48 h and cell viability were measured by MTT
assay. BML-275 inhibited cell survival in dose-dependent manner
(Fig. 2A). Next, we performed
clonogenic assay to determine the long-term growth inhibitory
effect of BML-275. Cells were treated with various concentrations
of BML-275 (0, 1, 3, 5 and/or 10 μM) for 1 day and
continuously cultured in fresh media for 14 days and colony
formation was measured by clonogenic assay. BML-275 significantly
inhibited colony formation in dose-dependent manner and at 10
μM BML-275 all of the tested pancreatic cancer cells showed
susceptibility to the AMPK inhibitor (Fig. 2B). MIA PaCa-2 cells showed
increased sensitivity to BML-275 and Panc-1 cells showed relatively
less sensitive to BML-275 among four pancreatic cancer cell lines
tested (Fig. 2A and B).
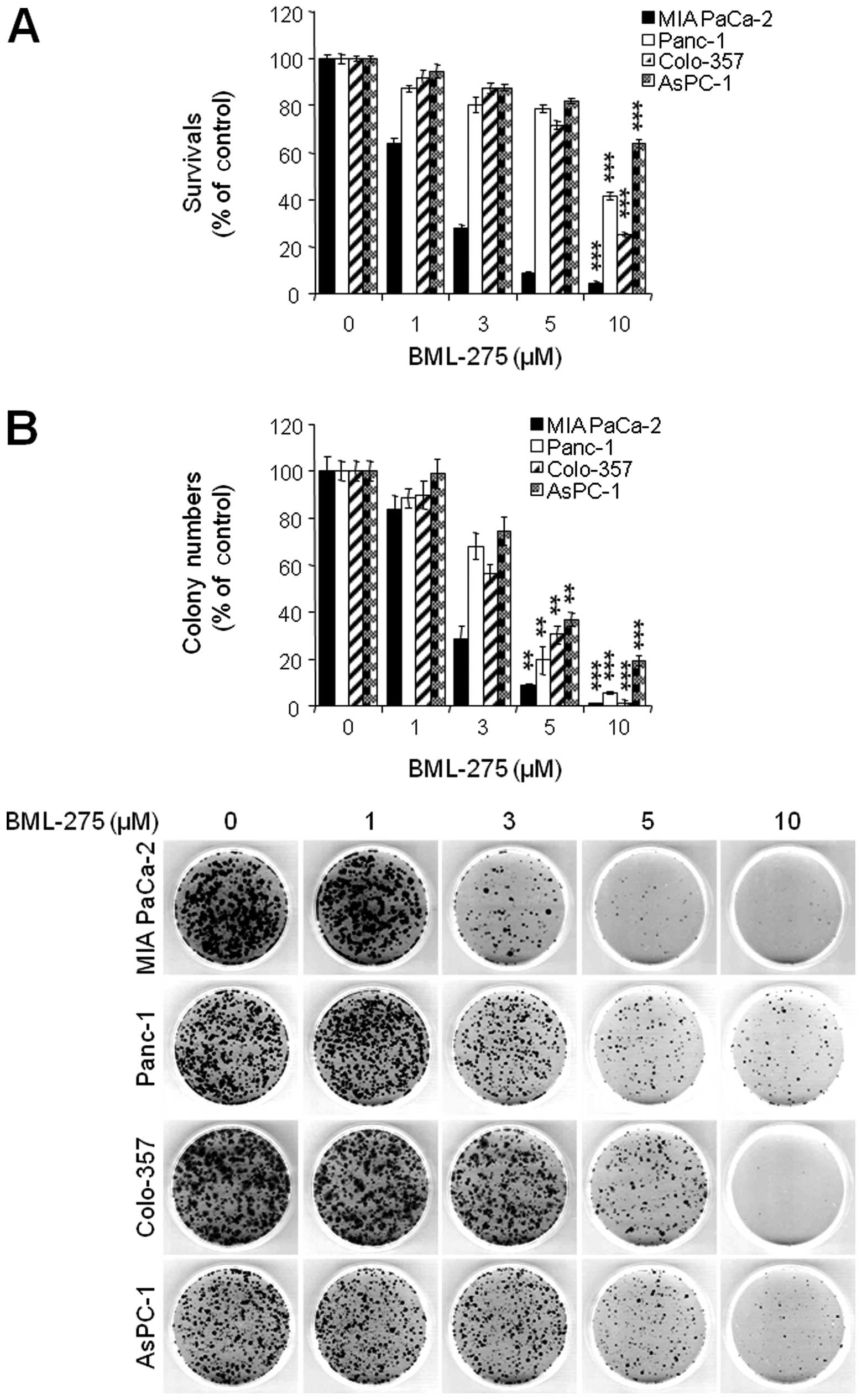 | Figure 2BML-275 inhibits cell viability in
human pancreatic cancer cells. (A) An MTT assay of MIA PaCa-2,
CFPAC-1, Panc-1 and AsPC-1 cells treated with various
concentrations of BML-275 (0, 1, 3, 5 and/or 10 μM) for 48 h
were used to determine cell viability. Error bars represent the
standard deviation. ***Represents statistically
significant difference with p-value <0.005 between 10 μM
BML-275 treated group and control group. (B) A clonogenic assay of
MIA PaCa-2, CFPAC-1, Panc-1 and AsPC-1 cells treated with BML-275
(0, 1, 3, 5 and/or 10 μM) for 24 h was used to determine the
long-term response. Colony numbers were counted and calculated as a
relative percentage (%) of the untreated control cells (upper) and
representative photograph of clonogenic assay results are shown
(lower). Experiments were repeated 3 times and similar results were
obtained. Error bars represent the standard deviation.
**Represents statistically significant difference with
p-value <0.01 between 5 μM BML-275 treated group and
control group and *** represents statistically
significant difference with p-value <0.005 between 10 μM
BML-275 treated group and control group. |
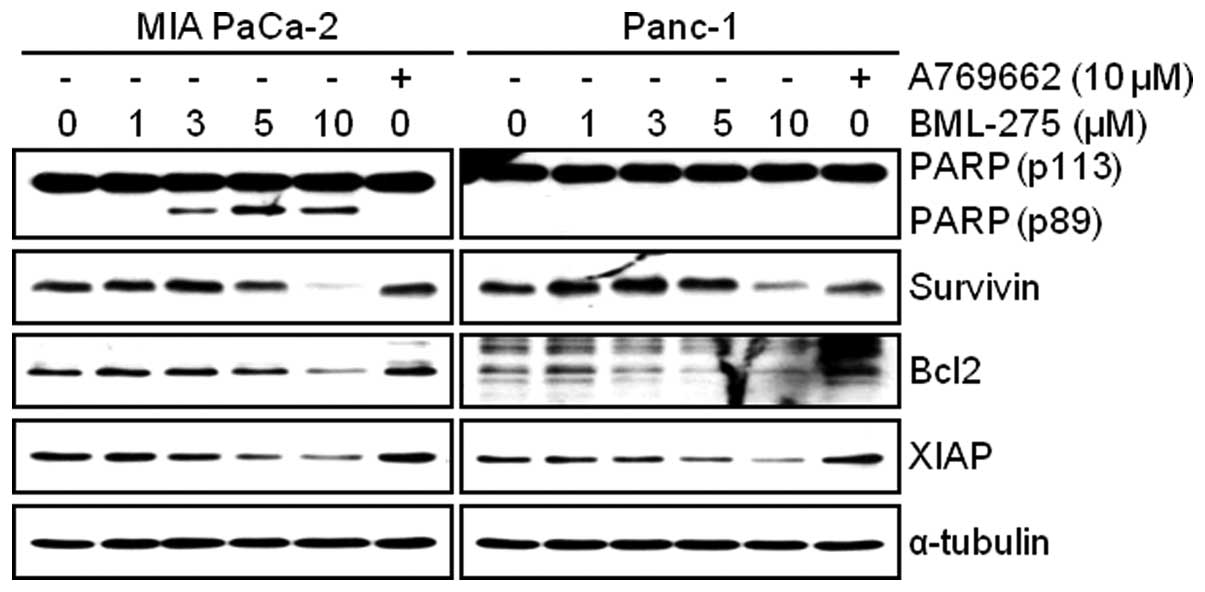
To investigate mechanism of apoptosis by BML-275
treatment, MIA PaCa-2 and Panc-1 cells were treated with various
concentrations of BML-275 or 10 μM A769662 for 24 h.
Apoptotic cell death was detected by WB analysis of a molecular
biomarker of apoptosis, PARP cleavage. On the contrary to cells
treated with A769662, cells treated with BML-275 showed an increase
of cleaved PARP in MIA PaCa-2 cells but not in Panc-1 cells
(Fig. 3). However, BML-275
treatment decreased the expression of anti-apoptotic proteins such
as Survivin, Bcl2 and XIAP in both cell lines (Fig. 3).
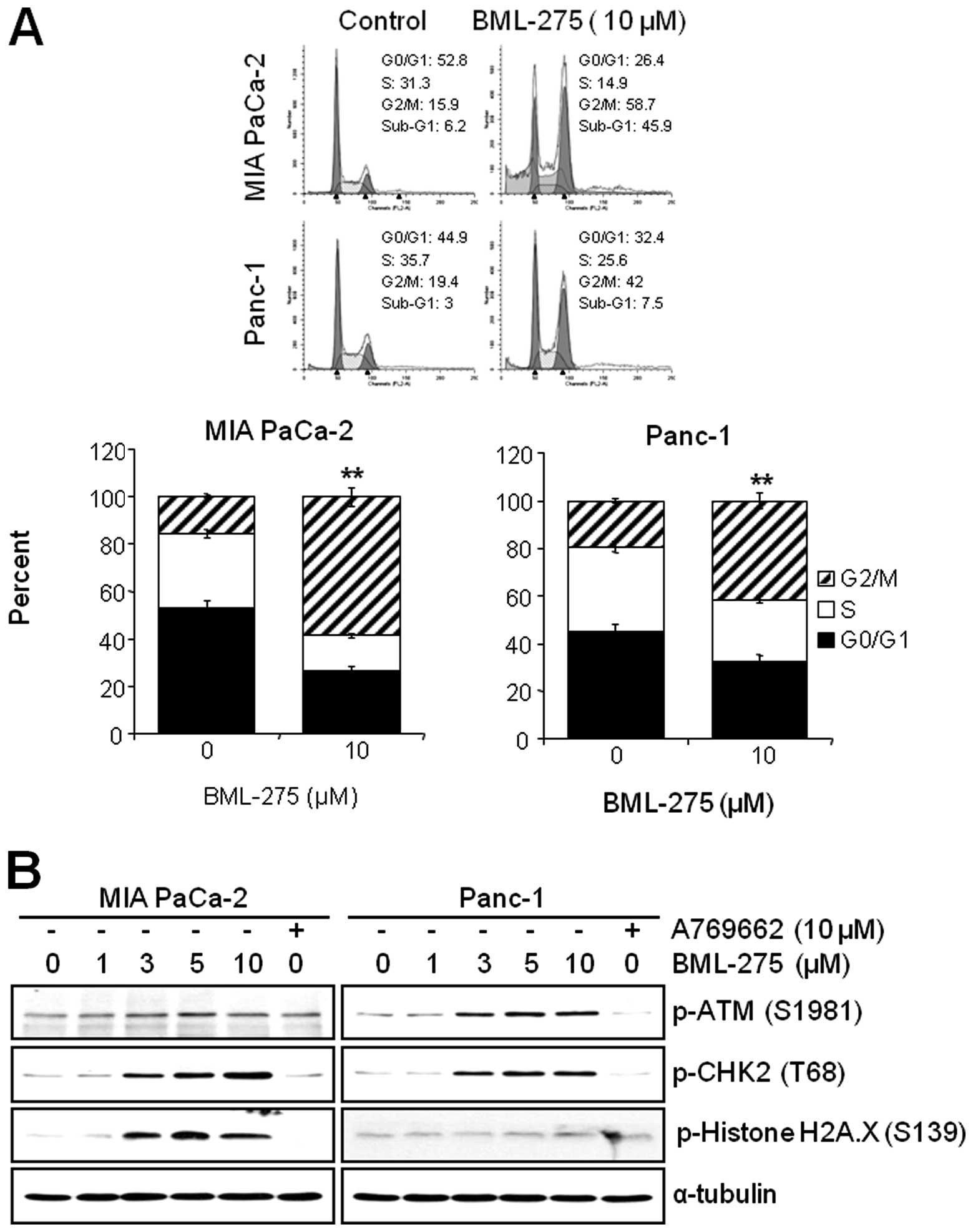
BML-275 induces G2/M arrest and
sub-G1
We next investigated if the pharmacological
inhibition of AMPK by BML-275 can affect the cell cycle progression
in pancreatic cancer cell lines. MIA PaCa-2 and Panc-1 cells were
treated with 10 μM BML-275 for 24 h and their cell cycle
profiles were assessed by FACS analysis. BML-275 treatment
significantly increased the cell population at G2/M-phase (from
15.9 to 58.7% in MIA PaCa-2 and from 19.4 to 42% in Panc-1) and
significantly decreased the cell population at G0/G1-phase (from
52.8 to 26.4% in MIA PaCa-2 and from 44.9 to 32.4% in Panc-1) and
S-phase (from 31.3 to 14.9% in MIA PaCa-2 cells and from 35.7 to
25.6% in Panc-1) (Fig. 4A).
Moreover, we also observed increase of sub-G1 populations. BML-275
increased the sub-G1 population in Panc-1 (from 3 to 7.5%) and more
significantly in MIA PaCa-2 (from 6.2 to 45.9%) (Fig. 4A).
DNA damage sensor CHK1/CHK2 plays a role in G2/M
checkpoint via the ataxia-telangiectasia mutated
(ATM)/ATM-RAD3-related (ATR) pathway. In order to further elucidate
the molecular mechanism leading to BML-275-mediated G2/M arrest, we
determined the activation of DNA damage signaling pathway.
Treatment of MIA PaCa-2 and Panc-1 cells with BML-275 for 24 h
increased the phosphorylation of ATM at Ser1981 and CHK2 at Thr68
in dose-dependent manner (Fig.
4B). These results coincide with cell cycle arrest in both cell
lines. However, the phosphorylation of Histone H2A.X at Ser139,
which is the molecular marker of DNA double-strand breaks, more
significantly increased in MIA PaCa-2 than in Panc-1 cells
(Fig. 4B). Increased levels of
CHK2 and H2A.X phosphorylation were more obvious in MIA PaCa-2
cells (Fig. 4B). On the contrary,
cells treated by 10 μM A769662 for 24 h did not induce the
phosphorylation levels of ATM, CHK2 or Histone H2A.X in either cell
line.
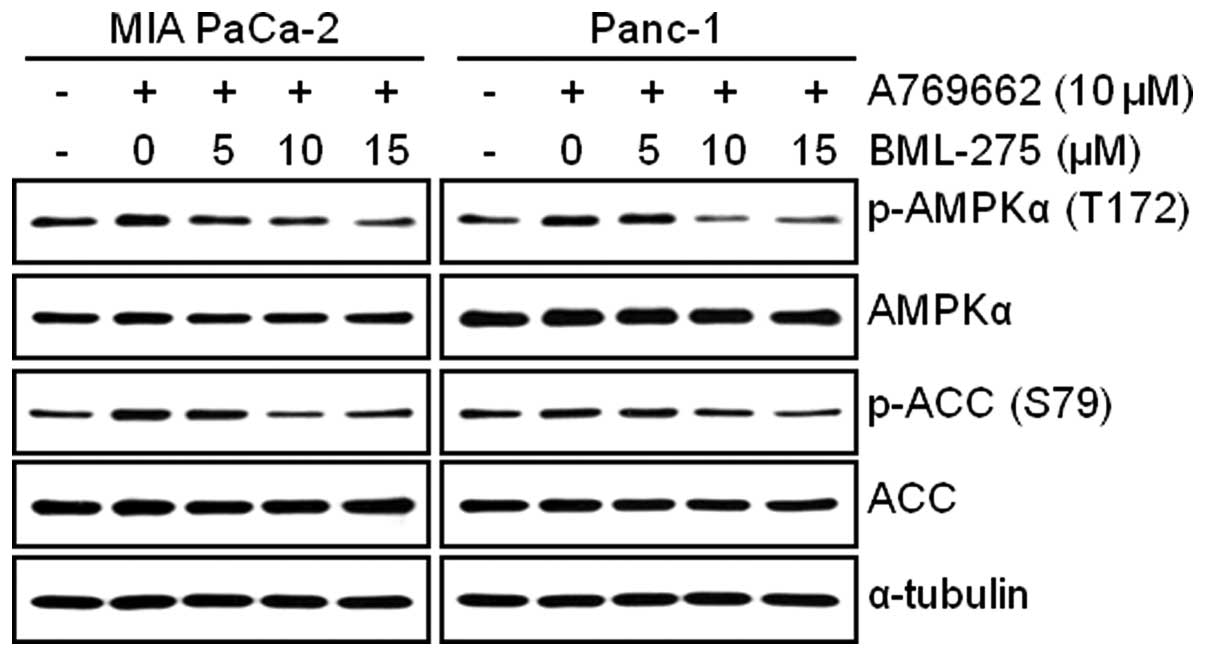
BML-275 decreases AMPKα activity in human
pancreatic cancer cells
In order to determine the decrease in cell survival
and increase in apoptotic cell death closely correlates with the
level of inhibition of AMPK activity, cells were pretreated with 10
μM A769662 for 6 h and administered with various
concentrations of BML-275 for 24 h. The treatment of 10 μM
A769662 for 6 h without BML-275 significantly activated
accumulation of phosphorylated levels of AMPKα and ACC in both cell
lines (Fig. 5). However, BML-275
treatment reduced the phosphorylation of AMPKα and ACC exerted by
A769662 in a dose-dependent manner (Fig. 5), suggesting that antitumor
effect(s) by BML-275 closely correlates with the level of
inhibition of AMPK activity in human pancreatic cancer cell
lines.
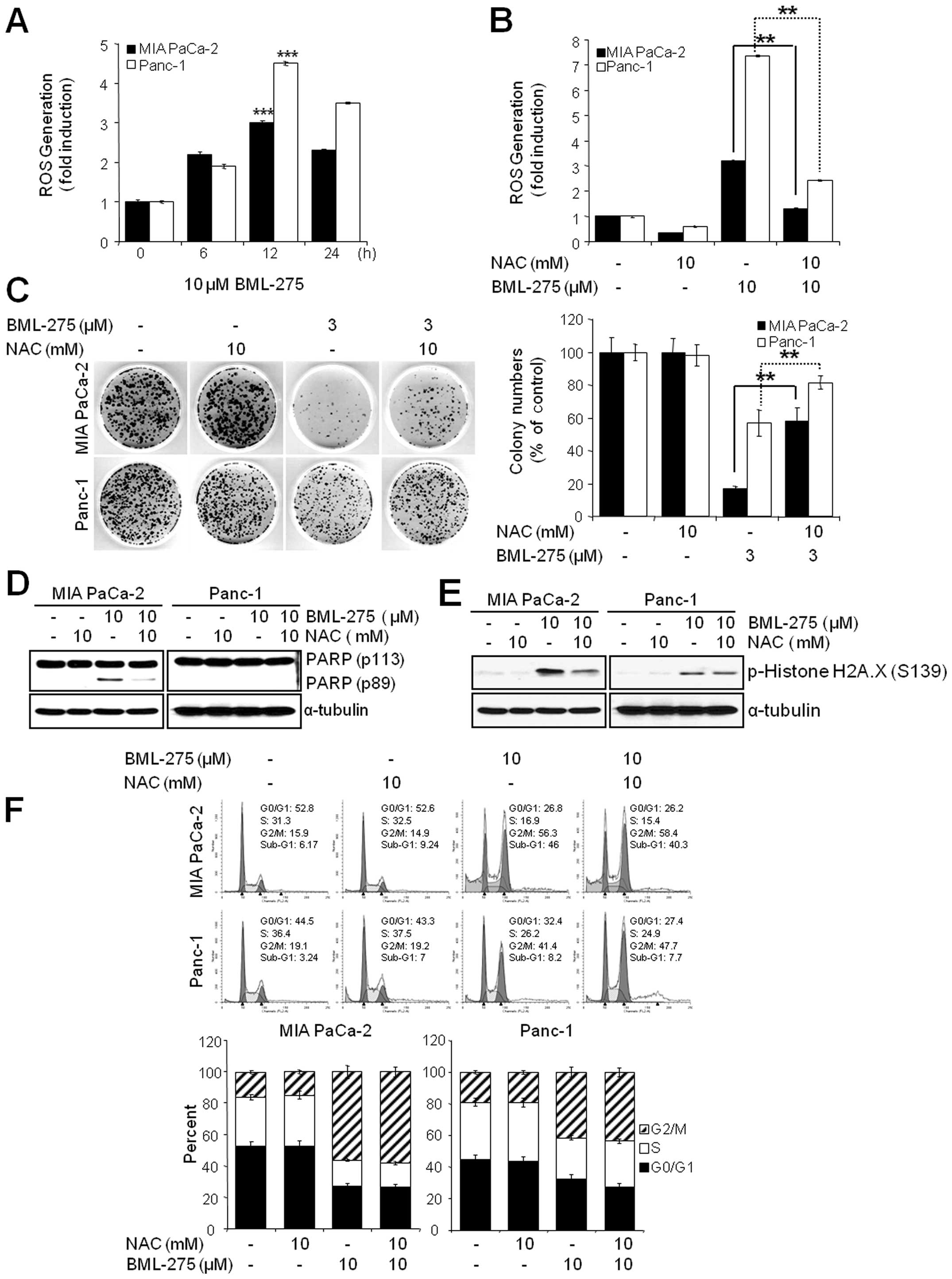
The generation of ROS by BML-275 is
critically required for the induction of cell death but not G2/M
arrest
Since oxidative stress is a potent inducer of
apoptosis, we next investigated if BML-275 could cause a generation
of ROS in pancreatic cancer cell lines. We determined ROS
generation by measuring the fluorescence of DCF which is formed by
the oxidation of DCFDA by peroxides. Our results demonstrated early
ROS generation by BML-275 in both cell lines (Fig. 6A). BML-275-induced ROS generation
was significantly diminished by incubation with the antioxidant
agent, NAC (Fig. 6B). NAC also
rescued BML-275-mediated inhibition of cell survival by MTT assay
(data not shown) and clonogenic assay (Fig. 6C). It also relieved the cleavage of
PARP by BML-275 treatment in MIA PaCa-2 cells (Fig. 6D). BML-275-mediated phosphorylation
of H2A.X at Ser139 also inhibited by NAC pretreatment (Fig. 6E). However, NAC administration did
not alleviate G2/M arrest induced by BML-275 treatment (Fig. 6F), suggesting that BML-275-mediated
G2/M arrest is ROS-independent at least in pancreatic cancer cell
lines used for this study.
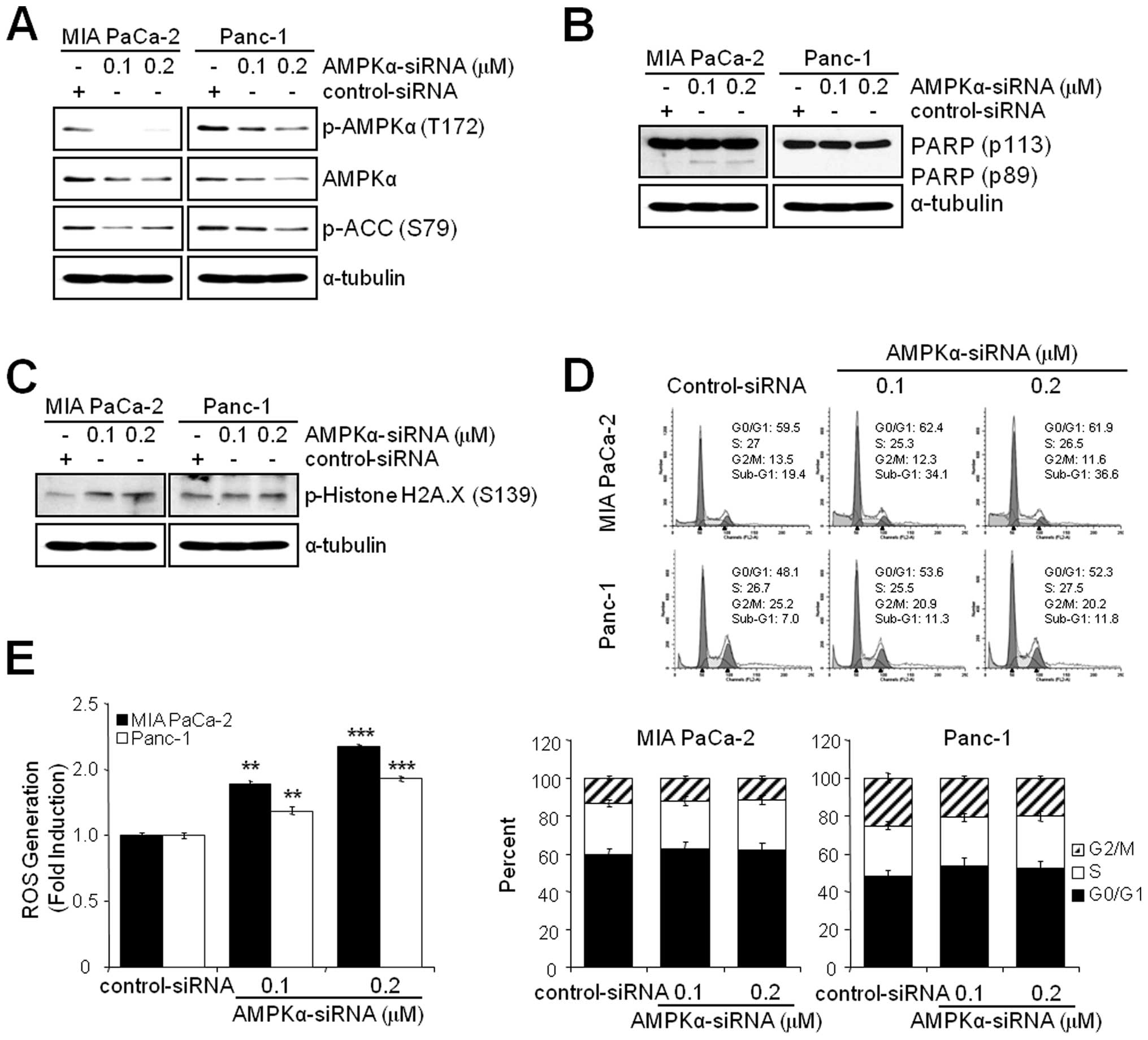
Knockdown of AMPKα induces ROS generation
and apoptosis but not G2/M arrest
Since the inhibition of AMPK by BML-275 induced DNA
damage, G2/M arrest and apoptosis in human pancreatic cancer cells,
MIA PaCa-2 and Panc-1 cells were transfected with control-siRNA or
AMPKα-siRNA to compare the effect(s) of BML-275 and knockdown of
AMPKα. Knockdown of AMPKα with concentration of 0.1 or 0.2
μM AMPKα-siRNA suppressed the level of total and
phosphorylated form of AMPKα and phosphorylated form of ACC in MIA
PaCa-2 and Panc-1 cells (Fig. 7A).
In addition, knockdown of AMPKα also induced apoptotic cell death
as evidenced by induction of PARP cleavage in MIA-PaCa-2 cells
(Fig. 7B) and accumulation of
sub-G1 cells in FACS analysis in MIA PaCa-2 cells (from 19.4% by
control to 34.1% by 0.1 μM AMPKα siRNA) but to a lesser
extent in Panc-1 cells (from 7.0% by control to 11.3% by 0.1
μM AMPKα-siRNA (Fig. 7D).
Knockdown of AMPKα in MIA PaCa-2 cells also induced phosphorylation
of H2A.X at Ser139 indicating DNA damage (Fig. 7C). The Panc-1 cells show resistance
to phosphorylation of H2A.X similarly to BML-275 treatment
(Fig. 4B). However, knockdown of
AMPKα activity fails to display a cell cycle arrest in G2/M-phase
in MIA PaCa-2 and Panc-1 cells (Fig.
7D). Finally, AMPKα knockdown induced ROS generation with
increasing concentration of AMPKα-siRNA (Fig. 7E). Taken together, in pancreatic
cancer cell lines, targeting of AMPKα is able to induce DNA damage,
ROS generation and apoptotic cell death but not G2/M arrest.
Discussion
In this study, we investigated the molecular
mechanism of antitumor effect(s) of BML-275, an AMPK inhibitor, in
human pancreatic adenocarcinoma. We found that: i) the levels of
total and phosphorylated form of AMPKα and ACC vary in several
different human pancreatic cancer cell lines; ii) BML-275 inhibits
cell proliferation in MIA PaCa-2, Panc-1, Colo-357 and AsPC-1
cells; iii) BML-275 induces DNA damage, apoptosis and G2/M arrest;
iv) the ROS generation by BML-275 is critically required for the
DNA damage and apoptosis but not G2/M arrest and v) knockdown of
AMPKα induces ROS generation, DNA damage and apoptosis but not G2/M
arrest. This is the first report showing that BML-275 induces DNA
damage, G2/M arrest and apoptosis in pancreatic cancer cell
lines.
AMPK is a survival factor for cancer cells. It is
involved in the augmentation of energy production through the
activation of glucose uptake, glycolysis and fatty acid oxidation
in response to ATP-depleting stresses (38). Solid tumors outgrowing the existing
vasculature are continuously exposed to a microenvironment in which
the supply of both oxygen and nutrition is limited. Previous
studies showed that AMPK is critical for cancer cell adaptation in
response to hypoxia or glucose deprivation (39–42).
The protective role of AMPK is not restricted to nutrient stress,
as this enzyme seems to play an important role in protecting tumor
cells from apoptosis induced by chemotherapeutic agents such as
doxorubicin, cisplatin and TRAIL (24–26).
In addition, pharmacological inhibition of AMPK by BML-275 induced
apoptotic cell death in myeloma, glioma, prostate cancer and breast
carcinoma cells (20–23). Moreover, transfection with
dominant-negative AMPK or AMPKα-siRNA was also sufficient to reduce
cell proliferation of BHK, HeLa and PC12 pheochromocytoma cells or
CWR22Rv1 and LNcaP prostate cancer cells (21,43).
Comparing with effective apoptosis inducing dose of BML-275 treated
in other cancer cell lines reported previously (20–23),
most of the pancreatic cancer cell lines responded to BML-275 with
different levels of responsiveness. Pancreatic cancer cell lines
with relatively high level of phosphorylated AMPK showed more
susceptibility to BML-275 treatment (MIA PaCa-2 and Colo-357), and
those with low phosphorylated AMPK showed relatively decreased
sensitivity (Panc-1 and AsPc-1).
Cancer cells usually exhibit increased levels on
intracellular ROS, which in turn can initiate various cycles
leading to further metabolic malfunction and ROS generation
(44,45). ROS cause oxidative damage to DNA,
proteins, lipids and other cellular components and therefore also
significant cellular stress (45).
A proposed therapeutic strategy against cancer is to treat cancer
cells with pharmacological agents that have pro-oxidant properties
which increase the intracellular ROS generation to a toxic
threshold that triggers cell death in the cancer cells without
harming normal cells (44).
Vuvicevic et al showed that BML-275 induces ROS generation
in glioma cell line, but AMPKα-siRNA treatment fails to induce ROS
generation and apoptosis (22). In
this study, an increased generation of ROS upon either BML-275 or
AMPKα-siRNA treatment was observed and the intracellular
accumulation of ROS seems to be one of critical factors in
BML-275-induced apoptosis. To verify this speculation, NAC,
scavenger of oxygen-free radicals, was challenged with BML-275. NAC
relieved BML-275 or AMPK-siRNA mediated ROS production and improved
cell viability based on the clonogenic assay, which suggested that
both chemical and genetic inhibitor regulate cell viability via
repressing AMPK activity.
The G2/M checkpoint plays an important role in
cellular response to genotoxic stimuli. The G2/M checkpoint
prevents cells from entering mitosis when DNA is damaged, providing
an opportunity for repair and stopping the proliferation of damaged
cells which help to maintain genomic stability (46). CHK1 and CHK2 kinases are activated
at G2-phase checkpoint by DNA damage or unreplicated chromosomal
DNA (47), and inactivate Cdc25C
through its phosphorylation (48,49).
Cdc25C was the protein phosphatase responsible for
dephosphorylating and activating Cdc2, a crucial step in regulating
the entry of all eukaryotic cells into the M-phase of the cell
cycle. In this study, BML-275 induces cell cycle arrest at
G2/M-phase possibly through the phosphorylation and activation of
CHK2 kinase. The pretreatment of NAC restores the generation of ROS
by BML-275 treatment in MIA PaCa-2 cell line, however, the cell
cycle arrest at G2/M phase cannot be relieved, suggesting unknown
effects of BML-275 or non-target effects may play a role in G2/M
arrest. Previously AMPKα-siRNA treatment was reported to induce
G2/M arrest in the absence of ROS generation and with no apparent
cell death in U251 glioma cells (22). However, in pancreatic cancer cell
line, the AMPKα-siRNA treatment induces generation of ROS and
apoptotic cell death but no apparent G2/M arrest. Thus, our finding
suggests that pancreatic cancer cells may be able to override the
cell cycle arrest (G2/M) in response to AMPK knockdown by siRNA. On
the other hand, the mechanism of DNA damage and cell death induced
by BML-275 seems to be via inhibition of AMPK activity followed by
stimulation of ROS production. Panc-1 is known as relatively more
resistant to various antitumor agents among several pancreatic
cancer cell lines (50–52). Our study also show panc-1 as more
resistant to apoptotic response (cell death and PARP cleavage) upon
the treatment of BML-275 and AMPKα-siRNA. Although we could not
demonstrate the mechanism of resistance of Panc-1 to BML-275
treatment, this may be due to its increased multidrug resistance
(MDR) gene products and/or constitutively activated cell surviving
signaling pathways that confer intrinsic drug resistance (50–54).
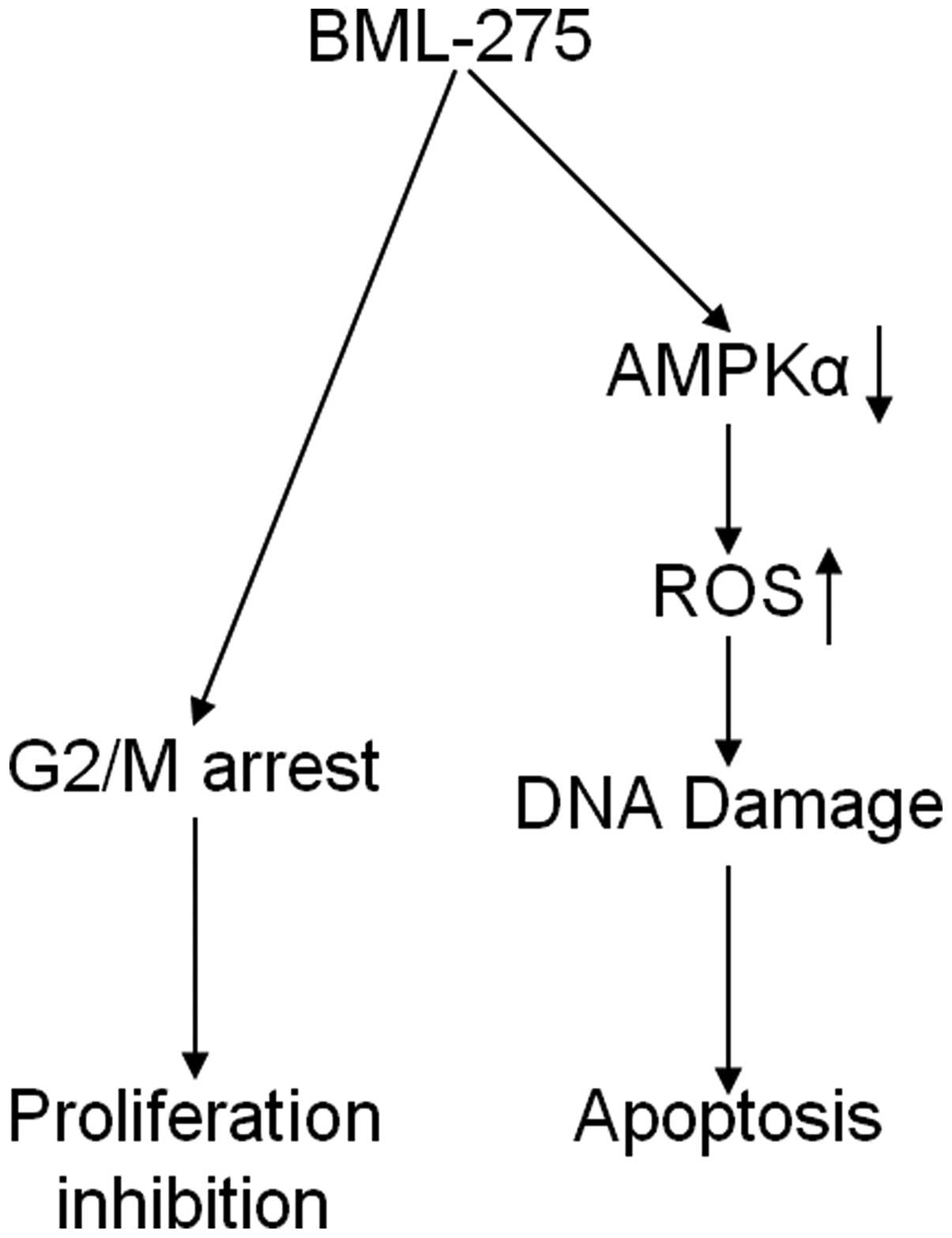
In conclusion, our findings implicate that BML-275
induces DNA damage and apoptosis through AMPK-dependent mechanism
and induces G2/M arrest through AMPK-independent mechanism
(Fig. 8). Although the molecular
mechanism of antitumor effect(s) by BML-275 requires further
investigation, this compound seems to be a novel potential
therapeutic agent to treat human pancreatic cancer.
Acknowledgements
IB was supported by National
Institutes of Health (1R03CA152530), the National Research
Foundation of Korea [R31-10069; World Class University (WCU)
program] and the Georgetown University Lombardi Comprehensive
Cancer Center (P30-CA051008).
References
|
1.
|
A JemalR SiegelJ XuE WardCancer
statistics, 2010CA Cancer J Clin60277300201010.3322/caac.20073
|
|
2.
|
MR KeighleyGastrointestinal cancers in
EuropeAliment Pharmacol
Ther18730200310.1046/j.0953-0673.2003.01722.x
|
|
3.
|
DG HardieAMP-activated/SNF1 protein
kinases: conserved guardians of cellular energyNat Rev Mol Cell
Biol8774785200710.1038/nrm224917712357
|
|
4.
|
DG HardieD CarlingThe AMP-activated
protein kinase - fuel gauge of the mammalian cells?Eur J
Biochem246259273199710.1111/j.1432-1033.1997.00259.x9208914
|
|
5.
|
DG HardieJW ScottDA PanER HudsonManagement
of cellular energy by the AMP-activated protein kinase systemFEBS
Lett546113120200310.1016/S0014-5793(03)00560-X12829246
|
|
6.
|
D CarlingThe AMP-activated protein kinase
cascade - a unifying system for energy controlTrends Biochem
Sci291824200410.1016/j.tibs.2003.11.00514729328
|
|
7.
|
DG HardieThe AMP-activated protein kinase
pathway - new players upstream and downstreamJ Cell
Sci11754795487200410.1242/jcs.0154015509864
|
|
8.
|
BB KahnT AlquierD CarlingDG
HardieAMP-activated protein kinase: ancient energy gauge provides
clues to modern understanding of metabolismCell
Metab11525200510.1016/j.cmet.2004.12.00316054041
|
|
9.
|
SA HawleyDA PanKJ MustardL RossJ BainAM
EdelmanBG FrenguelliDG HardieCalmodulin-dependent protein kinase
kinase-beta is an alternative upstream kinase for AMP-activated
protein kinaseCell
Metab2919200510.1016/j.cmet.2005.05.00916054095
|
|
10.
|
RL HurleyKA AndersonJM FranzoneBE KempAR
MeansLA WittersThe Ca2+/calmodulin-dependent protein
kinase kinases are AMP-activated protein kinase kinasesJ Biol
Chem2802906029066200515980064
|
|
11.
|
A WoodsK DickersonR HeathSP HongM
MomcilovicSR JohnstoneM CarlsonD
CarlingCa2+/calmodulin-dependent protein kinase
kinase-beta acts upstream of AMP-activated protein kinase in
mammalian cellsCell Metab221332005
|
|
12.
|
C CampàsJM LopezAF SantidriánM BarragánB
BellosilloD ColomerJ GilAcadesine activates AMPK and induces
apoptosis in B-cell chronic lymphocytic leukemia cells but not in T
lymphocytesBlood10136743680200312522004
|
|
13.
|
BA KefasY CaiK KerchhofsZ LingG MartensH
HeimbergD PipeleersM Van de CasteeleMetformin-induced stimulation
of AMP-activated protein kinase in beta-cells impairs their glucose
responsiveness and can lead to apoptosisBiochem
Pharmacol68409416200410.1016/j.bcp.2004.04.003
|
|
14.
|
M SaitohK NagaiK NakagawaT YamamuraS
YamamotoT NishizakiAdenosine induces apoptosis in the human gastric
cancer cells via an intrinsic pathway relawant to activation of
AMP-activated protein kinaseBiochem
Pharmacol6720052011200410.1016/j.bcp.2004.01.020
|
|
15.
|
R RattanS GiriAK SinghI
Singh5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside
inhibits cancer cell proliferation in vitro and in vivo via
AMP-activated protein kinaseJ Biol
Chem2803958239593200510.1074/jbc.M50744320016176927
|
|
16.
|
W ZhouWF HanLE LandreeJN ThupariML PinnT
BililignEK KimA VadlamudiSM MedghlchiR El MeskiniGV RonnettCA
TownsendFP KuhajdaFatty acid synthase inhibition activates
AMP-activated protein kinase in SKOV3 human ovarian cancer
cellsCancer
Res6729646971200710.1158/0008-5472.CAN-06-343917409402
|
|
17.
|
A IsakovicL HarhajiD StevanovicZ MarkovicM
Sumarac-DumanovicV StarcevicD MicicV TrajkovicDual antiglioma
action of metformin: cell cycle arrest and mitochondria-dependent
apoptosisCell Mol Life
Sci6412901302200710.1007/s00018-007-7080-417447005
|
|
18.
|
R OkoshiT OzakiH YamamotoK AndoN KoidaS
OnoT KodaT KamijoA NakagamaraH KizakiActivation of AMP-activated
protein kinase induces p53-dependent apoptotic cell death in
response to energetic stressJ Biol
Chem28339793987200810.1074/jbc.M70523220018056705
|
|
19.
|
TK SenquptaGM LeclercTT Hsieh-KinserGJ
leclercI SinghJC BarredoCytotoxic effect of
5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside (AICAR) on
childhood acute lymphoblastic leukemia (ALL) cells: implication for
targeted therapyMol Cancer646200710.1186/1476-4598-6-4617623090
|
|
20.
|
P BaumannS Mandl-WeberB EmmerichC StrakaR
SchmidmaierInhibition of adenosine monophosphate-activated protein
kinase induces apoptosis in multiple myeloma cellsAnticancer
Drugs18405410200710.1097/CAD.0b013e32801416b617351392
|
|
21.
|
HU ParkS SuyM DannerV DaileyY ZhangH LiDR
HydukeBT CollinsG GagnonB KallakuryD KumarML BrownA FornaceA
DritschiloSP CollinsAMP-activated protein kinase promotes human
prostate cancer cell growth and survivalMol Cancer
Ther8733741200910.1158/1535-7163.MCT-08-063119372545
|
|
22.
|
L VuvicevicM MisirkicK JanjetovicL
Harhaji-TrajkovicM PricaD StevanovicE IsenovicE SudarM
Sumarac-DumanovicD MicicV TrajkovicAMP-activated protein
kinase-dependent and-independent mechanisms underlying in vitro
antiglioma action of compound CBiochem
Pharmacol7716841693200910.1016/j.bcp.2009.03.00519428322
|
|
23.
|
J JinTD MullenQ HouJ BielawskiA BielawskaX
ZhangLM ObeidYA HannunYT HsuAMPK inhibitor compound C stiumulates
ceramine production and promotes BAx redistribution and apoptosis
in MCF-7 breast carcinoma cellsJ Lipid
Res5023892397200910.1194/jlr.M900119-JLR20019528633
|
|
24.
|
JH JangTJ LeeES YangS Min doYH KimSH KimYH
ChoiJW ParkKS ChoiTK KwonCompound C sensitizes caki renal cancer
cells to TRAIL-induced apoptosis through reactive oxygen
species-mediated down-regulation of c-FLIPL and Mcl-1Exp Cell
Res31621942203201010.1016/j.yexcr.2010.04.02820451517
|
|
25.
|
Q ZhuB ShenB ZhangW ZhangSH ChinJ JinDF
LiaoInhibition of AMP-activated protein kinase pathway sensitizes
human leukemia K562 cells to nontoxic concentration of
doxorubicinMol Cell
Biochem340275281201010.1007/s11010-010-0428-320339906
|
|
26.
|
HS KimJT HwangH YunSG ChiSJ LeeI KangKS
YoonWJ ChoeSS KimJ HaInhibition of AMP-activated protein kinase
sensitizes cancer cells to cisplatin-induced apoptosis via
hyper-induction of p53J Biol
Chem28337313742200810.1074/jbc.M70443220018079115
|
|
27.
|
K CollinsT JacksNP PavletichThe cell cycle
and cancerProc Natl Acad Sci
USA9427762778199710.1073/pnas.94.7.27769096291
|
|
28.
|
JK BuolamwiniCell cycle molecular targets
in novel anti-cancer drug discoveryCurr Pharm
Des6379392200010.2174/138161200340094810788588
|
|
29.
|
M HajduchL HavlieekJ VeselyR NovotnyV
MihalM StrnadSynthetic cyclin dependent kinase inhibitors. New
generation of potent anti-cancer drugsAdv Exp Med
Biol457341353199910.1007/978-1-4615-4811-9_3710500810
|
|
30.
|
J PinesFour-dimensional control of the
cell cycleNat Cell Biol1E73E79199910.1038/1104110559915
|
|
31.
|
WR TaylorGR StarkRegulation of the G2/M
transition by
p53Oncogene2018031815200110.1038/sj.onc.120425211313928
|
|
32.
|
S UedaH NakamuraH MasutaniT SasadaA
TakabayashiY YamaokaJ YodoiBaicalin induces apoptosis via
mitochondrial pathway as prooxidantMol
Immunol38781791200210.1016/S0161-5890(01)00115-811841838
|
|
33.
|
KB BeckmanBN AmesOxidative decay of DNAJ
Biol Chem2721963319636199710.1074/jbc.272.32.196339289489
|
|
34.
|
J CadetT DelatourT DoukiD GasparuttoJP
PougetJL RavanatS SauvaigoHydroxyl radicals and DNA base
damageMutal Res424921199910.1016/S0027-5107(99)00004-4
|
|
35.
|
MM VilenchikAG KnudsonEndogenous DNA
double-strand breaks:production, fidelity of repair and induction
of cancerProc Natl Acad Sci
USA1001287112876200310.1073/pnas.213549810014566050
|
|
36.
|
T FurukawaWP DuquidL RosenbergJ VialletDA
GallowayMS TsaoLong-term culture and immortalization of epithelial
cells from normal adult human pancreatic ducts transfected by the
E6E7 gene of human papilloma virus 16Am J
Pathol1481763177019968669463
|
|
37.
|
HQ DuongHJ KimHJ KangYS SeongI BaeZSTK474,
a PI3K inhibitor, suppresses proliferation and sensitizes human
pancreatic adenocarcinoma cells to gemcitabineOncol
Rep27182188201221993922
|
|
38.
|
DG HardieD CarlingM CarlsonThe
AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of
the eukaryotic cells?Annu Rev
Biochem67821855199810.1146/annurev.biochem.67.1.8219759505
|
|
39.
|
M LeeJT HwangHJ LeeSN JungI KangSG ChiSS
KimJ HaAMP-activated protein kinase activity is critical for
hypoxia-inducible factor-1 transcriptional activity and its target
gene expression under hypoxic conditions in DU145 cellsJ Biol
Chem2783965339661200310.1074/jbc.M306104200
|
|
40.
|
H YunM LeeSS KimJ HaGlucose deprivation
increases mRNA stability of vascular endothelial growth factor
through activation of AMP-activated protein kinase in DU145
prostate carcinomaJ Biol
Chem28099639972200510.1074/jbc.M412994200
|
|
41.
|
K KatoT OguraA KishimotoY MinegishiN
NakajimaM MiyazakiH EsumiCritical roles of AMP-activated protein
kinase in constitutive tolerance of cancer cells to nutrient
deprivation and tumor
formationOncogene2160826090200210.1038/sj.onc.120573712203120
|
|
42.
|
H EsumiK IzuishiK KatoK HashimotoY
KurashimaA KishimotoT OguraT OzawaHypoxia and nitric oxide
treatment confer tolerance to glucose starvation in a
5′-AMP-activated protein kinase dependent mannerJ Biol
Chem2773279132798200212091379
|
|
43.
|
MM ShawWK GurrRJ McCrimmonDF SchordererRS
Sherwin5′AMP-activated protein kinase alpha deficiency enhances
stress-induced apoptosis in BHK and PC12 cellsJ Cell Mol
Med112862982007
|
|
44.
|
D TrachoothamJ AlexandreP HuangTargeting
cancer cells by ROS-mediated mechanisms: a radical therapeutic
approach?Nat Rev Drug Discov8579591200910.1038/nrd280319478820
|
|
45.
|
J LuoNL SoliminiSJ ElledqePrinciples of
cancer therapy: oncogene and non-oncogene
addictionCell136823837200910.1016/j.cell.2009.02.02419269363
|
|
46.
|
M LobrichPA JeggoThe impact of a negligent
G2/M checkpoint on genomic instability and cancer inductionNat Rev
Cancer7861869200710.1038/nrc224817943134
|
|
47.
|
VA SmitsRH MedemaChecking out the G(2)/M
transitionBiochim Biophys
Acta1519112200110.1016/S0167-4781(01)00204-411406266
|
|
48.
|
T GotohK OhsumiT MatsuiH TakisawaT
KishimotoInactivation of the checkpoint kinase Cds1 is dependent on
cyclin B-Cdc2 kinase activation at the meiotic G2/M-phase
transition in xenopus oocytesJ Cell Sci11433973406200111591827
|
|
49.
|
SV SinghA Herman-AntosiewiczAV SinghKL
LewSK SrivastavaR KamathKD BrownL ZhangR
BaskaranSulforaphane-induced G2/M phase cell cycle arrest involves
checkpoint kinase 2-mediated phosphorylation of cell division cycle
25CJ Biol Chem2792581325822200410.1074/jbc.M31353820015073169
|
|
50.
|
W HuanwenL ZhiyongS XiaohuaR XinyuW KaiL
TonghuaIntrinsic chemoresistance to gemcitabine is associated with
constitutive and laminin-induced phosphorylation of FAK in
pancreatic cancer cell linesMol
Cancer8125200910.1186/1476-4598-8-12520021699
|
|
51.
|
JN KreutzerM RuzzeneB GuerraEnhancing
chemosensitivity to gemcitabine via RNA interfence targeting the
catalytic subunits of protein kinase CK2 in human pancreatic cancer
cellsBMC Cancer1040201010.1186/1471-2407-10-44020718998
|
|
52.
|
AV DanilovD NeupaneAS NagarajaEV
FeofanovaLA HumphriesJ DiRenzoM KorcDeltaNp63alpha-mediated
induction of epidermal growth factor receptor promotes pancreatic
cancer cell growth and chemoresistancePloS
One6e26815201110.1371/journal.pone.002681522053213
|
|
53.
|
W HagmannR JesnowskiJM LöhrInterdependence
of gemcitabine treatment, transporter expression, and resistance in
human pancreatic carcinoma cellsNeoplasia12740747201020824050
|
|
54.
|
Z YaoY OkabayashiY YutsudoT KitamuraW
OgawaM KasugaRole of Akt in growth and survival of Panc-1
pancreatic cancer
cellsPancreas244246200210.1097/00006676-200201000-0000611741181
|