Introduction
Numerous animal studies and clinical trials in
cancer have shown that ibuprofen reduces the incidence of and
mortality from cancer (1–3). However, adverse effects, such as
increased gastrointestinal (GI) ulceration, limit its potential for
long-term use. To reduce this side effect, different modifications
of ibuprofen have been sythesized and evaluated. These
modifications include guiacol ester (4), alkyl- or thio-ester (5), diethylcarbonate (6), 2-formylphenyl ester (7), N-hydroxymethyl-succinimide (8), β-D-glucopyranoside (9), polymerized-2-hydroxyethylmethacrylate
(10), PEG1000-linked chondroitin
(11), α-methyl, ethyl and propyl
glucopyranosides (12), cysteamide
(13), L-cysteine ethyl ester
(14) and NO-donating moieties
(15–17). Although most of these modifications
did result in a reduction of GI side effects, only a decreased or
similar effect of anti-inflammation or anticancer activity was
observed when compared with the parent compound ibuprofen. Recently
we developed a phospho-butanol-modified ibuprofen (designated
p-ibuprofen, hereinafter), which showed promising increased
anticancer activity in vitro and in xenograft tumor models
(18–20), elevated anti-inflammation in an
arthritis rat model (21), and
reduced GI toxic side effects. Xie et al(20) found that p-ibuprofen is minimally
metabolized by cultured cells, but extensively metabolized by mouse
liver microsomes, undergoing regioselective oxidation to produce
1-OH-p-ibuprofen and carboxyl-p-ibuprofen, which can be hydrolyzed
to the parent metabolites, 1-OH-ibuprofen and carboxyl-ibuprofen,
respectively. These results indicate that the anticancer effect of
p-ibuprofen may be different between in vitro and in
vivo situations. Therefore, additional in vivo studies
are necessary to evaluate the chemopreventive effect of p-ibuprofen
before it can be considered for human clinical trials.
Both COX-2 dependent and independent pathways may be
involved in the mechanism by which NSAIDs prevent cancer. It
remains to be fully elucidated whether p-ibuprofen suppresses
cancer growth by the COX-2 pathway or COX-2 independent pathways.
Previous studies (19–21) have suggested that oxidative stress
mediated apoptosis, reduced inflammatory cytokines, and inhibition
of NF-κB activation are involved in the mechanism of its action.
Wong et al(22) reported
that both p-ibuprofen and ibuprofen can be hydrolyzed by
carboxylesterases in the liver, and that the integrity of the drug
is critical for anticancer activity. p-Ibuprofen’s reduced GI
toxicity may be a result of the modification of the phospho-group
(with the COOH-group), which is known to account for the GI
toxicity of the conventional NSAIDs (23).
In this study, we tested the chemopreventive effect
of p-ibuprofen in a long-term use scenario using a chemical-induced
colon cancer model in rats. The acute and chronic toxicities of
p-ibuprofen were evaluated in rats. In additon, the effect of
p-ibuprofen on COX-2-dependent and -independent pathways, including
β-catening and NF-κB pathways, were studied in vitro.
Materials and methods
Drugs
Ibuprofen was purchased from Sigma (St. Louis, MO).
Phospho-butanol-ibuprofen was purchased from Chem-Master
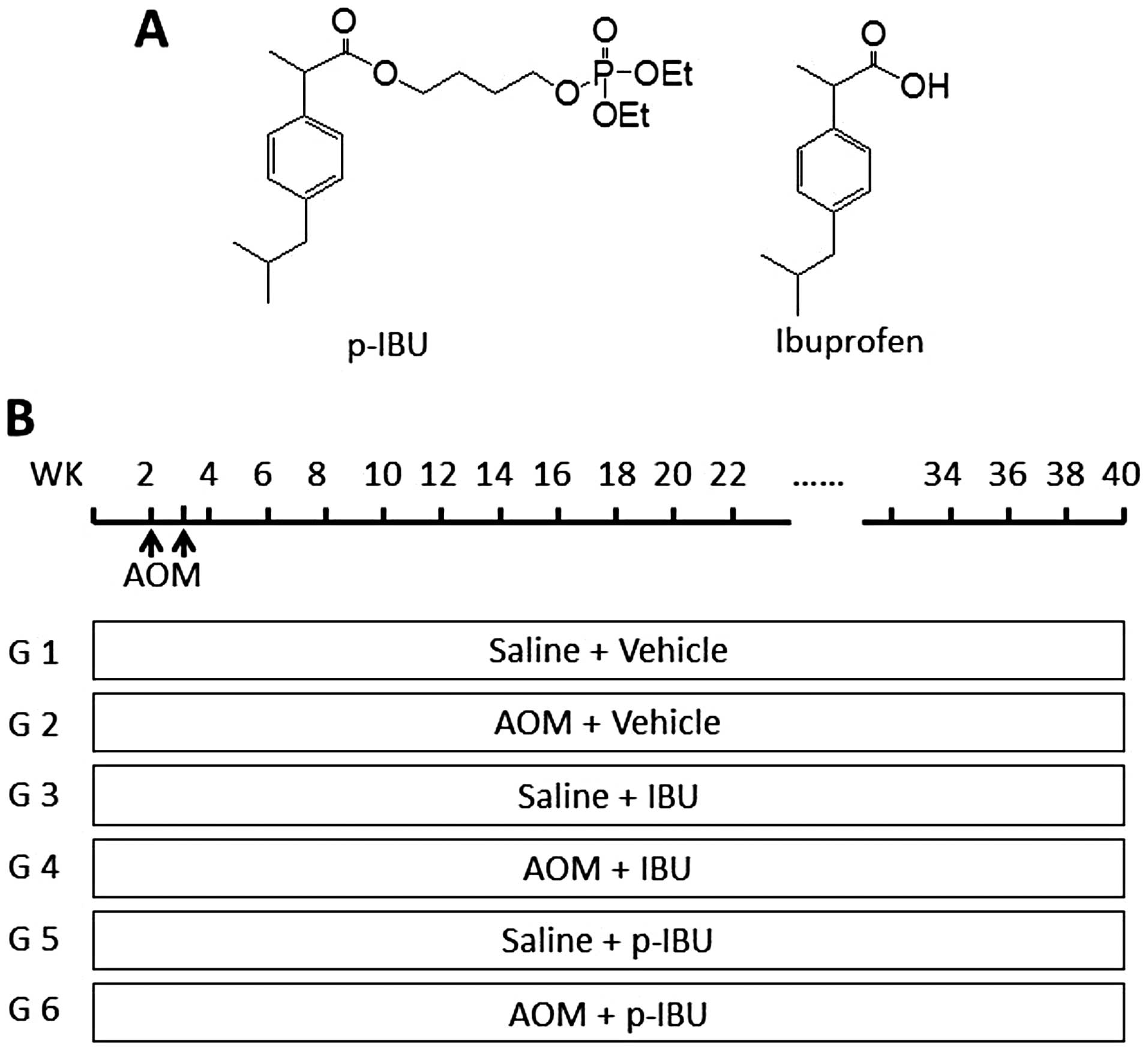
International Inc. (East Setauket, NY). The chemical structure is
shown in Fig. 1A. The purity of
synthetized drug p-ibuprofen is >99%.
Animal model and treatments
Fisher 344 male rats (135) (Harlan Sprague Dawley,
Indianapolis, IN), 3–4 weeks old with an initial average body
weight of 90 g, were acclimated for 1 week, divided into 6 groups:
groups 1, 3 and 5 were given saline as control, 15 rats per group;
and groups 2, 4 and 6 were given carcinogen, 30 rats per group.
Animals received either saline or carcinogen (azoxymethane, AOM, at
15 mg/kg) by subcutaneous injection, once a week for two weeks.
Drug administration was also initiated in rats via diet 10 days
before subcutaneous injections: vehicle for groups 1 and 2;
ibuprofen, 500 ppm, for groups 3 and 4; and phosphoibuprofen
(p-ibuprofen, the same hereinafter), 900 ppm (equal molar dose with
ibuprofen), for groups 5 and 6. All drugs were administered up
until the end of the experiment as shown in Fig. 1B. Animals were housed and
maintained according to the approved standards of Stony Brook
University Institutional Animal Care and Use Committee. Animals
were housed two per plastic cage with sawdust bedding and were kept
under standard laboratory conditions (room temperature, 22±2°C;
relative humidity, 50±5%; light/dark cycle 12/12 h). All animals
had access to food and tap water ad libitum. Rats were
observed daily and weighed once a week. Half of the rats from each
group were euthanized at week 20 and analyzed for aberrant crypt
foci (ACF). Blood was collected for pharmacokinetic (PK) analysis.
The remaining rats were euthanized at week 40 and colons were
dissected and analyzed for aberrant crypt foci (ACF) and tumors.
Heart, lung, liver, stomach and kidney were collected from animals
in the control groups and fixed in buffered-formalin for
histological analysis of potential toxic effects.
ACF analysis
For animals sacrificed at week 20, ACF were counted
in colon tissues as previously described (24). Briefly, the colons were removed,
rinsed with ice-cold phosphate-buffered saline (PBS), placed on
filter paper, opened longitudinally, and fixed in 10% buffered
formalin for 24 h. Then colon tissues were stained with 0.2%
methylene blue for 3 to 5 min. The number of ACF per colon was
determined by microscopic examination. ACF were distinguished from
surrounding normal crypts by increased size, thickened epithelial
cell lining, and enlarged cryptal area relative to surrounding
normal crypts as shown in Fig.
2A.
Tumor analysis
At week 40, the remaining animals were sacrificed by
CO2 asphyxiation. Colons were removed, opened
longitudinally, and rinsed with PBS. Tumors were counted and both
long-diameter by short-diameter were measured to calculate tumor
size. After measurements were recorded, one half of each colon
sample was frozen in liquid nitrogen and stored at −80°C for
further analyses. The remaining half of each colon was fixed in
buffered formalin for histopathology processing. Briefly, colon
tissue with tumors were equally cut into 10 pieces, and embedded in
paraffin blocks. Sections (4 μm) were stained with
hematoxylin and eosin to determine histopathology by pathologist,
or stained by immunohistochemistry and immunofluorescence for
mechanism study.
Toxicity
Thirty-five Fisher 344 rats (Harlan Sprague Dawley),
male, 8–9 weeks old, were divided into 5 groups (7 rats per group)
and treated with the following: group 1, vehicle; group 2,
ibuprofen 75 mg/kg of body weight; group 3, ibuprofen 670 mg/kg;
group 4, p-ibuprofen 135 mg/kg; and group 5, p-ibuprofen 1,215
mg/kg. The drugs were administered by gavage, once a day for 7
days. Animals were housed under standard conditions and euthanized
by CO2 asphyxiation 1 h after final drug administration.
Blood was collected for PK studies. Stomach, small intestine and
colon were checked for ulcers under magnification lens then fixed
with 10% buffered formalin for histology. Heart, liver, lung and
kidney were collected for toxicity analyses.
Pharmacokinetics and HPLC analysis
The blood samples collected from animals sacrificed
at week 20 and from the toxicity study with low dose treatment were
used for the PK study. Briefly, ibuprofen and its metabolites were
extracted by adding a 2-fold volume of acetonitrile. After
centrifugation for 10 min at 5,000 × g, the supernatants were
subjected to HPLC analysis. The HPLC system consisted of a Waters
Alliance 2695 Separations Module equipped with a Waters 2998
photodiode array detector (220 nm) (Waters, Milford, MA) and a
Thermo BDS Hypersil C18 column (150×4.6 mm, particle size 3
μm) (Thermo Fisher Scientific, Waltham, MA). The mobile
phase followed a gradient between buffer A [formic acid,
acetonitrile, H2O (95:4.9: 0.1 v/v/v)] and buffer B
(acetonitrile).
Prostaglandin E2 (PGE2) and
COX-2 in vitro
To evaluate the inhibitory effect of ibuprofen or
p-ibuprofen on COX-2, RAW 264.7 macrophages were pre-treated with
ibuprofen or p-ibuprofen, 130 μM, for 12 h, then incubated
with LPS, 100 ng/ml, overnight. The cells were collected for COX-2
measurement and analyzed by western blotting. Levels of
PGE2 in cell culture media were determined using a
commercially available immunoassay kit according to the
manufacturer’s instructions. Briefly, 1.5×106 Raw 264.7
macrophages were pre-incubated with ibuprofen or p-ibuprofen, 130
μM, for 12 h, followed by LPS (100 ng/ml) overnight. The
cultured media were collected to measure PGE2 level
using an ELISA kit (Cayman Chemical, Ann Arbor, MI).
Immunofluorescent double staining
Paraffin-embedded sections were deparaffinized,
rehydrated and microwave heated for 15 min in 0.01 mol/l citrate
buffer (pH 6.0) for antigen retrieval. Tissue sections were then
incubated with 5% donkey serum for 30 min, then treated with
primary antibodies, rabbit anti-phospho-p65, ser276 (Cell
Signaling, Danvers, MA) and mouse anti-β-catenin (Millipore,
Temecula, CA) or control IgG, at 4°C overnight. After washing with
PBS 3×5 min, the secondary donkey anti-mouse IgG and donkey
anti-rabbit IgG conjugated with fluorescents were added and
incubated at room temperature for 1 h. Slides were washed thrice
with PBS, mounted with media and observed under a fluorescence
microscope.
Cell culture
RAW 264.7 macrophage (mouse leukemic moncyte) and
HCT 116 colon cancer cell lines were purchased from American Type
Culture Collection (Manassas, VA) and cultured in DMEM or RPMI-1640
medium, respectively.
Western blot analysis
Whole cell extracts were obtained by lysing cells in
RIPA buffer [50 mM Tris-HCl (pH 7.4),150 mM NaCl, 1 mM
Na2EDTA, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1%
NP-40, 0,25% sodium deoxycholate and Protease Inhibitor Cocktail 2
(Sigma-Aldrich)]. Cytoplasmic and nuclear extracts were prepared
following a standard protocol (25); trypsinized cells were suspended in
lysis buffer to which NP-40 was added at a subsequent step (the
supernatant fraction represented the cytoplasmic extract); nuclei
were washed and centrifuged, followed by resuspension in extraction
buffer and pelleting. Protein extracts were analyzed by a
well-established standard western blot procedure (26). Rabbit anti-COX-2 antibody was
purchased from Cayman Chemical. Other primary antibodies are
indicated in the immunofluorescence staining.
Statistical analysis
Data are expressed as mean ± SEM and analyzed with
ANOVA. P≤0.05 was considered statistically significant.
Results
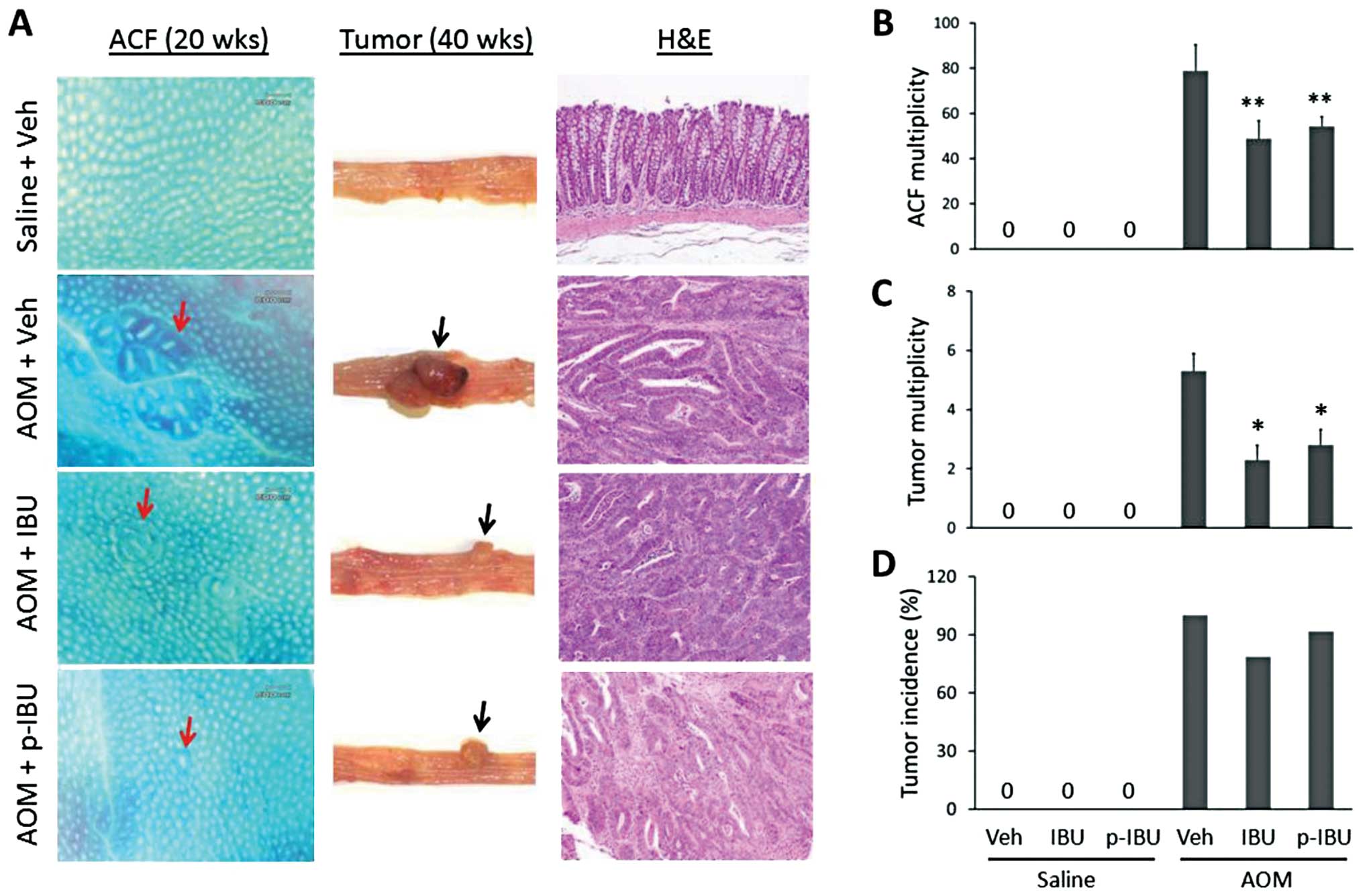
p-Ibuprofen prevents AOM-induced colon
cancer
AOM-induced ACF at week 20 and colon tumors at week
40 as shown in Fig. 2A. Histology
of tumors shows well-differentiated adenocarcinoma. As shown in
Table I and Fig. 2B, compared to vehicle treatment,
both p-ibuprofen or ibuprofen significantly reduced the
multiplicity of ACF by 31.2% (54.2±4.4 vs. 78.8±11.6, p<0.05),
or 37.9% (48.9±7.9 vs. 78.8±11.6, p<0.05), respectively.
However, no difference was observed between groups treated with
p-ibuprofen or ibuprofen (p>0.05). Similarly, as shown in
Table I and Fig. 2C, at week 40 treatment with either
p-ibuprofen or ibuprofen reduced the multiplicity of colon tumors
by 47.2% (2.8±0.52 vs. 5.3±0.59, p<0.01), or 56.6% (2.3±0.49 vs.
5.3±0.59, p<0.01), respectively. Again, a significant difference
(p>0.05) was not observed between groups treated with
p-ibuprofen and ibuprofen (Table I
and Fig. 2D). Furthermore,
pathological changes were not detected in heart, lung, liver,
stomach and kidney tissues. These results suggest that
phospho-modification did not significantly enhance the inhibitory
effect of ibuprofen in AOM-induced colon cancer in rats.
 | Table I.Inhibition of AOM-induced colonic ACF
and tumor multiplicity in rats by p-IBU. |
Table I.
Inhibition of AOM-induced colonic ACF
and tumor multiplicity in rats by p-IBU.
| Diet | Control
| IBU
| p-IBU
|
|---|
| Salinea | AOMa | Salinea | AOMa | Salinea | AOMa |
|---|
| 20 Weeks | | | | | | |
| Weight | 447±10.60 | 423±9.45 | 416±6.11 | 406±118.91 | 420±6.97 | 400±8.14 |
| ACFs | 0 | 78.8±11.6 | 0 | 48.9±7.9 | 0 | 54.2±4.4 |
| Tumors | 0 | 3.3±0.72 | 0 | 1.3±0.31 | 0 | 1.7±0.42 |
| 40 Weeks | | | | | | |
| Weight | 467±7.97 | 434±11.19 | 431±15.43 | 432±8.84 | 455±5.49 | 458±12.33 |
| ACFs | 0 | 34±5.17 | 0 | 35±4.05 | 0 | 45±3.68 |
| Tumors | 0 | 5.3±0.59 | 0 | 2.3±0.49 | 0 | 2.8±0.52 |
Phospho-modification of ibuprofen reduces
GI toxicity
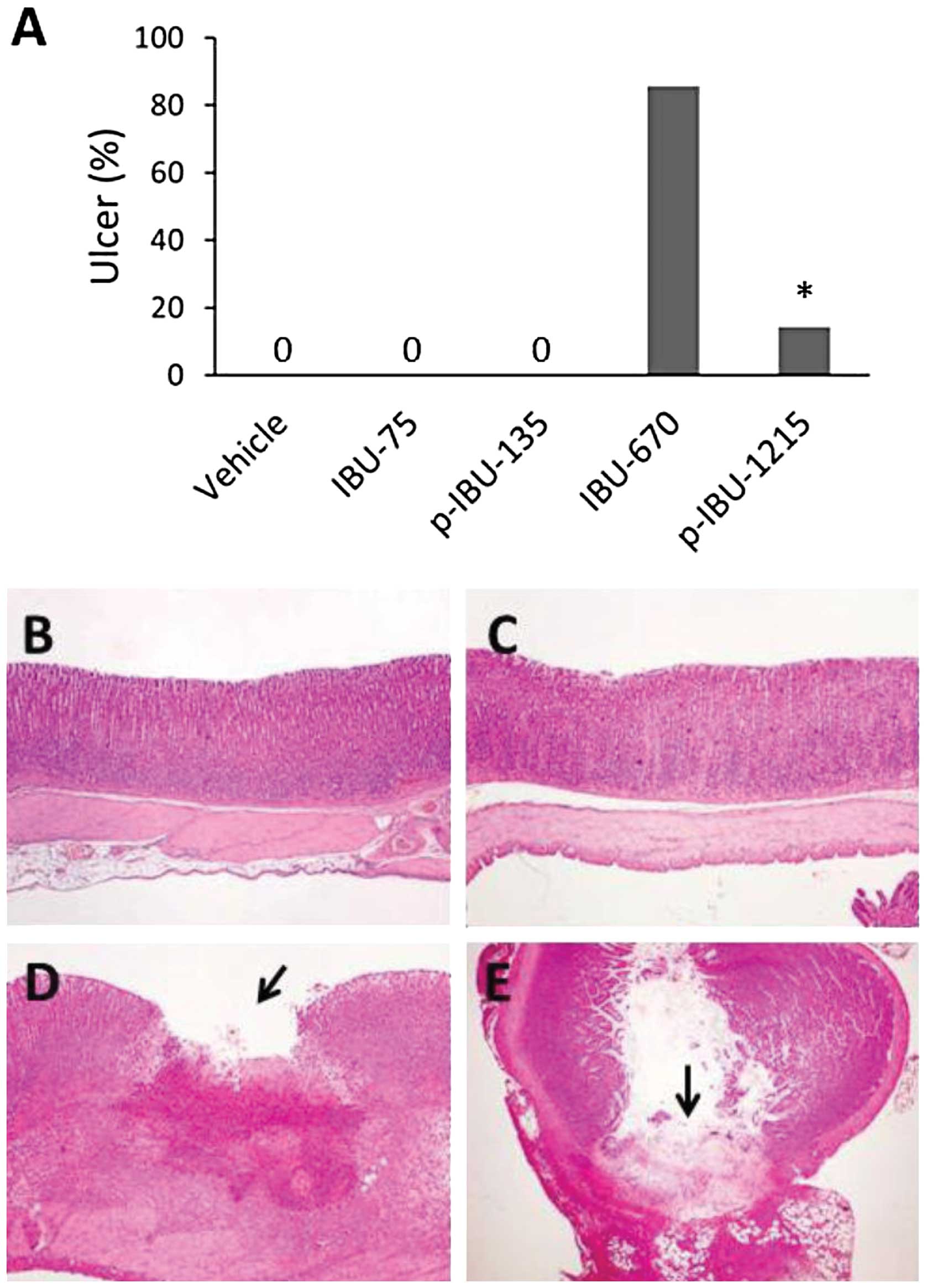
As shown in Fig.
3A, a high dose of ibuprofen (670 mg/kg) administered to rats
by gavage for 7 days led to stomach ulcerations in 85.7% (6 out of
7) of rats; whereas, an equal molar dose of p-ibuprofen (1,215
mg/kg) caused stomach ulceration in only 14.3% (1 out of 7) of
rats. The modification of ibuprofen with the phospho-moiety
remarkably reduced its GI side-effect (p<0.01). All animals with
stomach ulcers also exhibited ulcerations in the small intestine.
Histologically, the typical ulcer appeared with the hiatus of
mucosa and ulcerative tissue in the bottom of the ulcer (Fig. 3D). Some of the ulcers exhibited
inflammation and perforation (breaking through the wall) in both
stomach and intestine (Fig. 3B–E).
A low dose of both p-ibuprofen and ibuprofen did not lead to
ulceration in either stomach or intestine. There was no obvious
pathological change in heart, liver, lung and kidney. These results
demonstrate that, whereas the phospho-modification of ibuprofen did
not enhance ibuprofen’s inhibitory effect on AOM-induced colon
cancer in rats, p-ibuprofen significantly reduced the GI toxicity
of ibuprofen.
Pharmacokinetics
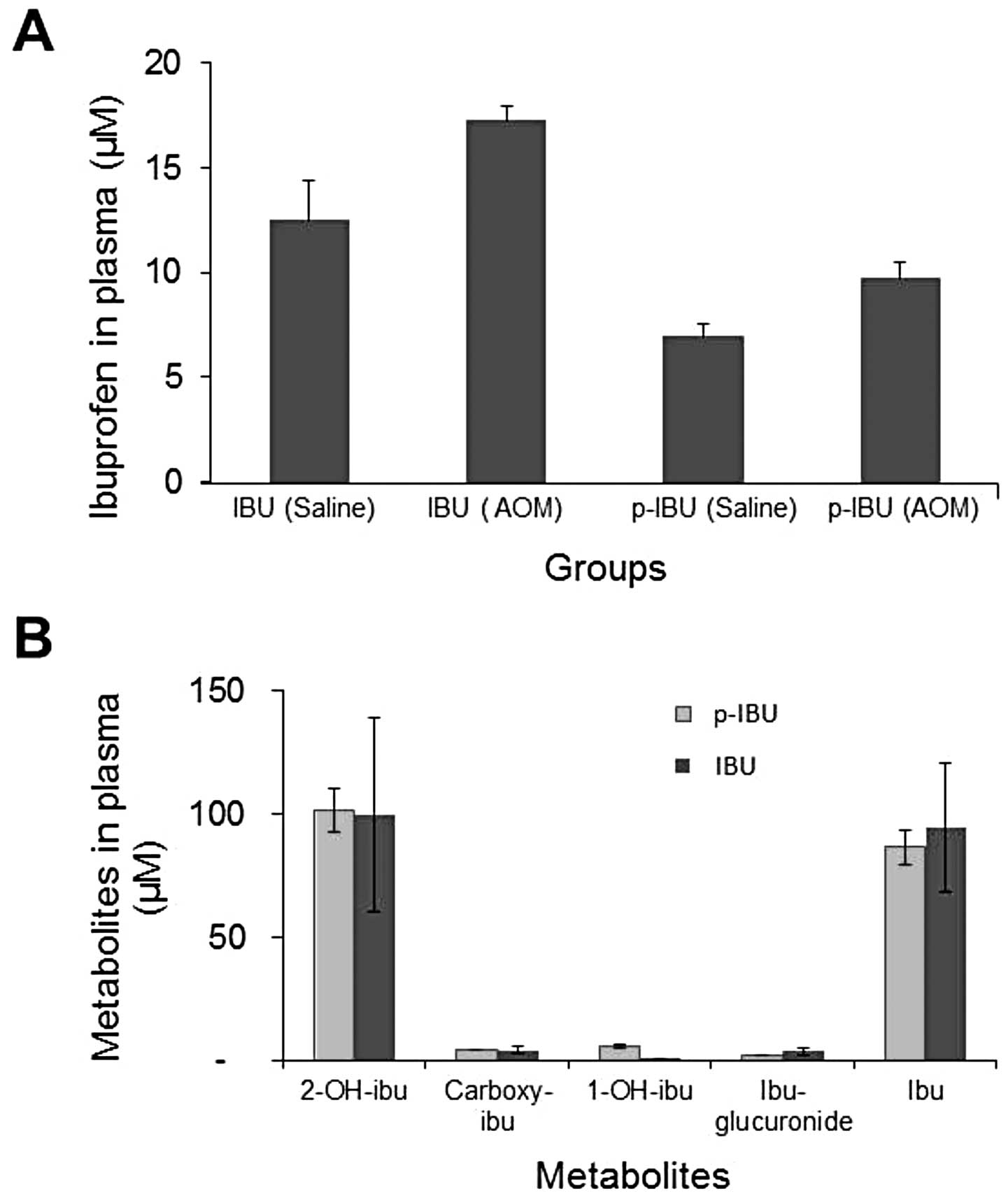
As shown in Fig.
4A, with continuous administration of ibuprofen or p-ibuprofen
in the diet for 20 weeks, ibuprofen can be detected in the plasma
of both treatment groups. The plasmic level of ibuprofen in animals
fed a diet with p-ibuprofen is lower than that of animals fed a
diet of ibuprofen. Both diet groups (p-ibuprofen and ibuprofen)
exhibited the tendency of a higher plasmic ibuprofen level in
AOM-treated animals as compared to saline control animals. However,
no statistical difference was noted. These trends were comparable
in the case where rats were on the same diet for 40 weeks. Intact
p-ibuprofen was not detectable in the plasma of animals fed a diet
consisting of or gavaged with p-ibuprofen. As seen in Fig. 4B, the metabolite of ibuprofen,
2-OH-ibuprofen, was detectable at a similar level as intact
ibuprofen in both groups fed a diet of p-ibuprofen (102 μM)
or ibuprofen (99 μM). Other metabolites were detected at
very low levels, including carboxy-ibuprofen (3 and 3 μM),
1-OH-ibuprofen (4 μM, undetectable) and
ibuprofen-glucuronide (1 and 1 μM); 20 weeks and 40 weeks,
respectively.
p-Ibuprofen inhibits COX-2 and
PGE2 in macrophages
We evaluated the classic pathway of NSAIDs-COX-2
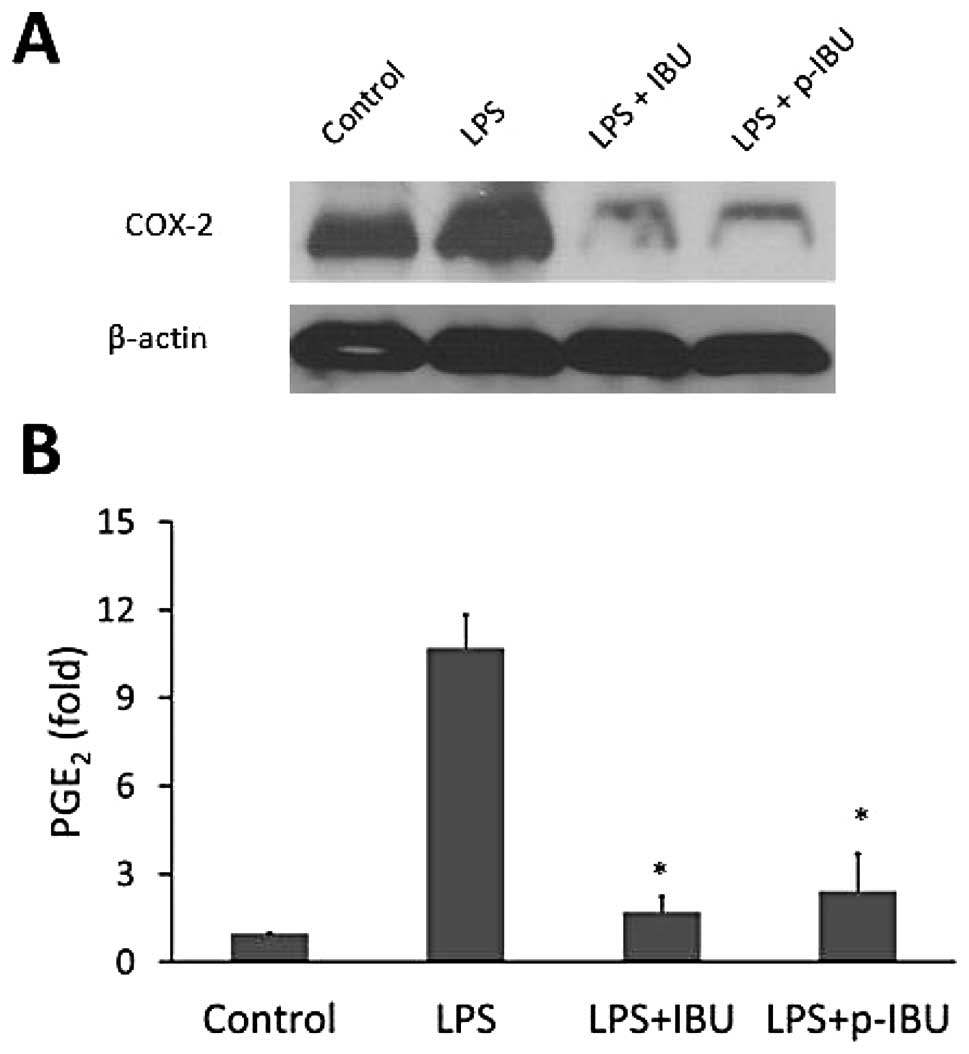
expression in macrophages. Our results showed that LPS highly
induced COX-2 levels and that this effect was completely blocked by
both ibuprofen and p-ibuprofen (Fig.
5A). These results suggest that phospho-modification did not
alter the property of ibuprofen in inhibiting COX-2 expression.
As seen in Fig. 5B,
both ibuprofen and p-ibuprofen significantly suppressed
PGE2 production by 84.1% (1.7±0.5 vs. 10.7±1.1,
p<0.01) and 77.4% (2.4±1.3 vs. 10.7±1.1, p<0.01) as compared
to LPS-treated control, respectively. However, no significant
difference between ibuprofen and p-ibuprofen (p>0.05) was
observed. This result is consistent with the observed inhibition of
COX-2, suggesting that phospho-modification does not decrease the
ability of ibuprofen to suppress PGE2 synthesis.
p-Ibuprofen suppresses β-catenin nuclear
translocation and NF-κB activation
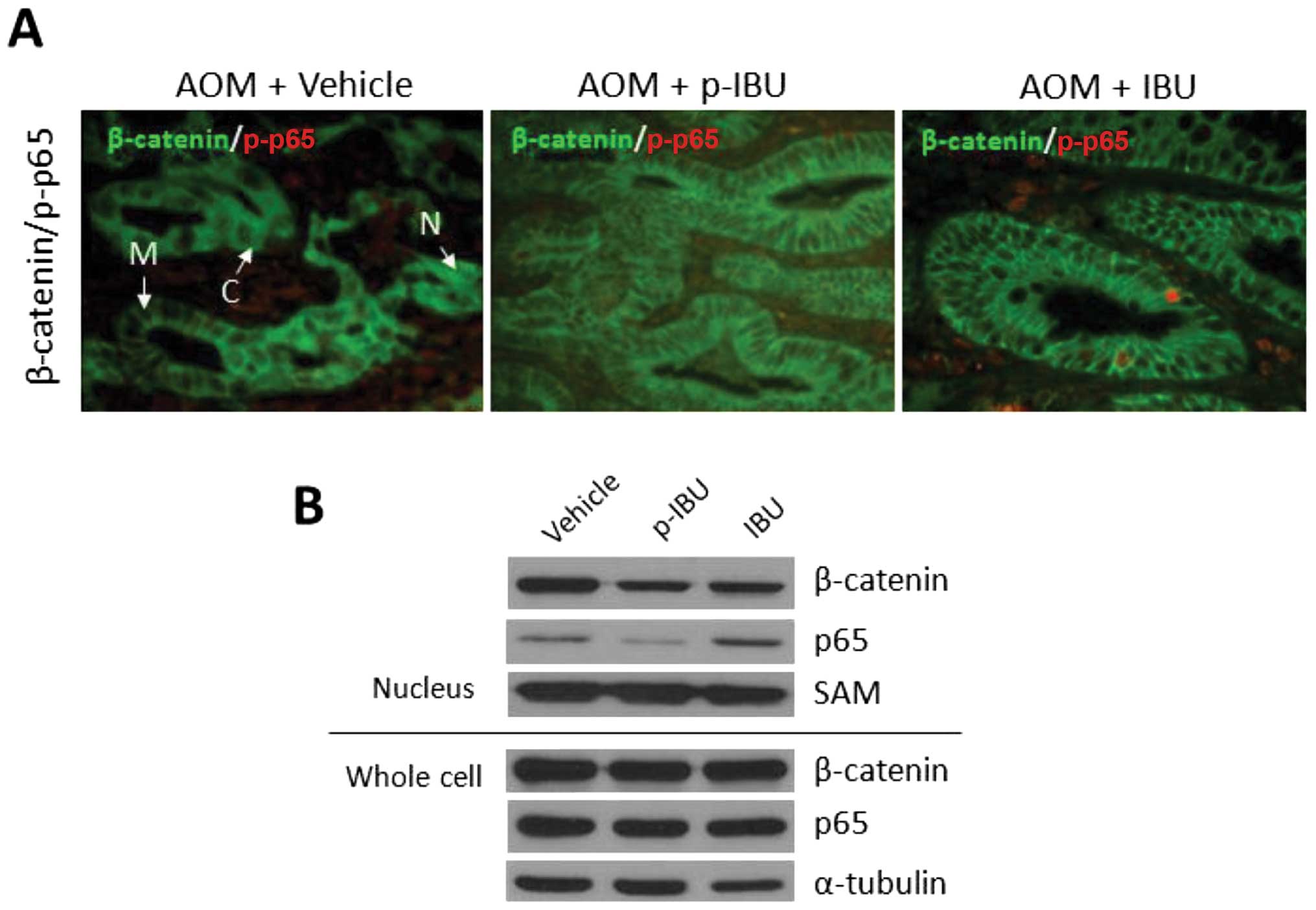
As shown in Fig.
6A, exposure to the carcinogen AOM induced β-catenin
cytoplasmic accumulation and nuclear translocation. These effects
were reduced by both p-ibuprofen and ibuprofen. The presence of
phosphorylated NF-κB subunit p65 indicates NF-κB activation. By
immunohistochemistry analysis (Fig.
6A), p-ibuprofen and not ibuprofen inhibited phosphorylation of
p65 (NF-κB activation). This in vivo observation was
confirmed by an in vitro study. As seen in Fig. 6B, treatment with both p-ibuprofen
and ibuprofen markedly decreased β-catenin levels in the nuclear
extracts of HCT116 cells as compared to control (vehicle-treated).
Interestingly, p-ibuprofen inhibited the nuclear translocation of
the NF-κB subunit p65 (NF-κB activation), but this inhibitory
effect was not observed for ibuprofen. This additional inhibitory
effect in NF-κB activation may explain why the lower plasmic level
of ibuprofen after treatment with p-ibuprofen exhibited a similar
tumor inhibitory effect with ibuprofen-treated group.
Discussion
The modification of existing NSAIDs is significant
for developing novel drugs in cancer prevention. To date, there
have been no reports indicating that modified ibuprofen possesses
increased anticancer activity and reduced GI toxic side effects.
For example, Shanbhag et al(27) modified ibuprofen by esterification
and amidation with various groups. These modified agents exhibited
less anti-inflammatory activity, but only
ibuprofen-O(CH2)2N(CH3)2HCl and
ibuprofen-NHCH2COOH showed decreased GI toxic side
effects when compated to the parent molecule. However, our group
developed a novel phospho-butanol-modified ibuprofen that exhibits
a markedly higher anti-inflammatory efficacy in
vivo(21) and anticancer
activity in vitro and in xenograft models (19, 20)
compared with its parent compound ibuprofen. Respectively, this
study showed that phospho-modified ibuprofen significantly reduced
the GI toxic side effect compared to the parent ibuprofen. In
addition, this compound inhibited AOM-induced colonic ACF and tumor
multiplicity in rats in an inhibitory manner similar to the parent
compound ibuprofen. This study also shows that this modification
significantly reduces the GI toxic side effect associated with the
unmodified parent, ibuprofen. p-Ibuprofen inhibits AOM-induced
colonic ACF and tumor multiplicity in rats with a potency that is
comparable to ibuprofen. As the PK results show, ibuprofen was
released into the blood of animals after administration with
p-ibuprofen. However, this level is slightly lower when compared to
direct ibuprofen administration. This may explain why p-ibuprofen
is able to retain the anticancer properties of ibuprofen.
For decades, the mechanisms by which NSAIDs prevent
cancer have focused on cyclooxygenase (COX) inhibition (28,29).
Recently, COX-2 has been shown to be upregulated in various
carcinomas and to have a central role in tumorigenesis (30,31).
The classical NSAIDs are not selective and inhibit both COX-1 and
COX-2, which results in the inhibition of prostaglandin and
thromboxane synthesis and reduced inflammation. This mode of action
also causes an adverse effect, irritation of the gastric mucosa, as
prostaglandins are considered to have a protective role in the
gastrointestinal tract. NSAIDs that have been engineered to
selectively inhibit COX-2, such as celecoxib and rofecoxib, cause
much less gastric irritation but may increase the risk of heart
attack and thrombosis as a result of the increase of thromboxane
unbalanced by prostacyclin. In the current study,
phospho-modification greatly reduced the GI ulcerogenicity of
ibuprofen even when a high dose (9 times regular dose) was used in
rats. This was considered to result from both the reduced
irritation of free carboxylic group and the inhibition of synthesis
of gastrointestinal PGE2 and PGI2 (32–34).
Consistent with these findings, our results also showed that, like
the parent ibuprofen, p-ibuprofen retained the ability to inhibit
COX-2 and PGE2 synthesis. We did not detect additional
toxic effect of p-ibuprofen in heart, lung, liver or kidney of rats
treated with both regular and high doses; thereby, suggesting that
p-ibuprofen is a potential novel drug for long-term use in cancer
prevention.
COX inhibition is not the only mechanism by which
NSAIDs act. Increasing evidence has shown that COX-independent
pathways are involved in the mechanism of action of NSAIDs,
especially in cancer prevention via inhibition of cell cycle
progression (35–38), induction of apoptosis (39,40),
anti-angiogenesis (41,42), the mTOR signaling pathway (43), direct gene alteration (44), oxidative phosphorylation in
mitochondria (45,46), the induction of nitro oxide
radicals (47), and the NF-κB
signaling pathway (26,48). Further investigation of
COX-independent pathways is necessary in order to gain a complete
understanding of the mechanism by which NSAIDs prevent cancer. In
this study, we show that both ibuprofen and phospho-modified
ibuprofen significantly inhibit β-catenin nuclear translocation
both in vivo and in vitro. This finding is consistent
with the results reported by Maier et al(49). Our preliminary results show that
p-ibuprofen significantly inhibits NF-κB activation in a human
colon cancer cell line. NF-κB, as a master transcriptional
regulator of inflammatory response, may be involved in the
mechanism of carcinogenesis (50,51).
Normally NF-κB is bound to an inhibitor, I-κB, in the cytoplasm. To
activate the signaling pathway, an I-κB kinase (IKK) is
phosphorylated and activated. The activated IKK degrades the NF-κB
inhibitor I-κB (canonical pathway) or p100 (non-canonical pathway)
and releases p65/p50 or RelB/p52 dimers (activated subunit forms of
NF-κB) to the nucleus. These proteins then regulate gene
transcription for cell survival, proliferation, and inflammation.
We will perform additional studies to explore how mechanistically
p-ibuprofen activates NF-κB signaling pathway.
In summary, phospho-modification of ibuprofen
remarkably reduces its GI toxic side effects while allowing it to
retain the anti-inflammatory and anticancer activities of its
parent compound. In addition to COX-2 mechanism, both ibuprofen and
phospho-modified ibuprofen may inhibit the β-catenin signaling
pathway and may suppress NF-κB activation in cancer cells. Taken
together, these results solidify our hypothesis that p-ibuprofen is
a potential effective novel drug for long-term use in colon cancer
prevention.
Acknowledgements
This study was supported by grants
K01CA106604 and R01CA140487. We would like to thank Dr Gang Xie for
is assistance with HPLC analysis.
References
|
1.
|
Johnson CC, Hayes RB, Schoen RE, Gunter MJ
and Huang WY; PLCO Trial Team: Non-steroidal anti-inflammatory drug
use and colorectal polyps in the Prostate, Lung, Colorectal, and
Ovarian Cancer Screening Trial. Am J Gastroenterol. 105:2646–2655.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Harris RE, Beebe-Donk J and Alshafie GA:
Similar reductions in the risk of human colon cancer by selective
and nonselective cyclooxygenase-2 (COX-2) inhibitors. BMC Cancer.
8:2372008. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Harris RE, Beebe-Donk J, Doss H and Burr
Doss D: Aspirin, ibuprofen, and other non-steroidal
anti-inflammatory drugs in cancer prevention: a critical review of
non-selective COX-2 blockade (Review). Oncol Rep. 13:559–583.
2005.
|
|
4.
|
Cioli V, Putzolu S, Rossi V and Corradino
C: A toxicological and pharmacological study of ibuprofen guaiacol
ester (AF 2259) in the rat. Toxicol Appl Pharmacol. 54:332–339.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Venuti MC, Young JM, Maloney PJ, Johnson D
and McGreevy K: Synthesis and biological evaluation of
omega-(N,N,N-trialkylammonium)alkyl esters and thioesters of
carboxylic acid nonsteroidal antiinflammatory agents. Pharm Res.
6:867–873. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Samara E, Avnir D, Ladkani D and Bialer M:
Pharmacokinetic analysis of diethylcarbonate prodrugs of ibuprofen
and naproxen. Biopharm Drug Dispos. 16:201–210. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Abordo EA, Bowden K, Huntington AP and
Powell SL: Prodrugs. Part 3. 2-Formylphenyl esters of indomethacin,
ketoprofen and ibuprofen and 6-substituted 2-formyl and
2-acylphenyl esters of aspirin. Farmaco. 53:95–101. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Mahfouz NM, Omar FA and Aboul-Fadl T:
Cyclic amide derivatives as potential prodrugs II:
N-hydroxymethylsuccinimide-/isatin esters of some NSAIDs as
prodrugs with an improved therapeutic index. Eur J Med Chem.
34:551–562. 1999. View Article : Google Scholar
|
|
9.
|
Kahns AH, Jensen PB, Mork N and Bundgaard
H: Kinetics of hydrolysis of indomethacin and indomethacin ester
prodrugs in aqueous solution. Acta Pharm Nord. 1:327–336.
1989.PubMed/NCBI
|
|
10.
|
Wang LF, Chiang HN and Wu PC: Kinetics and
hydrolysis mechanism of polymeric prodrugs containing ibuprofen,
ketoprofen, and naproxen as pendent agents. J Biomater Sci Polym
Ed. 13:287–299. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Peng YS, Lin SC, Huang SJ, et al:
Chondroitin sulfate-based anti-inflammatory macromolecular
prodrugs. Eur J Pharm Sci. 29:60–69. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Zhao X, Tao X, Wei D and Song Q:
Pharmacological activity and hydrolysis behavior of novel ibuprofen
glucopyranoside conjugates. Eur J Med Chem. 41:1352–1358. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Kourounakis PN, Tsiakitzis K, Kourounakis
AP and Galanakis D: Reduction of gastrointestinal toxicity of
NSAIDs via molecular modifications leading to antioxidant
anti-inflammatory drugs. Toxicology. 144:205–210. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Galanakis D, Kourounakis AP, Tsiakitzis
KC, et al: Synthesis and pharmacological evaluation of amide
conjugates of NSAIDs with L-cysteine ethyl ester, combining potent
antiinflammatory and antioxidant properties with significantly
reduced gastrointestinal toxicity. Bioorg Med Chem Lett.
14:3639–3643. 2004. View Article : Google Scholar
|
|
15.
|
Yeh RK, Chen J, Williams JL, et al:
NO-donating nonsteroidal antiinflammatory drugs (NSAIDs) inhibit
colon cancer cell growth more potently than traditional NSAIDs: a
general pharmacological property? Biochem Pharmacol. 67:2197–2205.
2004. View Article : Google Scholar
|
|
16.
|
Kashfi K, Ryan Y, Qiao LL, et al: Nitric
oxide-donating nonsteroidal anti-inflammatory drugs inhibit the
growth of various cultured human cancer cells: evidence of a tissue
type-independent effect. J Pharmacol Exp Ther. 303:1273–1282. 2002.
View Article : Google Scholar
|
|
17.
|
Williams JL, Borgo S, Hasan I, Castillo E,
Traganos F and Rigas B: Nitric oxide-releasing nonsteroidal
anti-inflammatory drugs (NSAIDs) alter the kinetics of human colon
cancer cell lines more effectively than traditional NSAIDs:
implications for colon cancer chemoprevention. Cancer Res.
61:3285–3289. 2001.
|
|
18.
|
Mattheolabakis G, Nie T, Constantinides PP
and Rigas B: Sterically stabilized liposomes incorporating the
novel anti-cancer agent phospho-ibuprofen (MDC-917): preparation,
characterization, and in vitro/in vivo evaluation. Pharm Res.
29:1435–1443. 2012. View Article : Google Scholar
|
|
19.
|
Sun Y, Huang L, Mackenzie GG and Rigas B:
Oxidative stress mediates through apoptosis the anticancer effect
of phosphononsteroidal anti-inflammatory drugs: implications for
the role of oxidative stress in the action of anticancer agents. J
Pharmacol Exp Ther. 338:775–783. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Xie G, Sun Y, Nie T, et al:
Phospho-ibuprofen (MDC-917) is a novel agent against colon cancer:
efficacy, metabolism, and pharmacokinetics in mouse models. J
Pharmacol Exp Ther. 337:876–886. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Huang L, Mackenzie G, Ouyang N, et al: The
novel phosphonon-steroidal anti-inflammatory drugs, OXT-328, MDC-22
and MDC-917, inhibit adjuvant-induced arthritis in rats. Br J
Pharmacol. 162:1521–1533. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Wong CC, Cheng KW, Xie G, et al:
Carboxylesterases 1 and 2 hydrolyze phospho-NSAIDs: relevance to
their pharmacological activity. J Pharmacol Exp Ther. 340:422–432.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Piazza GA, Keeton AB, Tinsley HN, et al: A
novel sulindac derivative that does not inhibit cyclooxygenases but
potently inhibits colon tumor cell growth and induces apoptosis
with antitumor activity. Cancer Prev Res (Phila). 2:572–580. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Bird RP: Observation and quantification of
aberrant crypts in the murine colon treated with a colon
carcinogen: preliminary findings. Cancer Lett. 37:147–151. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Natarajan K, Singh S, Burke TR Jr,
Grunberger D and Aggarwal BB: Caffeic acid phenethyl ester is a
potent and specific inhibitor of activation of nuclear
transcription factor NF-kappaB. Proc Natl Acad Sci USA.
93:9090–9095. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Williams JL, Ji P, Ouyang N, Liu X and
Rigas B: NO-donating aspirin inhibits the activation of NF-kappaB
in human cancer cell lines and Min mice. Carcinogenesis.
29:390–397. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Shanbhag VR, Crider AM, Gokhale R,
Harpalani A and Dick RM: Ester and amide prodrugs of ibuprofen and
naproxen: synthesis, anti-inflammatory activity, and
gastrointestinal toxicity. J Pharm Sci. 81:149–154. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Antonakopoulos N and Karamanolis DG: The
role of NSAIDs in colon cancer prevention. Hepatogastroenterology.
54:1694–1700. 2007.PubMed/NCBI
|
|
29.
|
Backlund MG, Mann JR and Dubois RN:
Mechanisms for the prevention of gastrointestinal cancer: the role
of prostaglandin E2. Oncology. 69(Suppl 1): 28–32. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Koki A, Khan NK, Woerner BM, et al:
Cyclooxygenase-2 in human pathological disease. Adv Exp Med Biol.
507:177–184. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Koch A, Gustafsson B, Fohlin H and
Sorenson S: Cyclooxygenase-2 expression in lung cancer cells
evaluated by immunocytochemistry. Diagn Cytopathol. 39:188–193.
2011.PubMed/NCBI
|
|
32.
|
Schoen RT and Vender RJ: Mechanisms of
nonsteroidal anti-inflammatory drug-induced gastric damage. Am J
Med. 86:449–458. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Mitchell JA and Warner TD:
Cyclo-oxygenase-2: pharmacology, physiology, biochemistry and
relevance to NSAID therapy. Br J Pharmacol. 128:1121–1132. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Wallace JL and Cirino G: The development
of gastrointestinal-sparing nonsteroidal anti-inflammatory drugs.
Trends Pharmacol Sci. 15:405–406. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Kardosh A, Blumenthal M, Wang WJ, Chen TC
and Schonthal AH: Differential effects of selective COX-2
inhibitors on cell cycle regulation and proliferation of
glioblastoma cell lines. Cancer Biol Ther. 3:55–62. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Narayanan BA, Condon MS, Bosland MC,
Narayanan NK and Reddy BS: Suppression of
N-methyl-N-nitrosourea/testosterone-induced rat prostate cancer
growth by celecoxib: effects on cyclooxygenase-2, cell cycle
regulation, and apoptosis mechanism(s). Clin Cancer Res.
9:3503–3513. 2003.
|
|
37.
|
Grosch S, Tegeder I, Niederberger E,
Brautigam L and Geisslinger G: COX-2 independent induction of cell
cycle arrest and apoptosis in colon cancer cells by the selective
COX-2 inhibitor celecoxib. FASEB J. 15:2742–2744. 2001.PubMed/NCBI
|
|
38.
|
Maier TJ, Schilling K, Schmidt R,
Geisslinger G and Grosch S: Cyclooxygenase-2 (COX-2)-dependent and
-independent anti-carcinogenic effects of celecoxib in human colon
carcinoma cells. Biochem Pharmacol. 67:1469–1478. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Arico S, Pattingre S, Bauvy C, et al:
Celecoxib induces apoptosis by inhibiting
3-phosphoinositide-dependent protein kinase-1 activity in the human
colon cancer HT-29 cell line. J Biol Chem. 277:27613–27621. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Hsu AL, Ching TT, Wang DS, Song X,
Rangnekar VM and Chen CS: The cyclooxygenase-2 inhibitor celecoxib
induces apoptosis by blocking Akt activation in human prostate
cancer cells independently of Bcl-2. J Biol Chem. 275:11397–11403.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Ostrowski J, Wocial T, Skurzak H and
Bartnik W: Do altering in ornithine decarboxylase activity and gene
expression contribute to antiproliferative properties of COX
inhibitors? Br J Cancer. 88:1143–1151. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Wei D, Wang L, He Y, Xiong HQ, Abbruzzese
JL and Xie K: Celecoxib inhibits vascular endothelial growth factor
expression in and reduces angiogenesis and metastasis of human
pancreatic cancer via suppression of Sp1 transcription factor
activity. Cancer Res. 64:2030–2038. 2004. View Article : Google Scholar
|
|
43.
|
Zhang YJ, Bao YJ, Dai Q, et al: mTOR
signaling is involved in indomethacin and nimesulide suppression of
colorectal cancer cell growth via a COX-2 independent pathway. Ann
Surg Oncol. 18:580–588. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Wang X, Baek SJ and Eling T: COX
inhibitors directly alter gene expression: role in cancer
prevention? Cancer Metastasis Rev. 30:641–657. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Petrescu I and Tarba C: Uncoupling effects
of diclofenac and aspirin in the perfused liver and isolated
hepatic mitochondria of rat. Biochim Biophys Acta. 1318:385–394.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Somasundaram S, Sigthorsson G, Simpson RJ,
et al: Uncoupling of intestinal mitochondrial oxidative
phosphorylation and inhibition of cyclooxygenase are required for
the development of NSAID-enteropathy in the rat. Aliment Pharmacol
Ther. 14:639–650. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Das S, Khan N, Mukherjee S, et al: Redox
regulation of resveratrol-mediated switching of death signal into
survival signal. Free Radic Biol Med. 44:82–90. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Vaish V, Tanwar L and Sanyal SN: The role
of NF-kappaB and PPARgamma in experimentally induced colorectal
cancer and chemoprevention by cyclooxygenase-2 inhibitors. Tumour
Biol. 31:427–436. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Maier TJ, Janssen A, Schmidt R,
Geisslinger G and Grosch S: Targeting the beta-catenin/APC pathway:
a novel mechanism to explain the cyclooxygenase-2-independent
anticarcinogenic effects of celecoxib in human colon carcinoma
cells. FASEB J. 19:1353–1355. 2005.PubMed/NCBI
|
|
50.
|
Paul S, DeCastro AJ, Lee HJ, et al:
Dietary intake of pterostilbene, a constituent of blueberries,
inhibits the beta-catenin/p65 downstream signaling pathway and
colon carcinogenesis in rats. Carcinogenesis. 31:1272–1278. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Onizawa M, Nagaishi T, Kanai T, et al:
Signaling pathway via TNF-alpha/NF-kappaB in intestinal epithelial
cells may be directly involved in colitis-associated
carcinogenesis. Am J Physiol Gastrointest Liver Physiol.
296:G850–G859. 2009. View Article : Google Scholar
|