Introduction
Head and neck squamous cell carcinoma (HNSCC) is one
of the most frequently occurring malignancies and a major cause of
morbidity and mortality (1,2).
Oral cancer is the most common among the HNSCCs and the most
frequently occurring oral cancer is squamous cell carcinoma (OSCC),
which accounts for more than 90% of all oral malignancies (3). The number of OSCC cases that occur
worldwide annually exceeds 300,000 (4). The mechanisms behind tumor
progression of OSCC are known to a limited extent, indicating a
clear need for comprehensive knowledge that can lead to more
specific and effective molecular target.
Microarray technology facilitates simultaneous
evaluation of thousands of genes in a specimen. The results of
microarray analysis provide researchers with high throughput
screening to study the roles played by specific genes in cancer
development and progression. We previously reported gene expression
profiling of OSCC to identify cancer-related genes (5,6).
The four and a half LIM domains (FHL) family
of genes is characterized by LIM domains, a term derived from the
first letters of three transcription factors: Lin-11, Isl-1 and
Mec-3. Because the LIM domains provide protein-protein binding
interfaces, the FHL genes play an important role in cellular
events such as focal adhesion and differentiation by repressing or
activating target protein (7).
FHL1 protein is highly expressed in skeletal muscle, where it
localizes to sarcomeres and sarcolemmas (8) and is expressed at intermediate levels
in cardiac muscle (9,10). The molecules associated with
intercellular adhesion might be inhibited during tumor progression
(11). Accordingly, the expression
of the FHL family member FHL1, located on human
chromosome Xq26, was downregulated in various types of malignancies
including lung, prostate and breast cancer (12).
Epigenetic alterations including hypermethylation of
the promoter regions are consistent and early events in neoplastic
progression. Such alterations are thought to contribute to the
neoplastic process by transcriptional silencing of tumor suppressor
gene expression. DNA methylation is reversible because it does not
alter the DNA sequence; however, it is heritable from cell to cell
(13). Epigenetic mechanisms have
emerged recently as major determinants of gene expression and are
implicated in the regulation of complex differentiation and
developmental processes, both under physiologic and pathological
conditions. In addition to hypermethylation of the promoter
regions, histone modification, particularly histone acetylation, is
a key factor in epigenetic regulation (14). Histone deacetylation, which is
associated with a repressed chromatin state, is tightly controlled
by two classes of enzymes, histone acetyltransferases and histone
deacetylases (HDAC) (15).
Inhibitors of HDAC display anti-cancer activities and are,
therefore, of growing clinical interest (16).
In the present study, we showed that FHL1 is
significantly downregulated in the OSCC-derived cell lines and
tissue samples from patients with OSCC and the methylation status
was significantly higher in OSCC specimens than in human normal
oral keratinocyte (HNOK) specimens. Treatment with the DNA
methyltransferase inhibitor, 5-aza-2′-deoxycytidine (5-aza-dC)
restored FHL1 mRNA expression in the OSCC-derived cell lines. In
contrast, no significant restoration of FHL1 expression was
observed when using sodium butyrate (NaB), a histone deacetylase
inhibitor and the FHL1 promoter region was significantly acetylated
in the OSCC-derived cell lines. Direct sequencing did not detect
any mutation in the entire coding region of the FHL1 gene.
These results suggested that downregulation of FHL1 expression in
OSCC-derived cell lines and HNOKs is correlated with DNA
hypermethylation of the FHL1 promoter region.
Materials and methods
Ethics statement
All patients provided informed consent after the
study protocol was explained. The institutional review board of
Chiba University approved the study.
OSCC-derived cell lines and tissue
specimens
The HSC-3, HO-1-N-1 and KON cell lines, derived from
human OSCCs, were purchased from the Human Science Research
Resources Bank (Osaka, Japan). The RIKEN BRC provided the Sa3,
HSC-2, HSC-4, HO-1-u-1 and Ca9-22 cell lines through the National
Bio-Resource Project of the MEXT (Ibaraki, Japan). Short tandem
repeat profiles confirmed cell identity. Primary cultured HNOKs
were obtained from three healthy donors and served as the normal
controls (17–21). All cells were grown in Dulbecco’s
modified Eagle’s medium (Sigma, St. Louis, MO) supplemented with
10% fetal bovine serum (Sigma) and 50 U/ml penicillin and
streptomycin (Sigma).
Tissue samples from 59 unrelated Japanese patients
with primary OSCCs who were treated at the Chiba University
Hospital were obtained during surgical resection. The resected
tissues were divided into two parts, one of which was frozen
immediately and stored at −80°C until RNA isolation and the second
was fixed in 20% buffered formaldehyde solution for pathologic
diagnosis and immunohistochemistry (IHC). Histopathologic analysis
of the tissues was performed according to the World Health
Organization criteria by the Department of Pathology, Chiba
University Hospital. Clinicopathologic staging was determined by
the tumor-node-metastases classification of the International Union
Against Cancer. All patients had OSCC that was confirmed
histologically and tumor samples were checked to ensure that tumor
tissue was present in >90% of the specimen.
Preparation of cDNA
Total RNA was isolated using TRIzol Reagent
(Invitrogen, Carlsbad, CA) according to the manufacturer’s
instructions. cDNA was generated from 5 μg of total RNA
using Ready-To-Go You-Prime First-Strand Beads (GE Healthcare,
Buckinghamshire, UK) and oligo(dT) primer (Hokkaido System Science,
Hokkaido, Japan), according to the manufacturer’s instructions.
mRNA expression analysis
Quantitative reverse-transcriptase-polymerase chain
reaction (qRT-PCR) was performed to evaluate the expression levels
of the target gene (FHL1) in OSCC-derived cell lines using
the Light Cycler®II 480 (Roche Diagnostics, Penzberg,
Germany). The primer sequences used to analyze FHL1 mRNA
expression were forward 5′-GAAGTGTGCTGGATG CAAGA-3′ and reverse
5′-GGGGGCTTCCTAGCTTTAGA-3′. The PCR reactions were carried out in a
final volume of 20 μl of a reaction mixture comprised of 0.4
μl (0.5 μM) of each primer, 0.2 μl (0.2
μM) of universal probe (Roche Diagnostics), 4 μl of
PCR grade water, 10 μl of 2x Probes Master (Roche
Diagnostics) and 5 μl of cDNA template according to the
manufacturer’s instructions. The qRT-PCR amplification was
performed according to the following cycle parameters: an initial
denaturation at 95°C for 5 min, 45 cycles of amplification at 95°C
(10 sec) for denaturation, 60°C (30 sec) for annealing and 72°C (1
sec) for extension. Following the amplification phase, a cooling
step was performed at 50°C for 30 sec. The transcript amounts for
the target genes were estimated from the respective standard curves
and normalized to the glyceraldehyde-3-phosphate dehydrogenase
(GAPDH) (forward 5′-GCTCTCTGCTCCTCCTGTTC-3′ and reverse
5′-ACGACCAAATCCGTTGACTC-3′) transcript amount determined in
corresponding samples. All amplifications were repeated three times
using cDNA prepared from three independent experiments.
Protein extraction
The cells were washed twice with cold
phosphate-buffered saline (PBS) and centrifuged briefly. The cell
pellets were incubated on ice for 30 min in a lysis buffer (7 M
urea, 2 M thiourea, 4% w/v CHAPS and 10 mM Tris pH 7.4) with a
proteinase inhibitor cocktail (Roche Diagnostics). The protein
concentration was measured with Bradford reagent (Bio-Rad,
Richmond, CA).
Western blot analysis
Protein extracts were electrophoresed on 4–12%
Bis-Tris gel, transferred to nitrocellulose membranes (Invitrogen)
and blocked for 1 h at room temperature in Blocking One (Nacalai
Tesque, Kyoto, Japan). The membranes were washed three times with
0.1% Tween-20 in Tris-buffered saline and incubated with 0.1
μg/ml rabbit anti-FHL1 polyclonal antibody (Aviva Systems
Biology, San Diego, CA) overnight at 4°C. The membranes were washed
again and incubated with a 1:2,500 of anti-rabbit IgG (H+L)
horseradish peroxidase (HRP) conjugate (Promega, Madison, WI) as a
secondary antibody for 1 h at room temperature. The proteins were
detected by SuperSignal Chemiluminescent substrate (Thermo,
Waltham, MA). Finally, the western blot analysis results were
visualized by exposing the membrane to a cooled CCD camera system,
Light-Capture II (ATTO, Tokyo, Japan). Signal intensities were
quantitated using the CS Analyzer version 3.0 software (ATTO).
IHC
IHC of 4-μm sections of paraffin-embedded
specimens was performed using rabbit anti-FHL1 polyclonal antibody
(Aviva Systems Biology). Briefly, after deparaffinization and
hydration, the slides were pretreated in 10 mM sodium citrate
buffer (pH 6.0) in a microwave oven for 5 min at 95°C. The
endogenous peroxidase activity was quenched by a 30-min incubation
in a mixture of 0.3% hydrogen peroxide solution in 100% methanol,
after which the specimens were blocked for 1 h at room temperature
with 1.5% blocking serum (Santa Cruz Biotechnology, Santa Cruz, CA)
in PBS before reaction with anti-FHL1 antibody (4.0 μg/ml)
at 4°C in a moist chamber overnight. Upon incubation with the
primary antibody, the specimens were washed three times in PBS and
treated with Envision System-HRP Labeled Polymer (Dako,
Carpinteria, CA) followed by color development in
3,3′-diaminobenzidine tetrahydrochloride (Dako). The slides were
then lightly counterstained with hematoxylin, dehydrated with
ethanol, cleaned with xylene and mounted. To quantify the state of
FHL1 protein expression in those components, we used IHC score
systems described previously (18,22–30).
Briefly, the stained cells were determined in at least five random
fields at magnification ×400 in each section. The intensity of the
FHL1 immunoreaction in the cells was scored as follows: 1+, weak;
2+, moderate; and 3+, intense. The cell number and the staining
intensity then were multiplied to produce an FHL1 IHC score. Cases
with a score exceeding 36.5 (±3 standard deviation) (SD) score for
normal tissue) were defined as FHL1-positive. The ±3 SD cutoff,
which statistically is 0.2% of the measurement and is expected to
fall outside this range, was used because it was unlikely to be
affected by a random experimental error produced by sample
manipulation (31). Two
independent pathologists, both of whom were masked to the patients’
clinical status, made these judgments.
Cell culture and 5-aza-2′-deoxycytidine
treatment
To assess reactivation of FHL1 gene
expression, OSCC derived cell lines were treated with different
concentrations (0 and 2 μM) of the DNA methyltransferase
inhibitor, 5-aza-dC (Wako, Osaka, Japan). On day 7, the cells were
harvested, total RNA was extracted and qRT-PCR was performed
(19).
Methylation-specific PCR
To determine if methylation of a CpG island of the
FHL1 promoter region contributes to the mRNA expression of FHL1,
DNA samples obtained from OSCC-derived cell lines were applied for
the methylation-specific PCR (MSP) assay. Bisulfite conversion was
carried out using the EZ DNA Methylation Direct kit (Zymo Research,
Orange, CA) according to the manufacturer’s instructions with the
following modifications. Briefly, 1×105 cells of genomic
DNA was denatured by the addition of digestion mixture, which
contained 13 μl of M-Digestion Buffer (Zymo Research), 6
μl PCR grade water and 1 μl Proteinase K (Zymo
Research). After incubation at 50°C for 20 min, the 20 μl
sample was added to 130 μl of CT Conversion Reagent (Zymo
Research) in a PCR tube. The tubes were placed in the thermal
cycler (Applied Biosystems, Foster City, CA) for 8 min at 98°C for
DNA denaturation and 3.5 h at 64°C for bisulfite conversion. The
modified DNA then was desalted, purified using Zymo-Spin IC Column
(Zymo Research) and eluted with 10 μl elution buffer.
Bisulfite modified DNA was used as a template and then MSP was
performed. The primers for MSP were designed using MethPrimer
software (http://www.urogene.org/methprimer) (32). The primer sequences were: FHL1
methylated forward, 5′-GTAAGTTATCGGGTTT CGAAGTC-3′; FHL1 methylated
reverse, 5′-ACAACCAAATA AAAATAACGTCTCG-3′; FHL1 unmethylated
forward, 5′-TGTAAGTTATTGGGTTTTGAAGTTG-3′; and FHL1 unmethylated
reverse 5′-AACCAAATAAAAATAACATCTC ACC-3′. The PCR reactions were
performed in a final volume of 25 μl containing 50 ng of
digested DNA, 0.3 μM of each specific primer, 0.25 μl
of 100X SYBR® Green I and 0.6 μl of MSP Enzyme
(Takara Bio, Shiga, Japan). PCR amplification was carried out at
95°C for 30 sec, 40 cycles at 98°C for 5 sec, 55°C for 30 sec and
72°C for 1 min. The amplified PCR products were separated on 10%
polyacrylamide gel and visualized by ethidium bromide after the
run. EpiScope® Methylated HeLa Genomic DNA (Takara Bio)
was used as a positive control for methylated alleles. HNOKs
genomic DNA was used as a negative control for unmethylated genes
(19,33).
Treatment with HDAC inhibitor and
chromatin immunoprecipitation
OSCC-derived cells were treated with a NaB, an HDAC
inhibitor, NaB (Wako). All OSCC-derived cell lines were plated at
50% confluence and treated with 5.0 mM of NaB. After a 24-h
incubation, total RNA was isolated from OSCC-derived cell lines and
qRT-PCR was performed.
Chromatin immunoprecipitation (ChIP) assays for
histone acetylation and histone methylation were performed using an
EpiScope ChIP Kit (anti-mouse IgG) (Takara Bio) according to the
manufacturer’s instructions. The OSCC-derived cell lines
(5×106 cells in a 10-cm dish) were cross-linked with 10
ml of 1% formaldehyde in medium for 5 min at room temperature and
then incubated in 2.1 ml Quenching Solution (Takara Bio) in medium
for 5 min. After rinsing cells with PBS, 1 ml of PBS was added to
harvest cells using a cell scraper. The cells were collected by
centrifugation (180 g, 3 min, 4°C) and resuspended in 1 ml
Cytoplasmic Lysis Buffer (Takara Bio) sheared with a Biomic 7040
Ultrasonic Processor (Seiko, Tokyo, Japan) to obtain 200–1,000-bp
fragments. After centrifugation (15,000 g, 3 min, 4°C) to remove
insoluble materials, 600 μl of dilution buffer (Takara Bio)
was added to the supernatant. For cross-linked ChIP, 200 μl
of Magnosphere™ anti-mouse IgG (Takara Bio) was washed with RIPA
Buffer-1 (Takara Bio), incubated with anti-acetyl histone H3 lysine
9 (4 μg) (H3K9ac), mouse monoclonal antibody (MAB Institute,
Hokkaido, Japan), anti-monomethyl histone H3 lysine 9 (4 μg)
(H3K9me), mouse monoclonal antibody (MAB Institute), or control
antibody (4 μg) (normal goat IgG, Santa Cruz) in a
2-μl proteinase inhibitor cocktail (Takara Bio), 20
μl 10X EASY Dilution (Takara Bio) and 54 μl RIPA
Buffer-1 overnight at 4°C with rotation. After two washings with
800 μl RIPA Buffer-1, an aliquot of ChIP input (10
μl) was incubated with Magnosphere anti-mouse IgG overnight
at 4°C with rotation. The beads were washed sequentially with 800
μl of RIPA Buffer-1, 800 μl of RIPA Buffer-2 and 800
μl of TE (Takara Bio) handled with a magnetic stand (Takara
Bio). After removing the TE, the beads were mixed with 100
μl chelate resin and incubated for 15 min at 95°C to reverse
cross-linking. The samples were treated with 1 μl Proteinase
K and 1 μl RNase A (Takara Bio) and incubated for 15 min at
65°C and boiled at 95°C for 10 min to deactivate the proteinase K.
After centrifugation (15,000 g, 1 min, 4°C) the supernatant was
collected. Each sample was analyzed by qRT-PCR and the results were
expressed as percentages of the input DNA. The primer sequences
were FHL1 promoter forward: 5′-TACTAAGGGGAGGGGTCGTC-3′ and FHL1
promoter reverse: 5′-GAGGTGGGAGCAACAAAGAC-3′ (34,35).
Mutation analysis
To screen the sequence variations of the FHL1
gene, direct DNA sequencing was performed as described previously
(10). Fragments encompassing each
of the six coding exons (exons 2–7) of FHL1 and their corresponding
splice junctions were amplified (primer sequences in Table I). Amplifications were performed
with FastStart Taq polymerase as described. Sequencing reactions
were carried out using BigDye V3.1 Cycle Sequencing kit (Applied
Biosystems) with the same forward primers as for PCR amplification
and sequencing products were electrophoresed on an ABI PRISM 3130xl
Genetic Analyzer (Applied Biosystems). PCR amplification was
carried out with an ABI 7500 Real-Time PCR system (Applied
Biosystems) under the following conditions: an initial denaturation
step at 95°C for 10 min, 40 cycles of 95°C for 15 sec and 60°C for
1 min and a final extension for 1 min.
 | Table I.Primers used in mutation
analysis. |
Table I.
Primers used in mutation
analysis.
| Exons | Forward primer | Reverse primer | Size (bp) |
|---|
| Exon 2 |
5′-ATGAGGGAGCCAGATCCAC-3′ |
5′-AGACATTGCCCCCGAGAG-3′ | 486 |
| Exon 3 |
5′-GTCCTCAGACCCCATGGAC-3′ |
5′-CTCAAGGTGGCTGCAGTG-3′ | 415 |
| Exon 4 |
5′-CCCAGGTGTAAGGGACTGTG-3′ |
5′-ACGTTAGCCCCACCATGA-3′ | 387 |
| Exon 5 |
5′-CATCCCCTCACCTCTGGA-3′ |
5′-TACGTCACAGGGCTGTCGT-3′ | 382 |
| Exon 6 |
5′-CCCATGGCTGTTGTGGAG-3′ |
5′-CAATAGCAGGGGGAGAAGG-3′ | 441 |
| Exon 7-1 |
5′-GCTTGTCGGTCTGTGAGTGG-3′ |
5′-ACTTTGCTGCAGGGTTGC-3′ | 484 |
| Exon 7-2 |
5′-AGGGGAAGAGTGGTCCTTCC-3′ |
5′-AGGGCAGAGTTCTGATGAGG-3′ | 426 |
Statistical analysis
The statistical significance of the FHL1 expression
levels was evaluated using Fisher’s exact test or Mann-Whitney U
test. P<0.05 was considered statistically significant. The data
are expressed as the mean ± standard error of the mean.
Results
Evaluation of FHL1 mRNA and protein
expression in OSCC-derived cell lines
To investigate mRNA and protein expression of FHL1
identified as a cancer-related gene in our previous microarray data
(6), we performed qRT-PCR and
western blot analyses using eight OSCC-derived cell lines (Sa3,
HSC-2, HSC-3, HSC-4, HO-1-u-1, HO-1-N-1, KON and Ca9-22). FHL1 mRNA
was significantly (Fig. 1,
*P<0.01) downregulated in all OSCC-derived cell lines
compared to the HNOKs. Representative results of the western blot
analysis are shown in Fig. 1. The
molecular weight of the FHL1 was 32 kDa. Mouse skeletal muscle
lysate was used as a positive control. A significant decrease in
FHL1 protein expression was observed in all OSCC-derived cell lines
compared to the HNOKs.
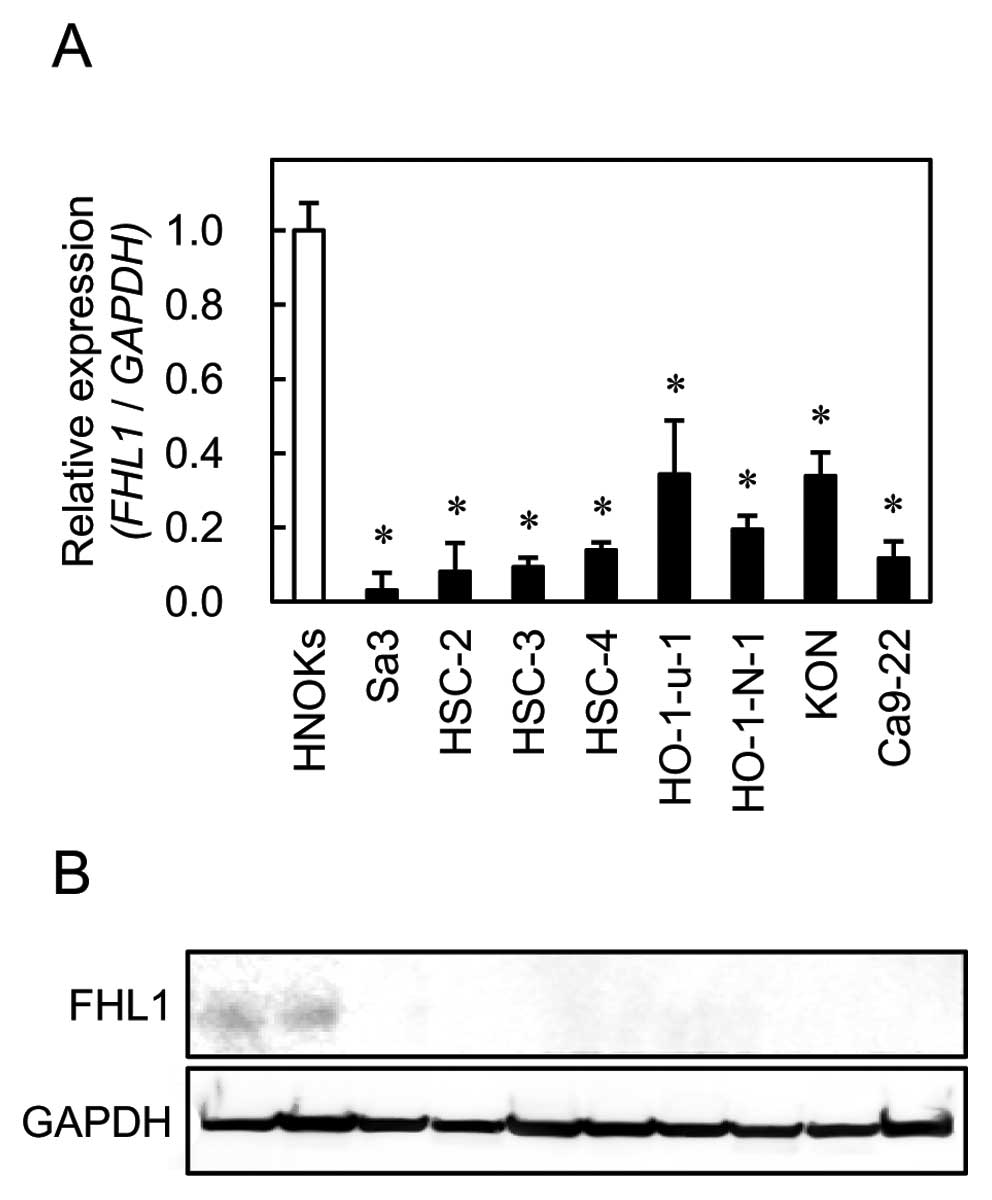 | Figure 1.Quantification of FHL1 mRNA
levels in OSCC-derived cell lines by qRT-PCR analysis (A). To
determine mRNA expression of FHL1 in OSCC, we performed
qRT-PCR analysis using eight OSCC-derived cell lines (Sa3, HSC-2,
HSC-3, HSC-4, Ca9-22, HO-1-u-1, HO-1-N-1 and Ca9-22) and HNOKs.
Significant downregulation of FHL1 mRNA is seen in all OSCC-derived
cell lines compared with that in the HNOKs. Data are expressed as
the means ± standard errors of the mean of values from three assays
(*P<0.01, Mann-Whitney U test). To investigate
protein expression of FHL1 in OSCC, we performed western
blot analysis using eight OSCC-derived cell lines (Sa3, HSC-2,
HSC-3, HSC-4, HO-1-u-1, HO-1-N-1, KON and Ca9-22) and HNOKs
(B). |
Evaluation of FHL1 expression in primary
OSCCs
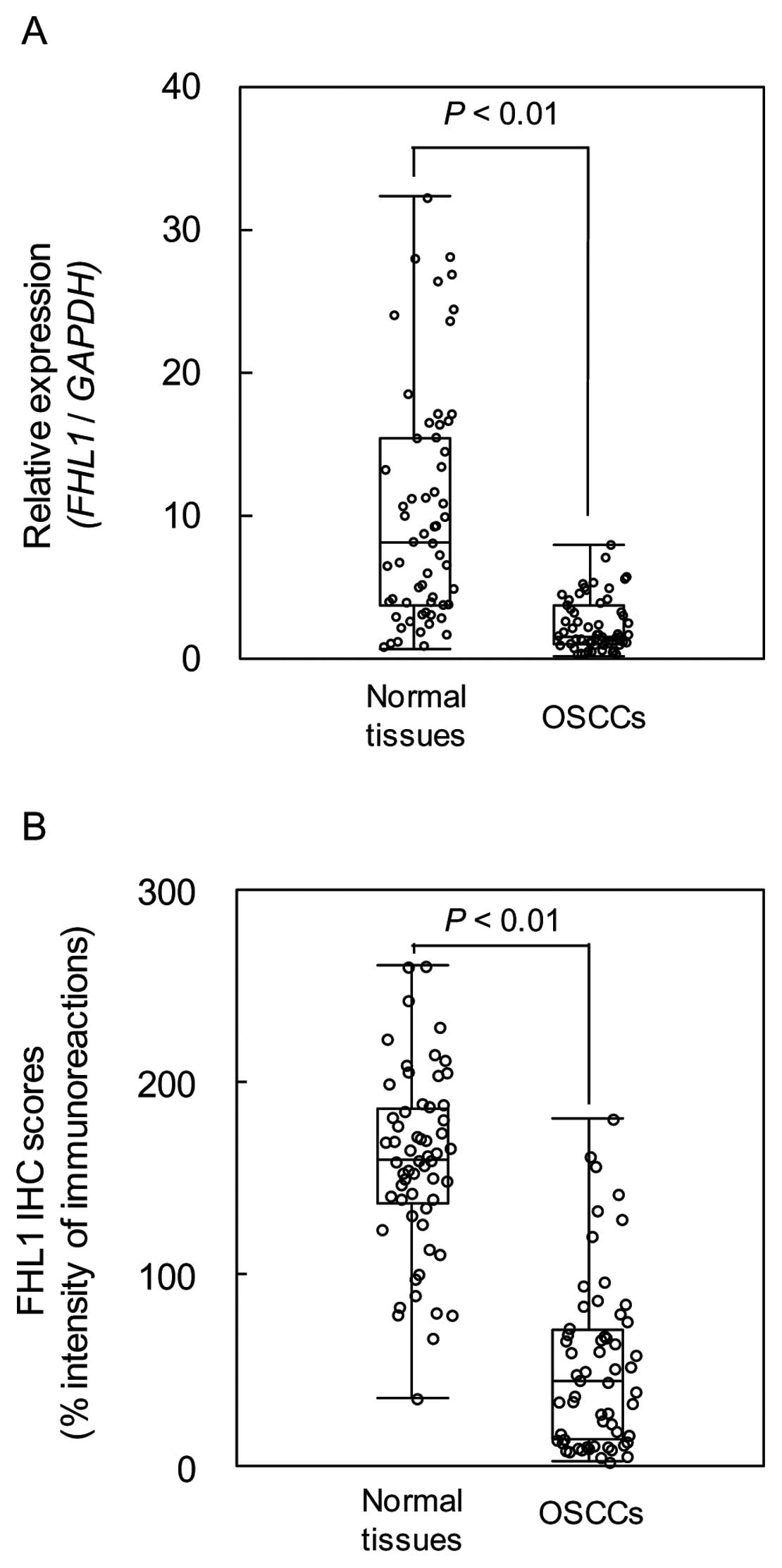
We measured the FHL1 mRNA expression levels in
primary OSCCs and paired normal oral tissues from 59 patients.
Similar to the data from the OSCC-derived cell lines, qRT-PCR
analysis showed that FHL1 mRNA expression was downregulated in 51
of 59 (86.4%) primary OSCCs compared to the corresponding normal
oral tissues (Fig. 2). The
relative mRNA levels in the normal oral tissues and primary OSCCs
ranged from 0.61 to 32.24 (median 8.02) and 0.09 to 7.81 (median
1.49), respectively (P<0.01). We analyzed FHL1 protein
expression in primary OSCCs using the IHC scoring system. The FHL1
IHC scores of normal oral tissues and OSCCs ranged from 36.5 to
260.8 (median 160.0) and 35.0 to 181.5 (median 45.0), respectively.
The IHC scores in primary OSCCs were significantly (P<0.01)
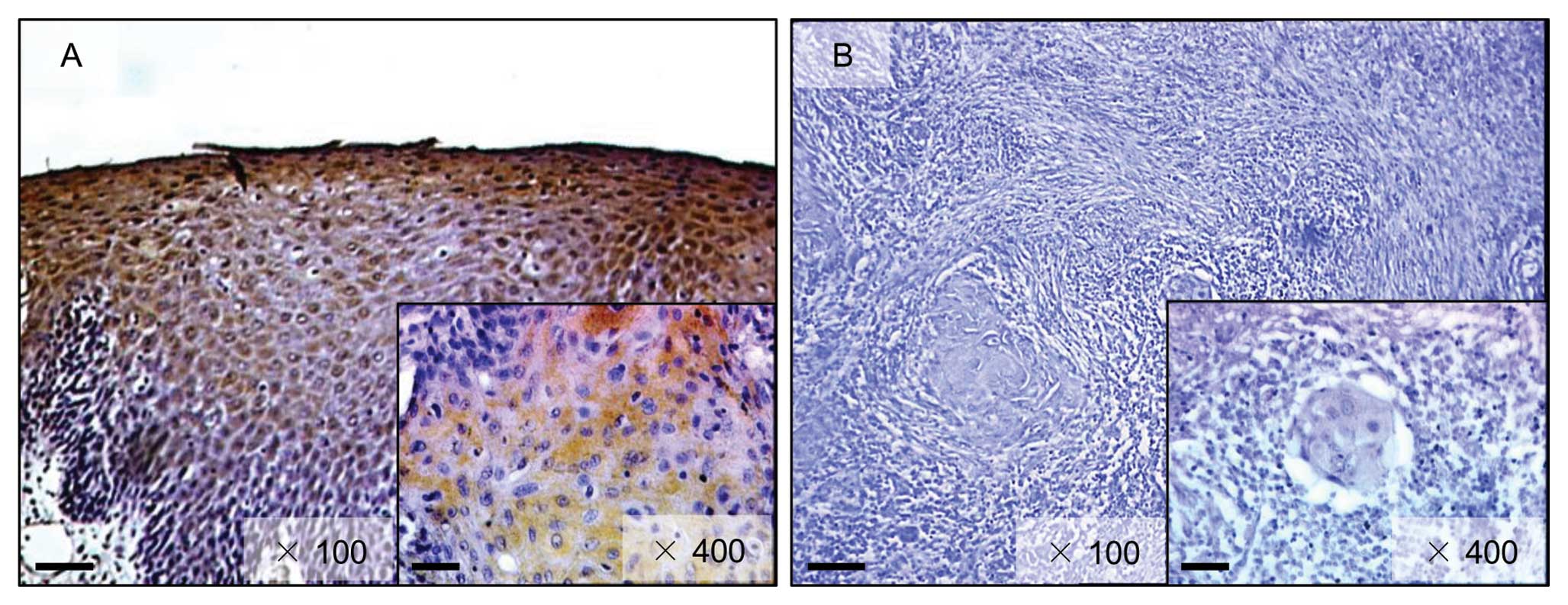
lower than in normal oral tissues (Fig. 2). Representative IHC results for
FHL1 protein in normal oral tissue and primary OSCCs are shown in
Fig. 3. Strong FHL1
immunoreactivity was detected in the cytoplasm of the basal cell
layers in normal tissue, whereas the OSCCs showed almost negative
immunostaining. The correlations between the clinicopathologic
characteristics of the patients with OSCC and the status of the
FHL1 protein expression level are shown in Table II. There was no significant
difference between the frequency of FHL1-negative cases and
clinicopathologic features.
| Table II.Correlation between the expression of
FHL1 and clinical classification in OSCCs. |
Table II.
Correlation between the expression of
FHL1 and clinical classification in OSCCs.
| Clinical
classification | Total | Results of
immunostaining No. of patients (%)
| P-value |
|---|
| FHL1(−) | FHL1(+) |
|---|
| Age at surgery
(years) | | | | |
| <60 | 16 | 8 (13.6) | 8 (13.6) | 0.615 |
| 60–70 | 13 | 8 (13.6) | 5 (8.5) | |
| >70 | 30 | 16 (27.1) | 14 (23.7) | |
| Gender | | | | |
| Male | 40 | 19 (32.2) | 21 (35.6) | 0.168 |
| Female | 19 | 13 (22.0) | 6 (10.2) | |
| T-primary tumor
size | | | | |
| T1 | 4 | 0 (0.0) | 4 (6.8) | 0.366 |
| T2 | 35 | 18 (30.5) | 17 (28.8) | |
| T3 | 7 | 6 (10.2) | 1 (1.7) | |
| T4 | 13 | 8 (13.6) | 5 (8.5) | |
| T1 + T2 | 39 | 18 (30.5) | 21 (35.6) | 0.103 |
| T3 + T4 | 20 | 14 (23.7) | 6 (10.2) | |
| N-regional lymph
node metastasis | | | | |
| N(+) | 22 | 12 (20.3) | 10 (16.9) | 0.593 |
| N(−) | 37 | 20 (33.9) | 17 (28.8) | |
| Stage | | | | |
| I | 3 | 0 (0.0) | 3 (5.1) | 0.259 |
| II | 22 | 13 (22.0) | 9 (15.3) | |
| III | 8 | 5 (8.5) | 3 (5.1) | |
| IV | 26 | 14 (23.7) | 12 (20.3) | |
| I + II | 25 | 13 (22.0) | 12 (20.3) | 0.797 |
| III + IV | 34 | 19 (32.2) | 15 (25.4) | |
| Histopathological
type | | | | |
| Well
differentiated | 38 | 19 (32.2) | 19 (32.2) | 0.137 |
| Moderately
differentiated | 17 | 10 (16.9) | 7 (11.9) | |
| Poorly
differentiated | 4 | 3 (5.1) | 1 (1.7) | |
| Tumor site | | | | |
| Tongue | 32 | 14 (23.7) | 18 (30.5) | 0.327 |
| Gingiva | 17 | 12 (20.3) | 5 (8.5) | |
| Buccal
mucosa | 4 | 2 (3.4) | 2 (3.4) | |
| Palate | 3 | 3 (5.1) | 0 (0.0) | |
| Oral floor | 3 | 1 (1.7) | 2 (3.4) | |
| Total | 59 | 32 | 27 | |
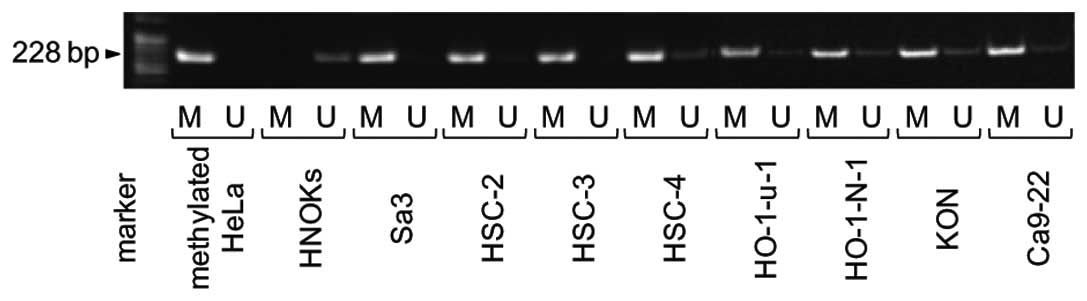
Methylation analysis
To investigate the mechanisms responsible for
downregulation of FHL1 expression, we analyzed the methylation
status of the FHL1 promoter region in the OSCC-derived cell lines.
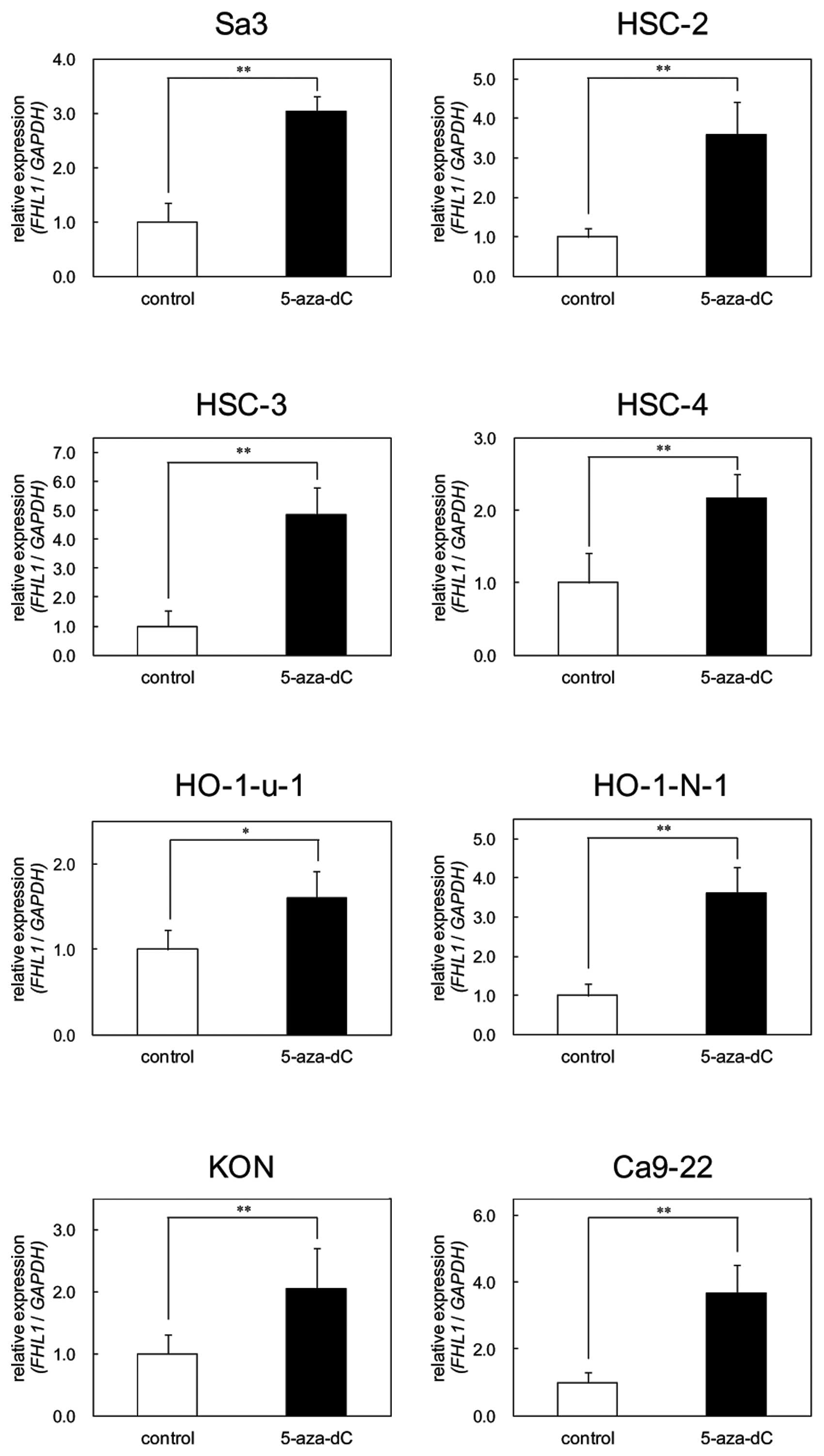
FHL1 promoter methylation was detected in all cell lines (Fig. 4). To further study the consequences
of loss of expression of FHL1 in association with hypermethylation
at the promoter region, the OSCC-derived cell lines, which showed
methylation and transcriptional inactivation of FHL1, were treated
with 5-aza-dC. Significantly (P<0.02) upregulated expression of
the FHL1 gene was observed after 5-aza-dC treatment in all
eight cell lines (Fig. 5).
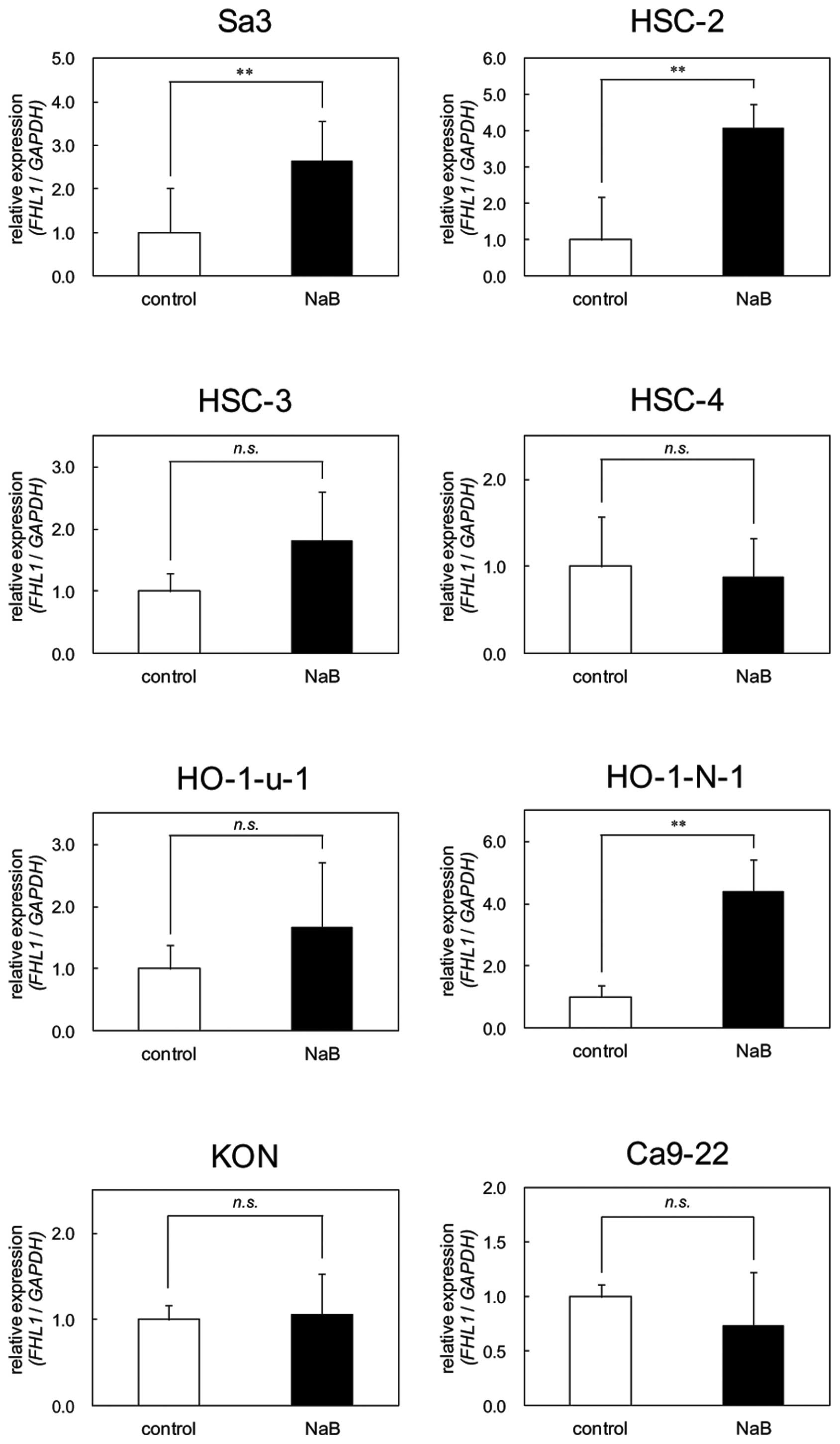
Effects of NaB on FHL1 expression
OSCC-derived cells were treated with NaB to assess
the effects of an HDAC inhibitor on FHL1 expression. In the Sa3,
HSC-2 and HO-1-N-1 cell lines, FHL1 was significantly (P<0.05)
upregulated after treatment with NaB for 24 h compared to the
untreated cells. There were no significant changes in the other
cell lines (Fig. 6).
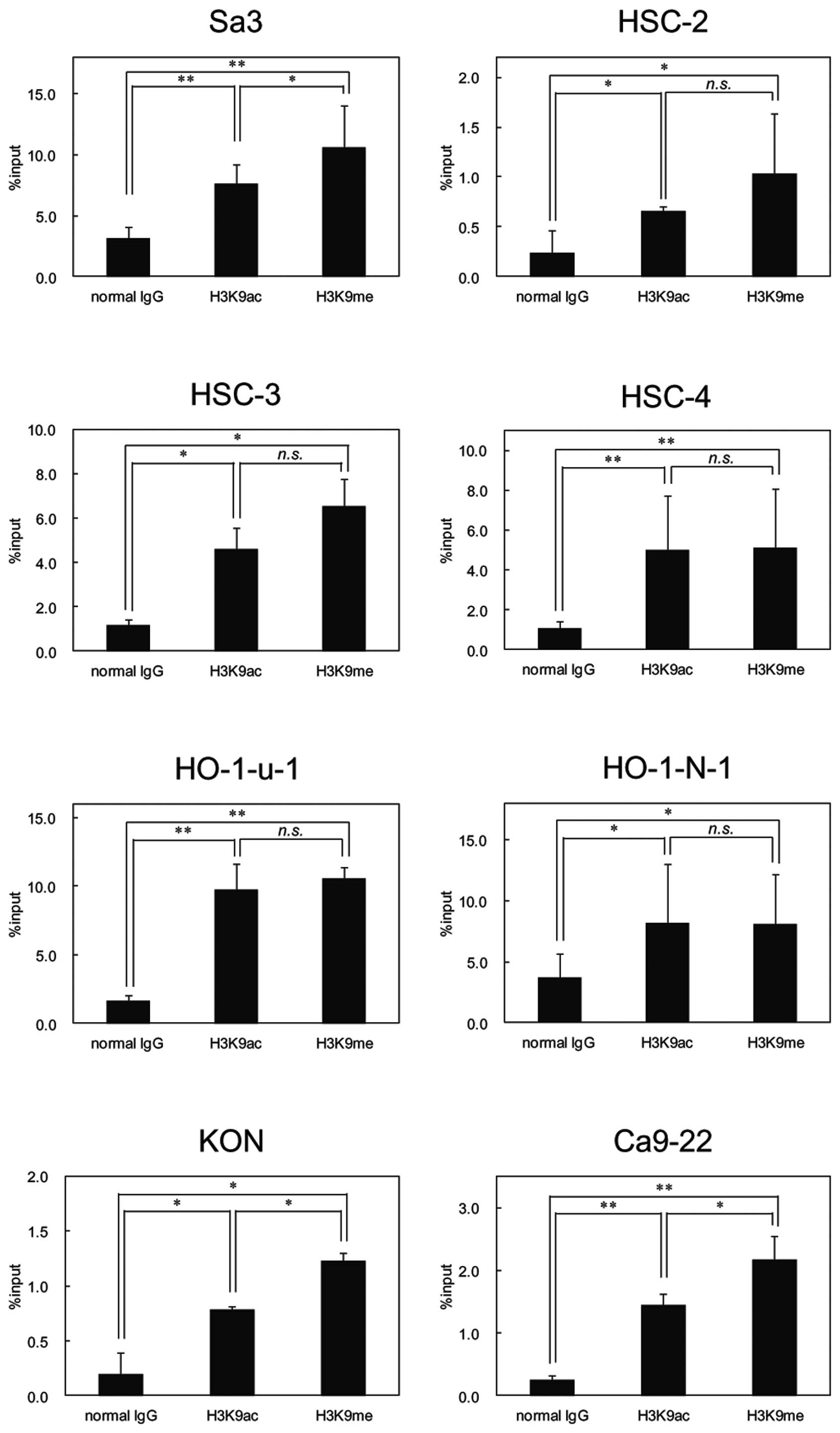
Histone 3 modification in OSCC cell
lines
Because FHL1 hypermethylation was detected in
OSCC-derived cell lines, we examined histone modification as
another mechanism responsible to FHL1 silencing in OSCCs. We
conducted ChIP of acetyl histone H3 at lysine 9 (H3K9ac) and
monomethyl histone H3 at lysine 9 (H3K9me) in all cell lines and
found significant enrichment of H3K9ac and H3K9me at the promoter
region of FHL1 in OSCC-derived cell lines (Fig. 7).
Mutational analysis
We screened DNA samples obtained from HNOKs and
eight OSCC-derived cell lines by direct sequencing. No band
shifting was detected in any exons of the FHL1 gene.
Discussion
We found that FHL1 was significantly downregulated
at the mRNA and protein level in the human OSCC-derived cell lines
and primary OSCCs. Treatment with DNA methyltransferase inhibitor
5-aza-dC restored FHL1 expression in all OSCC-derived cell lines.
The promoter region of FHL1 DNA methylation also was detected in
these cell lines. In contrast, no significant restoration of FHL1
expression was observed using NaB, an inhibitor of histone
deacetylase and the FHL1 promoter region was significantly
acetylated in all cell lines. Direct sequencing did not show any
mutation in the entire coding region of the FHL1 gene. In
contrast to mutational analyses, qRT-PCR analysis of FHL1 mRNA
showed frequent gene downregulation, indicating that other
mechanisms, such as DNA methylation and histone deacetylation, are
related to FHL1 gene silencing.
Although FHL1 downregulation has been reported in
several cancers, i.e., lung, breast and bladder (36–38),
the expression level and molecular function of FHL1 in OSCCs have
not been clarified. Since many tumor suppressor genes silenced by
DNA methylation have been identified in primary OSCC tissue
samples, we hypothesized that FHL1 expression is downregulated by
promoter hypermethylation and that FHL1 functions as a tumor
suppressor in OSCC-derived cell lines.
Hypermethylation of the promoter regions of various
genes has been recognized as one of the most frequent mechanisms
causing loss of gene function. Several studies have reported an
association between aberrant DNA methylation and carcinogenesis
(39–41). However, the association between
epigenetic changes and cancer etiology needs to be elucidated.
Several studies have reported a link between DNA
methylation and histone acetylation in which a family of
methyl-CpG-binding proteins is involved (42). Chromatin modifications are one of
the molecular mechanisms that mediate epigenetic regulation
(43). Of them, HDACs, the
chromatin-modifying enzymes, remove the acetyl groups, leading to
chromatin condensation and repression of transcription (44). A HDAC inhibitor also is involved in
the expression of several genes. In the present study, after
treatment of OSCC-derived cell lines with the HDAC inhibitor, NaB,
FHL1 transcription was restored in three of eight cell lines.
However, the ChIP assay showed that in OSCC-derived cell lines,
there was significantly enriched H3K9ac in the FHL1 promoter
region. These findings indicated that FHL1 expression in the
OSCC-derived cell lines was controlled by epigenetic silencing of
aberrant DNA methylation.
The present study suggests that downregulation of
FHL1 expression in OSCC-derived cell lines is correlated with CpG
hypermethylation of the FHL1 promoter region rather than histone
deacetylation or mutation. This agreed with our initial hypothesis
that the FHL1 promoter region is hypermethylated in OSCCs. In this
context, with accumulating knowledge of the mechanism of
inactivation of tumor suppressor genes, abnormal methylation at the
promoters of the tumor suppressor genes is another mechanism that
suppresses gene activity.
In conclusion, our results suggested that
downregulation of FHL1 during OSCC development and DNA methylation
might play a role in gene inactivation. Further studies with more
clinical samples will improve our ability to diagnose, prevent and
treat this neoplasm.
Acknowledgements
We thank Ms. Lynda C. Charters for
editing this manuscript.
References
|
1.
|
Lippman SM, Sudbø J and Hong WK: Oral
cancer prevention and the evolution of molecular-targeted drug
development. J Clin Oncol. 23:346–356. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Jemal A, Siegel R, Ward E, et al: Cancer
statistics, 2008. CA Cancer J Clin. 58:71–96. 2008. View Article : Google Scholar
|
|
3.
|
Bettendorf O, Piffkò J and Bànkfalvi A:
Prognostic and predictive factors in oral squamous cell cancer:
important tools for planning individual therapy? Oral Oncol.
40:110–119. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Kademani D: Oral cancer. Mayo Clin Proc.
82:878–887. 2007. View
Article : Google Scholar
|
|
5.
|
Tatsumoto T, Xie X, Blumenthal R, Okamoto
I and Miki T: Human ECT2 is an exchange factor for Rho GTPases,
phosphorylated in G2/M phases, and involved in cytokinesis. J Cell
Biol. 147:921–928. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Shinozuka K, Uzawa K, Fushimi K, et al:
Downregulation of carcinoembryonic antigen-related cell adhesion
molecule 1 in oral squamous cell carcinoma: correlation with tumor
progression and poor prognosis. Oncology. 76:387–397. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Brown S, McGrath MJ, Ooms LM, Gurung R,
Maimone MM and Mitchell CA: Characterization of two isoforms of the
skeletal muscle LIM protein 1, SLIM1. Localization of SLIM1 at
focal adhesions and the isoform slimmer in the nucleus of myoblasts
and cytoplasm of myotubes suggests distinct roles in the
cytoskeleton and in nuclear-cytoplasmic communication. J Biol Chem.
274:27083–27091. 1999.
|
|
8.
|
McGrath MJ, Mitchell CA, Coghill ID,
Robinson PA and Brown S: Skeletal muscle LIM protein 1 (SLIM1/FHL1)
induces alpha 5 beta 1-integrin-dependent myocyte elongation. Am J
Physiol Cell Physiol. 285:C1513–C1526. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Loughna PT, Mason P, Bayol S and Brownson
C: The LIM-domain protein FHL1 (SLIM 1) exhibits functional
regulation in skeletal muscle. Mol Cell Biol Res Commun. 3:136–140.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Chen DH, Raskind WH, Parson WW, et al: A
novel mutation in FHL1 in a family with X-linked scapuloperoneal
myopathy: phenotypic spectrum and structural study of FHL1
mutations. J Neurol Sci. 296:22–29. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Oku N, Sasabe E, Ueta E, Yamamoto T and
Osaki T: Tight junction protein claudin-1 enhances the invasive
activity of oral squamous cell carcinoma cells by promoting
cleavage of laminin-5 gamma2 chain via matrix metalloproteinase
(MMP)-2 and membrane-type MMP-1. Cancer Res. 66:5251–5257. 2006.
View Article : Google Scholar
|
|
12.
|
Sakashita K, Mimori K, Tanaka F, et al:
Clinical significance of loss of Fhl1 expression in human gastric
cancer. Ann Surg Oncol. 15:2293–2300. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Fackler MJ, McVeigh M, Mehrotra J, et al:
Quantitative multiplex methylation-specific PCR assay for the
detection of promoter hypermethylation in multiple genes in breast
cancer. Cancer Res. 64:4442–4452. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Fuks F: DNA methylation and histone
modifications: teaming up to silence genes. Curr Opin Genet Dev.
15:490–495. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Hassig CA and Schreiber SL: Nuclear
histone acetylases and deacetylases and transcriptional regulation:
HATs off to HDACs. Curr Opin Chem Biol. 1:300–308. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Huang L: Targeting histone deacetylases
for the treatment of cancer and inflammatory diseases. J Cell
Physiol. 209:611–616. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Endo Y, Uzawa K, Mochida Y, et al:
Sarcoendoplasmic reticulum Ca(2+) ATPase type 2 downregulated in
human oral squamous cell carcinoma. Int J Cancer. 110:225–231.
2004.
|
|
18.
|
Shimada K, Uzawa K, Kato M, et al:
Aberrant expression of RAB1A in human tongue cancer. Br J Cancer.
92:1915–1921. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Saito K, Uzawa K, Endo Y, et al: Plasma
membrane Ca2+ ATPase isoform 1 down-regulated in human
oral cancer. Oncol Rep. 15:49–55. 2006.
|
|
20.
|
Kasamatsu A, Uzawa K, Nakashima D, et al:
Galectin-9 as a regulator of cellular adhesion in human oral
squamous cell carcinoma cell lines. Int J Mol Med. 16:269–273.
2005.PubMed/NCBI
|
|
21.
|
Sakuma K, Kasamatsu A, Yamatoji M, et al:
Expression status of Zic family member 2 as a prognostic marker for
oral squamous cell carcinoma. J Cancer Res Clin Oncol. 136:553–559.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Tanaka C, Uzawa K, Shibahara T, Yokoe H,
Noma H and Tanzawa H: Expression of an inhibitor of apoptosis,
survivin, in oral carcinogenesis. J Dent Res. 82:607–611. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Kouzu Y, Uzawa K, Kato M, et al: WISP-2
expression in human salivary gland tumors. Int J Mol Med.
17:567–573. 2006.PubMed/NCBI
|
|
24.
|
Onda T, Uzawa K, Endo Y, et al: Ubiquitous
mitochondrial creatine kinase downregulated in oral squamous cell
carcinoma. Br J Cancer. 94:698–709. 2006.PubMed/NCBI
|
|
25.
|
Sakuma T, Uzawa K, Onda T, et al: Aberrant
expression of histone deacetylase 6 in oral squamous cell
carcinoma. Int J Oncol. 29:117–124. 2006.PubMed/NCBI
|
|
26.
|
Kato Y, Uzawa K, Yamamoto N, et al:
Overexpression of Septin1: possible contribution to the development
of oral cancer. Int J Oncol. 31:1021–1028. 2007.PubMed/NCBI
|
|
27.
|
Nomura H, Uzawa K, Yamano Y, et al:
Overexpression and altered subcellular localization of
autophagy-related 16-like 1 in human oral squamous-cell carcinoma:
correlation with lymphovascular invasion and lymph-node metastasis.
Hum Pathol. 40:83–91. 2009. View Article : Google Scholar
|
|
28.
|
Iyoda M, Kasamatsu A, Ishigami T, et al:
Epithelial cell transforming sequence 2 in human oral cancer. PloS
One. 5:e140822010. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Yamatoji M, Kasamatsu A, Kouzu Y, et al:
Dermatopontin: a potential predictor for metastasis of human oral
cancer. Int J Cancer. 130:2903–2911. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Lombardi DP, Geradts J, Foley JF, Chiao C,
Lamb PW and Barrett JC: Loss of KAI1 expression in the progression
of colorectal cancer. Cancer Res. 59:5724–5731. 1999.PubMed/NCBI
|
|
31.
|
Verburg FA, Wäschle K, Reiners C,
Giovanella L and Lentjes EG: Heterophile antibodies rarely
influence the measurement of thyroglobulin and thyroglobulin
antibodies in differentiated thyroid cancer patients. Horm Metab
Res. 42:736–739. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Li LC and Dahiya R: MethPrimer: designing
primers for methylation PCRs. Bioinformatics. 18:1427–1431. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Van der Auwera I, Van Laere SJ, Van den
Bosch SM, et al: Aberrant methylation of the adenomatous polyposis
coli (APC) gene promoter is associated with the inflammatory breast
cancer phenotype. Br J Cancer. 99:1735–1742. 2008.PubMed/NCBI
|
|
34.
|
Kimura H, Hayashi-Takanaka Y, Goto Y,
Takizawa N and Nozaki N: The organization of histone H3
modifications as revealed by a of specific monoclonal antibodies.
Cell Struct Funct. 33:61–73. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Wang LL, Qiu GR, Fu WN, Yuan ZW and Sun
KL: Transcriptional regulation of Fhl1 by estradiol in rat
myoblastocytes. Steroids. 75:368–372. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Shen Y, Jia Z, Nagele RG, Ichikawa H and
Goldberg GS: SRC uses Cas to suppress Fhl1 in order to promote
nonanchored growth and migration of tumor cells. Cancer Res.
66:1543–1552. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Fryknas M, Wickenberg-Bolin U, Goransson
H, et al: Molecular markers for discrimination of benign and
malignant follicular thyroid tumors. Tumour Biol. 27:211–220. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Li X, Jia Z, Shen Y, et al: Coordinate
suppression of Sdpr and Fhl1 expression in tumors of the breast,
kidney, and prostate. Cancer Sci. 99:1326–1333. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Yuan J, Luo RZ, Fujii S, et al: Aberrant
methylation and silencing of ARHI, an imprinted tumor suppressor
gene in which the function is lost in breast cancers. Cancer Res.
63:4174–4180. 2003.PubMed/NCBI
|
|
40.
|
Chen K, Sawhney R, Khan M, et al:
Methylation of multiple genes as diagnostic and therapeutic markers
in primary head and neck squamous cell carcinoma. Arch Otolaryngol
Head Neck Surg. 133:1131–1138. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Henken FE, Wilting SM, Overmeer RM, et al:
Sequential gene promoter methylation during HPV-induced cervical
carcinogenesis. Br J Cancer. 97:1457–1464. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Magdinier F and Wolffe AP: Selective
association of the methyl-CpG binding protein MBD2 with the silent
p14/p16 locus in human neoplasia. Proc Natl Acad Sci USA.
98:4990–4995. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Cheung P and Lau P: Epigenetic regulation
by histone methylation and histone variants. Mol Endocrinol.
19:563–573. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Hong S, Derfoul A, Pereira-Mouries L and
Hall DJ: A novel domain in histone deacetylase 1 and 2 mediates
repression of cartilage-specific genes in human chondrocytes. FASEB
J. 23:3539–3552. 2009. View Article : Google Scholar : PubMed/NCBI
|