Introduction
Hormone replacement therapy (HRT) containing either
estrogen alone or a combination of estrogen and a progestin such as
medroxyprogesterone acetate (MPA), is a commonly-used treatment for
menopause in women (1). Clinical
studies show that estrogen-based HRT is associated with increased
risk of uterine cancer, while there is an elevated risk of breast
cancer, metastasis and mortality in women undergoing
estrogen/progestin-based HRT (2–4). Our
recent studies show that progestins induce in vitro
expression of vascular endothelial growth factor (VEGF), a potent
angiogenic growth factor, in a subset of human breast cancer cells
that express mutant p53 (5,6) and
stimulate breast tumor growth in vivo(7,8).
Conversely, the anti-progestin RU-486 and anti-VEGF antibodies
inhibit secretion and function of VEGF in vitro and block
breast tumor growth in vivo(7,8).
These results support the hypothesis that pharmacological use of
progestins increases the risk of progesterone receptor
(PR)-dependent breast cancer in post-menopausal women by a
mechanism involving induction of VEGF. Though anti-progestins such
as mifepristone or RU-486 suppress progestin-dependent actions,
their use is limited due to a number of side effects that occur as
a result of cross-reactivity with other steroid receptors (9). Thus other means of controlling PR
mediated effects are being explored.
Hypoxia-inducible factor-1α (HIF-1α), is a basic
helix-loop-helix transcription factor that regulates expression of
VEGF and other genes that modulate growth, survival and metastasis
of tumor cells under conditions of hypoxia (10,11).
Under normoxic conditions, HIF-1α is rapidly degraded, but under
hypoxic conditions, HIF-1α dimerizes with and is stabilized by
HIF-1β, and the HIF-1α, β heterodimer actively regulates expression
of promoters containing its cognate response element,
hypoxia-response element (HRE) (12) that is also located in the VEGF
promoter. Several studies suggest that HIF-1α regulates expression
of VEGF in cancer cells (13,14).
3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole (YC-1) (15) was initially developed as a specific
HIF-1α inhibitor which interferes with binding of HIF-1α to
promoter regions of its target genes (10). However, YC-1 has been found to
possess additional properties; it is also a nitric oxide synthetase
(NOS)-independent activator of soluble guanylyl cyclase (sGC),
which has been used clinically to treat thrombosis and hypertension
(16), and it blocks NF-κB
activity in tumor cells (17).
Importantly, previous studies suggest that YC-1 may have
significant anti-tumor activity by targeting HIF-1α and NF-κB
transcription factors (18).
Recent studies in a bovine model have shown that
HIF-1α plays a role in PR mediated VEGF induction (13). With this in mind we used cell
culture techniques and rodent models to examine, both in
vitro and in vivo, the capacity of YC-1 as a HIF-1α
inhibitor which might be used to control progestin-stimulated
PR-dependent VEGF secretion and progression of progestin-dependent
breast cancer. Breast cancer cells were exposed to YC-1 in
vitro or in vivo and their growth and tumorigenic
properties examined. Unexpectedly we made the novel observation
that, both in vivo and in vitro, YC-1 downregulates
PR in human breast cancer cells. Furthermore, YC-1 arrests the
progression of progestin-dependent breast cancer cells in
vivo by preventing VEGF induction which depends upon progestin
activity.
Materials and methods
Materials
Human T47-D and BT-474 breast cancer cell lines were
obtained from ATCC (Manassas, VA).
3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole) (YC-1), was
purchased from Biomol International, LP (Plymouth Meeting, PA).
Phenol red-free DMEM/F12 medium, phosphate-buffered saline and
0.05% trypsin-EDTA were purchased from Invitrogen Corporation and
Life Technologies (Grand Island, NY). Fetal bovine serum (FBS) was
obtained from JRH Biosciences (Lenexa, KS).
Cell viability assay
T47-D and BT-474 human breast cancer cells were
exposed to YC-1 (1–100 μM) for 18 h and cell viability was
tested using the sulforhodamine B (SRB) assay (19,20).
Cell culture and VEGF ELISA
Cells were grown in DMEM/ F12 + 10% FBS for routine
culture. Media was replaced with DMEM/F12 containing 5%
dextran-coated charcoal (DCC)-treated serum for 24 h prior to
treatment of cells in triplicate with progestins and inhibitors for
a further 18 h. Media was collected and ELISA for VEGF performed
using a VEGF ELISA kit from R&D Systems, Inc. (Minneapolis, MN)
as previously described (21–23).
According to the manufacturer’s protocol, the minimum detectable
concentration of VEGF is less than 5 pg/ml, the intra-assay
precision has a coefficient of variance (CV %) between 3.5–6.5%,
while the CV % of the inter-assay ranges from 5.0 to 8.5%.
Bicinchoninic acid (BCA) protein
assay
BCA assay using bovine serum albumin as a standard
was used to determine total protein concentrations as described
before (21–23). All samples were analyzed in
duplicate.
Preparation of whole cell extract for
western blot analysis
Whole cell extracts were prepared with a nuclear
extract kit (Active Motif). Briefly, cells were grown in 100 mm
culture dish overnight and then treated with DMEM/F12 containing 5%
FBS-DCC serum for 24 h. Cells were then treated with 10 nM MPA
alone and in the presence of 100 μM YC-1, or YC-1 alone for
6 h. At the end of treatments, cells were washed with ice-cold PBS
containing phosphatase inhibitors (with kit), harvested by gentle
scraping with a cell lifter, and centrifuged for 5 min at 200 × g
at 4°C. Cell pellets were re-suspended in complete lysis buffer
(provided in the TransAm kit; 1 mM DTT and 1% protease inhibitor
cocktail was added prior to use) and incubated on ice for 30 min
with shaking. Samples were centrifuged at 14,000 × g for 15 min,
and the supernatant was transferred to a microcentrifuge tube,
aliquoted, and stored at −80°C.
Western blotting
Proteins (50 μg per lane) were separated in a
10% NuPAGE Bis-Tris Gel (Invitrogen, Carlsbad, CA). Electrophoresis
was performed at 120 V for 1.5 h using NuPAGE MOPS-SDS Running
Buffer. Separated proteins were transferred to polyvinylidene
difluoride membranes (Bio-Rad Laboratories, Hercules, CA) at 25 V
for 25 min (Dry transfer). The blots were blocked for 1 h at room
temperature (RT) in 5% non-fat dry milk in TBS containing 0.1%
Tween-20 (TBS-T), and incubated with anti-PR (1:200) and
anti-HIF-1α (1:150) antibodies overnight at 4°C. The blots were
washed 3 times with TBS-T and incubated with secondary antibody for
1 h at RT. The blots were then washed 7 times (8 min each) with
TBS-T and immunoreactive bands were visualized using an ECL plus
detection kit (Amersham, Pharmacia Biotech, Arlington Heights, IL).
Membranes were stripped and re-blotted for β-actin (Sigma, St.
Louis, MO), which was used as a control for protein loading.
RNA extraction and RT-PCR
Cells were treated with progestin (10 nM) ± 100
μM YC-1 for 6 h at 37°C. Ultraspec RNA reagent (1 ml)
(Biotecx Products, Oxon, UK) was added to each plate and RNA was
purified as described by the manufacturer. Pellets containing
purified RNA were re-suspended in DEPC treated water. RNA was DNase
treated prior to RT-PCR which was performed as described previously
(23). VEGF and β-actin primer
pairs were purchased from R&D Systems, Inc.; DNA sequences for
these are propriety. Forward 5′-3′: ATGAGAAGTATGACAACAGCC, and
reverse 5′-3′: TGAGTCCTTCCACGATACC were used for GAPDH. The
following cycle was employed; 60°C for 30 min for cDNA synthesis,
94°C for 2 min for denaturing and 40 cycles of 94°C for 15 sec,
50–55°C for 30 sec and 68°C for 1 min for amplification. Final
extension was for 7 min at 68°C.
DMBA-induced mammary tumors
Intact virgin female 40–45 day old Sprague-Dawley
rats (Harlan Sprague-Dawley, Indianapolis, IN) were housed
according to the guidelines of the Association for Assessment and
Accreditation of Laboratory Animal Care under conditions of 12-h
light/dark cycles and ad libitum access to food and water.
All surgical and experimental procedures were in accordance with
the ‘Guide for Care and Use of Laboratory Animals’ (NIH publication
85-23). Animals were given a single dose of 20 mg of DMBA in peanut
oil via gavage on day 0. On day 30, animals were anesthetized and
MPA pellets were implanted subcutaneously on the dorsal surface
(7). On day 68, YC-1 (3.75 mg/day)
(18), or vehicle, was
administered to animals via tail vein injection for 5 days. Animals
were palpated and tumors measured 2–3 times weekly. Mammary tumor
tissues were collected at the time of sacrifice for
immunohistochemistry (IHC) analysis. In order to determine blood
vessel perfusion within tumors, animals were injected with 0.5 mg
Texas red-tomato lectin conjugate 10 min prior to sacrificing, as
described (24).
Xenograft tumor study
Five to six week old female athymic nu/nu mice,
purchased from Harlan Sprague-Dawley, Inc., were housed in a
laminar airflow cabinet under specific pathogen-free conditions.
Nude mice were inoculated with 17-β-estradiol pellets (60-day timed
release, 1.7 mg) 24–48 h before implantation of T47-D or BT474
cells in both flanks as described previously (8). In this model tumor cells initially
grow but then spontaneously regress and the regression is rescued
with progestins (8). In the
experiments reported in this study, when tumors regressed to
approximately 50% of their peak volume following tumor cell
injection (6–10 days), MPA pellets (10 mg/60-day release) were
implanted. Once tumor volume reached 80–120 mm3, animals
received 10 daily treatments of YC-1 (600 μg/mouse, i.p.)
(18), or vehicle alone. Tumor
volume and animal weights were measured every 3 days. At the end of
the experiment, animals were sacrificed and tumors collected 2 h
following final treatment with YC-1 for IHC analysis.
Histology and immunohistochemisty
For both rat and mouse tumors, tissues were fixed
overnight in 4% paraformaldehyde and processed for paraffin
infiltration and embedding. Sections (5 μm) were mounted on
ProbeOn Plus microscope slides (Fischer Scientific, Inc.,
Pittsburgh, PA) and routinely stained with hematoxylin and eosin
(H&E) or prepared for immunohistochemical labeling. Prior to
immunohistochemistry, sections were dewaxed in xylene, rehydrated
through graded concentrations of ethanol, rinsed (wash buffer, Dako
Carpinteria, CA) prior to immersion and heated in 10 mmol/l citrate
buffer (pH 6.0) for 20 min for heat-induced epitope retrieval. This
tissue treatment was performed for PR, ERα, ERβ, CD34 and VEGF
immuno-labeling. Slides were cooled for 20 min, treated with 3%
H2O2 (to inactivate endogenous peroxidase
activity) and rinsed prior to incubation with 5% bovine serum
albumin for 20 min. Sections were then incubated for 60 min at room
temperature with each of the following antibodies: anti-PR antibody
[1:50 dilution of a rabbit anti-human PR polyclonal antibody
(A0098), Dako], anti-ERα [1:300 dilution of a rabbit anti-ERα
polyclonal antibody (sc-542), Santa Cruz Biotechnology, Inc., Santa
Cruz, CA], anti-ERβ [1:50 dilution of a mouse anti-ERβ monoclonal
antibody (MCA1974s) AbD Serotec, Raleigh, NC], anti-CD34 (1:100
dilution of a goat anti-CD34 polyclonal antibody), and an anti-VEGF
antibody [1:100 dilution of a rabbit anti-VEGF polyclonal antibody
(sc-152); Santa Cruz Biotechnology, Inc.]. Sections were then
washed, incubated for 30 min with a biotinylated secondary antibody
[rabbit anti-mouse IgG (Dako) for anti-ERβ labeled sections and a
rabbit anti-goat IgG (Dako) for the anti-CD34 probed sections] and
then for 30 min with a streptavidin-linked horseradish peroxidase
product (Dako). Sections for PR, ERα and VEGF were incubated with
EnVision, a horseradish peroxidase-labeled polymer conjugated to
anti-rabbit antibodies (Dako). Bound antibodies were visualized
following incubation with 3,3′-diaminobenzidine solution (0.05%
with 0.015% H2O2 in PBS; Dako) for 3–5 min.
Sections were counterstained with Meyer’s hematoxylin, dehydrated,
and coverslipped for microscopic examination.
Texas red conjugated-tomato lectin
Sections (8 μm) of frozen tumors were made
using a cryostat. After rinsing, sections were incubated with 4%
paraformaldehyde, rinsed again, and mounted with DAPI (Vectashield
Hardset with DAPI, Vector Lab, Burlingame, CA) and coverslipped. In
order to visualize Texas red-labeled tomato lectin staining,
samples were imaged with a 590 nm bandpass filter at 1/2 second
exposure.
Statistical analysis
For VEGF analysis, data were analyzed by one-way
ANOVA. Data were checked for normality and homogeneity of variance.
Since a number of analyses did not satisfy these assumptions, data
were ranked and ANOVA performed as outlined by Conover and Iman
(25). Fisher’s protected least
significant difference (LSD) was performed to determine treatment
differences, as suggested by Chew (26). All significance was based on the
ranked transformations; however treatment values were presented as
actual values. For DMBA studies, a difference in the growth curve
of tumor size was compared via t-test. For nude mice studies,
points along the tumor volume curve were compared via one-way
ANOVA. FoveaoPro 3.0® software analysis was used to
determine positive staining by area in immunohistochemical studies.
One-way ANOVA was used to statistically compare VEGF, CD34, and PR
staining differences among experimental groups. For all statistical
comparisons, p<0.05 was regarded as statistically
significant.
Results
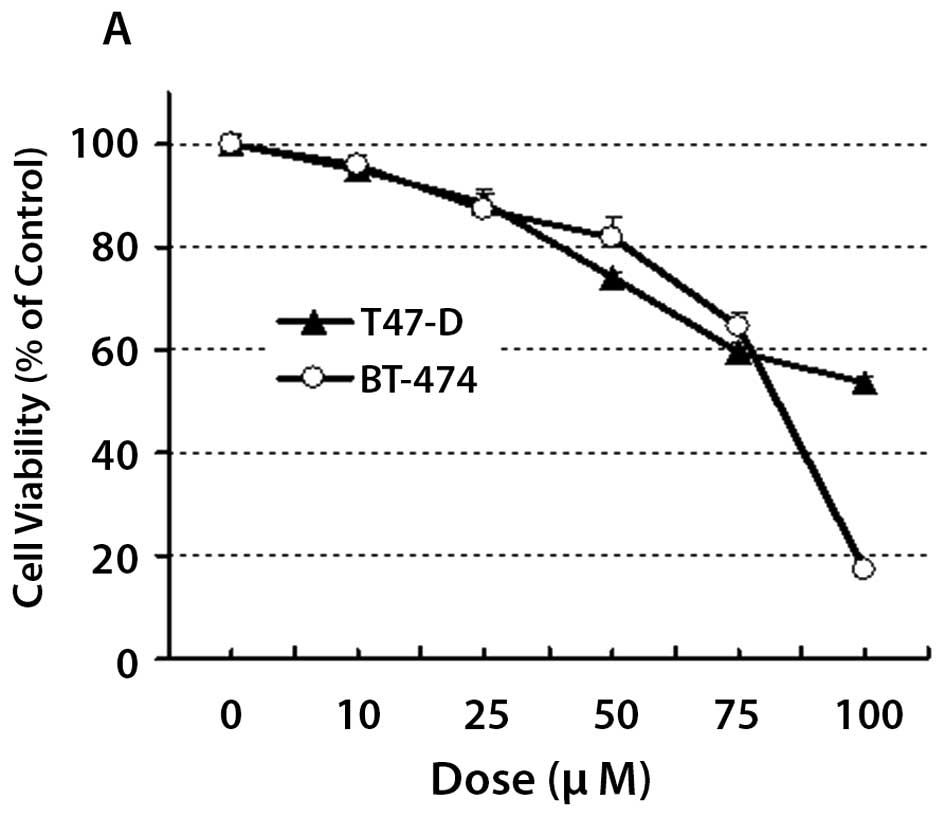
YC-1 reduces cell viability of human
breast cancer cells in a concentration dependent manner
T47-D and BT-474 cells were treated with increasing
doses of YC-1 for 18 and cell viability monitored using the SRB
assay (Fig. 1A). YC-1 effectively
reduced cell viability in a concentration-dependent manner in a
similar fashion to its effect on prostate cancer cells (17). In keeping with a previous study
(17), we used a dose of 100
μM YC-1 for subsequent in vitro analysis that
involved 6–18 h exposure to the drug. Since YC-1 was extremely
potent against BT-474 cells at this concentration, we performed
subsequent in vitro studies using T47-D cells in an effort
to study the molecular mechanisms underlying the effects of
YC-1.
YC-1 inhibits progestin-stimulated
secretion of VEGF from T47-D cells
To determine whether YC-1 plays a role in
progestin-stimulated secretion of VEGF from human breast cancer
cells, T47-D cells were incubated for 30 min with 10- or 100
μM YC-1 or 1 μM anti-progestin RU-486 [anti-progestin
(21–23)]. Following this initial incubation,
10 nM MPA (most commonly used synthetic progestin in HRT) or 10 nM
progesterone was added for 18 h and VEGF secreted into the culture
medium was quantified by ELISA. As reported previously by us
(21–23), expression of VEGF was 3- to 4-fold
higher in MPA and progesterone-treated T47-D cells compared with
controls; VEGF induction was completely inhibited by a 100-fold
excess of RU-486, indicating the involvement of PR (Fig. 1B). Interestingly, while 10
μM was without effect, 100 μM YC-1 completely
inhibited the induction of VEGF by progestin in T47-D cells in the
time frame tested (Fig. 1B and
data not shown). YC-1 also reduced basal levels of VEGF secretion
from T47-D breast cancer cells, most likely by inhibiting HIF-1α,
which is the major transcription factor required for VEGF
induction, though this remains to be proven. Importantly, YC-1 also
blocked the effect on VEGF secretion of other synthetic progestins
used in HRT, including norethindrone and norgestrel (Fig. 1C).
YC-1 inhibits progression of
MPA-dependent human breast cancer xenografts in nude mice
We previously developed a mouse xenograft model for
identifying and characterizing factors that promote or prevent
progestin-accelerated breast cancer (8). In the present study we used this
model to examine the effects of YC-1 on growth of MPA-stimulated
T47-D xenograft tumors. In this experimental design (Fig. 2A), xenograft tumors were allowed to
grow to a volume of 80–120 mm3, after which
tumor-bearing mice were treated with YC-1 [600 μg/mouse/day
(18)] for 10 days. Tumor volume
and animal weight were measured every third day throughout the
experiment. Fig. 2A shows clearly
that YC-1 inhibited growth and caused regression of T47-D xenograft
tumors. At the chosen dose level, YC-1 was well tolerated and no
signs of toxicity were detected in YC-1-treated mice (data not
shown). Similar experiments conducted using nude mice bearing
xenograft tumors derived from tamoxifen-resistant Her-2-neu
enriched BT-474 breast cancer cells (27) showed that YC-1 strongly and rapidly
inhibited growth of BT-474 tumor xenografts that were exposed to
the natural hormone progesterone (data not shown).
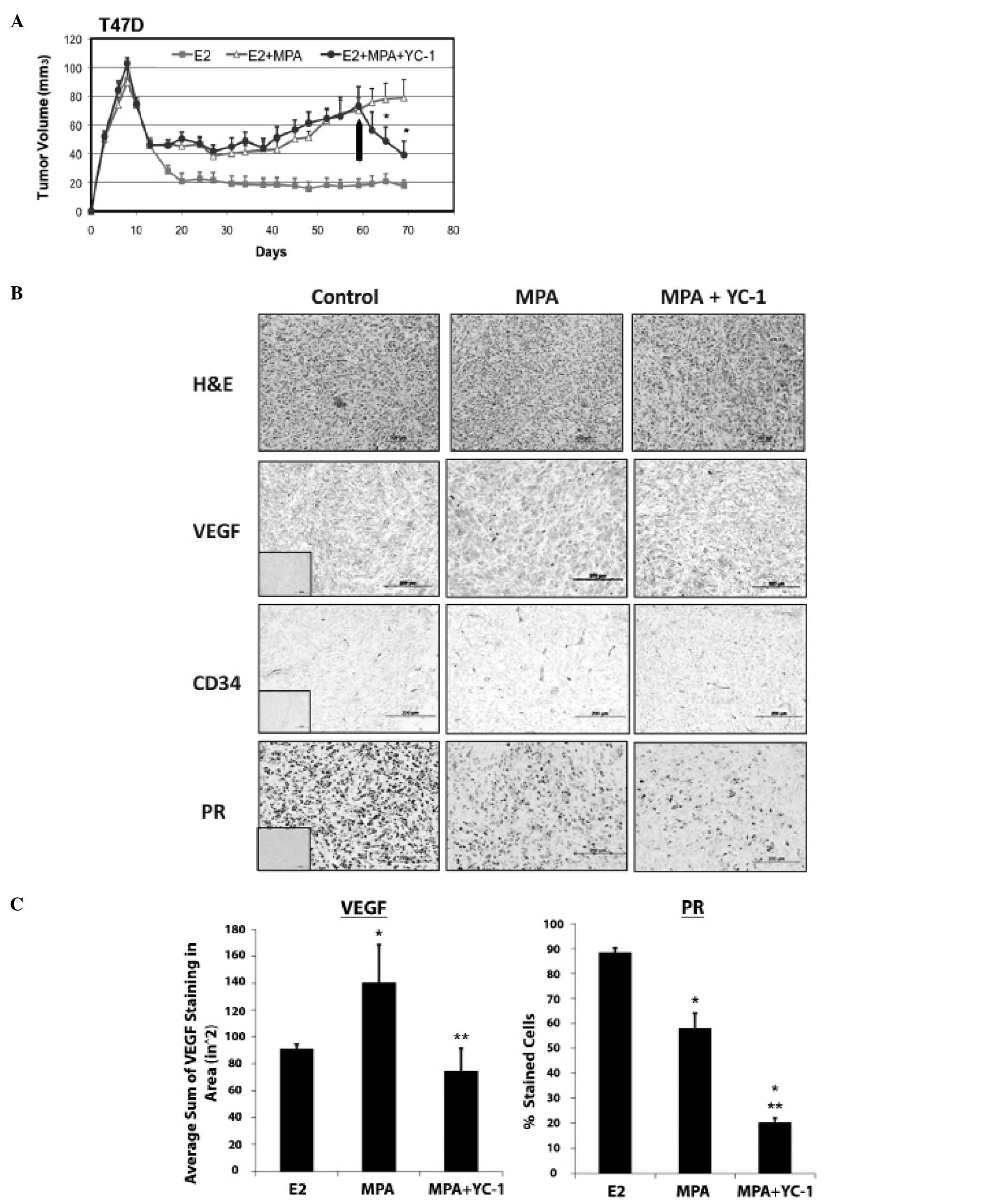 | Figure 2.YC-1 inhibits progression of
MPA-dependent breast cancer xenograft tumors in nude mice. (A),
Xenograft tumors derived from T47D-cells were grown as described in
Materials and methods. Tumor bearing mice were treated with YC-1 or
vehicle alone (i.p., arrow) as described in Materials and methods
(n=5, 10 tumors per group). *Significantly different
from controls as analyzed by one-way ANOVA, p<0.05. (B),
Immunohistochemical analysis of MPA-dependent, T47D-derived tumors.
H&E, VEGF, CD34 and PR expression were analyzed in control
(mice containing E2 pellets), untreated (E2 + MPA +
vehicle) and treated (E2 + MPA + YC-1) tumors. Collected
tumor tissues were sectioned and immunostained for the proteins
shown, as described in Materials and methods. Insets represent
negative controls with no primary antibody staining for each
antibody. Original magnification, ×20. (C), Quantification of VEGF
and PR immunostaining from tissues analyzed in (B). Four fields
from each tumor section were analyzed to control for variation due
to cellularity (control, n=3 tumors; MPA + vehicle, n=3 tumors; MPA
+ YC-1, n=4 tumors). VEGF staining (left panel): positive VEGF
staining was quantified as the number of VEGF-positive pixels in
different fields using FoveaoPro 3.0 analysis software. Error bars
represent SEM. *Statistical significance compared to
control (p<0.001; ANOVA). **Statistical significance
compared to MPA + vehicle (p<0.001; ANOVA). PR staining (right
panel) percentage of positive cells in 6 fields of each section was
counted. Error bars represent SEM (*p<0.001). |
Immunohistochemical studies were performed on
MPA-exposed T47-D xenograft tumors from YC-1 and vehicle-treated
mice. Tissues for these studies were isolated from animals
sacrificed 2 h after the final injection with YC-1. As expected,
VEGF staining was higher in tumors from MPA-treated mice, and
exposure to YC-1 reduced VEGF expression by a statistically
significant amount (Fig. 2B and
C). Although the difference was not statistically significant,
YC-1 also appeared to reduce expression of CD34 in tumors from
MPA-treated mice (Fig. 2C; 16±2.3,
n=3, MPA + YC-1 vs. 20±5.5, n=5, MPA); this finding is consistent
with the possibility that YC-1 inhibits angiogenesis in T47-D
xenograft tumors under the chosen experimental conditions.
YC-1 reduces PR levels in vivo
Immunohistochemical analysis of PR expression was
also carried out on T47-D xenograft tumors from YC-1 treated mice.
Fig. 2B and C (right panel) shows
that PR levels were significantly lower in tumors from mice treated
with MPA or MPA/YC-1 than in tumors from control animals treated
with only E2 [all animals were administered E2 by implant to
facilitate initial tumor formation; however E2 alone does not lead
to tumor growth under the conditions used for growing P-dependent
tumors (8,27)]. The difference in PR expression
between each of these three groups was statistically significant,
suggesting that different mechanisms are responsible for repression
of PR by YC-1 and MPA. Additional studies showed that the
expression of ERα and ERβ is not modulated by YC-1 or MPA in the
context of this mouse xenograft tumor model (data not shown).
YC-1 reduces PR levels in vitro and
inhibits progestin-dependent transcription of the VEGF gene
In order to confirm in vivo observations that
YC-1 downregulates VEGF and PR, we conducted in vitro assays
and examined the effect of YC-1 on transcription of VEGF mRNA in
MPA-treated cells. For this study, T47-D cells were incubated for
30 min in the presence of 100 μM YC-1 or 1 μM RU-486,
followed by 6 h with 10 nM MPA (23). YC-1 and RU-486 completely blocked
progestin-stimulated VEGF transcription (Fig. 3A). The effect of YC-1 on levels of
PR and HIF-1α was also determined in treated and untreated T47-D
cells. Fig. 3B demonstrates that
exposure to YC-1 for 6 h ± MPA resulted in reduced expression
and/or stability of PR in T47-D cells. A similar effect was
observed in cells exposed to progestins which activate PR [coupled
to receptor degradation (30) and
stimulates secretion of VEGF (21–23)]. PR expression was lower in those
cells treated with a combination of MPA and YC-1 compared with
cells treated with only YC-1, suggesting different mechanisms of
action. Such an effect was also observed in vivo (see
above). YC-1 did not promote degradation of HIF-1α in T47-D breast
cancer cells under the conditions employed, indicating that PR loss
did not occur as a result of a generalized cytotoxic effect of
YC-1. β-actin was used as loading control.
YC-1 inhibits progression of DMBA-induced
MPA-driven mammary tumors and reduces expression of VEGF in tumor
cells
Our previous studies showed that MPA stimulates
expression and secretion of VEGF and accelerates the development of
DMBA-induced mammary tumors in female Sprague-Dawley rats (7). In the present study we sought to
determine whether YC-1 inhibits mammary tumor progression in this
model system. DMBA was administered to female Sprague-Dawley rats
(45–50 days old), MPA pellets (25 mg, 60-day release) were
implanted subcutaneously on day 28 and tumors were allowed to
develop. On day 68, tumor-bearing animals were treated with YC-1
(3.75 mg/rat/day) as described in Materials and methods. YC-1
administration continued for 5 consecutive days; control animals
were treated with vehicle (Fig.
4A). At the commencement of YC-1 treatment tumors ranged in
size from 2 to 100 mm3 (tumors grow at different rates
in this animal model system and new tumors develop at various times
resulting in variations in size). Animals were weighed to monitor
drug toxicity. Tumors were measured and palpated daily during the
course of treatment and 2–3 times per week after the cessation of
daily injections until the end of the study. As expected a
continuous increase in tumor size occurred in vehicle-treated
animals, while YC-1 inhibited growth and progression of tumors with
an original size up to 100 mm3, the largest size of
tumor treated (Fig. 3A). The
effect of YC-1 lasted for 20 days after the last injection of the
drug, at which point the experiment was terminated and tumors
removed for immunohistochemical analysis. Based on animal weight,
YC-1 was not toxic at this dose (data not shown).
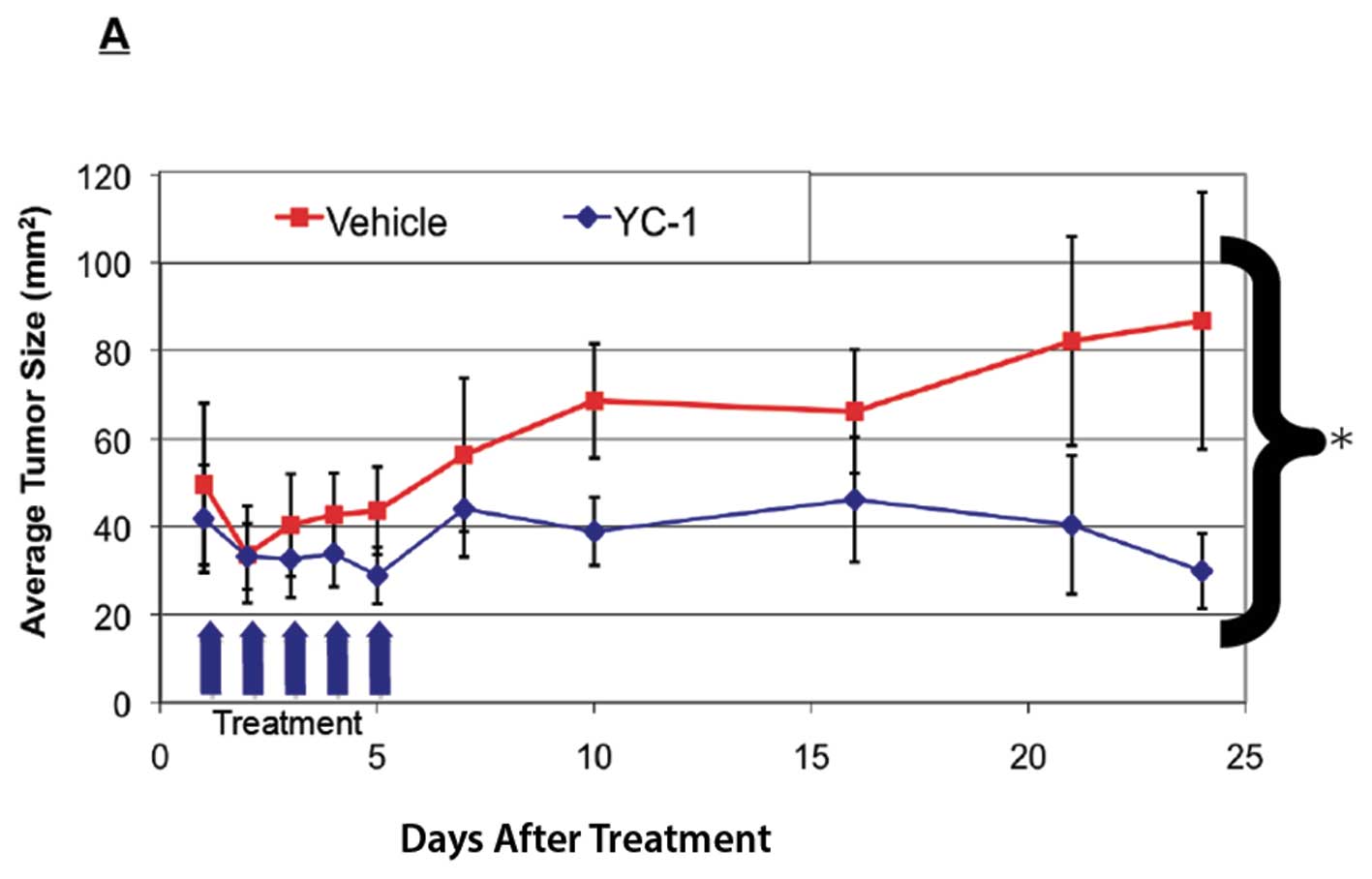 | Figure 4.(A), YC-1 suppresses
progestin-induced progression of DMBA-induced mammary tumors in
rats. Sprague-Dawley rats were treated with 20 mg DMBA via gavage.
After 4 weeks, 25 mg/60-day timed release pellets of MPA were
implanted as described in Materials and methods. Beginning on day
68, 3.75 mg YC-1, or vehicle, was administered to animals for a
further 5 days via tail vein injection. Error bars represent SEM,
n=3 animals for control group (7 tumors) and 4 animals in the YC-1
treatment group (9 tumors). *Significant difference
between control and YC-1 treated group as analyzed by t-test,
p<0.05. (B), Immunohistochemical analysis of tumor tissues from
DMBA animal studies shown in (A). Collected tumor tissues were
sectioned and immunostained for the designated protein as described
in Materials and methods. Insets represent no antibody control.
Original magnification, ×20. Bar represents 50 μm except in
figures containing Tomato lectin staining, where bar represents 100
μm. |
Although the histology of DMBA-induced mammary
tumors from animals administered YC-1 did not differ greatly from
those given vehicle, immunohistochemical analysis showed a
significant reduction in VEGF staining in tumors obtained from YC-1
treated animals (Fig. 4B, upper
panel), together with a corresponding decrease in CD34 staining
(Fig. 4B, middle panel),
suggesting a reduced number of blood vessels. The reduction in
CD34-positive blood vessels was not statistically significant;
however, blood vessels in tumors collected from YC-1 treated rats
were also smaller in diameter, further suggesting a reduced blood
supply compared with tumors from control animals not administered
YC-1. This was confirmed by a significantly reduced rate of blood
perfusion in tumors from YC-1-treated rats (Fig. 4B, lower panel; see Materials and
methods for experimental details). As noted above, YC-1 had no
effect on the levels of expression of PR, ERα or ERβ in
DMBA-induced mammary tumors in rats (data not shown). However,
receptor expression was examined at just one time point, twenty
days after the last injection of YC-1, which may allow PR to
recover. Consequently additional studies of hormone receptor
expression in YC-1 treated DMBA-initiated rats are needed.
Discussion
Recently a number of studies and clinical trials
have shown that HRT which contains a progestin component is
associated with a higher risk of metastatic breast cancer than HRT
lacking progestin (1–4). Anti-progestins, such as RU-486,
cannot be used to mitigate this risk, because of their lack of
specificity and severe adverse effects (9). Because a large number of
postmenopausal women who are already at significant breast cancer
risk are routinely exposed to progestin-containing HRT, there is an
urgent need to develop viable alternative strategies that can be
rapidly developed for clinical use, to lower the risk of or even
prevent breast cancer. Here we report the results of preclinical
studies in which we evaluated the potential of YC-1, a recently
developed anticancer agent, to prevent the progestin-dependent
progression of human breast cancer.
Progestins induce VEGF in breast cancer cells
(29) in a PR-dependent manner and
cause tumor progression and metastasis in animal models (8,27).
VEGF expression within tumor cells is under the control of a number
of essential transcription factors, one of the most important being
HIF-1α, which regulates survival genes, including VEGF (14,15).
Higher levels of HIF1α and VEGF are associated with increased risk
of tumor metastasis (27,30). Interestingly, recent studies also
provide evidence that HIF-1α is involved in hormone mediated
regulation of VEGF (13,31). Thus HIF-1α appears to play a
central role in the regulation of VEGF by steroid hormones in
reproductive tissues, though a direct role of HIF-1α in nuclear
receptor mediated events remains to be established. Since the
induction of VEGF by progestins is well established in breast
cancer cells (8,9,21–23,29)
and it is possible that one mechanism through which this occurs
involves HIF-1α, we undertook studies using both
naturally-occurring and synthetic progestins and found that YC-1
inhibits induction of VEGF in both T47-D and BT-474 cells.
Synthetic progestins which are used in various formulations
worldwide as HRT all induce VEGF and have the potential to promote
tumor metastasis (27). Although
previous studies have shown that YC-1 inhibits angiogenesis
(32), ours is the first to
demonstrate that it blocks progestin-dependent angiogenesis. We
propose therefore that YC-1 could be a useful drug which might be
used clinically to prevent progestin-dependent tumor growth and
metastasis. To lend further support to the notion that YC-1 might
prove effective against progestin-dependent breast disease, we
found that it also suppressed PR levels in breast cancer cells,
both in vitro and in vivo. The effect of YC-1 could
therefore be 2-fold; inhibition of HIF-1α and downregulation of PR,
making it a uniquely suitable small molecule drug for combating
those forms of breast cancer which are largely dependent on the
actions of progestins to stimulate the production of VEGF. Further
studies are required to determine the molecular mechanism through
which YC-1 brings about the loss of PR, since it is possible that
HIF-1α regulates PR and YC-1 shuts down PR synthesis due to loss of
HIF-1α activity.
This study demonstrates that YC-1 effectively
inhibits the growth of progestin-accelerated xenograft tumors
derived from T47-D or BT-474 cells in mice and DMBA-induced tumors
in rats, supporting the notion that it possesses significant
potential as an anti-breast cancer agent in vivo. Although
the exact mechanism by which YC-1 exerts its in vivo effects
has not yet been established, we propose that YC-1, by inhibiting
progestin-mediated VEGF-dependent angiogenesis, reduces blood flow
to xenograft tumors in these experimental model systems.
Alternatively, YC-1-mediated downregulation of PR could play a key
role in the anti-tumor effectiveness of this HIF-1α inhibitor, a
possibility which remains to be tested. Since ER levels were not
affected by YC-1, it is likely that combination therapy using YC-1
and an anti-estrogen might be more effective than therapy using
either compound alone, another scenario that requires further
study.
In the Sprague-Dawley rat DMBA-induced
progestin-accelerated tumor model, YC-1 prevented the progression
of progestin-dependent tumors and inhibited expression of VEGF, but
did not downregulate PR. However, since these tumors were analyzed
several days after the end of YC-1 treatment it is possible that PR
was re-expressed in tissues over time. While there was no
significant reduction in the number of blood vessels in tumors from
YC-1-treated animals, blood vessels were smaller and had reduced
capacity for perfusion (Fig. 4B),
a phenomenon we also observed in other experimental models
(24,33). It therefore appears that YC-1
inhibits tumor growth in the DMBA-induced progestin-accelerated
tumor primarily by downregulating VEGF. However, additional
analysis of the kinetics of PR expression during studies such as
these is warranted.
In conclusion, this study provides strong evidence
that YC-1, a potent HIF-1α inhibitor, may be a useful pharmacologic
agent which might be used to treat and possibly prevent
PR-dependent breast cancer as a result of promoting PR loss. This
could be especially valuable in a clinical context to mitigate the
increased breast cancer risk associated with progestin-containing
HRT.
Acknowledgements
This work was supported by NIH grants
R56CA-86916 and 1F31CA130167; COR award from College of Veterinary
Medicine, and Research Funds from RADIL, University of Missouri.
S.M.H. is the Zalk Missouri Professor of Tumor Angiogenesis.
References
|
1.
|
Ross R, Paganini-Hill A and Pike MC:
Effect of hormone replacement therapy on breast cancer risk:
estrogen versus estrogen plus progestin. J Natl Cancer Inst.
92:328–332. 2000. View Article : Google Scholar
|
|
2.
|
Schairer C, Lubin J, Troisi R, Sturgeon S,
Brinton L and Hoover R: Menopausal estrogen and estrogen progestin
replacement therapy and breast cancer risk. JAMA. 283:485–491.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Li CI, Malone KE, Porter PL, Weiss NS,
Tang MT, Cushing-Haugen KL and Daling JR: Relationship between long
durations and different regimens of hormone therapy and risk of
breast cancer. JAMA. 289:3254–3263. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Chlebowski RT, Anderson GL, Gass M, Lane
DS, Aragaki AK, Kuller LH, Manson JE, Stefanick ML, Ockene J, Sarto
GE, Johnson KC, Wactawski-Wende J, Ravdin PM, Schenken R, Hendrix
SL, Rajkovic A, Rohan TE, Yasmeen S and Prentice RL; WHI
Investigators: Estrogen plus progestin and breast cancer incidence
and mortality in postmenopausal women. JAMA. 304:1684–1692. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Liang Y, Wu J, Stancel GM and Hyder SM:
p53-dependent inhibition of progestin-induced VEGF expression in
human breast cancer cells. J Steroid Biochem Mol Biol. 93:173–182.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Liang Y and Hyder SM: Proliferation of
endothelial and tumor epithelial cells by progestin-induced
vascular endothelial growth factor from human breast cancer cells:
paracrine and autocrine effects. Endocrinology. 146:3632–3641.
2005. View Article : Google Scholar
|
|
7.
|
Benakanakere I, Besch-Williford C, Schnell
J, Brandt S, Ellersieck MR, Molinolo A and Hyder SM: Natural and
synthetic progestins accelerate
7,12-dimethylbenz[a]anthracene-initiated mammary tumors and
increase angiogenesis in Sprague-Dawley rats. Clin Cancer Res.
12:4062–4071. 2006.PubMed/NCBI
|
|
8.
|
Liang Y, Besch-Williford C, Brekken RA and
Hyder SM: Progestin-dependent progression of human breast tumor
xenografts: a novel model for evaluating antitumor theraputics.
Cancer Res. 67:9929–9936. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Horwitz K: The molecular biology of
RU-486. Is there a role for anti-progestins in the treatment of
breast cancer? Endocr Rev. 12:146–163. 1992.PubMed/NCBI
|
|
10.
|
Otrock ZK, Hatoum HA, Awada AH, Ishak RS
and Shamseddine AI: Hypoxia-inducible factor in cancer
angiogenesis: structure, regulation and clinical perspectives. Crit
Rev Oncol Hematol. 70:93–102. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Carmeliet P, Dor Y, Herbert J-M, Fukumura
D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R,
Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D and
Keshet E: Role of HIF-1[alpha] in hypoxia-mediated apoptosis, cell
proliferation and tumour angiogenesis. Nature. 394:485–490.
1998.
|
|
12.
|
Forsythe JA, Jiang BH, Iyer NV, Agani F,
Leung SW, Koos RD and Semenza GL: Activation of vascular
endothelial growth factor gene transcription by hypoxia-inducible
factor 1. Mol Cell Biol. 16:4604–4613. 1996.PubMed/NCBI
|
|
13.
|
Shimizu T and Miyamoto A: Progesterone
induces the expression of vascular endothelial growth factor (VEGF)
120 and Flk-1, its receptor, in bovine granulose cells. Anim Reprod
Sci. 102:228–237. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Semenza GL: HIF-1: using two hands to flip
the angiogenic switch. Cancer Metastasis Rev. 19:59–65. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Chun YS, Yeo EJ, Choi E, Teng CM, Bae JM,
Kim MS and Park JW: Inhibitory effect of YC-1 on the hypoxic
induction of erythropoietin and vascular endothelial growth factor
in Hep3B cells. Biochem Pharmacol. 61:947–954. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Tulis DA, Durante W, Peyton KJ, Chapman
GB, Evans AJ and Schafer AI: YC-1, a benzyl indazole derivative,
stimulates vascular cGMP and inhibits neointima formation. Biochem
Biophys Res Commun. 279:646–652. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Huang YT, Pan SL, Guh JH, Chang YL, Lee
FY, Kuo SC and Teng CM: YC-1 suppresses constitutive nuclear
factor-kappaB activation and induces apoptosis in human prostate
cancer cells. Mol Cancer Ther. 4:1628–1635. 2005. View Article : Google Scholar
|
|
18.
|
Yeo E-J, Chun YS, Cho YS, Kim J, Lee J-C,
Kim M-S and Park J-W: YC-1: A potential anticancer drug targeting
hypoxiainducible factor 1. J Natl Cancer Inst. 95:516–525. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Rubinstein LV, Shoemaker RH, Paull KD,
Simon RM, Tosini S, Skehan P, Scudiero DA, Monks A and Boyd MR:
Comparison of in vitro anticancer-drug-screening data generated
with a tetrazolium assay versus a protein assay against a diverse
panel of human tumor cell lines. J Natl Cancer Inst. 82:1113–1118.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Skehan P, Storeng R, Scudiero D, Monks A,
McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S and Boyd MR:
New colorimetric cytotoxicity assay for anticancer-drug screening.
J Natl Cancer Inst. 82:1107–1112. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Carroll CE, Ellersieck MR and Hyder SM:
Curcumin inhibits MPA-induced secretion of VEGF from T47-D human
breast cancer cells. Menopause. 15:570–574. 2008. View Article : Google Scholar
|
|
22.
|
Hyder S, Murthry L and Stancel GM:
Progestin regulation of vascular endothelial growth factor in human
breast cancer cells. Cancer Res. 58:392–395. 1998.PubMed/NCBI
|
|
23.
|
Mafuvadze B, Benakanakere I and Hyder SM:
Apigenin blocks induction of vascular endothelial growth factor
mRNA and protein in progestin-treated human breast cancer cells.
Menopause. 17:1055–1063. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Benakanakere I, Besch-Williford C,
Ellersieck MR and Hyder SM: Regression of progestin-accelerated
7,12-dimethylbenz[a] anthracene-induced mammary tumors in
Sprague-Dawley rats by p53 reactivation and induction of massive
apoptosis: a pilot study. Endocr Relat Cancer. 16:85–98.
2009.PubMed/NCBI
|
|
25.
|
Conover W and Iman RL: Rank
transformations as a bridge between parametric and nonparametric
statistics. Am Stat. 35:124–129. 1981.
|
|
26.
|
Chew V: Comparison among treatment means
and analysis of variance. Agricultural Research Service, USDA
(ARS/H/6 Report); Washington, DC: pp. 32–35. 1977
|
|
27.
|
Liang Y, Benakanakere I, Besch-Williford
C, Hyder RS, Ellersieck MR and Hyder SM: Synthetic progestins
induce growth and metastasis of BT-474 human breast cancer
xenografts in nude mice. Menopause. 17:1040–1047. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Dennis AP, Lonard DM, Nawaz Z and O’Malley
BW: Inhibition of the 26S proteasome blocks progesterone
receptor-dependent transcription through failed recruitment of RNA
polymerase II. J Steroid Biochem Mol Biol. 94:337–346. 2005.
View Article : Google Scholar
|
|
29.
|
Hyder SM, Chiappetta C and Stancel GM:
Pharmacological and endogenous progestins induce vascular
endothelial growth factor expression in human breast cancer cells.
Int J Cancer. 92:469–473. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Rundqvist H and Johnson RS: Hypoxia and
metastasis in breast cancer. Curr Top Microbiol Immunol.
345:121–139. 2010.PubMed/NCBI
|
|
31.
|
Kazi A, Jones JM and Koos RD: Chromatin
immunoprecipitation analysis of gene expression in the rat uterus
in vivo: estrogen-induced recruitment of both estrogen receptor α
and hypoxia-inducible factor 1 to the vascular endothelial growth
factor promoter. Mol Endocrinol. 19:2006.–2019. 2005.
|
|
32.
|
Pan SL, Guh JH, Peng CY, Wang SW, Chang
YL, Cheng FC, Chang JH, Kuo SC, Lee FY and Teng CM: YC-1
[3-(5′-hydroxymethyl-2′-furyl)-1-benzyindazole] inhibits
endothelial cell functions induced by angiogenic factors in vitro
and angiogenesis in vivo models. J Pharmacol Exp Ther. 314:35–42.
2005.
|
|
33.
|
Liang Y, Besch-Williford C, Benakanakere
I, Thorpe PE and Hyder SM: Targeting mutant p53 protein and the
tumor vasculature: an effective combination therapy for advanced
breast tumors. Breast Cancer Res Treat. 125:407–420. 2010.
View Article : Google Scholar : PubMed/NCBI
|