Introduction
Giant cell tumor of bone (GCT) is a destructive
neoplasm of uncertain etiology that affects the epiphyseal ends of
long bones in young adults. The tumor causes severe bone
destruction in the vicinity of major skeletal joints necessitating
complex reconstructive surgery to eradicate the tumor and save the
joint. Despite aggressive therapy, this tumor tends to recur
locally, eventually requiring surgical measures of increasing
complexity, resulting in significant morbidity and disadvantages to
these young patients. Histologically this tumor epitomizes the
model of bone destruction brought about by pathological processes.
GCT exhibits three histological different cell types (1): the multinucleated osteoclast-like
giant cells, the spindle-shaped stromal-like cells and the
round-shaped macrophage-like cells. GCT stromal cells (GCTSCs) are
the primary neoplastic cells of this tumor and are the only
proliferating cell component in long-term culture (2), which recruits osteoclast-like giant
cells that eventually mediate the bone destruction. GCTSCs are able
to proliferate unlimitedly in cell culture and express early
osteoblastic differentiation markers such as collagen type I, bone
sialoprotein and osteonectin proteins (3). The oncogenesis of GCT and what drives
the neoplastic stromal cells to proliferate extensively and pause
at an early differentiation stage of preosteoblast lineage remains
unknown.
Telomeric associations (TAS) have been reported as
one of the most common genetic aberrations in GCT (4–6). It
has been proposed that telomeric instability is responsible for a
large degree of intra-tumor heterogeneity and the telomere serves
as a precursor lesion to subsequent clonal structural aberrations
of chromosome 11 in GCT. Altered telomerase (hTERT) activity and
inactivation of an alternative telomere lengthening mechanism were
found in GCT, suggesting that TAS alone does not contribute to the
development of genetic instability in GCT (7). The cause of genetic instability in
GCT remains obscure. In a recent study it was shown that p63
expression is significantly higher in GCT than many other primary
bone tumors (8). Another report
demonstrated the expression of TAp63 isoform but not DNp63 in GCT
mononuclear cells (9).
p63 gene encodes two primary transcripts, TAp63 and
DNp63, which are controlled by two separate promoters, P1 and P2.
TAp63 is generated from the P1 promoter and contains the
transactivation domain (TAD), the DNA binding domain (DBD) and the
oligomerization domain (OD). In contrast, DNp63 is generated from
the P2 promoter and does not have an amino terminal TAD. The
DN-terminal variants are generally regarded as dominant negative
versions of p53 family members, as they can occupy promoter-binding
sites but fail to transactivate gene expression (10,11).
Independent of their N-terminal status, p63 exists as a variety of
C-terminal splicing variants, most commonly known as three
variants, α, β and γ (12). In
addition, two more C-terminus p63 variants have been discovered,
the variant δ derived from the skipping of exons 12 and 13, and the
variant ε generated by a premature transcriptional termination in
intron 10 (13). p63 is required
for limb and skin development (14,15).
It is also involved in modulation of gene expression associated
with apoptosis, cell proliferation and inhibition of tumor
progression in p53-dependent signaling pathways. Moreover, p63 can
also regulate gene expression in p53-independent pathways for more
specific genes that are associated with development, epithelial
terminal differentiation and cell adhesion. Since there is growing
evidence that p63 is involved in oncogenesis through different
mechanisms, this study aimed to understand the specific role of p63
in cell proliferation and oncogenesis of GCTSCs.
Materials and methods
Cell culture
GCT specimens were collected from the patients
underwent surgical excision of the tumor at Prince of Wales
Hospital. All protocols were approved by the local institutional
ethics committee. Primary culture of GCTSCs was established as
described previously (16). In
brief, freshly obtained GCT tissues were chopped in DMEM containing
10% FBS and 100 U/ml penicillin (PSN). The resultant cell
suspension was transferred to culture flasks and cultured at 37°C
in a humidified atmosphere of 5% CO2 and 95% air.
Culture medium was changed every 2–3 days and upon reaching
confluence, the cells were subcultured. The stromal phenotype of
GCTSCs was verified by immunofluorescent staining using mouse
anti-human STRO-1 monoclonal antibody (MAB4315; Chemicon
International, Temecula, CA, USA). Mesenchymal stem cells (MSC)
obtained from bone marrow of healthy donors were used as the
control cells for evaluation in this study. GCTSC has been reported
to be of the MSC origin (17,18).
Bone marrow aspirated from the iliac crests was collected and the
mononuclear cells were isolated by Ficoll gradient centrifugation
method. The cells were rinsed twice with PBS and cultured in DMEM
containing 2% FBS and 100 U/ml penicillin (PSN). The cells were
then cultured at 37°C in a humidified atmosphere of 5%
CO2 and 95% air.
Immunohistochemistry
GCT specimens collected from the patients were
fixed, embedded and processed for tissue sectioning. For
immunohistochemical staining of p63, tissue sections (5 μm)
were de-waxed in xylene and then rehydrated in sequentially diluted
ethanol followed by PBS. Endogenous peroxidase activity was then
blocked with 0.3% hydrogen peroxide in absolute methanol for 20
min. Tissue sections were then incubated with 5% normal goat-serum
in 1% BSA-PBS for 30 min to block non-specific IgG binding.
Thereafter, primary antibody p63 (Abcam, Cambridge, UK) was applied
at a dilution of 1:500 in 1% BSA-PBS for incubation at 4°C
overnight. After stringent washing twice in PBS, a secondary
antibody was used for further incubation and a streptavidin-biotin
complex system (ABC reagent, Dako, Ely, Cambridgeshire, UK) with
diaminobenzidene (DAB) as chromogen was used for color development.
Slides were finally counterstained with hematoxylin and examined
under a light microscope.
p63 knockdown by siRNA transfection
GCTSCs were plated into 6-well plates at a density
of 1.5×105 cells/well or 96-well plates at 7,000
cells/well depending on the types of assessment to be performed
following siRNA transfection. The cells were kept in
antibiotic-free medium for 24 h before transfection. The cells were
then transfected with a mixture of OptiMEM, 5 μl of
lipofectamine/well (Lipofectamine™ RNAiMAX Transfection Reagent,
Invitrogen, Grand Island, NY, USA) and either scramble siRNA
(Stealth RNAi™ siRNA Negative Controls, Invitrogen) or p63 siRNA
(TP63 Stealth Select RNAi™ siRNA, Invitrogen) at a final
concentration of 50 nM for 48 to 72 h. The sequences of these
siRNAs are available from the manufacturer. The cells were then
subjected to further assessments as follows.
Bromodeoxyuridine (BrdU) incorporation
assay and cell cycle analysis
Cell proliferation rate at 72 h after transfection
was measured by BrdU incorporation using the Cell Proliferation
ELISA, BrdU (colorimetric) kit (Roche Diagnostics, Germany)
according to the manufacturer’s instructions. In a parallel
experiment for cell cycle analysis, the transfected cells were
trypsinized, washed twice with PBS and fixed in 70% ethanol. The
fixed cells were washed with PBS, incubated with 100 mg/ml RNase at
37°C for 30 min, stained with PI (50 mg/ml) and analyzed on a
FACScan flow cytometer (Becton-Dickinson, USA). The percentages of
cells in different phases of the cell cycle were analyzed using the
ModFit LT version 3.0 (Verity Software House, Topsham, ME,
USA).
mRNA extraction and real-time PCR
The cells were harvested at 48 h after transfection
for RNA extraction with TRIzol® reagent (Gibco-BRL,
Grand Island, NY, USA). Extracted RNA was reverse transcribed into
first-strand cDNA using QuantiTect Reverse Transcription kit
(Qiagen, Valencia, CA, USA). The amount of cDNA used for the
amplification of the target genes were normalized by human GAPDH
gene. Primer sequences have been designed using Primer Express from
Applied Biosystems (Branchburg, NJ, USA). The ABI 7500 fast
real-time PCR system and power SYBR-Green PCR Master mix (Applied
Biosystems) were used to perform 40 cycles of PCR with each PCR
performed in triplicate.
Protein extraction and western blot
analysis
Total cell lysates from the transfected cells were
denatured by boiling in Laemmli sample buffer and loaded onto a
gradient SDS-polyacrylamide gel (30 μg protein/lane). After
being resolved by electrophoresis, proteins were transferred to an
Immobilon-P membrane (Millipore, Bedford, MA, USA) via
electroblotting. The membrane was blocked with 5% non-fat milk in
TBST and incubated overnight at 4°C with specific primary
antibodies. p63, CDC25C and CDC2 antibodies (Abcam, Cambridge, UK)
were used, followed by incubation with an anti-mouse IgG secondary
antibody conjugated with horseradish peroxidase (Pierce, Rockford,
IL, USA). Sites of antibody-antigen reaction were visualized by
using the SuperSignal West Pico Chemiluminescent Substrate kit
(Pierce) and recorded by a Kodak 440CF Image Station.
Chromatin immunoprecipitation (ChIP)
assay
Chromatin immunoprecipitation assay was performed
using ChampionChIP One-Day Kit (SABiosciences, Valencia, CA, USA)
according to manufacturer’s instruction with few modifications as
described below. Briefly, GCTSCs were seeded in 100-mm dish and
harvested until 80–90% confluence. Chromatins were prepared and
fragmented by sonication. Fragmented chromatin was pre-cleared with
protein A beads and normal mouse IgG (Santa Cruz Biotechnology,
Santa Cruz, CA, USA) at 4°C for 2 h. Ten microliters pre-cleared
lysate was saved up as input fraction. Two immunoprecipitation
reactions were set up with 1 ml pre-cleared lysate and 4 mg normal
mouse IgG or anti-p63 antibody (Abcam). Reactions were incubated at
4°C overnight and followed by adding 60 μl of protein A
beads and incubated at 4°C for further 1 h. After a number of
washing steps, reverse cross-linking and purification of pulldown
chromatins, as well as input fraction, were performed as described
in manufacturer’s manual. The immunoprecipitated material was
amplified using primers specific for CDC2 promoter:
5′-AGCCTCTTTCTCTC CCCTCATAGA-3′ (forward), 5′-AGAGGCATGCATTTAGAG
ACTG-3′ (reverse) or CDC25 promoter: 5′-ATTCGGCCCTCC CAACCTCTGT-3′,
5′-GTCTTCGCCTGTGTCCGATCCC-3′ (reverse) which contain the
p53-reponsive elements (p53-REs).
Results
p63 expression in GCT
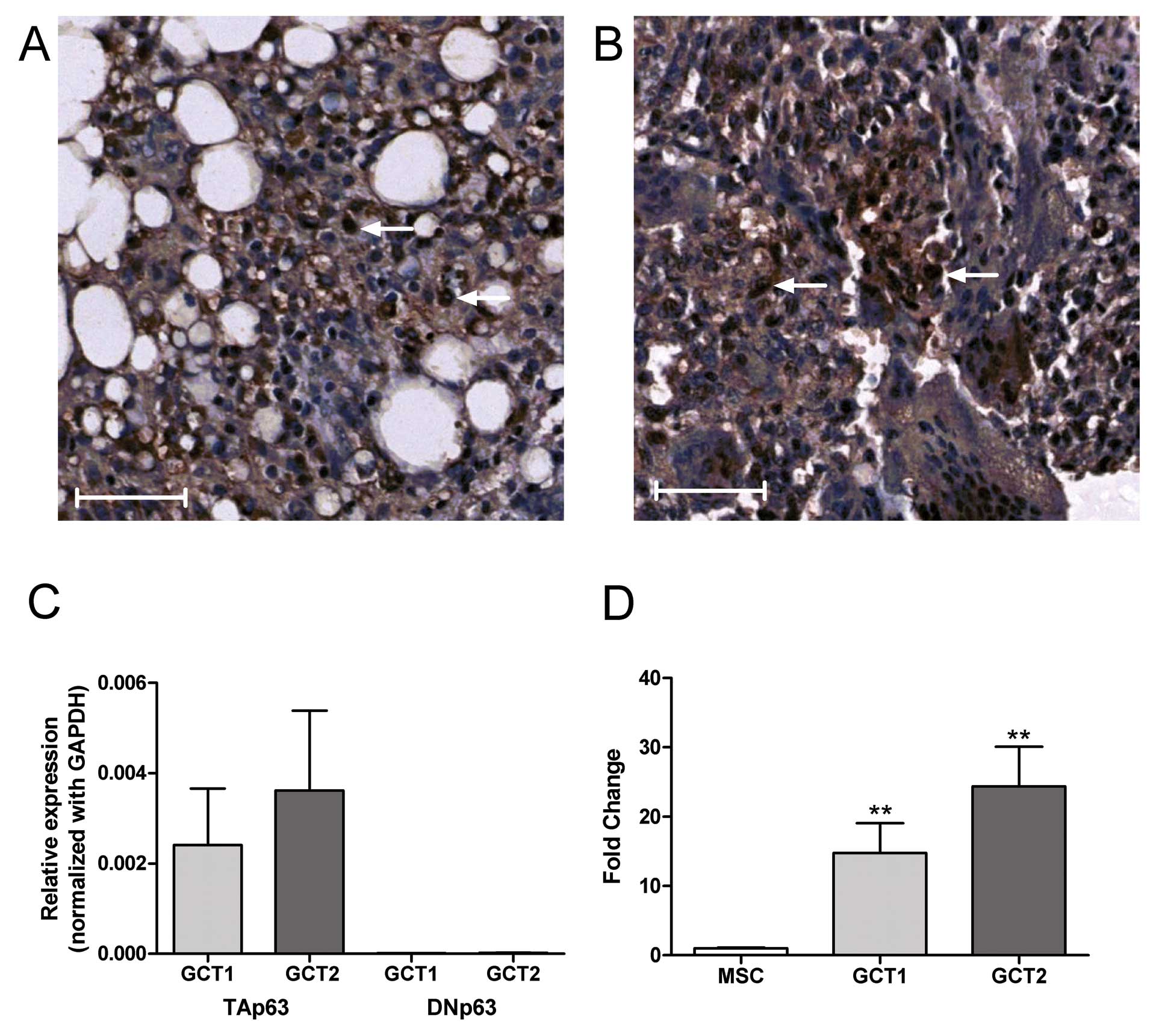
By immunohistochemical staining, p63 expression was
found to be mainly localized in the nuclei of mononuclear cells in
GCT specimens (Fig. 1A and B). By
quantitative real-time PCR analysis, TAp63 isoform, but not DNp63
was detected in GCTSCs from primary culture (Fig. 1C). As compared to bone
marrow-derived MSCs, the expression of TAp63 mRNA in GCTSC was
significantly higher (>15-fold) (Fig. 1D). In order to determine the
functional role of endogenous p63 in GCTSC, we used a RNA-mediated
interference (siRNA) approach to knockdown p63 in GCTSCs and
assessed the morphological changes and cell proliferation status
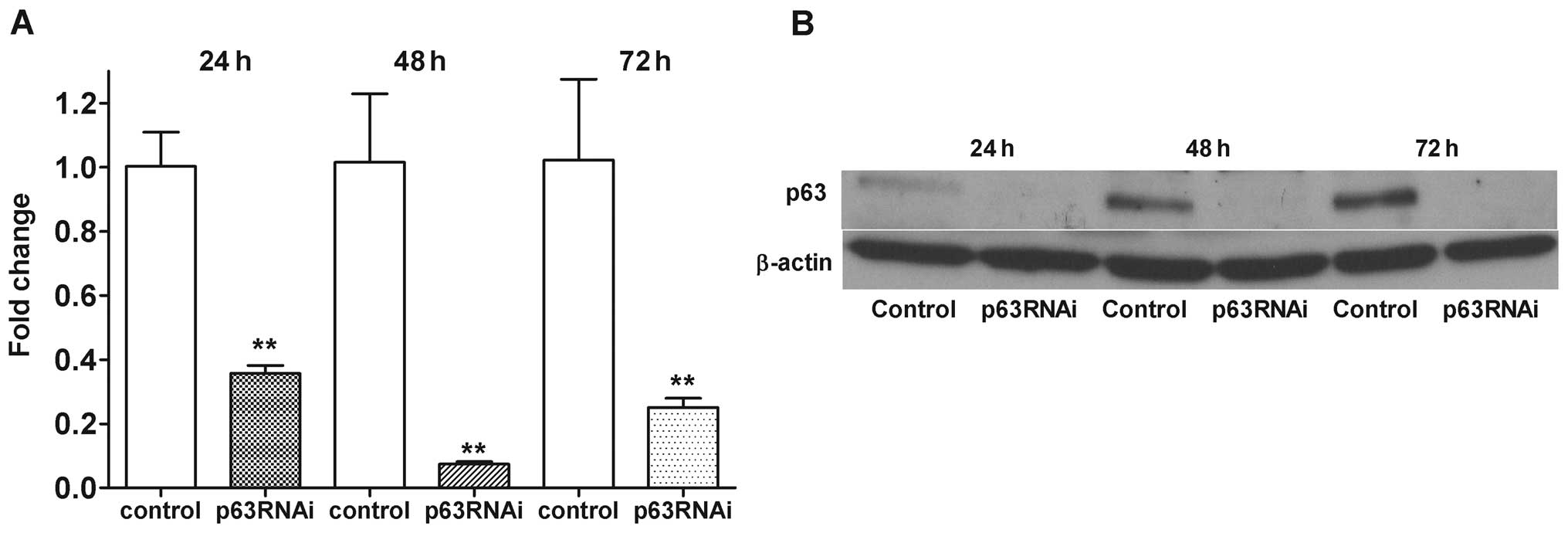
associated with p63 suppression in GCTSCs. The capability of p63
siRNA to abrogate the endogenous p63 expression in GCTSCs at 24–72
h was confirmed by real-time PCR (Fig.
2A) and western blot analysis (Fig. 2B). The level of p63 mRNA expression
was reduced to 0.07-fold of the control at 48 h.
Knockdown of p63 inhibited cell
proliferation and induced cell-cycle arrest
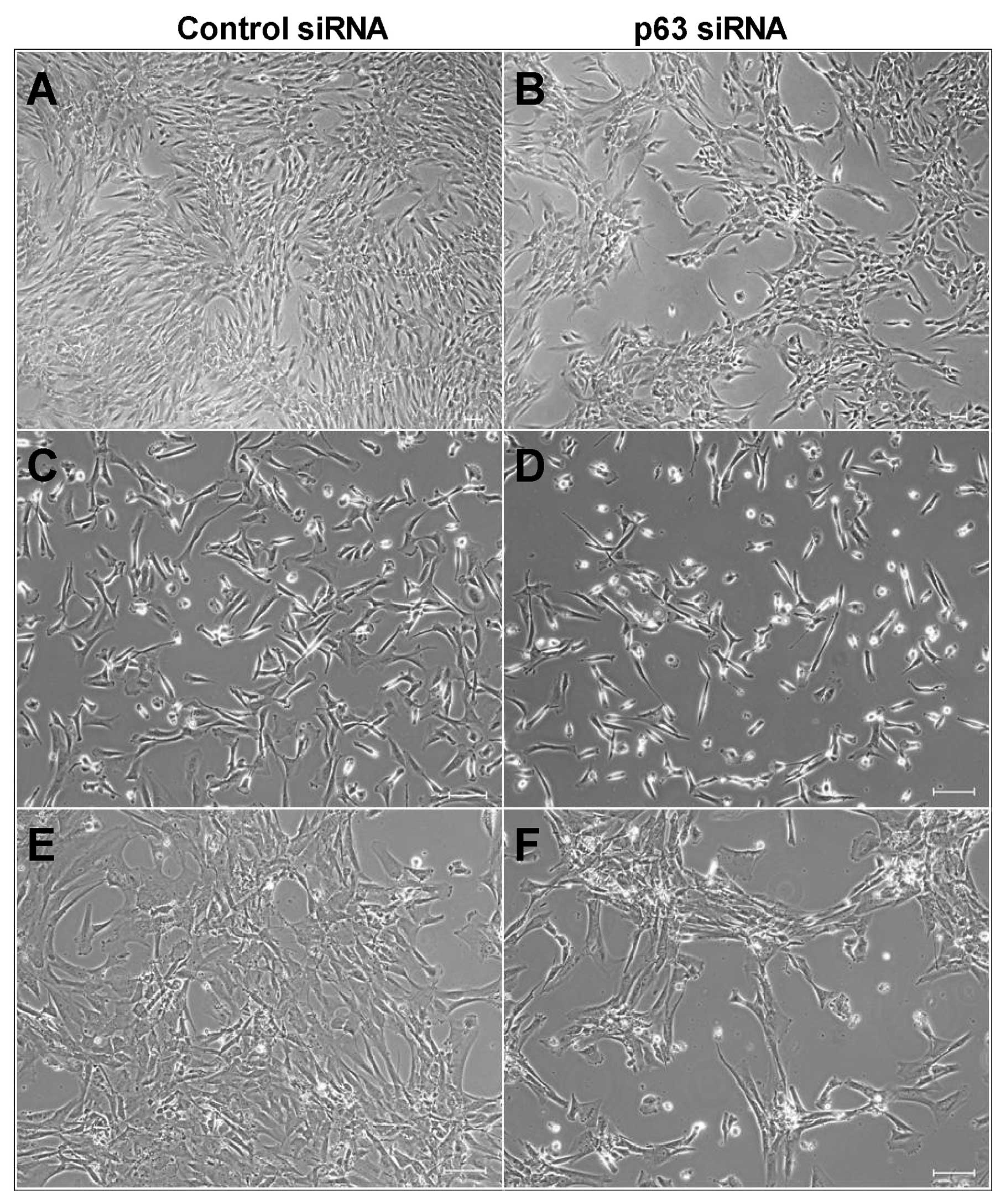
Suppression of p63 greatly reduced cell density in
both MSCs and GCTSCs at 72 h after siRNA transfection (Fig. 3). Further examination of cell
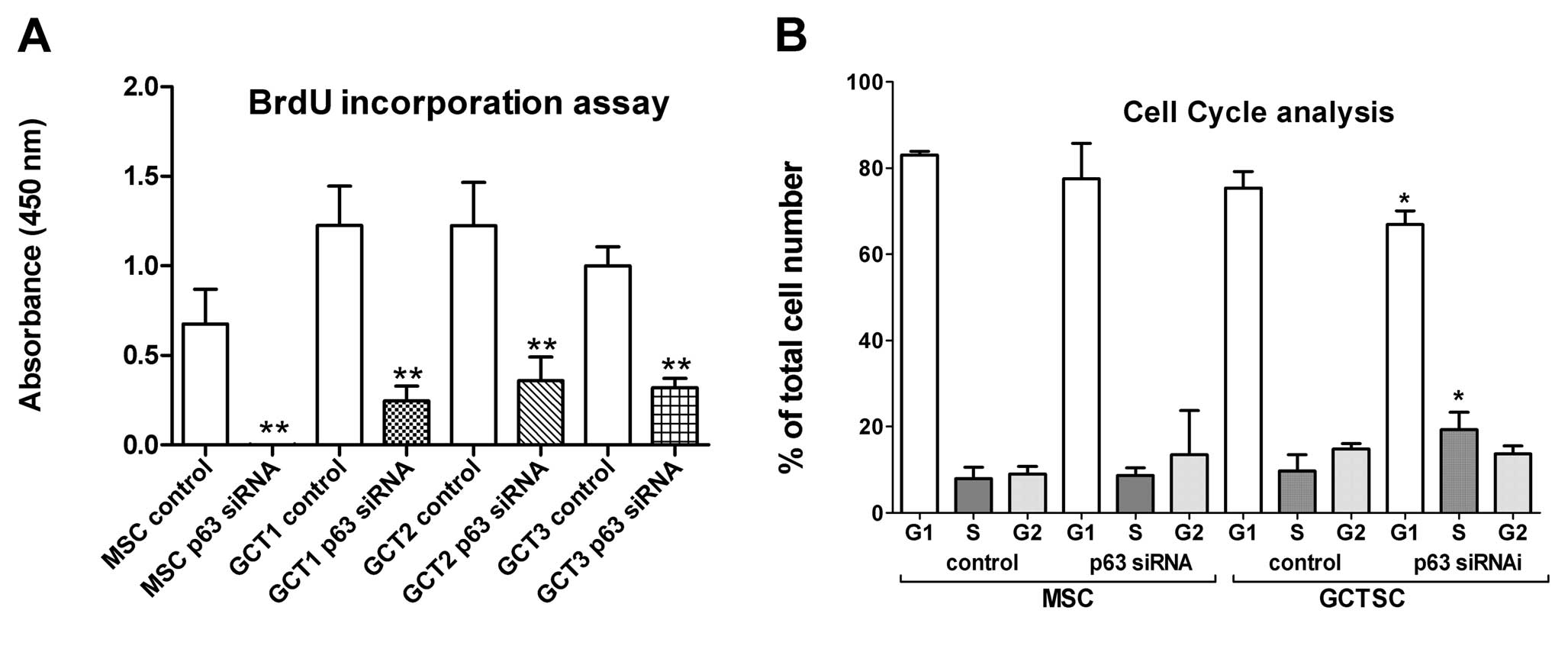
proliferation and apoptosis by BrdU assay and Annexin V/PI staining
followed by flow cytometry analysis, respectively, showed that p63
knockdown significantly inhibited cell proliferation in both MSCs
and GCTSCs (Fig. 4A). It was noted
that p63 siRNA completely ceased the cell proliferation in MSCs but
inhibited 68 to 80% of cell proliferation in GCTSCs. No significant
cell apoptosis was observed after p63 suppression in both MSCs and
GCTSCs (data not shown). In addition, suppression of p63 led to
cell cycle arrest at S-phase in GCTSCs. The percentage of cells in
S-phase was significantly increased at 72 h after p63 siRNA
transfection (Fig. 4B). Knockdown
of p63 in MSCs increased the cell population at G2-phase, but not
to a significant level.
Suppression of p63 reduced cell cycle
progression-associated gene expression
Further examination of cell cycle
progression-associated genes including ADA, CCND3, POLD2, CDC25C
and CDC2 were performed, the selection was on the basis of their
reported function associated with transcriptional regulation of the
cell cycle (19). The mRNA
expression of CDC2 and CDC25C in p63-siRNA-transfected cells were
found substantially reduced after 24–72 h treatment. This finding
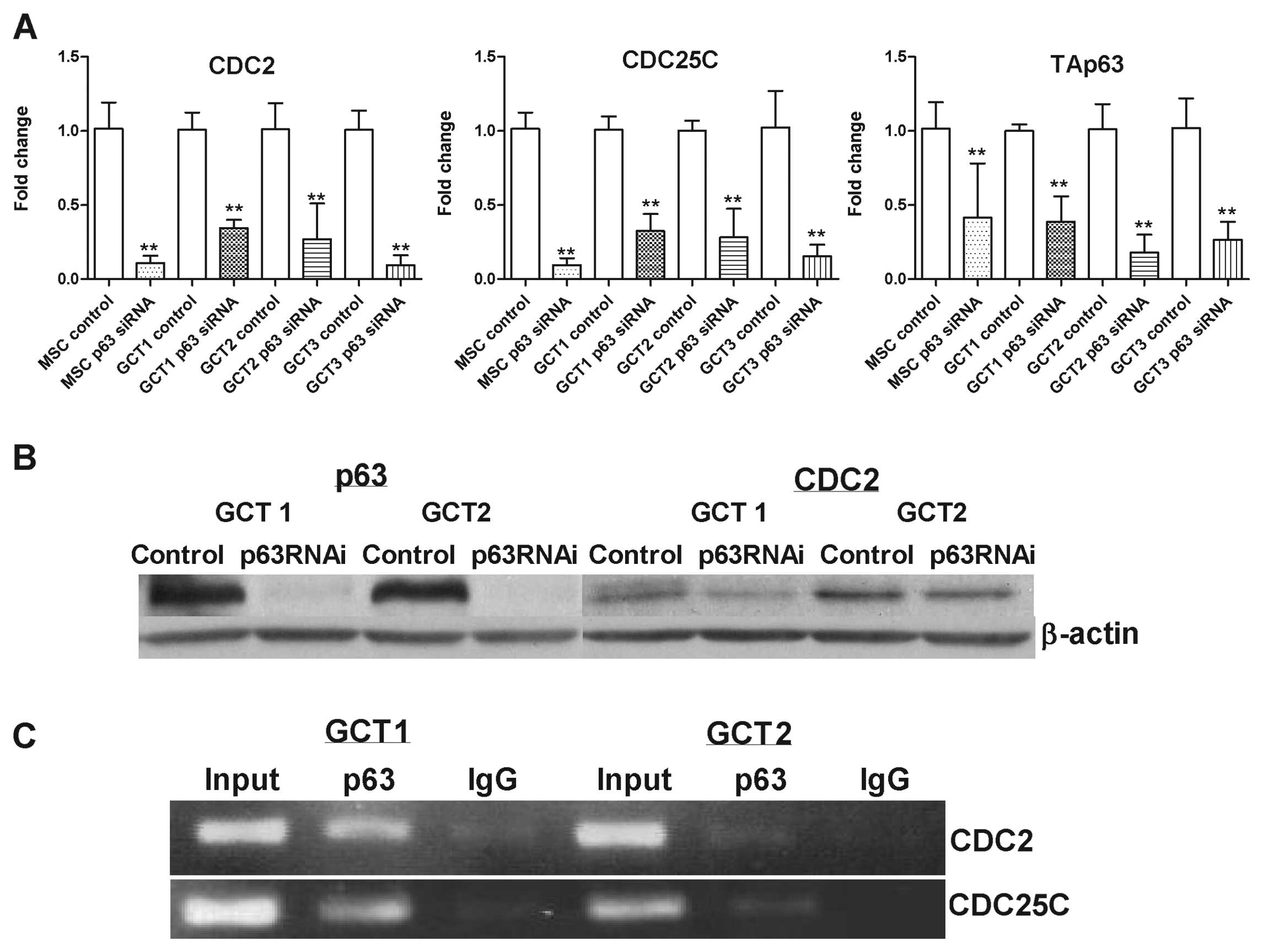
was verified in three GCTSC cell lines as well as MSCs (Fig. 5A). p63 siRNA-mediated
downregulation of CDC2 at protein level was confirmed by western
blot analysis (Fig. 5B), however,
CDC25C protein was not detected in GCTSCs in the assay (data not
shown).
In vivo binding of p63 to the regulatory
regions of CDC2 and CDC25C genes
A chromatin immunoprecipitation assay was performed
to correlate the transcription of CDC2 and CDC25C genes with the
in vivo recruitment of p63 on their regulatory regions.
Cross-linked chromatin from two GCTSC cell lines were
immunoprecipitated with a specific anti-p63 antibody (Fig. 5C). p63 was bound to the p53-RE
present in the regulatory region of CDC2 and CDC25C genes.
Discussion
In the present study, we confirmed p63 expression in
the mono-nuclear cells in GCT, which is consistent with the data
reported elsewhere (7,8). By quantitative real-time PCR
analysis, we showed a higher level (>15-fold) of TAp63
expression in GCTSCs than that in MSCs. Furthermore, we observed a
75% knockdown of p63 gene in GCTSCs by siRNA transfection at 72 h,
which led to a greatly reduced cell proliferation rate to 20 to 32%
of the control in three GCTSC cell lines examined. Suppression of
endogenous p63 induced cell cycle arrest at S-phase in GCTSCs.
These findings suggest that p63 may regulate specific genes
associated with cell cycle progression. Thus a number of cell cycle
associated genes regulating the tumor suppressor activity of p53
were further investigated. It was found that the mRNA expression of
CDC2 and CDC25C was substantially reduced by p63-siRNA transfection
for 24–72 h. By ChIP assay, p63 was found to be recruited on the
regulatory regions of both CDC2 and CDC25C genes which contain the
p53-REs in GCTSCs. The in vivo binding of p63 to both CDC2
and CDC25C p53-REs in GCTSC suggests that p63 may promote GCT
progression by inactivating the tumor suppressor activity of
p53.
TAp63 was also found to inactivate the tumor
suppressor activity of p53 in thyroid cancer and promotes cancer
progression (20). There has been
growing evidence that p63 is involved in oncogenesis through
different mechanisms. For example, p63 mediates survival in
squamous cell carcinoma by suppression of p73-dependent apoptosis
(21); it also contributes to
tumori-genesis by conferring a proliferative potential on cancer
cells, allowing increased self-renewal by transactivating target
genes responsible for cell division such as the adenosine deaminase
gene (22). In addition, an
imbalance in the expression of TA and DN isoforms for the benefit
of DNp63 variants has been reported in squamous cell carcinoma of
the nasopharynx (23), skin
(24), lung (25), bladder (26) and oesophagus (27). The overexpression of DNp63 in many
cancers (28,29) provides evidence that DNp63 may be a
true oncogene in several tumor types. Nevertheless, a previous
report showing TAp63 expression in gastric cancer suggests that
TAp63 may also be involved in tumor progression (30). The detailed mechanisms of p63 in
GCT tumorigenesis, however, remain obscure at the molecular level;
further studies on the regulation of gene transcription of CDC2 and
CDC25C and inactivation of p53 are necessary to further validate
our hypothesis.
In summary, this study demonstrated that p63
regulated cell proliferation through two cell cycle related genes,
CDC2 and CDC25C, by binding to their p53-REs in human GCTSCs. This
provides a potential molecular rationale for treating GCT through
targeting p63 gene. Knockdown of the overexpressed p63 by gene
delivery for inhibiting the neoplastic stromal cell proliferation
may provide a novel therapeutic strategy for GCT. The significant
advantage of the gene therapy is that it may reduce the
proliferation rate of the neoplastic GCTSCs and attenuate the
aggressiveness of the GCT, resulting in less bone destruction. This
may enable clinicians to scale back the extent of surgery, thereby
preventing the unnecessary sacrifice of major bones and joints.
Indeed, our group has recently pioneered the minimally invasive
approach towards GCT treatment. This gives a major clinical benefit
to GCT patients as it reduces the need for major surgery and
enables the delivery of potential adjuvant drugs directly into the
tumor cavity (31).
Acknowledgements
This study was supported by the Direct
Grant for Research 2010/2011, CUHK (ref. no. 2010.1.060).
References
|
1.
|
Goldring SR, Schille AL, Mankin HJ, Dayer
JM and Krane SM: Characterization of cells from human giant cell
tumors of bone. Clin Orthop Relat Res. 204:59–75. 1986.PubMed/NCBI
|
|
2.
|
Zheng MH, Robbins P, Xu J, Huang L, Wood
DJ and Papadimitriou JM: The histogenesis of giant cell tumor of
bone: a model of interaction between neoplastic cells and
osteoclasts. Histol Histopathol. 16:297–307. 2001.PubMed/NCBI
|
|
3.
|
Huang L, Teng XY, Cheng YY, Lee KM and
Kumta SM: Expression of preosteoblast markers and Cbfa-1 and
Osterix gene transcripts in stromal tumor cells of giant cell tumor
of bone. Bone. 34:393–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Sciot R, Dorfman H, Brys P, et al:
Cytogenetic-morphologic correlations in aneurismal bone cyst, giant
cell tumor of bone and combined lesions. Mod Pathol. 13:1206–1210.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Schwartz HS, Eskew JD and Butler MG:
Clonality studies in giant cell tumor of bone. J Orthop.
20:387–390. 2002.PubMed/NCBI
|
|
6.
|
Sawyer JR, Goosen LS, Binz RL, Swanson CM
and Nicholas RW: Evidence for telomeric fusions as a mechanism for
recurring structural aberrations of chromosome 11 in giant cell
tumor of bone. Cancer Genet Cytogenet. 159:32–36. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Forsyth RG, Boeck G, Bekaert S, et al:
Telomere biology in giant cell tumour of bone. J Pathol.
214:555–563. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Lee CH, Espinosa I, Jensen KC, et al: Gene
expression profiling identified p63 as a diagnostic marker for
giant cell tumor of the bone. Mod Pathol. 21:531–539. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Dickson BC, Li SQ, Wunder JS, et al: Giant
cell tumor of bone express p63. Mod Pathol. 21:369–375. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Müller M, Schleithoff ES, Stremmel W,
Melino G, Krammer PH and Schilling T: One, two, three-p53, p63, p73
and chemosensitivity. Drug Resist Updat. 9:288–306. 2006.PubMed/NCBI
|
|
11.
|
Murray-Zmijewski F, Lane DP and Bourdon
JC: p53/p63/p73 isoforms: an orchestra of isoforms to harmonize
cell differentiation and response to stress. Cell Death Differ.
13:962–972. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Ghioni P, Bolognese F, Duijf PH, Van
Bokhoven H, Mantovani R and Guerrini L: Complex transcriptional
effects of p63 isoforms: identification of novel activation and
repression domains. Mol Cell Biol. 22:8659–8668. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Mangiulli M, Valletti A, Caratozzolo MF,
Tullo A, Sbisà E, Pesole G and D’Erchia AM: Identification and
functional characterization of two new transcriptional variants of
the human p63 gene. Nucleic Acids Res. 37:6092–6104. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Moll UM and Slade N: p63 and p73: roles in
development and tumor formation. Mol Cancer Res. 2:371–386.
2004.PubMed/NCBI
|
|
15.
|
Hermanns P and Lee B: Transcriptional
dysregulation in skeletal malformation syndromes. Am J Med Genet.
106:258–271. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Lau CP, Huang L, Tsui SK, Ng PK, Leung PY
and Kumta SM: Pamidronate, farnesyl transferase, and geranylgeranyl
transferase-I inhibitors affects cell proliferation, apoptosis, and
OPG/RANKL mRNA expression in stromal cells of giant cell tumor of
bone. J Orthop Res. 29:403–413. 2011. View Article : Google Scholar
|
|
17.
|
Wulling M, Delling G and Kaiser E: The
origin of the neoplastic stromal cell in giant cell tumor of bone.
Hum Pathol. 34:983–993. 2003.PubMed/NCBI
|
|
18.
|
Robinson D, Segal M and Nevo Z: Giant cell
tumor of bone. The role of fibroblast growth factor 3 positive
mesenchymal stem cells in its pathogenesis. Pathobiology.
70:333–342. 2002.PubMed/NCBI
|
|
19.
|
Lefkimmiatis K, Caratozzolo MF, Merlo P,
et al: p73 and p63 sustain cellular growth by transcriptional
activation of cell cycle progression genes. Cancer Res.
69:8563–8571. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Malaguarnera R, Mandarino A, Mazzon E, et
al: The p53-homologue p63 may promote thyroid cancer progression.
Endocr Relat Cancer. 12:953–971. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Rocco JW, Leong CO, Kuperwasser N, DeYoung
MP and Ellisen LW: p63 mediates survival in squamous cell carcinoma
by suppression of p73-dependent apoptosis. Cancer cell. 9:45–56.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Sbisà E, Mastropasqua G, Lefkimmiatis K,
Caratozzolo MF, D’Erchia AM and Tullo A: Connecting p63 to cellular
proliferation: the example of the adenosine deaminase target gene.
Cell Cycle. 5:205–212. 2006.PubMed/NCBI
|
|
23.
|
Crook T, Nicholls JM, Brooks L, O’Nions J
and Allday MJ: High level expression of deltaN-p63: a mechanism for
the inactivation of p53 in undifferentiated nasopharyngeal
carcinoma. Oncogene. 19:3439–3444. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Senoo M, Tsuchiya I, Matsumura Y, et al:
Transcriptional dysregulation of the p73L/p63/ p51/p40/KET gene in
human squamous cell carcinomas: expression of Delta Np73L a novel
dominant-negative isoform and loss of expression of the potential
tumor suppressor p51. Br J Cancer. 84:1235–1241. 2001. View Article : Google Scholar
|
|
25.
|
Hibi K, Trink B, Patturajan M, et al: AIS
is an oncogene amplified in squamous cell carcinoma. Proc Natl Acad
Sci USA. 97:5462–5467. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Park BJ, Lee SJ, Kim JI, et al: Frequent
alteration of p63 expression in human primary bladder carcinomas.
Cancer Res. 60:3370–3374. 2000.PubMed/NCBI
|
|
27.
|
Taniere P, Martel-Planche G, Saurin JC,
Lombard-Bohas C, Berger F, Scoazec JY and Hainaut P: TP53 mutations
amplification of P63 and expression of cell cycle proteins in
squamous cell carcinoma of the oesophagus from a low incidence area
in Western Europe. Br J Cancer. 85:721–726. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Zaika AI, Kovalev S, Marchenko ND and Moll
UM: Overexpression of the wild-type p73 gene in breast cancer
tissues and cell lines. Cancer Res. 59:3257–3263. 1999.PubMed/NCBI
|
|
29.
|
Zaika AI, Slade N, Erster SH, et al:
DeltaNp73, a dominant-negative inhibitor of wild-type p53 and
TAp73, is up regulated in human tumors. J Exp Med. 196:765–780.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Tannapfel A, Schmelzer S, Benicke M, et
al: Expression of the p53 homologues p63 and p73 in multiple
simultaneous gastric cancer. J Pathol. 195:163–170. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Wong KC, Kumta SM, Tse LF, Ng EW and Lee
KS: Navigation Endoscopic Assisted Tumor (NEAT) surgery for benign
bone tumors of the extremities. Comput Aided Surg. 15:32–39. 2010.
View Article : Google Scholar : PubMed/NCBI
|