Introduction
Hepatocellular carcinoma (HCC) is one of the most
common malignant tumors worldwide and most HCC arises from chronic
liver disease, which is associated with liver cirrhosis.
Etiological factors for hepatocarcinogenesis include chronic
hepatitis B or hepatitis C virus infection, long-term exposure to
aflatoxin B1 in food, alcohol addiction and hemochromatosis. The
precise molecular mechanisms for its development beyond initiation
have not been elucidated, but as for many other tumors, the
development and progression of HCC is a multistep process and
activation of oncogenes and inactivation of tumor suppressor genes
caused by genetic or epigenetic aberrance are involved in
carcinogenesis (1). Among these,
epigenetic inactivation of tumor suppressor genes by
hypermethylation of CpG islands in promoter sequences plays an
important role (2–4). Yoshikawa et al(6) compared genomic NotI
restriction fragments between normal and HCC tissues by restriction
landmark genomic scanning (RLGS) analysis and isolated several
aberrantly methylated genes, such as suppressor of cytokine
signaling-1 (SOCS-1), SOCS-3 and apoptotic
speck protein-like (ASCL) (5–10).
Here, we demonstrate that the Delta-like 3
(DLL3) gene was aberrantly methylated in its CpG island in
HCC cells. DLL3 is a member of Delta/Serrate/Lag2 (DSL) ligands for
Notch receptors and plays a role in Notch signaling. Five DSL
ligands (DLL1, 3, 4; and Jagged 1 and 2) and four Notch receptors
have been identified in mammals (11). Notch signaling is an evolutionarily
conserved signaling pathway essential for embryonic development and
regulates cellular processes such as differentiation,
proliferation, survival and apoptosis in various cell types
(12,13). However, DLL3 is the most
structurally divergent DSL ligand and function of DLL3 in Notch
signaling is complicated by conflicting reports (14–16).
DLL3 is expressed throughout the presomitic mesoderm and is
localized to the rostral somatic compartments. Homozygous
disruptions of Notch1 and DLL3 result in severe abnormalities in
somitogenesis (17–19) and mutations in the human DLL3
homolog cause recessive skeletal abnormalities in spondylocostal
dysostosis (20–22). The role of DLL3 in carcinogenesis
has not been reported.
In this study, we sought to examine the silencing of
DLL3 by methylation and to characterize its roles in HCC. Our data
indicate that DLL3 is silenced by methylation and DLL3 expression
is associated with cell growth suppression in HCC. Our findings
confirm a novel function of DLL3 in hepatocarcinogenesis.
Materials and methods
Cell lines
Human HCC cell lines HuH1, HuH4 and HuH7 were
purchased from the Japanese Culture Collection. HuH2 and Kim1 were
gifts from the Department of Pathology, The Cancer Institute and
the Japanese Foundation for Cancer Research. Hep3B and Li-7 were
obtained from the cell resource center for Biomedical Research
Institute of Development, Aging and Cancer, Tohoku University. FLC4
was a gift from Dr Seishi Nagamori. Cells were maintained in
RPMI-1640 medium (Sigma-Aldrich, St. Louis, MO, USA) containing 10%
fetal bovine serum (Invitrogen, Carlsbad, CA, USA) at 37°C under a
5% CO2 atmosphere. For DLL3-reactivation study, cells
were treated with 1 μM 5-Aza-2′-deoxycytidine (5-Aza-dC) alone for
4 days or with 1 μM 5-Aza-dC for 4 days and 300 nM trichostatin A
(TSA) for 1 day.
Reverse transcription-PCR analysis
Total RNA of HCC cell lines was isolated using an
RNeasy mini kit (Qiagen, Hilden, Germany), and cDNA was synthesized
using the Superscript Preamplification System (Invitrogen). An
aliquot of cDNA was subjected to amplification using Taq
polymerase (Takara, Shiga, Japan). The primer sequences were
CGAGCTGCAGAT CCACTCT and CGCCTCACATTCGTCCTC. The reaction was
carried out for 35 cycles of denaturation at 94°C for 40 sec,
annealing at 62°C for 40 sec and extension 72°C for 180 sec. An
aliquot of PCR product was analyzed by 1.5% agarose gel
electrophoresis, followed by ethidium bromide staining.
Methylation-specific PCR (MSP)
Bisulfite modification of genomic DNA was performed
as described previously (23). The
methylation-specific primer sequences for DLL3 were
CGGGATTATTTACGTATGATTTC [nucleotides (nt) 103,584-103,606 in
AC011500] and CCGACCCCAAAAA ACCAAAAACG (nt 103,686-103,708). The
unmethylation-specific primer sequences were TGTGGGATTATTTA
TGTATGATTTT (nt 103,582-103,606) and CCCAACCCCA AAAAACCAAAAACA (nt
103,686-103,709). An aliquot of bisulfite-modified DNA was
amplified by PCR. PCR was carried out with preheating at 94°C for
120 sec and 80°C for 30 sec, followed by 30 cycles of 94°C for 40
sec, 60°C for 40 sec and 72°C for 60 sec. An aliquot of PCR product
was analyzed by 4.0% agarose gel electrophoresis.
Construction of expression vector
A full-length DLL3 cDNA was isolated from human
liver RNA (BD Sciences, Rockville, MD, USA) by PCR and the product
was cloned into the EcoRI site of a pcDNA 3.1/HisB vector
(Invitrogen). A clone, designated pcDNA3-DLL3, showed an in-frame
ligation and correct sequence.
Colony formation assay
Cells (5.0×104 for HuH2,
1.0×105 for Kim1) were transfected with 4 μg of either
pcDNA-DLL3 or pcDNA3.1 backbone vector. Colonies were selected in
the presence of G418 (1,000 μg/ml for HuH2, 300 μg/ml for Kim1) for
4 weeks and colonies were photographed after staining.
Flow cytometry analysis of cell
death
HuH2 cells were transfected with either pcDNA-DLL3
or pcDNA3.1 backbone vector. After 48 h, cell apoptosis was
analyzed with an Annexin V-FITC kit (Bender MedSystems, Burlingame,
CA, USA) along with PI according to the manufacturer’s protocol.
Briefly, both adhered and floating cells (5×105/ml) were
resuspended in binding buffer and incubated with Annexin V-FITC for
10 min at room temperature. Cells were then washed with PBS and
incubated with PI (final concentration 1 μg/ml) solution and DNA
contents of the cells were measured with a flow cytometer (Beckman
Coulter, Brea, CA, USA).
TUNEL analysis
TUNEL assay was performed using a kit (Roche
Biochemicals Inc., Mannheim, Germany) according to manufacturer’s
protocol. Briefly, cells were plated on 18×18 mm coverslips placed
in a 6-well plate and transfected with 1 μg of either pcDNA-DLL3 or
pcDNA3.1 backbone vector. After 48 h, cells were fixed with 4%
paraformaldehyde, and permeabilized with 0.1% Triton X-100 after
blocking endogenous peroxidase. TUNEL reaction mixture was added to
the cells and cells were incubated with converter-POD before adding
substrate solution. Over 3,000 cells were counted from 15 randomly
selected fields under a microscope.
Measurement of single-stranded DNA
DNA in apoptotic cells is sensitive to formamide and
denatured DNA was detected with a monoclonal antibody against
single-stranded DNA with an ApoStrand™ ELISA apoptosis detection
kit (Enzo Life Sciences, Plymouth Meeting, PA, USA) according to
the manufacturer’s protocol. Briefly, 3.0×103 or
4.0×103 cells were plated in a 96-well microplate and
transfected with 50 ng of either pcDNA-DLL3 or pcDNA3.1 backbone
vector. After 48 h, cells were fixed and dried to attach cells to
the plate surface. Cells were then treated with formamide and
heated at 56°C for 30 min and were then incubated with antibody
mixture for 30 min after blocking non-specific binding sites. After
washing, peroxidase substrate was added to each well and absorbance
was measured at 405 nm with an ELISA reader (Corona Electric Co.
Ltd., Ibaragi, Japan).
Western blot analysis
Expression of cleaved Notch I was detected by
western blot analysis. Cells were seeded onto 10-cm dishes and
transfected with 4 μg of either pcDNA-DLL3 or pcDNA3.1 backbone
vector. After 48 h, cells were solubilized in lysis buffer [20 mM
Tris-HCl pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% deoxycholic acid, 0.1%
sodium dodecyl sulfate containing complete protease inhibitor
cocktail (Roche Diagnostic GmbH, Mannheim, Germany)], followed by
centrifugation at 14,000 rpm for 15 min at 4°C. Supernatants (20
μg) were resolved by electrophoresis and were transferred to
Immobilon-P membrane (Millipore, Billerica, MA, USA). Cleaved Notch
was detected by probing membrane with antibody against cleaved
Notch (Cell Signaling Technology, Beverly, MA, USA). Horseradish
peroxidase-labeled anti-rabbit IgG was used as a secondary antibody
and chemiluminescent reaction was carried out using ECL plus
western blotting detection reagents (GE Healthcare UK,
Buckinghamshire, UK). Signals were detected with a LAS-3000 lumino
image analyzer (Fuji Photo Film, Tokyo, Japan).
Results
mRNA expression and methylation status of
DLL3 in HCC cells
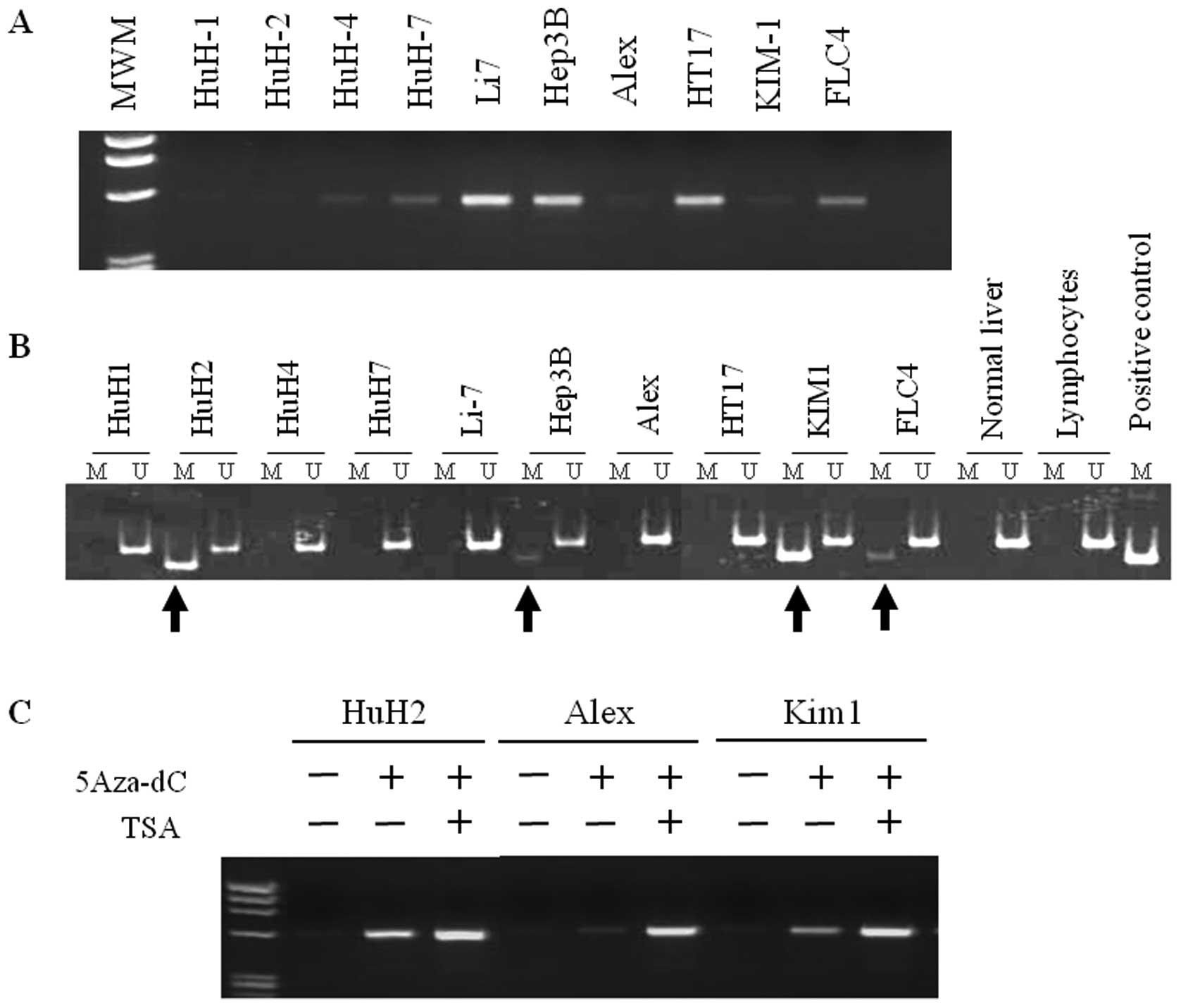
We first analyzed DLL3 mRNA expression in 10 HCC
cell lines. As shown in Fig. 1A,
an amplified band was clearly detected in 6 cell lines (HuH4, HuH7,
Li7, Hep3B, HT17 and FLC4) and a faint band was detected in Alex
and Kim1 cells. No mRNA expression was observed in HuH1, HuH2 cells
in addition to normal liver.
Methylation status of the DLL3 CpG islands
was then analyzed by MSP. A primer set was designed in exon1, which
lies within the DLL3 CpG islands. As shown in Fig. 1B, apparent methylation of
DLL3 was detected in four (HuH2, Hep3B, Kim1 and FLC4) cell
lines among 10 cell lines tested with RT-PCR. Aberrant methylation
of DLL3 was not detected in normal liver tissue or
lymphocytes.
We next analyzed whether a demethylating agent,
5-Aza-2′-deoxycytidine (5-Aza-dC), and a histone deacetylase
inhibitor, trichostatin A (TSA), can reactivate DLL3 expression in
HuH1, HuH2, HuH4, Alex and Kim1 cells. As shown in Fig. 1C, DLL3 expression was reactivated
by 5-Aza-dC treatment in all cell lines tested. Although no
methylation was detected in HuH1 and Alex cells with MSP, a clear
amplified band was detected in these cells after treatment of
5-Aza-dC. Moreover, a robust effect was obtained by additional
treatment with TSA in HuH2, Alex and Kim1 cells. These results
suggest that expression of DLL3 is frequently suppressed or
silenced in association with DNA methylation in HCC cells.
Growth suppression by DLL3
restoration
Colony formation assay was performed in order to
investigate the effects of DLL3 overexpression on cell growth. As
shown in Fig. 2, overexpression of
DLL3 markedly suppressed colony formation in both HuH2 and Kim1
cells, in which DLL3 was silenced in association with DNA
methylation. This suggests that DLL3 has cell growth activity in
HCC cells.
Induction of cell death by DLL3
expression
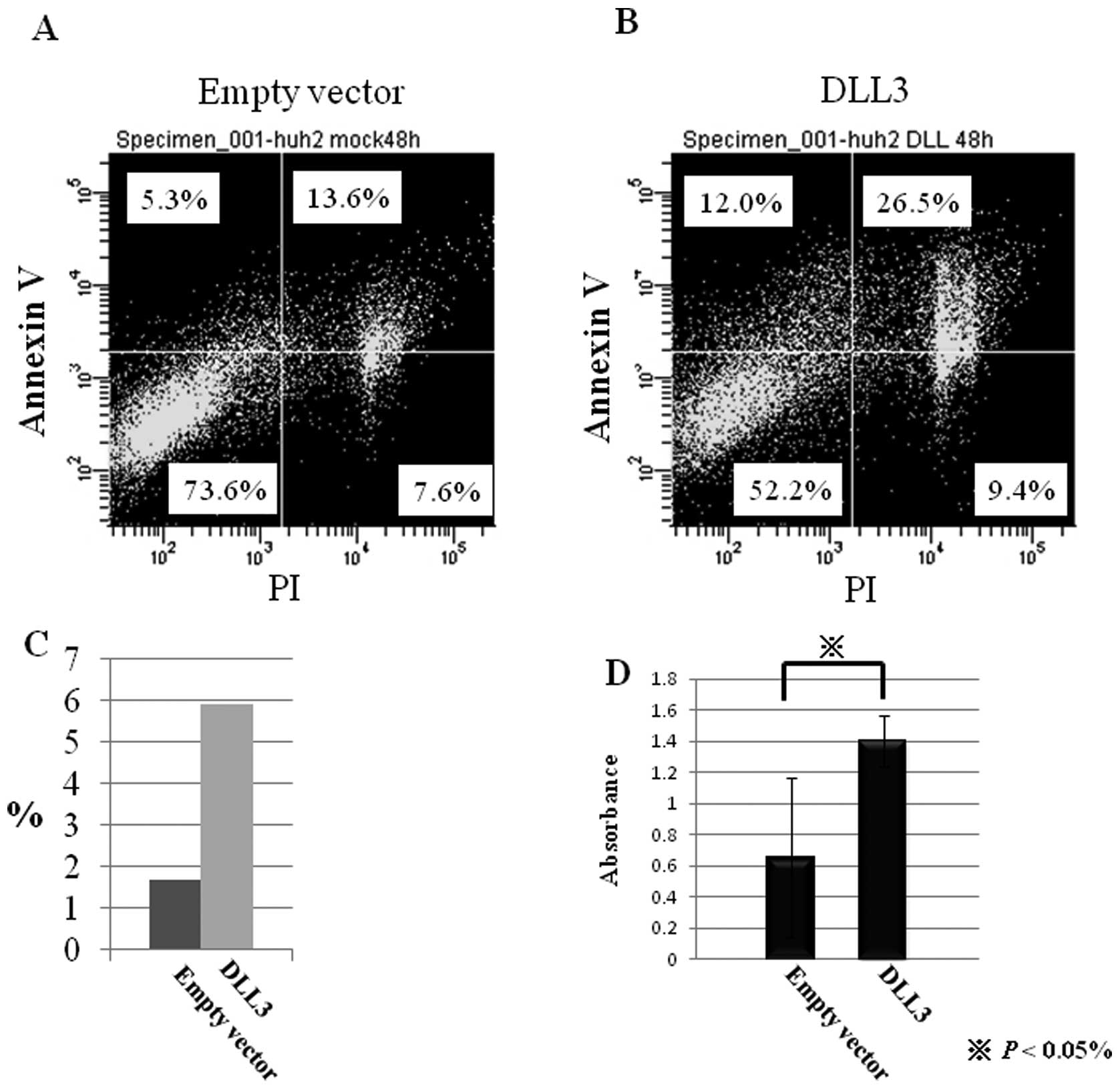
Flow cytometric analysis was performed in order to
investigate the effects of DLL3 overexpression on cell death. Of
the cells transfected with backbone vector, 21.2 and 18.9% were
positive for PI and Annexin V, respectively (Fig. 3A and B). On the other hand, 35.9
and 38.5% of DLL3-transfected cells were positive for PI and
Annexin V, respectively. These results suggest that overexpression
of DLL3 induced cell death in HuH2 cells chiefly via apoptosis.
Induction of apoptosis by DLL3
expression
In order to confirm the apoptotic effects of DLL3 on
HuH2 cells, apoptotic cells were detected by the TUNEL method. As
shown in Fig. 3C, the number of
TUNEL-positive cells increased by transfection of DLL3. The
percentage of TUNEL-positive cells in mock-transfected cells and
DLL3-transfected cells was 1.68 and 5.93%, respectively.
Furthermore, the amount of single-stranded DNA was significantly
increased by transfection of DLL3 in HuH2 cells (Fig. 3D). These data support the notion
that restoration of DLL3 induces apoptosis in HuH2 cells in which
DLL3 is silenced by DNA methylation.
Effects of DLL3 on Notch1 activation
Notch1 is a transmembrane protein and the
carboxy-terminal Notch1 fragment is released upon binding to a
ligand. The resulting activated cytosolic fragment translocates to
the nucleus, where it activates transcription. As shown in Fig. 4, western blot analysis showed no
significant differences in cleaved Notch1 expression upon
overexpression of DLL3.
Discussion
HCC develops as a result of aberrant genetic and
epigenetic events, similarly to other cancers (1). Mutations in tumor suppressor genes,
such as p53, β-catenin and Axin, are detected in 20–30% of
HCC samples (24,25). The epigenetic pathway involves
hypomethylation of the HCC genome causing genomic instability,
hypermethylation of CpG islands in promoter sequences leading to
inactivation of the genes, and histone modification affecting
chromatin conformation. In HCC, aberrant promoter hypermethylation
associated with gene silencing is observed in genes such as
SOCS-1, SOCS-3, ASCL,
p16INK4a, Ras association domain family
1A (RASSF1A), placental glutathione S transferase
P1 (GSTP1) and E-cadherin(8–10,26–29).
Yoshikawa et al(6) analyzed genomic NotI restriction sites
in human HCC and found aberrantly methylated genes, including
SOCS-1, SOCS-3, ASCL and DLL3, from
multiple aberrant NotI sites. SOCS-1, known as JAB and SS-1,
switches cytokine signaling ‘off’ by means of its direct
interaction with Janus kinase (JAK). The authors demonstrated that
restoration of SOCS-1 suppressed growth rate and
anchorage-independent growth of cells in which SOCS-1 is
methylation-silenced and JAK2 was constitutively activated
(8). SOCS-3, which is
methylation-silenced in HCC, negatively regulates cell growth and
cell migration by enhancing JAK/STAT and FAK signaling (9). The restoration of ASCL in
methylation-silenced HCC cells induces growth suppression by the
induction of apoptosis (10).
In this study, we found that DLL3 is also silenced
in HCC cells by aberrant promoter methylation and that the
restoration of DLL3 in methylation-silenced HuH2 cells leads to
cell growth suppression by induction of apoptosis. We detected
apoptosis by the TUNEL method and expression of Annexin V, an early
marker for apoptosis. About 5.9% of the DLL3-transfected cells were
positive for TUNEL staining, whereas 38.5% of the transfected cells
were positive for Annexin V expression. It is possible that
methodological differences explain why the ratio of apoptotic cells
differed between the two experiments; for detection of Annexin V
expression, both adherent and floating cells were subjected to flow
cytometry, whereas TUNEL staining was carried out using only
adherent cells on the cover slip. In both experiments, apoptotic
cells were detected at 3.5 and 2.0-fold higher levels when compared
to mock-treated cells. Moreover, the amount of single-stranded DNA
was significantly increased in DLL3-transfected cells, and this
suggests apoptotic effects for DLL3 in HCC.
DLL3 is a member of the DSL ligands (Delta, Serrate
and Lag2), which are type 1 cell surface proteins having multiple
tandem epidermal growth factor (EGF) repeats in their
extra-cellular domains. In addition, Delta and Serrate proteins
contain a conserved cysteine-rich region known as the DSL domain in
their extracellular portion. The DSL domain, flanking N-terminal
domain, and the first two EGF repeats are required for binding to
Notch (30,31). DLL3 is the shortest among the three
DLL ligands, with only six EGF-like repeats compared with the eight
repeats identified in DLL1 and DLL4. The functions of DLL3 in Notch
signaling have been complicated by conflicting reports.
Dunwoodie et al(14) reported that injection of
DLL3 RNA into Xenopus oocytes is able to inhibit
primary neuron formation, as observed in ectopic expression of
constitutively active Notch1, suggesting that DLL3 is able
to activate Notch signaling. However, Ladi et al reported
that DLL3 does not bind or activate any of the known mammalian
Notch receptors when presented in trans, although DLL3 inhibited
ligand-induced Notch signaling when coexpressed with Notch at the
cell surface in cis(15).
In addition, Geffers et al recently reported that DLL3 does
not activate Notch in D. melanogaster nor repress
DLL1-mediated Notch activation in vivo. They also
demonstrated that endogenous DLL3 predominantly resides in the
Golgi apparatus and is virtually absent from the cell surface,
whereas DLL1 is located on the cell surface (16). These results strongly suggest that
DLL3 differs functionally from other DSL ligands.
Our western blot analysis demonstrated that
overexpression of DLL3 does not increase the expression of cleaved
Notch1 in HuH2 cells, thus suggesting that cell growth suppression
is induced via a Notch-independent pathway. However, it is not
clear how DLL3 induced apoptosis in HCC cells, its Golgi
localization may be the key to understanding the novel function of
DLL3, including cell growth suppression. In summary, we found that
restoration of DLL3 expression induced apoptosis in HuH2 cells via
a Notch1-independent pathway.
References
|
1
|
Herath NI, Leggett BA and MacDonald GA:
Review of genetic and epigenetic alterations in
hepatocarcinogenesis. J Gastroenterol Hepatol. 21:15–21. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yang B, Guo M, Herman JG and Clark DP:
Aberrant promoter methylation profiles of tumor suppressor genes in
hepatocellular carcinoma. Am J Pathol. 163:1101–1107. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhu JD: DNA methylation and hepatocellular
carcinoma. J Hepatobiliary Pancreat Surg. 13:265–273. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee S, Lee HJ, Kim JH, Lee HS, Jang JJ and
Kang GH: Aberrant CpG island hypermethylation along multistep
hepatocarcinogenesis. Am J Pathol. 163:1371–1378. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nagai H, Ponglikitmongkol M, Mita E,
Ohmachi Y, Yoshikawa H, Saeki R, Yumoto Y, Nakanishi T and
Matsubara K: Aberration of genomic DNA in association with human
hepatocellular carcinomas detected by 2-dimensional gel analysis.
Cancer Res. 54:1545–1550. 1994.PubMed/NCBI
|
|
6
|
Yoshikawa H, Monte DL, Nagai H, Wands JR,
Matsubara K and Fujiyama A: Chromosomal assignment of human genomic
NotI restriction fragments in a two-dimentional electrophoresis
profile. Genomics. 31:28–35. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yoshikawa H, Nagai H, Oh KS, Tamai S,
Fujiyama A, Nakanishi T, Kajiyama G and Matsubara K: Chromosomal
assignment of aberrant NotI restriction DNA fragments in primary
hepatocellular carcinoma. Gene. 197:129–135. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yoshikawa H, Matsubara K, Qian GS, Jackson
P, Groopman JD, Manning JE, Harris C and Herman JG: SOCS-1, a
negative regulator of the JAK/STAT pathway, is silenced by
methylation in human hepatocellular carcinoma and shows
growth-suppression activity. Nat Genet. 28:29–35. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Niwa Y, Kanda H, Shikauchi Y, Saiura A,
Matsubara K, Kitagawa T, Yamamoto J, Kubo T and Yoshikawa H:
Methylation silencing of SOCS-3 promotes cell growth and migration
by enhancing JAK/STS and FAK signaling in human hepatocellular
carcinoma. Oncogene. 24:6406–6417. 2005.PubMed/NCBI
|
|
10
|
Kubo T, Yamamoto J, Shikauchi Y, Niwa Y,
Matsubara K and Yoshikawa H: Apoptotic speck protein-like, a highly
homologous protein to apoptotic speck protein in the pyrin domain,
is silenced by DNA methylation and induces apoptosis in human
hepatocellular carcinoma. Cancer Res. 64:5172–5177. 2004.
View Article : Google Scholar
|
|
11
|
D’Souza B, Miyamoto A and Weinmaster G:
The many facets of Notch ligands. Oncogene. 27:5148–5167. 2008.
|
|
12
|
Bray SJ: Notch signaling: a simple pathway
becomes complex. Nat Rev Mol Cell Biol. 7:678–689. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fiuza UM and Arias AM: Cell and molecular
biology of Notch. J Endocrinol. 194:459–474. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dunwoodie SL, Henrique D, Harrison SM and
Beddington RSP: Mouse Dll3: a novel divergent Delta gene which may
complement the function of other Delta homologues during early
pattern formation in the mouse. Development. 124:3065–3076.
1997.
|
|
15
|
Ladi E, Nichols JT, Ge W, Miyamoto A, Yao
C, Yang LT, Boulter J, Sun YE, Kintner C and Weinmaster G: The
divergent DSL ligand Dll3 does not activate Notch signaling but
cell autonomously attenuates signaling induced by other DSL
ligands. J Cell Biol. 170:983–992. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Geffers I, Serth K, Chapman G, Jaekel R,
Schuster-Gossler K, Cardes R, Sparrow DB, Kremmer E, Dunwoodie S,
Klein T and Gossler A: Divergent functions and distinct
localization of the Notch ligands DLL1 and DLL3 in vivo. J Cell
Biol. 30:465–476. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Conlon RA, Reaume AG and Rossant J: Notch1
is required for the coordinate segmentation of somites.
Development. 121:1633–1645. 1995.PubMed/NCBI
|
|
18
|
Kusumi K, Sun ES, Kerrebrock AW, Bronson
RT, Chi DC and Bulotsky MS: The mouse pudgy mutation disrupts Delta
homologue Dll3 and initiation of early somite boundaries. Nat
Genet. 19:274–278. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dunwoodie SL, Clements M, Sparrow DB, Sa
X, Conlon RA and Beddington RS: Axial skeletal defects caused by
mutation in the spondylocostal dysplasia/pudgy gene Dll3 are
associated with disruption of the segmentation clock within the
presomitic mesoderm. Development. 129:1795–1806. 2002.
|
|
20
|
Bulman MP, Kusumi K, Frayling TM, McKeown
C, Garrette C, Lander ES, Krumlauf R, Hattersley AT, Ellard S and
Turnpenny PD: Mutations in the human delta homologues, DLL3, cause
axial skeletal defects in spondylocostal dysostosis. Nat Genet.
24:438–441. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sparrow DB, Clements M, Withington SL,
Scott AN, Novotny J, Sillence D, Kusumi K, Beddington RS and
Dunwoodie SL: Diverse requirements for N signaling in mammals. Int
J Dev Biol. 46:365–374. 2002.
|
|
22
|
Turnpenny PD, Whittock N, Duncan J,
Dunwoodie S, Kusumi K and Ellard S: Novel mutations in DLL3, a
somitogenesis gene encoding a ligand for the N signaling pathway,
cause a consistent pattern of abnormal vertebral segmentation in
spondylocostal dysostosis. J Med Genet. 40:333–339. 2003.
View Article : Google Scholar
|
|
23
|
Herman JG, Graff JR, Myohanen S, Nelkin BD
and Baylin SB: Methylation-specific PCR: a novel PCR assay for
methylation status of CpG islands. Proc Natl Acad Sci USA.
93:9821–9826. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lunn RM, Zhang YJ, Wang LY, Chen CJ, Lee
PH, Lee CS, Tsai WY and Santella RM: p53 mutations, chronic
hepatitis B virus infection, and aflatoxin exposure in
hepatocellular carcinoma in Taiwan. Cancer Res. 57:3471–3477.
1997.PubMed/NCBI
|
|
25
|
De la Costa A, Romagnolo B, Billuart P,
Renard CA, Buendia MA, Soubrane O, Fabre M, Chelly J, Beldjord C,
Kahn A and Perret C: Somatic mutations of the beta-catenin gene are
frequent in mouse and human hepatocellular carcinomas. Proc Natl
Acad Sci USA. 95:8847–8851. 1998.PubMed/NCBI
|
|
26
|
Matsuda Y, Ichida T, Matsuzawa J, Sugimura
K and Asakura H: p16(INK4) is inactivated by extensive CpG
methylation in human hepatocellular carcinoma. Gastroenterology.
116:394–400. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Schagdarsurengin U, Wikens L, Steinemann
D, Flemming P, Kreipe HH, Pfeifer GP, Schlegelberger B and Dammann
R: Frequent epigenetic inactivation of the RASSF1A gene in
hepatocellular carcinoma. Oncogene. 22:1866–1871. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tchou JC, Lin X, Freije D, Isaacs WB,
Brooks JD, Rashid A, De Marzo AM, Kanai Y, Hirohashi S and Nelson
WG: GSTP1 CpG island DNA hypermethylation in hepatocellular
carcinomas. Int J Oncol. 16:663–676. 2000.PubMed/NCBI
|
|
29
|
Liu J, Lian Z, Han S, Waye MMY, Wang H, Wu
MC, Wu K, Ding J, Arbuthnot P, Kew M, Fan D and Feitelson MA:
Downregulation of E-cadherin by hepatitis B virus X antigen in
hepatocellular carcinoma. Oncogene. 25:1008–1017. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Parks AL, Stout JR, Shepard SB, Klueg KM,
Dos Santos AA, Parody TR, Voskova M and Muskavitch AT:
Structure-function analysis of delta trafficking, receptor binding
and signaling in Drosophila. Genetics. 174:1947–1961. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shimizu K, Chiba S, Kumano K, Hosoya N,
Takahashi T, Kanda Y, Hamada Y, Yazaki Y and Hirai H: Mouse Jagged1
physically interacts with Notch2 and other Notch receptors.
Assessment by quantitative methods. J Biol Chem. 274:32961–32969.
1999. View Article : Google Scholar : PubMed/NCBI
|