Introduction
The cytoskeleton is comprised of actin filaments,
intermediate filaments and microtubules. Microtubules are dynamic
structures that are important for a range of cellular functions,
such as intracellular trafficking, cell movement and division,
where they are involved in chromosome segregation. α- and β-tubulin
heterodimers polymerise into a hollow tube denoted microtubule
(1,2). The stability of the microtubule is
regulated through GTP-hydrolysis of β-tubulin; binding of GTP
allows polymerisation, but within the microtubule GTP can be
hydrolysed to GDP (1,2). Microtubules constantly polymerise and
depolymerise, a process termed dynamic instability. Since
micro-tubules play an essential role in chromosome segregation,
drugs that interfere with microtubule function prevent the cell
from mitosis. A number of microtubule targeting drugs are used in
the clinic to treat human cancers, including oesophageal cancer.
Examples are the vinca alkaloids that destabilise microtubules by
binding close to the GTP-binding region in β-tubulin (3), and the taxanes which bind to
polymerised microtubules and stabilise the GDP-bound form of
β-tubulin by forcing them into a configuration resembling the
GTP-bound state (4).
Growth factors and their receptors have been shown
to be of pivotal significance for the occurrence and development of
cancer. The human epidermal growth factor receptor (HER) family is
comprised of four members, i.e. EGFR (HER1, ErbB1), HER2 (ErbB2,
Neu), HER3 (ErbB3) and HER4 (ErbB4) (5). These receptors are tyrosine kinases
that are activated by ligand-induced dimerisation. There are
several ligands for the receptors in the HER family, and these have
different binding specificities, resulting in formation of homo- or
heterodimeric receptor complexes. HER family members are commonly
activated in human cancer cells by different mechanisms including
autocrine stimulation, mutations or overexpression (6). Dysregulated and improper receptor
activation leads to induction of signals that promote
proliferation, survival, migration and angiogenesis, events that
are all central for tumour development and progression.
Over-activated EGFR is recognised as an important mechanism in
several types of cancer, including colorectal cancer (7), head and neck cancer (8), and non-small cell lung cancer
(9) and has become a target of
interest in the treatment of these tumours.
There are data indicating possible interactions
between EGFR and the microtubule system. Gao et al
demonstrated that histone deacetylase 6 (HDAC6), a
microtubule-associated deacetylase, associates with the endosomal
compartments and controls EGFR trafficking and degradation
(10). This is consistent with
data from Deribe et al showing that HDAC6 negatively
regulates EGFR endocytosis and degradation by controlling the
acetylation status of α-tubulin and subsequently receptor
trafficking along microtubules (11).
Oesophageal carcinoma is the seventh most common
cause of cancer-related death in the Western world (12). The standard treatment for localised
oesophageal carcinoma includes a combination of radiation and
chemotherapy, sometimes followed by surgery. Preoperative
chemoradiotherapy or chemotherapy (13) has been demonstrated to give a
significant survival benefit. However, patients with advanced
metastatic disease that are treated with palliative chemotherapy
have a poor prognosis with a median survival time of less than one
year. The 5-year survival rate of all diagnosed patients is only
around 15%. Thus, there is an urgent need to improve current
therapies. In oesophageal carcinoma patients, EGFR has been
reported to be commonly overexpressed (14) and the overexpression is correlated
to lymph node metastasis, vascular invasion and shorter survival
(15–17). The EGF and TGF ligands function as
mitogens for oesophageal tumour cells (18) and activation of EGFR signalling has
been implicated in metastasis via modulation of cell adhesion,
angiogenesis, invasion and migration.
In the current study we have investigated the
possible interaction of anti-microtubule drugs and the EGFR
signalling system in human oesophageal cancer cells. Treatment with
the drugs led to inhibition of proliferation of the cells.
Additionally, microtubule destabilising agents were also shown to
inhibit EGFR phosphorylation. These effects could be inhibited by
simultaneous addition of the protein tyrosine phosphatase inhibitor
sodium orthovanadate, suggesting that disruption of the microtubule
network leads to release or activation of a tyrosine phosphatase.
This study shows that microtubule targeting drugs have other
effects beyond interfering with the mitotic spindle.
Materials and methods
Cell culturing and counting
The human ESCC (oesophageal squamous cell carcinoma)
cell lines Kyse30, Kyse70, Kyse140, Kyse150, Kyse180, Kyse410,
Kyse450, Kyse510 and Kyse520 (Deutsche Sammlung von Mikroorganismen
und Zellkulturen GmbH, Braunschweig, Germany) were cultivated in
RPMI-1640 medium, supplemented with 10% fetal bovine serum (FBS), 2
mM L-glutamine, 50 units of penicillin and 50 μg
streptomycin/ml (Sigma-Aldrich, St. Louis, MO, USA). Cells were
split twice a week by incubation in 13–20 μl/cm2
5X Trypsin-EDTA Solution (Sigma-Aldrich) in 37°C until detachment
from the surface. Cells were used for further experiments when
70–90% confluent. Cell counting was performed using a
Coulter® Z2 Particle count and size analyser.
Cell growth analysis using resazurin
assay
Kyse70 and Kyse140 cells were seeded in 96-well
plates at a concentration of 5,000 cells per well and were allowed
to grow overnight. Docetaxel, podophyllotoxin (PPT) and vincristine
are from Sigma-Aldrich. Five or seven different concentrations of
docetaxel, podo phyllotoxin (PPT) or vincristine were then added to
the cell medium. After 24 and 48 h, resazurin (Alamar Blue,
Sigma-Aldrich) was added at a concentration of 440 μM to
each well followed by incubation in the dark for 1 h at room
temperature. Wells containing only culture medium served as blank.
The analysis of fluorescence (560EX nm/590EM nm) using a Wallac
Victor Multilabel Counter (Wallac, Turku, Finland) was followed by
calculations of relative numbers of viable cells expressed as
percentages of untreated cells. Resazurin detects cell viability by
converting the reagent to a fluorescent indicator in response to
metabolically active cells (19,20).
The resazurin assay is quantitative with respect to time and dose,
and separate experiments showed a linear correlation between the
number of viable cells and the emitted light (data not shown). Each
experiment was performed three times.
Microtubule staining
Kyse70 and Kyse140 cells were grown on coverslips
overnight and then treated with 5 μM of either docetaxel,
PPT or vincristine for 24 h. Cells were fixed in 2% formaldehyde
and permeabilised with 0.2% Triton X-100. Coverslips were blocked
with 10 mM glycine at room temperature for 1 h, incubated with
primary mouse anti-α-tubulin antibody (Sigma-Aldrich), followed by
incubation with a secondary polyclonal goat anti-mouse antibody
labelled with FITC (Dako, Glostrup, Denmark). The nuclei were
stained with DAPI. Coverslips were mounted on object slides using
Fluoromount-G (Southern Biotechnology Associates, Birmingham, AL,
USA). Microtubule staining was visualised using a Zeiss
immunofluorescence microscope at x40 magnification.
Immunoprecipitation and western blot
analysis
Subconfluent Kyse70 and Kyse140 cells were treated
with different concentrations (0.5, 5 and 10 μM) of PPT,
vincristine or docetaxel for 24 and 48 h in starvation medium
containing 0.1% FBS. Subsequently, cells were stimulated with 100
ng/ml EGF (Chemicon, Temecula, CA, USA) or IGF-1 (R&D Systems,
Minneapolis, MN, USA) for 5 min and washed with ice-cold
phosphate-buffered saline before lysis. Cell lysates were prepared
according to Lennartsson et al(21). Briefly, total protein concentration
was determined using the BCA Protein Assay Kit (Pierce, Rockford,
IL, USA). Total cell lysate (TCL) were submitted to
SDS-polyacrylamide gel electrophoresis (SDS-PAGE). For
immunoprecipitation, antibodies against IGF-1Rβ were added to each
lysate at a concentration of 1 μg/ml. Protein A-Sepharose
was added in order to collect immunocomplexes. After washing of the
beads, samples were boiled in reducing sample buffer and subjected
to SDS-PAGE. Separated proteins were electrotransferred to
polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA,
USA), membranes blocked using 5% BSA, and then incubated with
primary antibody overnight at 4°C. Antibodies used were anti-EGFR,
anti-IGF-1Rβ and anti-Akt1/2 rabbit polyclonal antibodies (Santa
Cruz Biotechnology, Santa Cruz, CA, USA), anti-phosphotyrosine
mouse monoclonal antibodies PY99 (Santa Cruz Biotechnology),
phospho-specific anti-Erk and phospho-specific anti-Akt antibodies
(Cell Signalling Technology, Beverly, MA, USA), anti-PTP1B mouse
antibodies (BD Biosciences, San Jose, CA, USA), anti-β-actin mouse
monoclonal antibodies (Sigma-Aldrich) and anti-Erk2 rabbit serum
(Ludwig Institute for Cancer Research, Uppsala, Sweden). Anti-PTPε
rabbit serum was from Dr A. Elson (The Weizmann Institute of
Science, Israel). EGF was from Chemicon and IGF-1 from R&D
Systems. After washing, membranes were incubated with horseradish
peroxidase-conjugated anti-mouse or anti-rabbit IgG antibodies
(Amersham Biosciences, Uppsala, Sweden) and proteins visualised
using ECL western blotting detection systems (Roche Applied
Science, Indianapolis, IN, USA) on a cooled charge-coupled device
(CCD) camera (Bio-Rad Life Science, Hercules, CA, USA). The images
were analysed using the software Quantity One.
siRNA interference
siRNA was employed to knockdown PTPε and PTP1B
expression. Anti-PTPε siRNA targets sequence: GCGAACAGGUACAUUCAUA;
anti-PTP1B siRNA targets sequence: GGAGAAAGGUUCGUUAAAA;
non-targeting siRNA was used as a control (target sequence
CGTACGCGGA ATACTTCGA). To downregulate PTP1B and PTPε expression,
different concentrations of siRNA for anti-PTP1B and anti-PTPε were
incubated with Kyse70 and Kyse140 cells for 48 h. Levels of
knockdown were tested after measuring protein levels by
immunoblotting. Meanwhile, different concentrations of microtubule
targeting drugs were added to the cell cultures for 24 h.
Sodium orthovanadate treatment
Subconfluent Kyse70 and Kyse140 cells were treated
with 10 μM PPT, vincristine or docetaxel for 24 h. Before
cell lysis, sodium orthovanadate (Na3VO4) was
added in the medium at a concentration of 1 mM for 1 h, followed by
stimulation with 100 ng/ml EGF for 5 min. Total cell lysates were
used for immunoblotting analysis.
Results
Podophyllotoxin (PPT), vincristine and
docetaxel interact with the microtubule network and affect survival
of ESCC cells
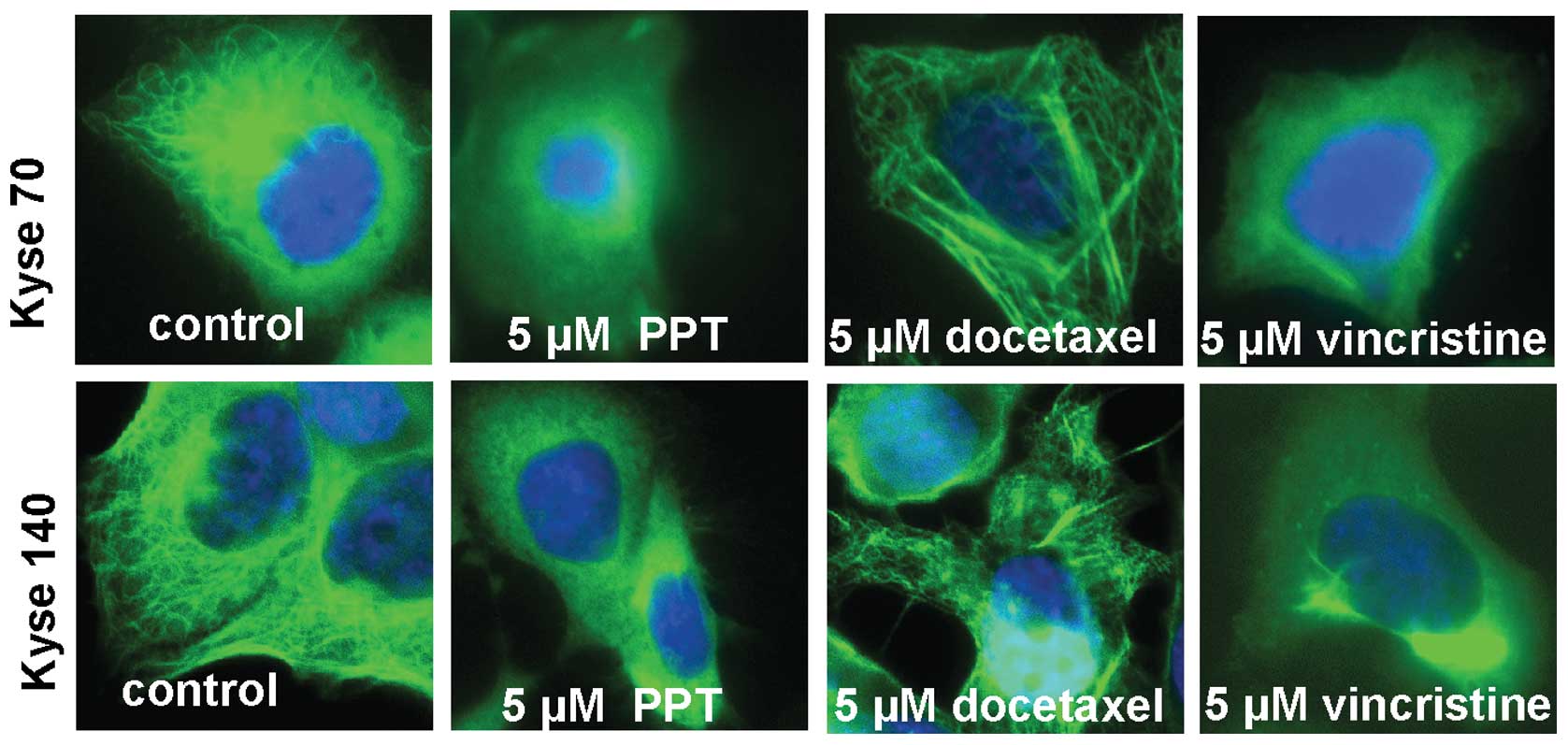
After 24-h treatment, the microtubule destabilising
drugs PPT (5 μM) and vincristine (5 μM) disrupted the
microtubule network while the microtubule stabilising drug
docetaxel (5 μM) stabilised the network (Fig. 1) in oesophageal cancer cells. After
24-h drug treatment, we did not observe nuclear pyknosis and
massive apoptotic bodies in either Kyse70 or Kyse140 cells under
microscope following DAPI staining. Treatment with PPT, docetaxel
and vincristine in Kyse70 and Kyse140 cells resulted in
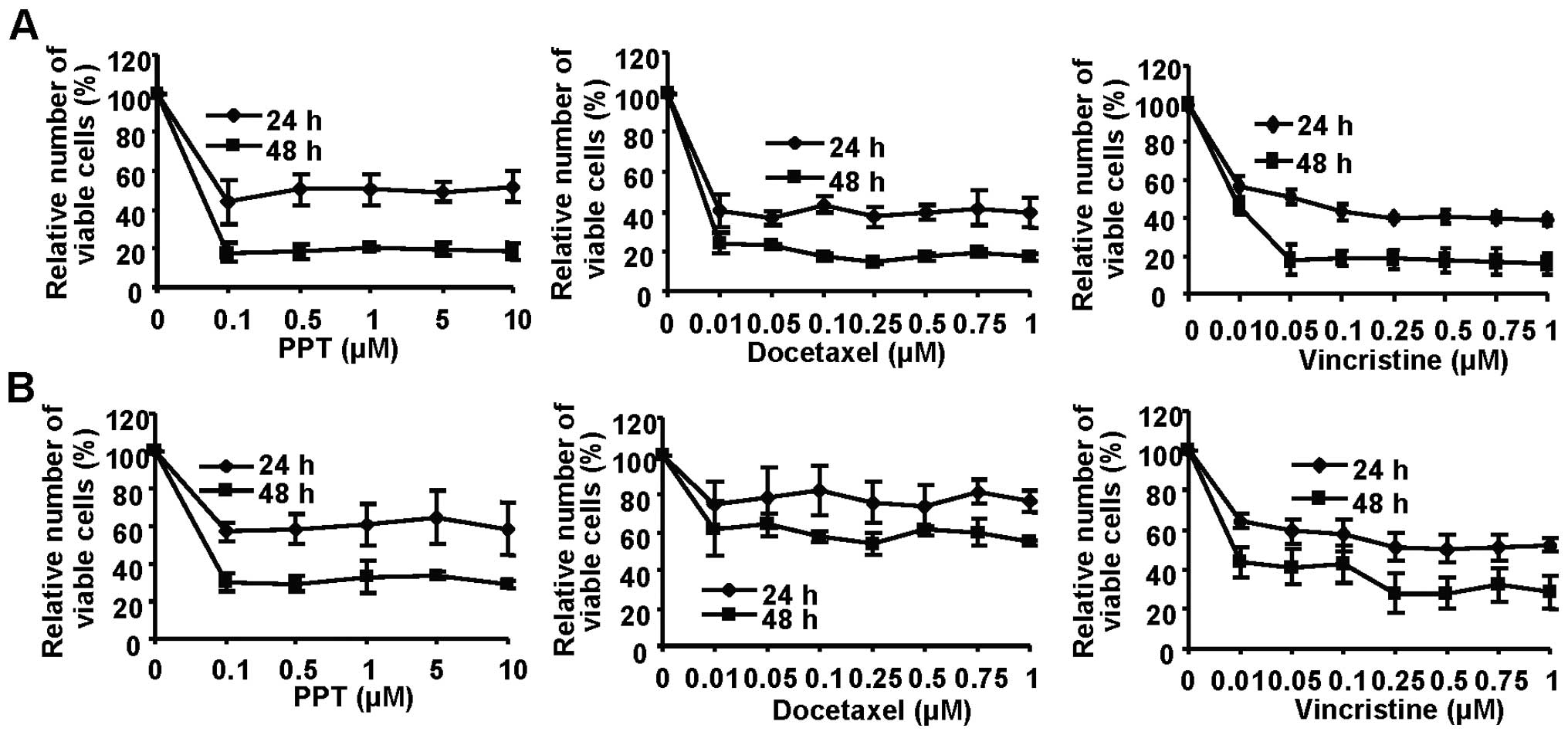
time-dependent plateau-shaped dose-response curves in a
proliferation assay, consistent with tubulin interaction and cell
cycle arrest. Maximal inhibition (>80%) of Kyse70 (Fig. 2A) was achieved at low
concentrations (0.1 μM PPT, 0.1 μM docetaxel and 0.05
μM vincristine at 48 h), while Kyse140 cells appeared
slightly less sensitive (Fig.
2B).
Microtubule destabilising drugs decrease
EGFR phosphorylation and downstream signalling in ESCC cells
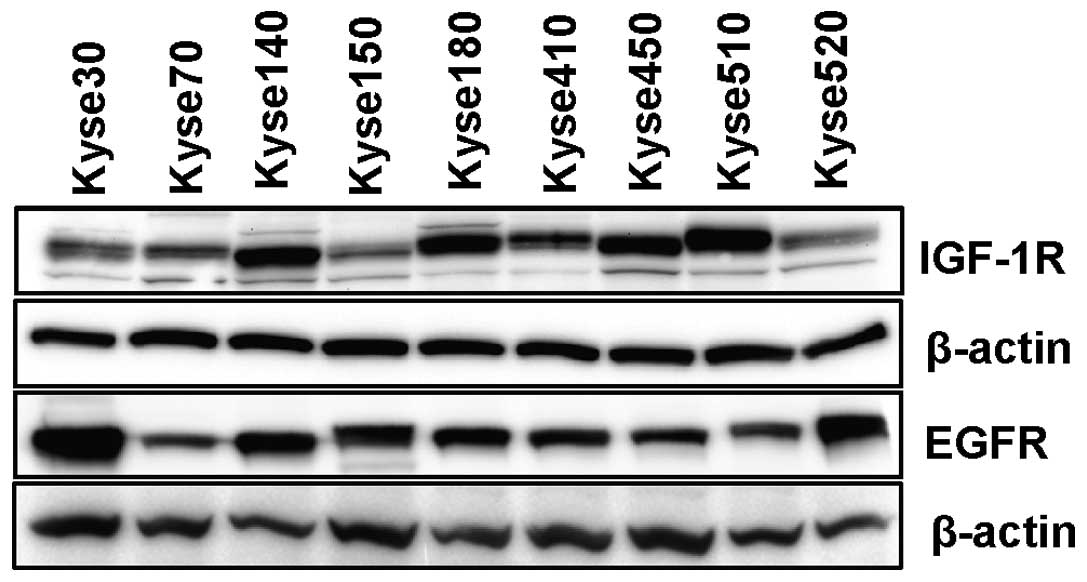
It has been reported that EGFR is commonly
overexpressed in oesophageal carcinoma (14). Variable but detectable levels of
EGFR were observed in all 9 oesophageal carcinoma cell lines used
in this study (Fig. 3). The
possible interactions between EGFR and microtubules were
investigated by treatment of Kyse70 and Kyse140 cells with
increasing concentrations of micro tubule targeting drugs for 24 or
48 h, followed by 5 min EGF stimulation. The concentrations used in
this study are comparable to or slightly higher than the clinically
achievable peak plasma concentrations in patients (22,23).
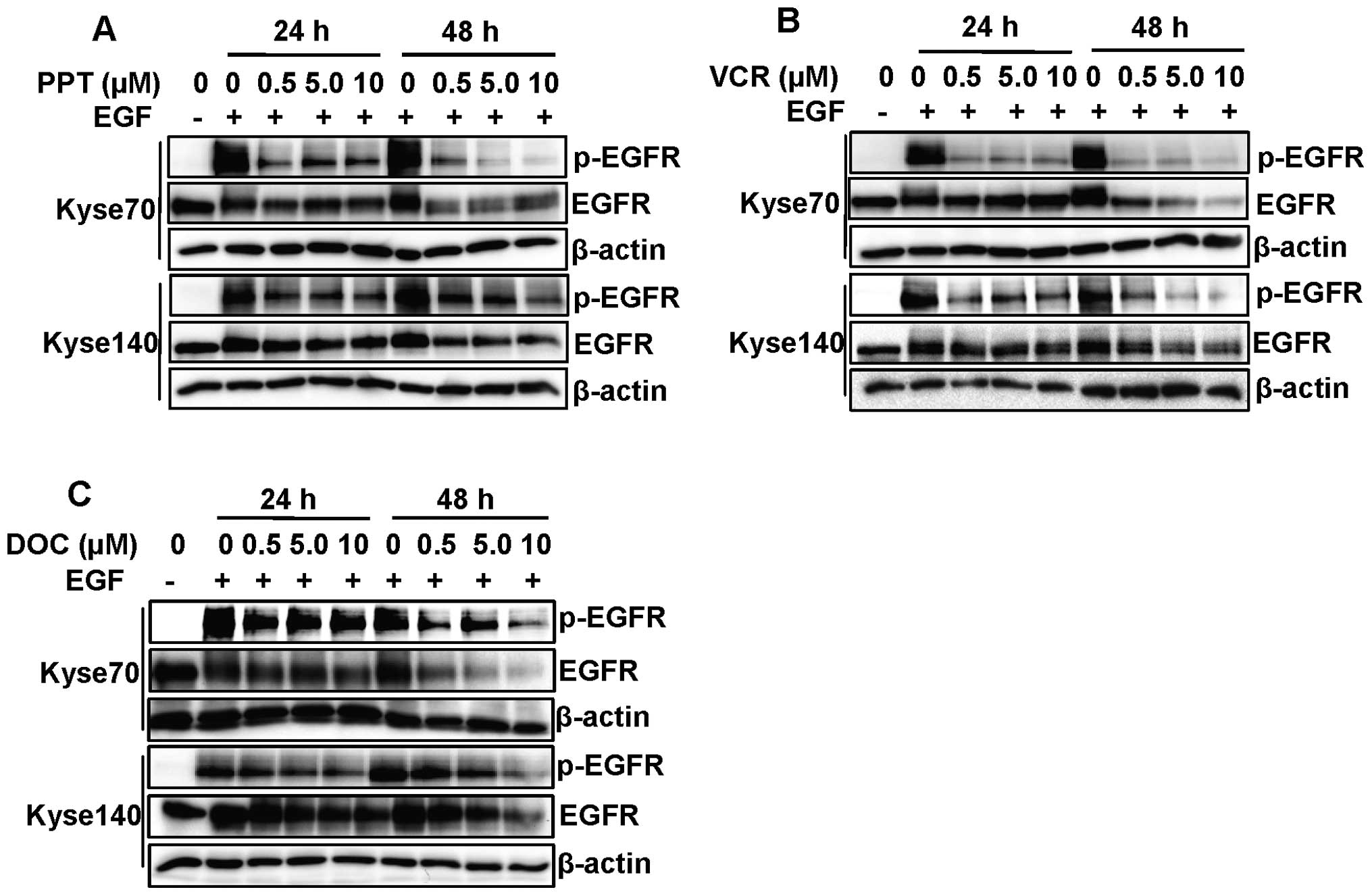
The microtubule destabilising agents PPT (Fig. 4A) and vincristine (Fig. 4B) inhibited EGFR tyrosine
phosphorylation after 24 h treatment at all concentration where we
did not observe the inhibition of EGFR expression. However, the
microtubule stabilising agent docetaxel had a minor inhibitory
effect on EGFR phospho rylation after 24 h treatment (Fig. 4C). After 48 h of drug treatment
with PPT, vincristine and docetaxel, a substantial inhibition of
EGFR expression and phosphorylation could be observed, indicating a
possible indirect effect due to the cytotoxicity of microtubule
targeting drugs (Fig. 4).
Dephosphorylation was observed already after 24 h of treatment
whereas EGFR downregulation was most pronounced after 48 h,
indicating that inhibition of phosphorylation and receptor
downregulation are two distinct events.
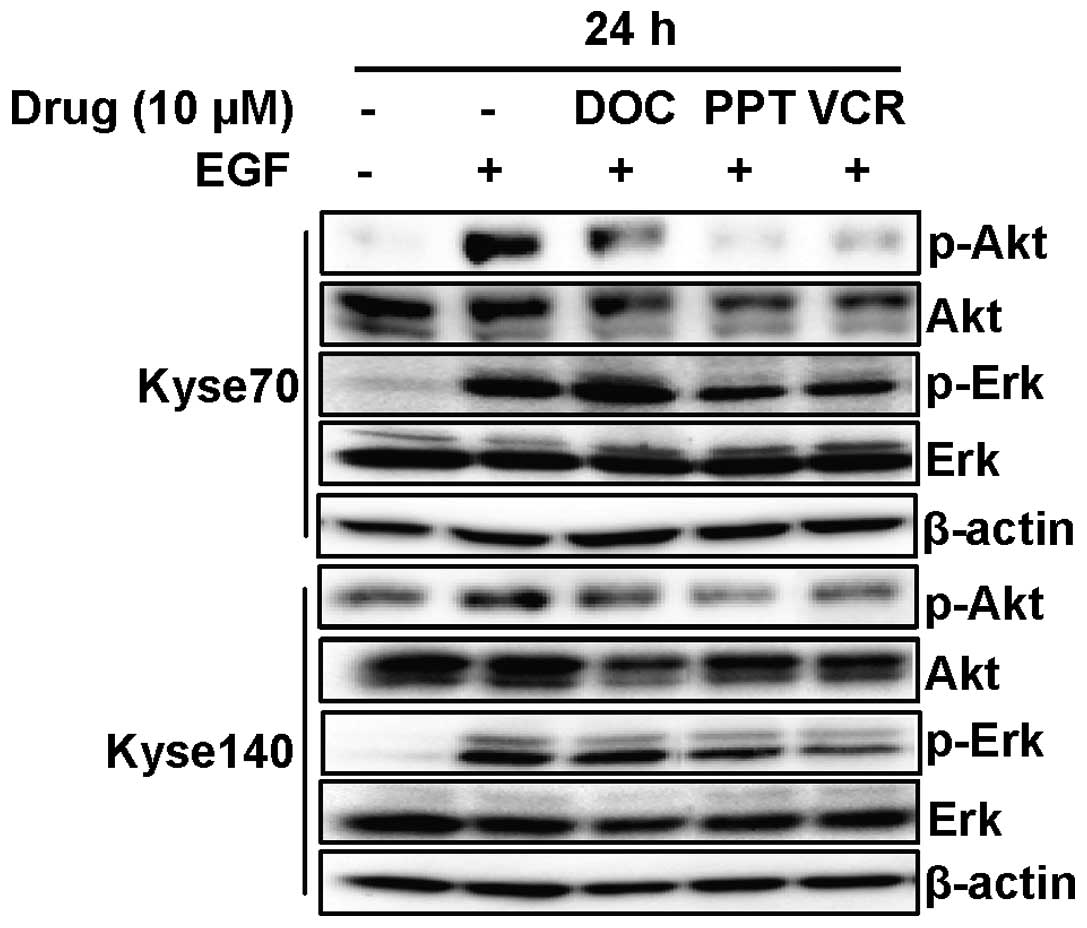
To investigate if decreased phospho-EGFR also
resulted in decreased signalling downstream of EGFR, cells were
treated with microtubule targeting drugs for 24 h thus avoiding
EGFR degradation. Docetaxel, PPT and vincristine downregulated
phosphorylation as well as protein levels of Akt (Fig. 5) in both Kyse70 and Kyse140 cells.
Moreover, the microtubule destabilising agents PPT and vincristine
inhibited phosphorylation of Erk but did not cause downregulation
of total Erk protein levels. Docetaxel had no effect on the
activation of Erk in either cell line.
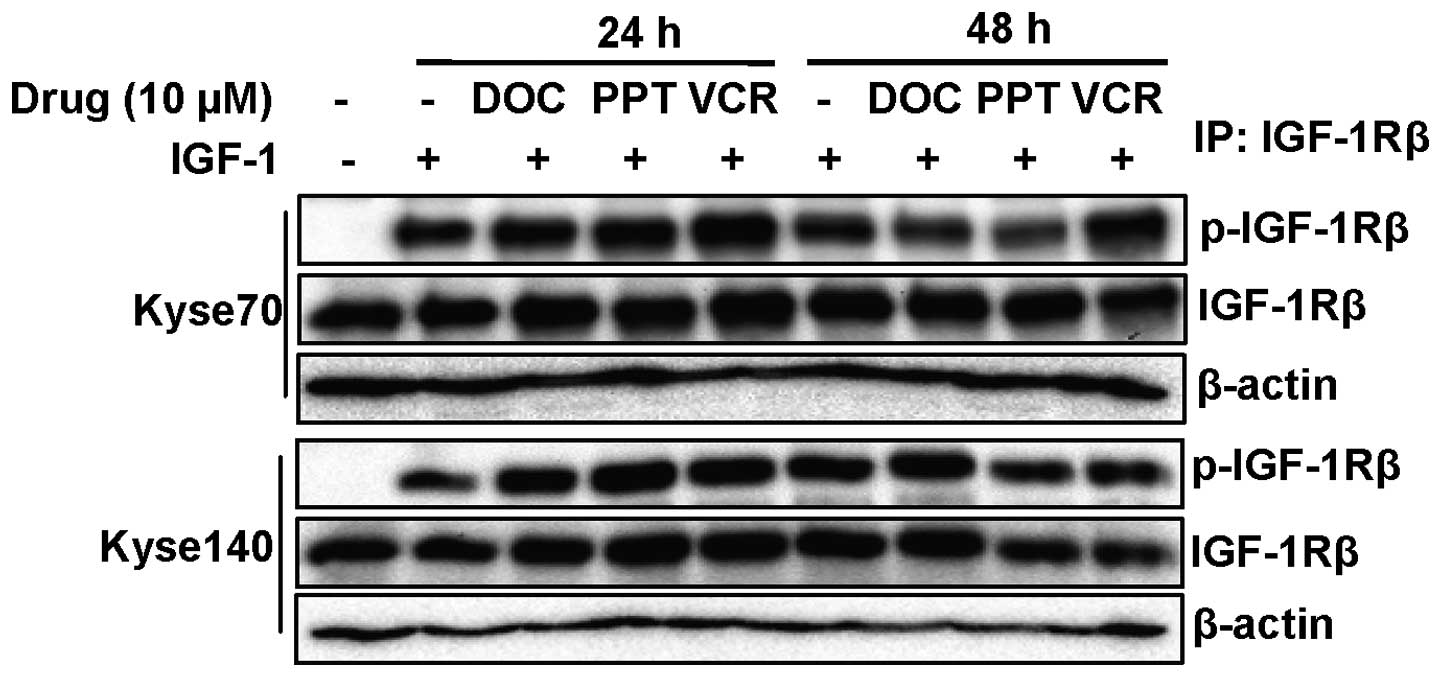
Microtubule targeting drugs demonstrated
no inhibition of phosphorylation or expression of IGF-1R in ESCC
cells
IGF-1R, which is becoming an important target in the
treatment of cancer, has been found to be significantly
overexpressed in oesophageal squamous cell carcinoma tissue
compared with adjacent normal tissue (24). We found that IGF-1R was expressed
in all nine ESCC cell lines (Fig.
3). To test whether microtubule targeting drugs can affect
IGF-1R, we treated cells with increasing concentrations of
microtubule targeting drugs (0.5, 5 and 10 μM) for 24 or 48
h, followed by 5 min of IGF-1 stimulation. We were not able to
observe any effect either on ligand-induced IGF-1R phosphorylation
or on IGF-1R expression (Fig. 6).
Thus, it appears that disruption of the microtubule system
selectively inhibits EGFR function over that of IGF-1R, indicating
that EGFR downregulation is not caused by general drug
cytotoxicity.
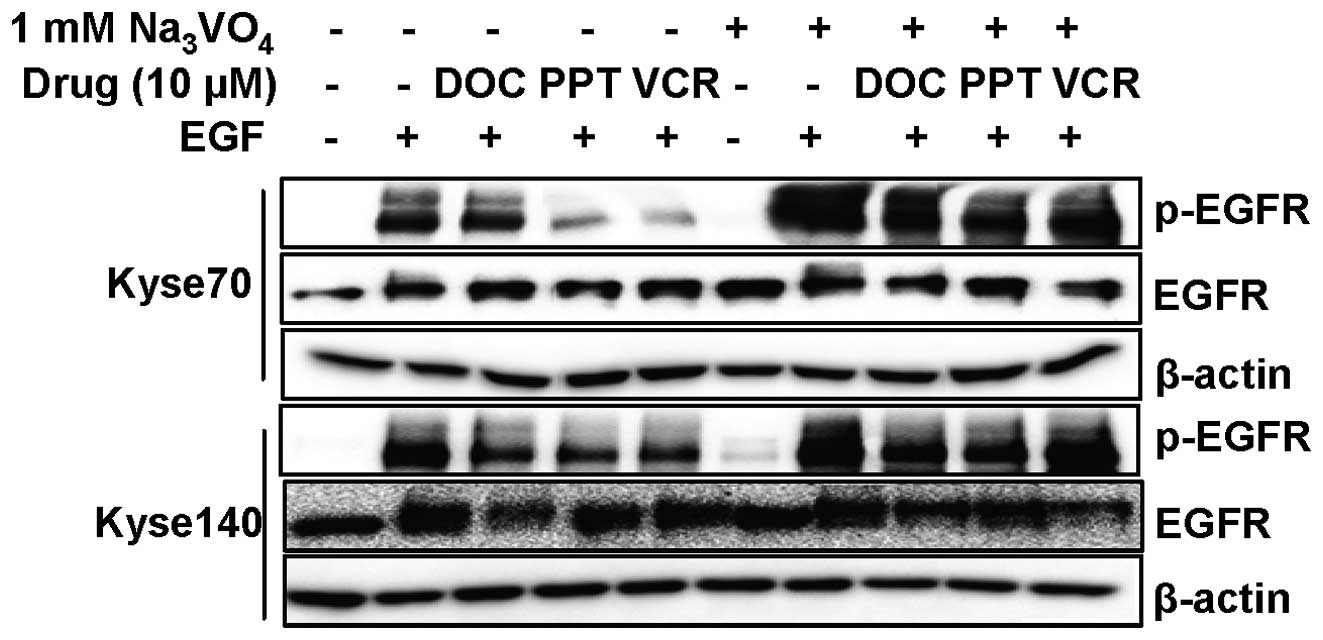
Reduced EGFR phosphorylation induced by
microtubule destabilising agents can be reversed by treatment with
a tyrosine phosphatase inhibitor
EGFR dephosphorylation caused by microtubule
destabilising agents was not correlated with protein downregulation
for all drugs and cell lines. In addition, the EGFR
dephosphorylation occurred before substantial receptor
downregulation could be seen, suggesting that these effects are
independent of each other (Fig.
4). To further explore the mechanism of EGFR dephosphorylation
we treated cells for 24 h, to avoid major EGFR downregulation, with
microtubule targeting drugs in the presence or absence of the
tyrosine phosphatase inhibitor sodium orthovanadate
(Na3VO4). As can be seen in Fig. 7, sodium orthovanadate treatment to
a large extent abolished EGFR dephosphorylation. Thus, one
possibility is that disruption of the microtubule network releases
or activates a tyrosine phosphatase that can dephosphorylate EGFR
but not IGF-1R.
PTP1B or PTPε downregulation can not
reverse EGFR dephosphorylation induced by microtubule disrupting
agents
The tyrosine phosphatase PTPε co-localises with
microtubules in cells and its binding to tubulin can inhibit its
activity; conversely disrupting microtubule structures increased
PTPε activity (25). PTP1B has
been reported to interact with endocytosed EGFR and promote its
dephosphorylation, and this complex is disrupted by sodium
othovanadate (26,27). To further explore which protein
tyrosine phosphatase is involved in EGFR dephosphorylation induced
by microtubule disruption, RNAi was employed to downregulate PTP1B
and PTPε expression in Kyse70 and Kyse140 cells as shown in
Fig. 8A and B. Neither PTP1B nor
PTPε downregulation could reverse the effect of the microtubule
disrupting drugs on EGFR dephosphorylation (Fig. 8C and D). Furthermore, we tested if
PTPε downregulation could affect the EGFR dephosphorylation induced
by micro-tubule targeting agents in the A431 cell line which is
originated from epidermoid carcinoma and known to overexpress EGFR.
PPT dephosphorylated EGFR in both A431 moc (cytPTPε negative) and
A431 (cytPTPε overexpressed) cells (Fig. 8E), proving also in other cells than
ESCC cells that PTPε downregulation can not reverse EGFR
dephosphorylation induced by micro-tubule targeting agents.
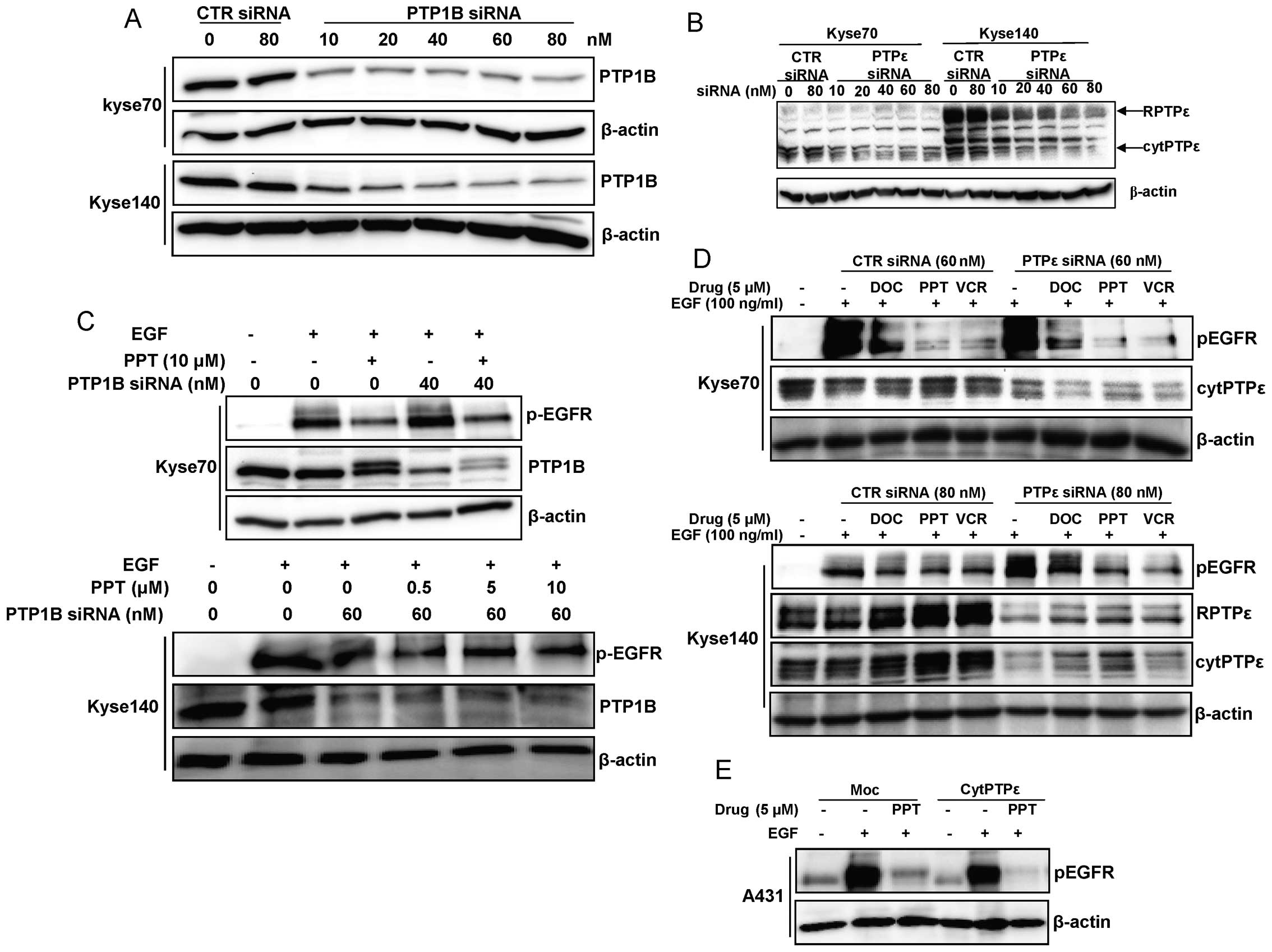 | Figure 8.PTP1B or PTPε downregulation could not
reverse EGFR dephosphorylation induced by microtubule disrupting
agents. (A and B) The anti-PTPε antibody recognizes both cytPTPε
and RPTPε, which can be distinguished according to their
electrophoretic mobility: cytPTPε, 70 kDa; RPTPε, 110 kDa. PTP1B
and PTPε was downregulated by specific siRNA transfected using
siLentFect™ from Bio-Rad in Kyse70 and Kyse140 cells. For each
experiment, non-targeting siRNA was used as a control. β-actin was
used as a loading control for each blot. (C) Kyse70 and Kyse140
cells were grown to sub-confluency, starved and treated with 10
μM PPT for 24 h to avoid EGFR degradation. Meanwhile,
specific siRNA was added to downregulate PTP1B expression. After
stimulation with EGF, western blotting was performed on total cell
lysates to detect phosphorylated EGFR as well as PTP1B. β-actin was
used as a loading control for each blot. (D) Kyse70 and Kyse140
cells were grown to sub-confluency, starved and treated with 5
μM vincristine (VCR), docetaxel (DOC) or PPT for 24 h to
avoid EGFR degradation. Meanwhile, PTPε expression was
downregulated using siRNA. After stimulation with EGF, western
blotting was performed on total cell lysates to detect
phosphorylated EGFR as well as PTPε. β-actin was used as a loading
control. (E) A431 moc (cytPTPε negative) and A431 (cytPTPε
overexpressed) cells were grown to sub-confluency, starved and
treated with 5 μM PPT for 24 h to avoid EGFR degradation.
After stimulation with EGF, western blotting was performed on total
cell lysates to detect phosphorylated EGFR. Each experiment has
been repeated at least three times. Non-targeting siRNA was used in
all experiments as a control. |
Discussion
Microtubules are involved in various cellular
functions, including cell adhesion, movement, replication and
division. Microtubule inhibition can interfere with chromosome
segregation during mitosis and disrupt cell signalling, hence
promoting cell cycle arrest and cell death (28,29).
In the present study, we have investigated the cytotoxic effects of
the microtubule targeting drugs docetaxel, vincristine, and PPT in
oesophageal carcinoma cell lines. As expected, microtubule
targeting drugs disrupted the microtubule network and inhibited
cell survival in oesophageal carcinoma cells. Surprisingly, we also
found that disruption of the microtubule network was associated
with dephosphorylation of EGFR and subsequent reduced activation of
Akt and Erk. Co-treatment with a tyrosine phosphatase inhibitor
diminished this effect, suggesting that disruption of the
microtubule network leads to exposure of EGFR to an active tyrosine
phosphatase. Neither 24 nor 48 h of drug treatment had any effect
on IGF-1R phosphorylation or stability, suggesting some degree of
receptor selectivity and that the EGFR downregulation is not due to
general toxicity.
In both Kyse70 and Kyse140 cells, phosphorylation of
Akt was inhibited by docetaxel, vincristine and PPT. However, the
degradation of total Akt protein after drug treatment may partially
explain the dephosphorylation of Akt. Compared to Akt, Erk protein
levels were not affected by drug treatment, while phosphorylation
of Erk was inhibited by vincristine and PPT but not docetaxel in
both Kyse70 and Kyse140 cells. This suggests that Akt protein
levels may be more easily affected by a general drug response and
that microtubule destabilisation, but not stabilisation, affects
downstream signalling of EGFR. The observed dephospho rylation of
EGFR could be reversed by a tyrosine phosphatase inhibitor,
suggesting that a tyrosine phosphatase is activated following
microtubule disruption. It is possible that microtubule
destabilising agents activate a phosphatase that is selective for
EGFR over IGF-1R in ESCC cells. Protein tyrosine phosphatases
(PTPs) strictly control receptor tyrosine kinase (RTK)
phosphorylation and downstream signalling. Several PTPs have been
reported to dephosphorylate tyrosine residues of EGFR and regulate
signalling, including T-cell PTP (TCPTP), Src homology
phosphotyrosine phosphatase 1 and 2 (SHP1 and 2), PTP1B, PTPN9,
density-enhanced phosphatase-1 (DEP-1), RPTPσ and RPTPκ (30–35).
So far only PTPε and PTP1B have been reported to interact with the
microtubule system (25–27). However, using siRNA to downregulate
PTP1B and PTPε expression in ESCC cells, we found no evidence
supporting that PTP1B or PTPε downregulation could influence the
effect of the microtubule disrupting drugs on EGFR
dephosphorylation (Fig. 8C and D).
Elucidating which PTP(s) is important for regulation of EGFR
phospho rylation in ESCC cells following disruption of the
microtubule network is subject for future studies.
The additional mechanism of action of tubulin
inhibitors on EGFR signalling suggested in the present work may
have clinical impact on the selection of drug combinations for the
treatment of oesophageal cancer as well as other cancer types.
Several clinical studies involving EGFR targeted therapies in
oesophageal cancer have been performed, including the antibody
cetuximab as well as the tyrosine kinase inhibitors erlotinib
(Tarceva®) and gefitinib (Iressa®). Although
not yet a standard of care, the results from these studies suggest
that treatment with EGFR targeted therapies, alone or in
combination with chemotherapy and/or radiotherapy, is feasible with
promising clinical activity (36,37).
Ongoing and future clinical trials involving EGFR targeted
therapies and anti-tubulin acting chemotherapy in combination is
recommended to consider the potential interactions between these
treatments, both with respect to clinical efficacy of the treatment
and to the selection of appropriate biomarkers.
We demonstrated that microtubule targeting drugs
inhibited the survival of oesophageal cancer cells involving a
reduction of tyrosine phosphorylation and activation of EGFR, and
that this effect is reversible by inhibition of tyrosine
phosphatases using sodium orthovanadate. Thus, we propose that in
addition to the previously described mechanisms of action for
microtubule disrupting chemotherapeutics, these drugs may also lead
to EGFR dephosphorylation and downregulation of EGFR-induced
signalling. These findings may have a clinical impact on the
selection of chemotherapeutic drug combinations for the treatment
of oesophageal cancer as well as other cancer types.
Acknowledgements
The authors are grateful to Dr Markus
Dagnell at Cancer Center, Karolinska Institutet, for providing A431
moc (cytPTPε negative) and A431 (cytPTPε overexpressed) cells. We
also thank Dr Ari Elson from Weizmann Institute of Science for
providing anti-PTPε antibody.
References
|
1.
|
Li H, DeRosier DJ, Nicholson WV, Nogales E
and Downing KH: Microtubule structure at 8 Å resolution. Structure.
10:1317–1328. 2002.
|
|
2.
|
Mitchison T and Kirschner M: Dynamic
instability of microtubule growth. Nature. 312:237–242. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Lobert S, Vulevic B and Correia JJ:
Interaction of vinca alkaloids with tubulin: a comparison of
vinblastine, vincristine, and vinorelbine. Biochemistry.
35:6806–6814. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Burkhart CA, Berman JW, Swindell CS and
Horwitz SB: Relationship between the structure of taxol and other
taxanes on induction of tumor necrosis factor-alpha gene expression
and cytotoxicity. Cancer Res. 54:5779–5782. 1994.PubMed/NCBI
|
|
5.
|
Bazley LA and Gullick WJ: The epidermal
growth factor receptor family. Endocr Relat Cancer. 12:S17–S27.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Normanno N, Bianco C, Strizzi L, et al:
The ErbB receptors and their ligands in cancer: an overview. Curr
Drug Targets. 6:243–257. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Arnold D and Seufferlein T: Targeted
treatments in colorectal cancer: state of the art and future
perspectives. Gut. 59:838–858. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Bonner JA, Harari PM, Giralt J, et al:
Radiotherapy plus cetuximab for squamous-cell carcinoma of the head
and neck. N Engl J Med. 354:567–578. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Pao W and Chmielecki J: Rational,
biologically based treatment of EGFR-mutant non-small-cell lung
cancer. Nat Rev Cancer. 10:760–774. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Gao YS, Hubbert CC and Yao TP: The
microtubule-associated histone deacetylase 6 (HDAC6) regulates
epidermal growth factor receptor (EGFR) endocytic trafficking and
degradation. J Biol Chem. 285:11219–11226. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Deribe YL, Wild P, Chandrashaker A, et al:
Regulation of epidermal growth factor receptor trafficking by
lysine deacetylase HDAC6. Sci Signal. 2:ra842009.PubMed/NCBI
|
|
12.
|
Devesa SS, Blot WJ and Fraumeni JF Jr:
Changing patterns in the incidence of esophageal and gastric
carcinoma in the United States. Cancer. 83:2049–2053. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Gebski V, Burmeister B, Smithers BM, Foo
K, Zalcberg J and Simes J: Survival benefits from neoadjuvant
chemoradiotherapy or chemotherapy in oesophageal carcinoma: a
meta-analysis. Lancet Oncol. 8:226–234. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Jankowski J HD, Hopwood D and Wormsley KG:
Expression of epidermal growth factor, transforming growth factor
alpha and their receptor in gastro-oesophageal diseases. Dig Dis.
11:1–11. 1993.
|
|
15.
|
Gibault L, Metges JP, Conan-Charlet V, et
al: Diffuse EGFR staining is associated with reduced overall
survival in locally advanced oesophageal squamous cell cancer. Br J
Cancer. 93:107–115. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Kim LW, Tsung-Teh W, In Seon C, et al:
Expression of epidermal growth factor receptor in esophageal and
esophagogastric junction adenocarcinomas. Cancer. 109:658–667.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Wei Q, Chen L, Sheng L, Nordgren H, Wester
K and Carlsson J: EGFR, HER2 and HER3 expression in esophageal
primary tumours and corresponding metastases. Int J Oncol.
31:493–499. 2007.PubMed/NCBI
|
|
18.
|
Oku K, Tanaka A, Yamanishi H, et al:
Effects of various growth factors on growth of a cloned human
esophageal squamous cancer cell line in a protein-free medium.
Anticancer Res. 11:1591–1595. 1991.PubMed/NCBI
|
|
19.
|
Ahmed SA, Gogal RM Jr and Walsh JE: A new
rapid and simple non-radioactive assay to monitor and determine the
proliferation of lymphocytes: an alternative to
[3H]thymidine incorporation assay. J Immunol Methods.
170:211–224. 1994.PubMed/NCBI
|
|
20.
|
Nociari MM, Shalev A, Benias P and Russo
C: A novel one-step, highly sensitive fluorometric assay to
evaluate cell-mediated cytotoxicity. J Immunol Methods.
213:157–167. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Lennartsson J, Wardega P, Engstrom U,
Hellman U and Heldin CH: Alix facilitates the interaction between
c-Cbl and platelet-derived growth factor beta-receptor and thereby
modulates receptor down-regulation. J Biol Chem. 281:39152–39158.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
McLeod HL, Kearns CM, Kuhn JG and Bruno R:
Evaluation of the linearity of docetaxel pharmacokinetics. Cancer
Chemother Pharmacol. 42:155–159. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Sethi VS, Jackson DV Jr, White DR, et al:
Pharmacokinetics of vincristine sulfate in adult cancer patients.
Cancer Res. 41:3551–3555. 1981.PubMed/NCBI
|
|
24.
|
Nemoto T, Ohashi K, Akashi T, Johnson JD
and Hirokawa K: Overexpression of protein tyrosine kinases in human
esophageal cancer. Pathobiology. 65:195–203. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Sines T, Granot-Attas S, Weisman-Welcher S
and Elson A: Association of tyrosine phosphatase epsilon with
microtubules inhibits phosphatase activity and is regulated by the
epidermal growth factor receptor. Mol Cell Biol. 27:7102–7112.
2007. View Article : Google Scholar
|
|
26.
|
Flint AJ, Tiganis T, Barford D and Tonks
NK: Development of ‘substrate-trapping’ mutants to identify
physiological substrates of protein tyrosine phosphatases. Proc
Natl Acad Sci USA. 94:1680–1685. 1997.
|
|
27.
|
Eden ER, White IJ, Tsapara A and Futter
CE: Membrane contacts between endosomes and ER provide sites for
PTP1B-epidermal growth factor receptor interaction. Nat Cell Biol.
12:267–272
|
|
28.
|
Poruchynsky MS, Wang EE, Rudin CM,
Blagosklonny MV and Fojo T: Bcl-xL is phosphorylated in malignant
cells following microtubule disruption. Cancer Res. 58:3331–3338.
1998.PubMed/NCBI
|
|
29.
|
Blagosklonny MV, Giannakakou P, el-Deiry
WS, et al: Raf-1/bcl-2 phosphorylation: a step from microtubule
damage to cell death. Cancer Res. 57:130–135. 1997.PubMed/NCBI
|
|
30.
|
Tarcic G, Boguslavsky SK, Wakim J, et al:
An unbiased screen identifies DEP-1 tumor suppressor as a
phosphatase controlling EGFR endocytosis. Curr Biol. 19:1788–1798.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Xu Y, Tan L-J, Grachtchouk V, Voorhees JJ
and Fisher GJ: Receptor-type protein-tyrosine phosphatase-kappa
regulates epidermal growth factor receptor function. J Biol Chem.
280:42694–42700. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Suarez Pestana E, Tenev T, Gross S,
Stoyanov B, Ogata M and Bohmer FD: The transmembrane protein
tyrosine phosphatase RPTPsigma modulates signaling of the epidermal
growth factor receptor in A431 cells. Oncogene. 18:4069–4079.
1999.PubMed/NCBI
|
|
33.
|
Keilhack H, Tenev T, Nyakatura E, et al:
Phosphotyrosine 1173 mediates binding of the protein-tyrosine
phosphatase SHP-1 to the epidermal growth factor receptor and
attenuation of receptor signaling. J Biol Chem. 273:24839–24846.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Agazie YM and Hayman MJ: Development of an
efficient ‘substrate-trapping’ mutant of Src homology
phosphotyrosine phosphatase 2 and identification of the epidermal
growth factor receptor, Gab1, and three other proteins as target
substrates. J Biol Chem. 278:13952–13958. 2003.
|
|
35.
|
Yuan T, Wang Y, Zhao ZJ and Gu H:
Protein-tyrosine phosphatase PTPN9 negatively regulates ErbB2 and
epidermal growth factor receptor signaling in breast cancer cells.
J Biol Chem. 285:14861–14870. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Ku GY and Ilson DH: Esophagogastric
cancer: targeted agents. Cancer Treat Rev. 36:235–248. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Ekman S, Dreilich M, Lennartsson J, et al:
Esophageal cancer: current and emerging therapy modalities. Expert
Rev Anticancer Ther. 8:1433–1448. 2008. View Article : Google Scholar : PubMed/NCBI
|