Introduction
Hepatocellular carcinoma (HCC) is the fifth most
common malignancy in men and the eighth most common in women
worldwide. It is estimated to cause approximately half a million
deaths annually (1). Several risk
factors for HCC have been reported, including infection with
hepatitis B and C viruses, dietary intake of afratoxin and alcohol
consumption. However, the molecular pathogenesis of HCC remains
poorly understood.
MicroRNAs (miRNAs) are ∼22 nucleotide non-coding
RNAs that function as endogenous silencers of target genes.
Currently, more than 1,000 miRNAs have been identified in the human
genome (the miRBase database) and each miRNA is predicted to
control hundreds of gene targets. miRNAs are expressed in a
tissue-specific manner and play important roles in development,
cell proliferation, apoptosis and oncogenesis (2–5).
Dysregulation of miRNAs in cancer has been repeatedly described
(6–8). HCC is no exception and various
HCC-specific miRNA signatures have been described (9).
DNA methylation of CpG islands within the promoter
regions of tumor suppressor genes is known to inhibit
transcriptional initiation and thereby silence these genes. Growing
evidence indicates that some tumor-suppressive miRNAs are also
epigenetically silenced by promoter DNA methylation in cancer
(10), suggesting the diagnostic
and therapeutic potential of these miRNAs.
In the present study, we aimed to identify miRNA
genes that are silenced by DNA hypermethylation in HCC. We screened
for genes with promoter DNA hypermethylation using a genome-wide
methylation microarray analysis and found that miR-335,
which is harbored within an intron of its protein-coding host gene
MEST, is downregulated by aberrant promoter hypermethylation
in HCC.
Materials and methods
Cell lines and primary tumors
The following 21 HCC cell lines were examined: HLE,
HLF, PLC/PRF/5, Li7, Huh7, Hep3B, SNU354, SNU368, SNU387, SNU398,
SNU423, SNU449, SNU475, JHH-1, JHH-2, JHH-4, JHH-5, JHH-6, JHH-7,
Huh1 and HepG2 (11).
Paired tumor and non-tumor tissues were obtained
from 32 HCC patients who underwent surgery. All specimens were
immediately frozen in liquid nitrogen and were stored at −80°C
until further use. Genomic DNA and total RNA were isolated using
the Puregene DNA isolation kit (Gentra, Minneapolis, MN) and TRIzol
reagent (Invitrogen, Carlsbad, CA), respectively. Twenty tumor
samples were available for DNA methylation analyses and 32 paired
tumor and non-tumor samples were available for microRNA and mRNA
analyses. This study was approved by the ethics committees and
conducted in accordance with the Declaration of Helsinki. Informed
consent was obtained from each patient.
Methylation array analysis
We performed a genome-wide DNA methylation analysis
called microarray-based integrated analysis of methylation by
isoschizomers (MIAMI), as previously described (12–14).
The complete experimental procedure can be obtained at http://grc.dept.med.gunma-u.ac.jp/~gene/image/MIAMI%20Protocol%20V4.pdf.
Changes in methylation were judged by assessing the differences in
methylation-sensitive HpaII cleavage and
methylation-insensitive MspI cleavage between samples. We
used a custom microarray, which contains ∼38,000 probes chosen from
the Agilent promoter array, on an eArray system (http://earray.chem.agilent.com/earray/).
TaqMan miRNA assay
Reverse transcription (RT) reactions and real-time
quantitative polymerase chain reactions (PCR) were performed using
the TaqMan MicroRNA RT kit (Applied Biosystems, Darmstadt,
Germany), TaqMan MicroRNA Assays (Applied BioSystems) and ABI PRISM
7300 Fast Real-time PCR system (Applied Biosystems), according to
the manufacturer’s instructions. RNU6B was used as an
endogenous control for miRNA levels.
Drug treatment
Cells were treated with 1 or 5 μM of
5-aza-2′-deoxycytidine (5-aza-dCyd; Sigma-Aldrich, St. Louis, MO)
for 4 days or 50 ng/ml of trichostatin A (TSA; Wako, Osaka, Japan)
for 1 day. In assessing drug synergy, cells were cultured in the
presence of 1 or 5 μM of 5-aza-dCyd for 4 days and were then
treated for an additional 24 h with 50 ng/ml of TSA.
Methylation analysis
Methylation status was examined by
methylation-specific PCR (MSP), bisulfite PCR followed by
restriction enzyme digestion [combined bisulfite and restriction
analysis (COBRA)] (15) and
bisulfite sequencing analysis, as previously described (16). The primers used are listed in
Table I. Briefly, for MSP, genomic
DNA was treated with sodium bisulfite using an EZ DNA Methylation
kit (Zymo Research, Orange, CA) and subjected to PCR using specific
primer sets. For COBRA, genomic DNA was treated with sodium
bisulfite and subjected to PCR. The PCR products were digested with
BstUI, which recognizes sequences unique to the methylated
alleles, but cannot recognize unmethylated alleles and the digested
products were electrophoresed on 3% agarose gels and stained with
ethidium bromide. Methylation levels were calculated as the ratio
of the gray scale value of the methylated band to that of the
combined methylated and unmethylated bands. The gray scale value
was obtained by scanning the gel with Adobe Photoshop CS3 Extended
software (Adobe Systems Inc., San Jose, CA, USA). For
bisulfite-sequencing, the PCR products were cloned and then
sequenced. CpGenome universal unmethylated and methylated DNA
(Chemicon, Billerica, MA) served as controls for unmethylated and
methylated DNA, respectively.
 | Table I.Sequences of PCR primers used in the
study. |
Table I.
Sequences of PCR primers used in the
study.
| Purpose | Gene | Forward primer | Reverse primer |
|---|
| Methylation
specific primer |
miR-335/MEST | Methylation
specific primer |
5′-TTGTAATAGGTGGCGTTGAC-3′ |
5′-ACTCGAAACTAAAACGTCGC-3′ |
| Unmethylation
specific primer |
5′-TTTTTGTAATAGGTGGTGTTGAT-3′ |
5′-ACTCAAAACTAAAACATCACCAA-3′ |
|
miR-101-2/RCL1 | Methylation
specific PCR |
5′-GATTGGTAATTTTCGCGTC-3′ |
5′-GCGCTACCATTAATCCGTA-3′ |
| Unmethylation
specific primer |
5′-GATTGGTAATTTTTGTGTT-3′ |
5′-ACACTACCATTAATCCATA-3′ |
| Real-time
quantitative RT-PCR | MEST | |
5′-CGCAGGATCAACCTTCTTTC-3′ |
5′-CATCAGTCGTGTGAGGATGG-3′ |
Real-time quantitative RT-PCR
We quantified mRNA using a real-time fluorescence
detection method, as previously described (11). Real-time quantitative PCR
experiments were performed with the LightCycler system using
FastStart DNA Master Plus SYBR Green I (Roche Diagnostics,
Penzberg, Germany), according to the manufacturer’s protocol. The
primers used are listed in Table
I. The endogenous control for mRNA was GAPDH.
Statistical analysis
Spearman’s rank correlation test, Wilcoxon
signed-rank test and Mann-Whitney U test were performed using SPSS
15.0 software (SPSS, Inc., Chicago, IL). P-values of <0.05 were
considered significant.
Results
Genome-wide DNA methylation profiles in
HCC
To identify miRNA genes that are silenced by DNA
hypermethylation in HCC, we compared DNA methylation profiles
between three HCC cell lines (SNU449, Li-7 and PLC/PRF/5) and one
normal liver tissue using the MIAMI method. The microarray covers
approximately 38,000 probes (corresponding to promoter regions of
about 14,000 genes), which include 411 probes for miRNA (167 miRNA
genes). MIAMI analyses revealed that 575 probes (484 genes) were
hypermethylated and 350 probes (277 genes) were hypomethylated
similarly in the three HCC cell lines compared to normal liver. The
hypermethylated genes included eight miRNA genes (miR-let-7b,
miR-101-2, miR-122a, miR-146b, miR-149, miR-200b, miR-335 and
miR-497). Therefore, further analysis was focused on these
eight miRNA genes. The strategy and partial results are shown in
Fig. 1.
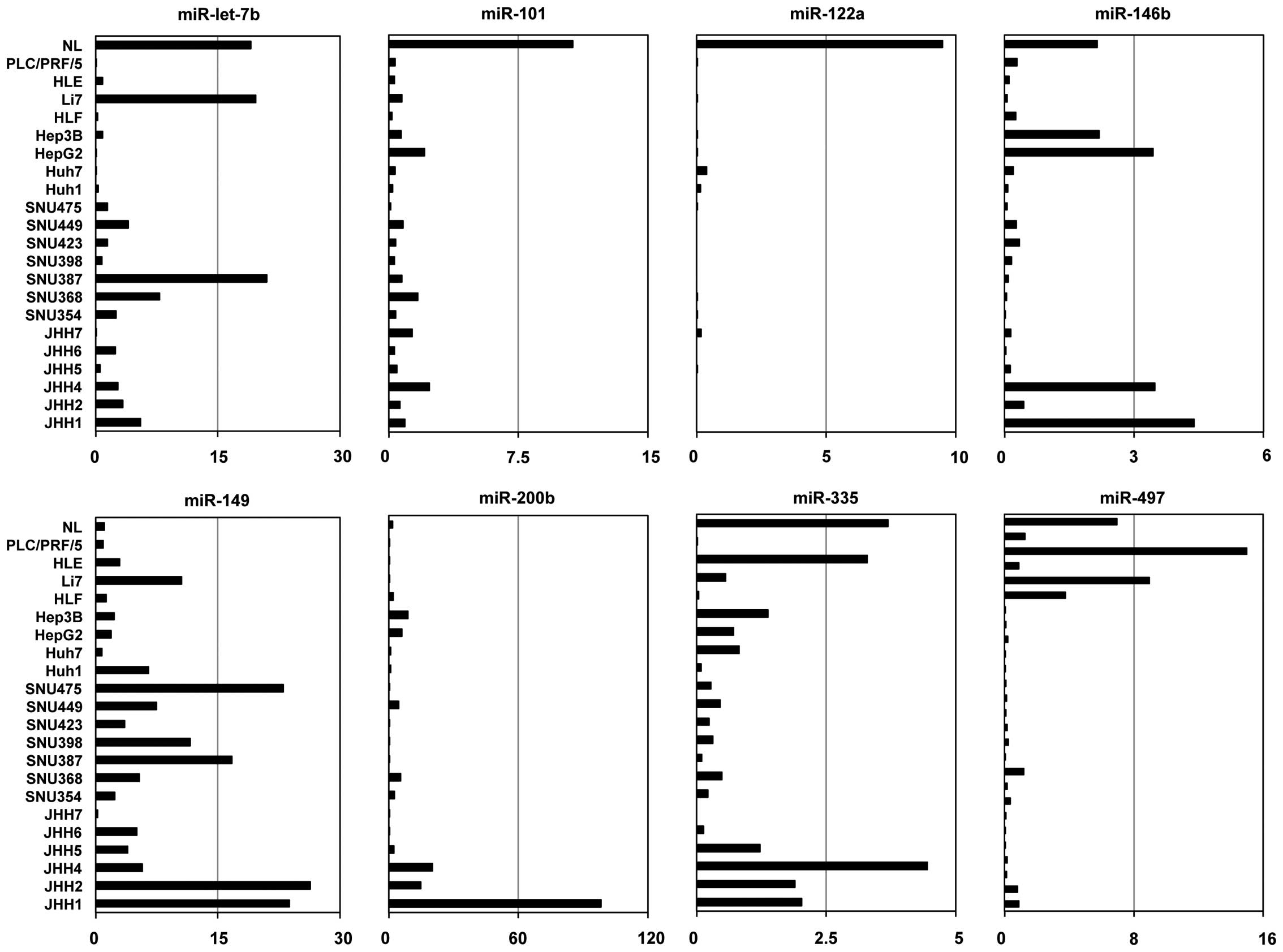
Expression of candidate miRNAs in HCC
cell lines
We analyzed the expression levels of the eight
miRNAs in 21 human HCC cell lines and normal liver using TaqMan
miRNA PCR. Expression levels of six miRNAs (miR-let-7b,
miR-101-2, miR-122a, miR-146b, miR-335 and miR-497), but
not two of the miRNAs (miR-149 and miR-200b), were
lower in more than half of the 21 cell lines than normal liver
(Fig. 2).
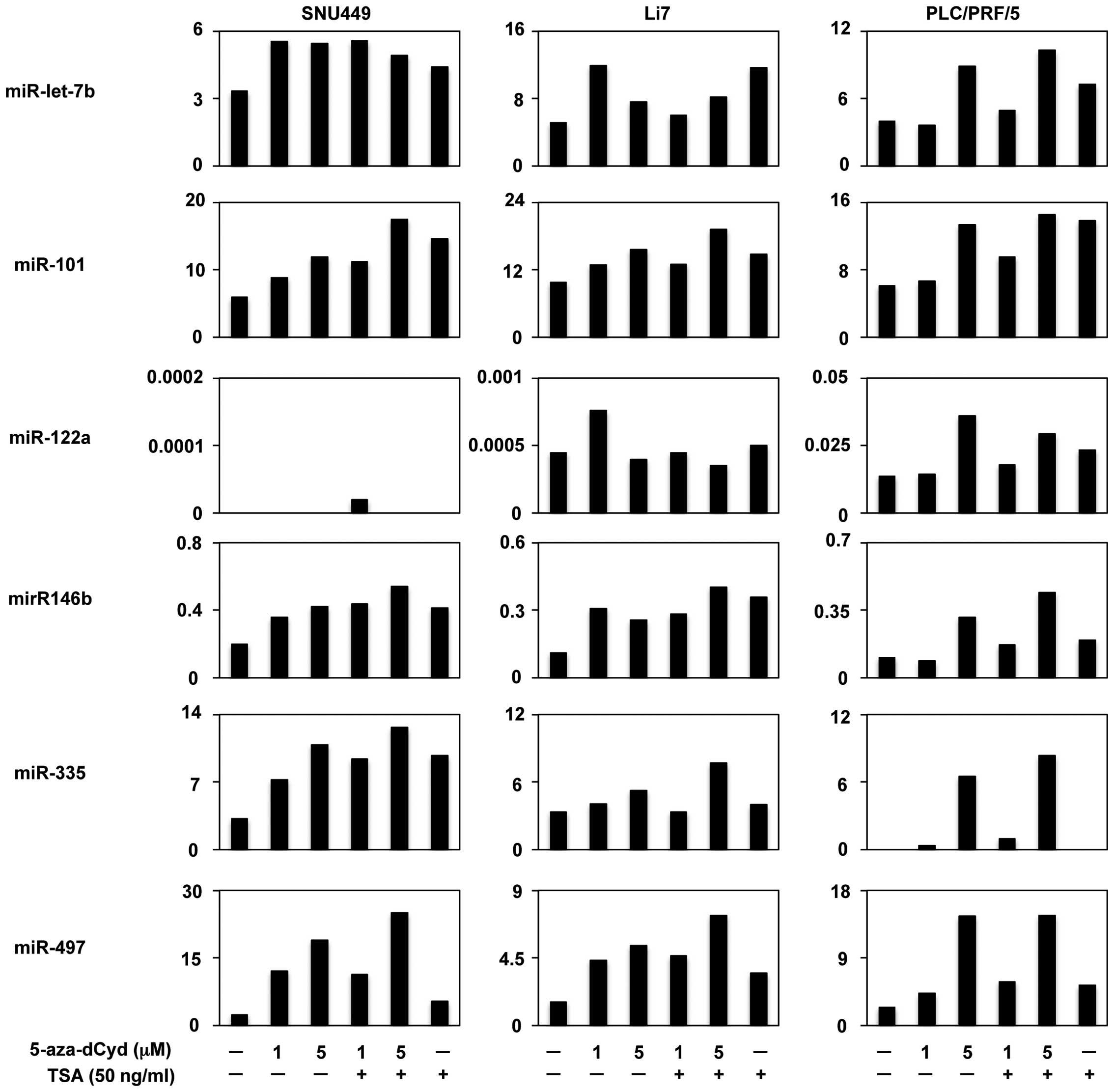
Restoration of miRNA expression by the
methyltransferase inhibitor
We then assessed the effects of demethylation on the
expression of the six candidate miRNAs. Three HCC cell lines
(SNU449, Li7 and PLC/PRF/5) were treated with 5-azadCyd, a
methyltransferase inhibitor and miRNA expression levels were
assayed with TaqMan miRNA PCR. Expression of four miRNAs
(miR-101-2, miR-146b, miR-335 and miR-497), but not
two of the miRNAs (miR-let-7b and miR-122a), were
restored with 5-aza-dCyd treatment in all three HCC cells (Fig. 3), suggesting that aberrant DNA
methylation suppressed the expression of these four miRNAs.
Additionally, it was observed that treatment with a histone
deacetylase inhibitor, TSA, enhanced the expression of these four
miRNAs by 5-aza-dCyd in all three cell lines (Fig. 3). These findings suggest that
histone deacetylation may also contribute to the transcriptional
repression of these four miRNAs.
Methylation of miR-335/MEST in HCC
cells
About half of all miRNA genes are encoded in the
introns of protein-encoding genes and subsequently excised from a
primary transcript in common with protein coding genes, so-called
host genes (10,17–19).
Thus, these miRNA genes are more likely to be susceptible to
transcriptional repression by aberrant DNA methylation of CpG
islands located in the host genes. Of the selected four genes
(miR-101-2, miR-146b, miR-335 and miR-497), we
identified that miR-101-2 and miR-335 are intronic
miRNAs using the human genome browser at UCSC (February 2009).
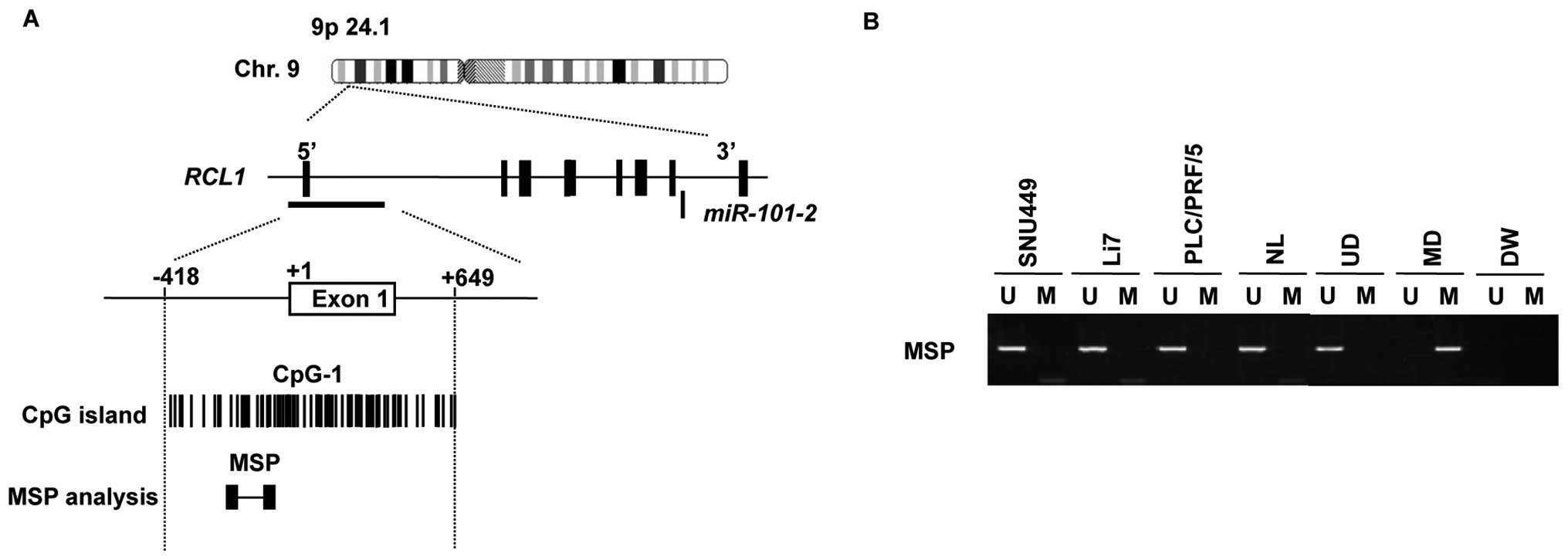
miR-101-2 and miR-335 are located within the introns
of RNA terminal phosphate cyclase-like 1 gene (RCL1)
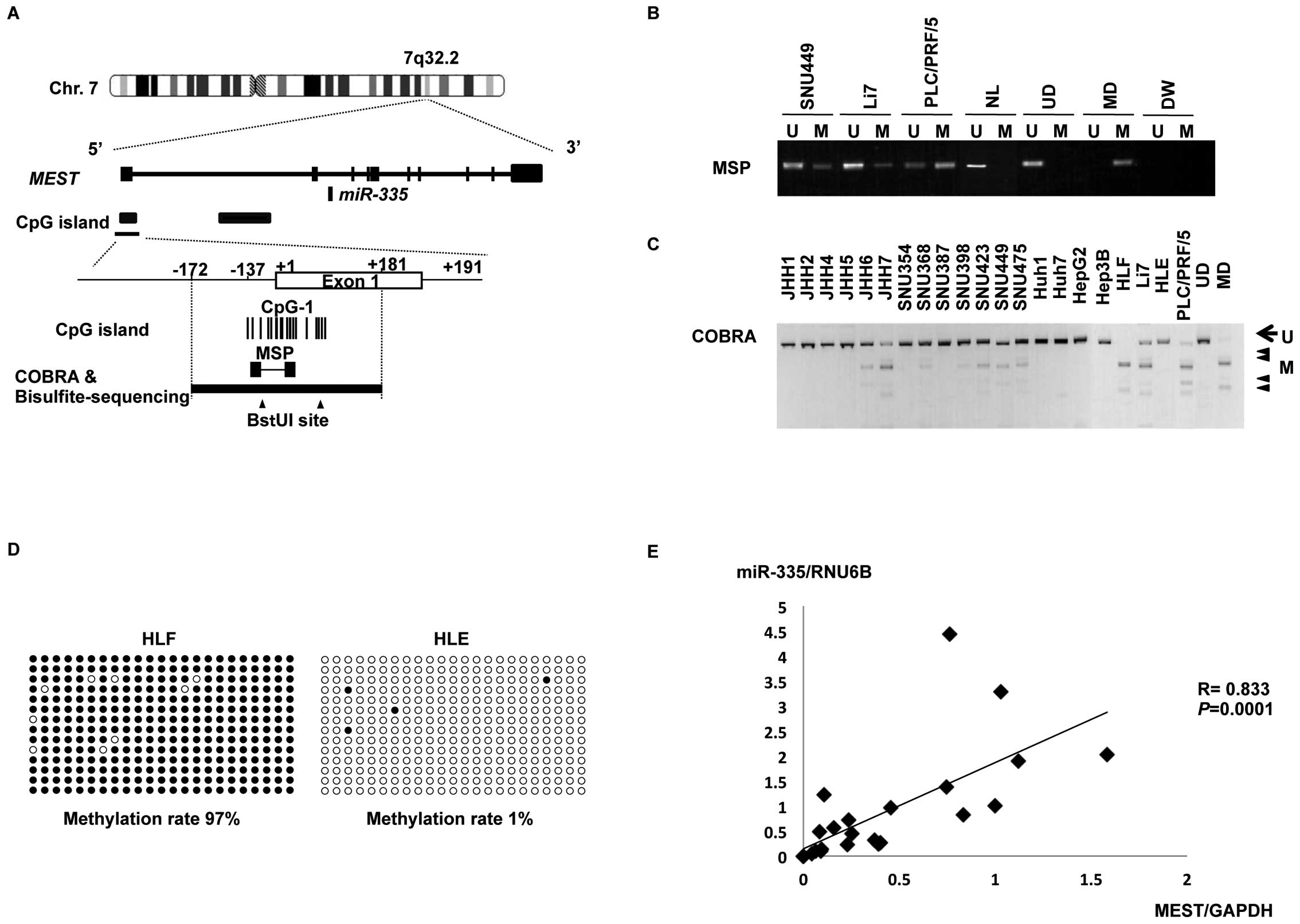
(Fig. 4A) and mesoderm specific
transcript homolog gene (MEST) (Fig. 5A), respectively. We also found CpG
islands around the transcription start sites of
miR-101-2/RCL1 and miR-335/MEST genes using
the genome database of the European Bioinformatics Institute.
However, no CpG islands were found around miR-146b or
miR-497.
Therefore, we assessed the methylation status of the
CpG islands of miR-101-2/RCL1 and miR-335/MEST via
MSP in three HCC cells (SNU449, Li7 and PLC/PRF/5) and normal
liver. MSP analyses indicated that the CpG island of
miR-101-2/RCL1 was not methylated in these HCC cells
(Fig. 4B), whereas aberrant DNA
methylation within the CpG island of miR-335/MEST was
evident in all three HCC cells (Fig.
5B).
To confirm and quantify the methylation status of
miR-335/MEST, we assayed DNA methylation levels of the
miR-335/MEST CpG island using the COBRA technique, which
involves bisulfite PCR followed by restriction enzyme digestion, in
21 HCC cell lines. COBRA analyses (Fig. 5C) revealed that the
miR-335/MEST CpG island was hypermethylated in three
cell types (JHH7, HLF and PLC/PRF/5) that lack the expression of
miR355 (Fig. 2), partly
methylated in eight (JHH6, SNU368, SNU398, SNU423, SNU449, SNU475,
Huh7 and Li7) with reduced expression of miR355 (Fig. 2) and unmethylated in the remaining
10 cell lines, including HLE. Consistent with the results of COBRA,
further analysis of the PCR products with bisulfite-sequencing
showed that the CpG island was hypermethylated in HLF cells
(methylation rate, 97%) and hypomethylated in HLE cells
(methylation rate, 1%) (Fig. 5D).
Taken together, these data suggest that the
miR-335/MEST CpG island was hypermethylated in some
HCC cells. The physical relationship between miR-335,
MEST, the CpG island and the primers used for MSP and COBRA
are shown in Fig. 5A.
The expression levels of miR-335
significantly correlated with those of MEST in 21 HCC cell
lines (Spearman’s rank correlation test, r=0.83; P=0.0001)
(Fig. 5E), supporting the notion
that the intronic miR-335 is co-expressed with its host
gene, MEST, under the control of the host gene promoter.
Methylation and reduced expression of
miR-335 in primary HCC tumors
To determine whether the methylation of the
miR-335/MEST CpG island observed in HCC cell lines also
occurs in primary human HCC, we assessed the methylation status of
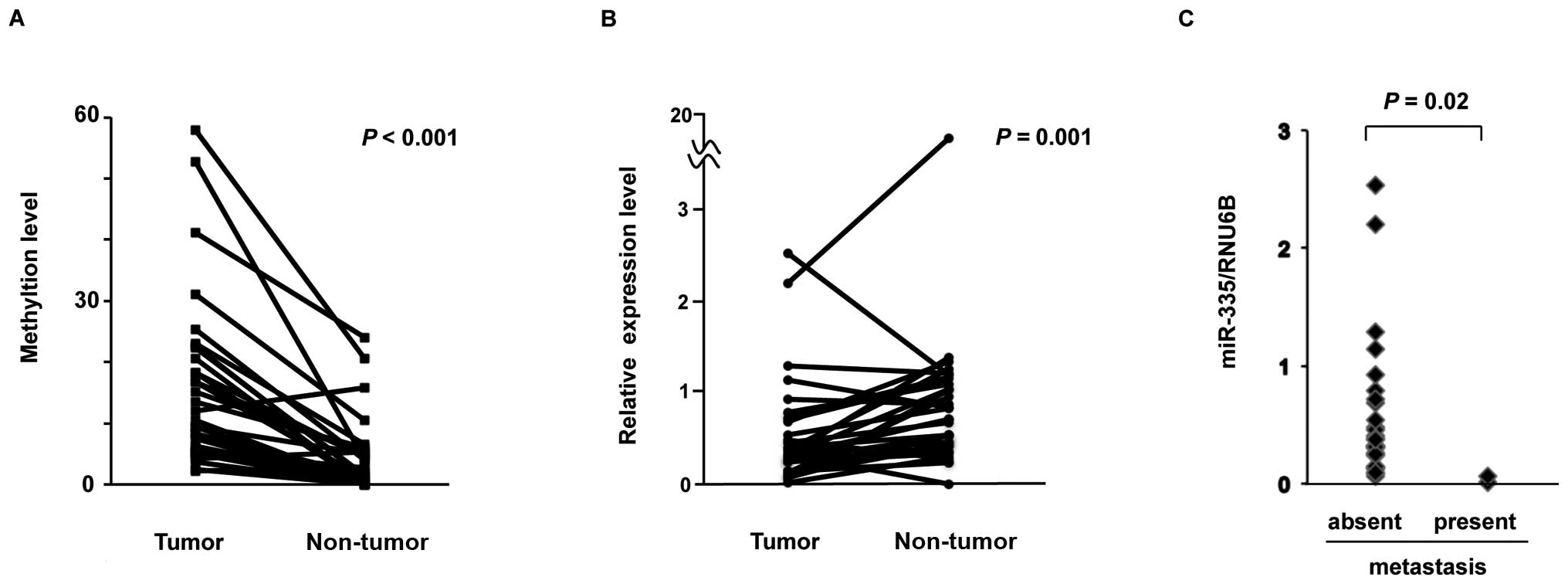
miR-335/MEST in paired tumor and non-tumor tissues from 20
patients with primary HCC by using COBRA. Methylation of
miR-335/MEST was observed in all 20 HCC tumors and in
15 of the 20 non-tumor liver tissues. Although methylation of
miR-335/MEST was found in both HCC tumors and
non-tumor tissues, the level of miR-335/MEST
methylation was significantly higher in 18 (90%) out of 20 tumors,
compared to their non-tumor tissue counterparts (Wilcoxon
signed-rank test, P<0.001) (Fig.
6A).
To investigate whether the reduced expression of
miR-335 observed in HCC cells was relevant in primary HCC
tumors, we analyzed the expression of miR-335 in paired
tumor and non-tumor tissues from 32 HCC patients via TaqMan miRNA
PCR. The expression level of miR-335 was significantly lower
in 25 (78%) out of 32 tumors, compared to their non-tumor tissue
counterparts (Wilcoxon signed-rank test, P= 0.001) (Fig. 6B). Taken together, these findings
suggest that the expression of miR-335 was frequently
reduced by aberrant DNA methylation in primary HCCs.
Since miR-335 was identified as a metastasis
suppressor miRNA in breast cancer by Tavazoie et al(20), we examined the relationship between
the expression levels of miR-335 and the presence of distant
metastasis in these 32 primary HCCs. The expression of
miR-335 was significantly lower in HCC tumors with distant
metastasis than in those without distant metastasis (Mann-Whitney U
test, P=0.02) (Fig. 6C),
suggesting that a reduced expression of miR-335 may be
association with distant metastasis in HCC, as well as in breast
cancer.
Discussion
This is the first report that miR-335 is
downregulated in HCC via aberrant promoter hypermethylation, which
was demonstrated through a number of approaches. First, we screened
for genes with promoter DNA methylation in HCC cell lines using
MIAMI, a powerful method for genome-wide profiling of promoter
methylation in the human genome (12–14)
and found eight miRNA genes that were possibly methylated in HCC
cells. Further methylation analyses, including MSP, COBRA,
bisulfite-sequencing and drug treatment with 5-aza-dCyd and TSA,
combined with expression analyses, narrowed down the candidate
methylated miRNA genes and confirmed that the
miR-335/MEST CpG island was hypermethylated in some
HCC cells. In primary HCCs, the level of miR-335/MEST
methylation was significantly higher and the expression of
miR-335 was significantly lower in tumors compared to their
non-tumor tissue counterparts, suggesting that the expression of
miR-335 was reduced by aberrant DNA methylation in primary
HCCs. Furthermore, our results suggest that a reduced expression of
miR-335 may be associated with distant metastasis in
HCC.
DNA hypermethylation of CpG islands within promoter
regions is known to be an epigenetic aberration leading to the
inactivation of tumor-suppressive miRNA in cancer, which is similar
to that of many classical tumor-suppressor genes. To date, 19
intergenic miRNA genes, which are located in the non-coding regions
between genes and 42 intronic (intragenic) miRNA genes, which are
harbored within introns of their protein-coding host genes, have
been identified as tumor-suppressive miRNA (10). Of these, miR-335 was
reported to suppress metastasis and migration by targeting SOX4 and
tenascin C and inhibit tumor initiation in breast cancer (20,21).
The transcription of miR-335 was shown to be co-regulated
with MEST by promoter hypermethylation in breast cancer
cells (21). Furthermore, it was
demonstrated that miR-335 regulates Rb1 and controls cell
proliferation in a p53-dependent manner (22). Recent studies have shown that
miR-335 orchestrates cell proliferation, migration and
differentiation in human mesenchymal stem cells (23), as well as inhibits growth and
invasion of malignant astrocytoma cells (24). Further work will be aimed at
elucidating the role of miR-335 in the carcinogenesis and
metastasis of HCC.
References
|
1.
|
Bosch FX, Ribes J, Cléries R and Díaz M:
Epidemiology of hepatocellular carcinoma. Clin Liver Dis.
9:191–211. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
He L and Hannon GJ: MicroRNAs: small RNAs
with a big role in gene regulation. Nat Rev Genet. 5:522–531. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Xu P, Guo M and Hay BA: MicroRNAs and the
regulation of cell death. Trends Genet. 20:617–624. 2004.
View Article : Google Scholar
|
|
6.
|
Calin GA, Dumitru CD, Shimizu M, Bichi R,
Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, Rassenti L,
Kipps T, Negrini M, Bullrich F and Croce CM: Frequent deletions and
downregulation of micro-RNA genes miR15 and miR16 at 13q14 in
chronic lymphocytic leukemia. Proc Natl Acad Sci USA.
99:15524–15529. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Michael MZ, SM OC, van Holst Pellekaan NG,
Young GP and James RJ: Reduced accumulation of specific miRNAs in
colorectal neoplasia. Mol Cancer Res. 1:882–891. 2003.
|
|
8.
|
Takamizawa J, Konishi H, Yanagisawa K,
Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y,
Mitsudomi T and Takahashi T: Reduced expression of the let-7 miRNAs
in human lung cancers in association with shortened postoperative
survival. Cancer Res. 64:3753–3756. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Borel F, Konstantinova P and Jansen PL:
Diagnostic and therapeutic potential of miRNA signatures in
patients with hepatocellular carcinoma. J Hepatol. 56:1371–1383.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Kozaki KI and Inazawa J: Tumor-suppressive
microRNA silenced by tumor-specific DNA hypermethylation in cancer
cells. Cancer Sci. 103:837–845. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Zen K, Yasui K, Nakajima T, Zen Y, Zen K,
Gen Y, Mitsuyoshi H, Minami M, Mitsufuji S, Tanaka S, Itoh Y,
Nakanuma Y, Taniwaki M, Arii S, Okanoue T and Yoshikawa T: ERK5 is
a target for gene amplification at 17p11 and promotes cell growth
in hepatocellular carcinoma by regulating mitotic entry. Genes
Chromosomes Cancer. 48:109–120. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Hatada I, Fukasawa M, Kimura M, Morita S,
Yamada K, Yoshikawa T, Yamanaka S, Endo C, Sakurada A, Sato M,
Kondo T, Horii A, Ushijima T and Sasaki H: Genome-wide profiling of
promoter methylation in human. Oncogene. 25:3059–3064. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Hatada I, Morita S, Kimura M, Horii T,
Yamashita R and Nakai K: Genome-wide demethylation during neural
differentiation of P19 embryonal carcinoma cells. J Hum Genet.
53:185–191. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Hatada I, Namihira M, Morita S, Kimura M,
Horii T and Nakashima K: Astrocyte-specific genes are generally
demethylated in neural precursor cells prior to astrocytic
differentiation. PLoS One. 3:e31892008. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Xiong Z and Laird PW: COBRA: a sensitive
and quantitative DNA methylation assay. Nucleic Acids Res.
25:2532–2534. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Dohi O, Takada H, Wakabayashi N, Yasui K,
Sakakura C, Mitsufuji S, Naito Y, Taniwaki M and Yoshikawa T:
Epigenetic silencing of RELN in gastric cancer. Int J Oncol.
36:85–92. 2010.PubMed/NCBI
|
|
17.
|
Rodriguez A, Griffiths-Jones S, Ashurst JL
and Bradley A: Identification of mammalian microRNA host genes and
transcription units. Genome Res. 14:1902–1910. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Kim YK and Kim VN: Processing of intronic
microRNAs. EMBO J. 26:775–783. 2007. View Article : Google Scholar
|
|
19.
|
Saini HK, Griffiths-Jones S and Enright
AJ: Genomic analysis of human microRNA transcripts. Proc Natl Acad
Sci USA. 104:17719–17724. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Tavazoie SF, Alarcon C, Oskarsson T, Padua
D, Wang Q, Bos PD, Gerald WL and Massague J: Endogenous human
microRNAs that suppress breast cancer metastasis. Nature.
451:147–152. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Png KJ, Yoshida M, Zhang XH, Shu W, Lee H,
Rimner A and Chan TA: MicroRNA-335 inhibits tumor reinitiation and
is silenced through genetic and epigenetic mechanisms in human
breast cancer. Genes Dev. 25:226–231. 2011. View Article : Google Scholar
|
|
22.
|
Scarola M, Schoeftner S, Schneider C and
Benetti R: miR-335 directly targets Rb1 (pRb/p105) in a proximal
connection to p53-dependent stress response. Cancer Res.
70:6925–6933. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Tome M, Lopez-Romero P, Albo C, Sepulveda
JC, Fernandez-Gutierrez B, Dopazo A, Bernad A and Gonzalez MA:
miR-335 orchestrates cell proliferation, migration and
differentiation in human mesenchymal stem cells. Cell Death Differ.
18:985–995. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Shu M, Zheng X, Wu S, Lu H, Leng T, Zhu W,
Zhou Y, Ou Y, Lin X, Lin Y, Xu D, Zhou Y and Yan G: Targeting
oncogenic miR-335 inhibits growth and invasion of malignant
astrocytoma cells. Mol Cancer. 10:592011. View Article : Google Scholar : PubMed/NCBI
|