Introduction
Statins inhibit β-hydroxy-β-methylglutaryl CoA
(HMG-CoA) reductase, which converts HMG-CoA to mevalonate. They are
effective cholesterol-lowering drugs and exhibit anti-cancer
effects by inducing apoptosis and cell cycle arrest (1). Moreover, the inhibition of the
mevalonate pathway by statins causes perturbation of the
endoplasmic reticulum (ER) and stress. In response to ER
dysfunction, cells combat the stress and restore ER homeostasis by
means of the unfolded protein response (UPR), which includes
ER-associated degradation and control of translation (2,3).
Among various ER responses, eIF2α phosphorylation primarily
protects cells from stress by attenuating global translation and
specifically upregulating chaperone proteins, although under
prolonged and severe stress it leads to apoptosis (4). Statin-induced eIF2α phosphorylation
has been shown to protect macrophages from hypoxia-induced cell
death (5); however,
lovastatin-induced eIF2α phosphorylation has been shown to lead to
apoptosis in human head and neck squamous cell carcinoma (6). Elucidating the role of eIF2α
phosphorylation induced by statins may lead to the development of
novel protective and therapeutic approaches against
hypercholesterolemia and cancer.
Under various stress conditions, the tumor
suppressor p53 plays a pivotal role in the execution of ER
stress-induced apoptosis via the activation of the BH3-only
proteins, such as Puma and Noxa, in a transcription-dependent
manner (7) and via a
transcription-independent pathway; it activates members of the
pro-apoptotic Bcl-2 family, such as Bax, Bid and Bak, or their
translocation to the mitochondrial membrane (8). In our previous study, we demonstrated
that simvastatin induced apoptosis in cancer cells by stabilizing
p53 and stimulating its translocation with Bax to the mitochondria,
resulting in the release of cytochrome c(9). However, the mechanisms by which the
ER stress response, particularly eIF2α phosphorylation, is linked
to the p53-mediated mitochondrial apoptotic pathway in
statin-induced apoptosis, have not been investigated.
In the present study, we investigated the molecular
link between eIF2α phosphorylation in the ER stress response and
the p53 transcription-independent mitochondrial apoptotic pathway
in the statin-induced apoptosis of MethA fibrosarcoma cells. We
report that the eIF2α phosphorylation on serine 51 (Ser51) of the
ER stress response attenuates cell death by inhibiting the
stabilization of p53 and its translocation to the mitochondria in
statin-induced apoptosis.
Materials and methods
Cells and reagents
Mouse MethA fibrosarcoma cells were maintained in
RPMI-1640 (Invitrogen, Carlsbad, CA, USA) supplemented with 5%
fetal bovine serum, 100 U/ml penicillin and 10 μg/ml
streptomycin at 37°C and 5% CO2. Simvastatin and
lovastatin (MSD Korea, Ansan, Korea) were reconstituted in absolute
ethanol and stored at −20°C. Mevalonolactone, farnesyl
pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP) were
purchased from Sigma-Aldrich (St. Louis, MO, USA). Salubrinal,
tumnicamycin and SP600125 were obtained from Calbiochem (San Diego,
CA, USA) and anti-tubulin antibody (T5186) from Sigma-Aldrich.
Antibodies against p53, Bax, protein kinase RNA-like endoplasmic
reticulum kinase (PERK), phospho-PERK, eIF2α, phosphoeIF2α,
CCAAT/enhancer-binding protein homologous protein (CHOP)/GADD153,
BiP/78 kDa glucose-regulated protein (Grp78), HRP-conjugated goat
anti-mouse antibody and HRP-conjugated goat anti-rabbit antibody
were supplied by Santa Cruz Biotechnology (Santa Cruz, CA, USA),
while antibodies against phospho-JNK, total JNK and heat-shock
protein (HSP) 60 were obtained from BD Biosciences (San Diego, CA,
USA).
Cell fractionation and western blot
analysis
Cell fractionation was performed with a Mitochondria
Isolation kit (Pierce, Rockford, IL, USA) according to the
manufacturer’s instructions. For western blot analysis, cells were
harvested, washed with ice-cold PBS and lysed in RIPA buffer [10 mM
Tris (pH 7.4), 150 mM NaCl, 0.5% NP-40, 0.1% deoxycholate, 1 mM
PMSF, 2 mM sodium fluoride and 1 mM sodium orthovanadate] for 15
min. Samples (3–30 μg) were then subjected to SDS-PAGE and
western blot analysis was performed using primary antibodies and
HRP-conjugated secondary antibodies, followed by detection with
West-Pico Chemiluminescent Substrates (Pierce) in the dark.
Knockdown experiments and site-directed
mutagenesis
siRNAs directed against eIF2α and CHOP were
purchased from Santa Cruz Biotechnology and transiently transfected
into MethA cells using Lipofectamine 2000 (Invitrogen). Stable
clones were selected in the presence of 4 μg/ml puromycin
and screened by western blot analysis or RT-PCR. Substitution of
the residue serine 51 of eIF2α with alanine (Ala) was performed
using the QuikChange™ Site-Directed Mutagenesis kit (Stratagene, La
Jolla, CA, USA). Briefly, wild-type eIF2α was amplified from MethA
cDNA by PCR and cloned into the EcoRI and XhoI sites
of pBluescript SK(+) vector (Stratagene). The mutant form of eIF2α
was amplified from wild-type eIF2α/pBluescript SK(+) with Pfu
polymerase (Intron, Seongnam, Korea) using the following
mutagenesis primers and PCR conditions: forward, 5′-gcg aat tca tgc
cgg ggc taa gtt gta g-3′; reverse, 5′-cgc tcg agt taa tct tca gct
ttg gct t-3′; 18 cycles of 30 sec at 95°C, 60 sec at 55°C and 10
min at 68°C. The PCR product was DpnI (Promega)-treated and
transformed into DH5α competent cells and the substitution was
confirmed by DNA sequencing. The wild-type and mutant forms of
eIF2α were subcloned into the EcoRI and XhoI sites of
the pCMV-tag2b vector (Stratagene) and transfected into the MethA
cells. Stable clones of the eIF2α wild-type and mutant forms were
selected in the presence of 4 mg/ml neomycin and screened by
western blot analysis.
Measurement of subgenomic content and
mitochondrial membrane potential (MMP)
To analyze subgenomic content, cells were incubated
under the indicated conditions, washed with PBS, fixed with 70%
ice-cold ethanol at 4°C for 1 h and stained with 50 μg/ml
propidium iodide (PI) (Sigma-Aldrich) containing 100 μg/ml
RNase (Sigma-Aldrich) at 37°C for 30 min. DNA content was analyzed
by a FACSCalibur (Becton-Dickinson, San Jose, CA, USA). To analyze
MMP, cells were washed with PBS and stained with 500 μl of 4
μg/ml rhodamine-123 solution at 37°C for 20 min. They were
then incubated with 0.1 mg/ml PI stock solution for 3 min.
Incorporation of rhodamine-123 and PI was analyzed by a
FACSCalibur. The green fluorescence of rhodamine-123 and the red
fluorescence of PI were collected over the range of FL1 and FL2,
respectively. All data were calculated using CellQuest software
(Becton-Dickinson).
Immunofluorescence
MethA cells were plated (1×105 cells) on
poly-L-lysine-coated coverslips (BD Biosciences) and treated with
the indicated reagents for 12 or 36 h. Following incubation, the
cells were washed with PBS, stained with MitoTracker Red (Molecular
Probes, Eugene, OR, USA) for 20 min, washed twice with PBS, fixed,
and permeabilized with Cytofix/Cytoperm solution (BD Biosciences)
for 20 min at 4°C. The cells were washed with PBS and blocked with
PBS containing 1% BSA for 20 min. They were then incubated with
primary antibodies (anti-p53 and anti-Bax) diluted in 1% BSA/PBS
for 1 h at RT. After washing with PBS 5 times, cells were incubated
with anti-rabbit conjugated Alexa 488 (Molecular Probes) for 1 h at
RT and stained with 1 μg/ml Hoechst dye (Molecular Probes)
for 10 min. After washing with PBS, the coverslips were mounted
using Vectashield HardSet (Vector Laboratories, Burlingame, CA,
USA) on glass slides and analyzed under a laser scanning microscope
(LSM 5; Carl Zeiss, Oberkochen, Germany).
Results
Statins induce apoptosis and ER stress
response by depletion of the isoprenyl products of the mevalonate
pathway
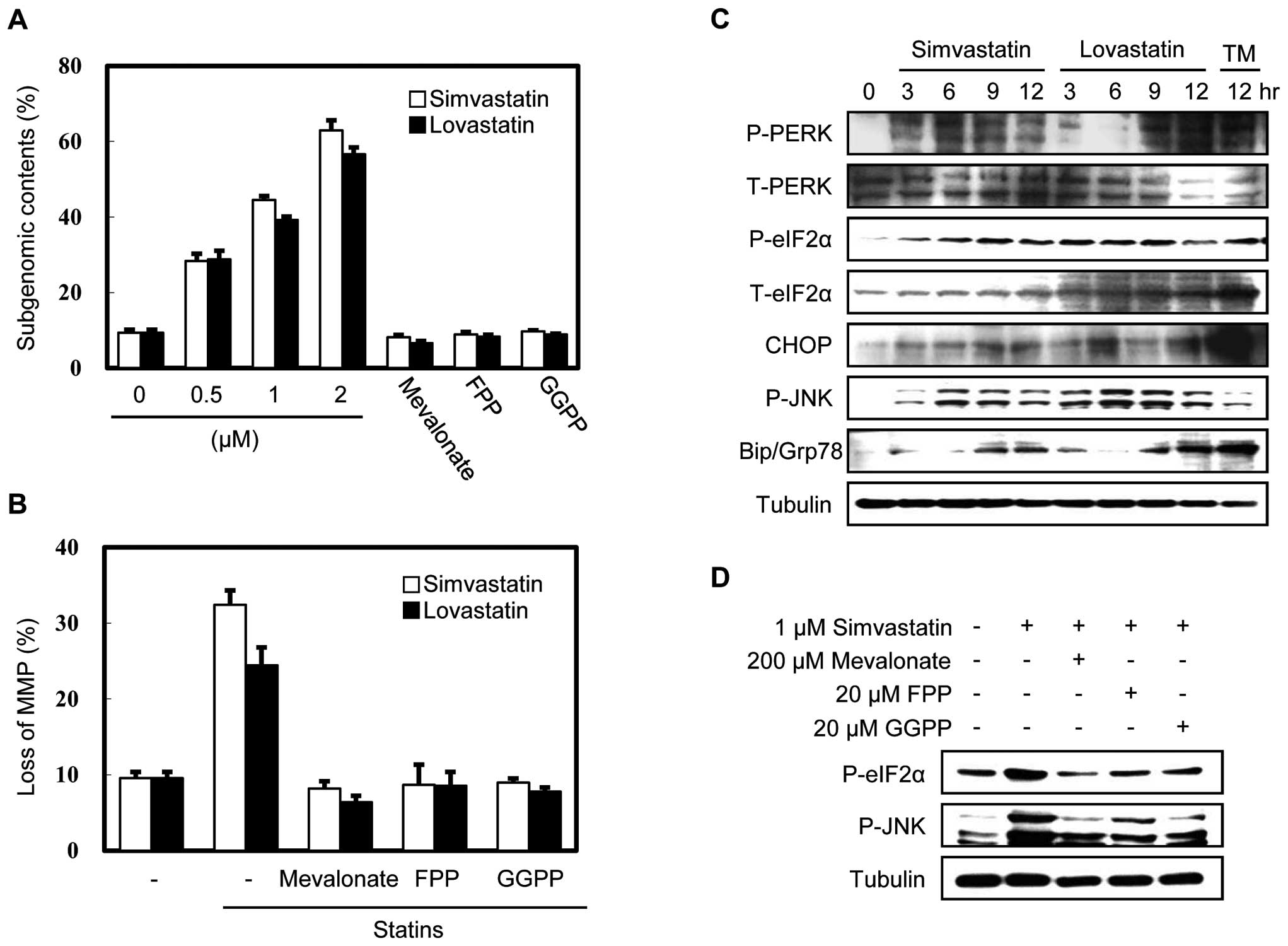
To evaluate the effect of statins on subgenomic
content and MMP, we treated MethA fibrosarcoma cells with
simvastatin (a natural statin) or lovastatin (a synthetic statin)
for 24 h and analyzed DNA fragmentation and changes in MMP. As
shown in Fig. 1, both simvastatin
and lovastatin induced DNA fragmentation in a dose-dependent manner
and disrupted MMP, while these effects were completely inhibited by
supplementation with downstream products of the mevalonate pathway,
such as mevalonate, FPP and GGPP. Supplementation with squalene,
the direct precursor of cholesterol, was ineffective, indicating
that the level of cholesterol was not critical for apoptosis (data
not shown), but that the key factor was the depletion of
isoprenoids of the mevalonate pathway. To investigate whether the
statin-induced apoptosis was related to ER stress, we examined
several cardinal indicators of ER stress in statin-treated MethA
cells by immunoblot analysis (Fig.
1C). We discovered that the CHOP and BiP/Grp78 protein
expression was induced and that the phosphorylation of PERK, eIF2α
and JNK was increased. These results indicate that statins induce a
general ER stress response during apoptosis.
To determine which downstream products of the
mevalonate pathway are critical for statin-induced ER stress, we
examined the effects of mevalonate, FPP and GGPP supplementation on
simvastatin-treated MethA cells. As shown in Fig. 1D, these downstream products of the
mevalonate pathway reduced the phosphorylation of eIF2α and JNK,
suggesting that the depletion of isoprenoids induced by statins
disrupts ER homeostasis.
Inhibition of eIF2α dephosphorylation
opposes statin-induced apoptosis
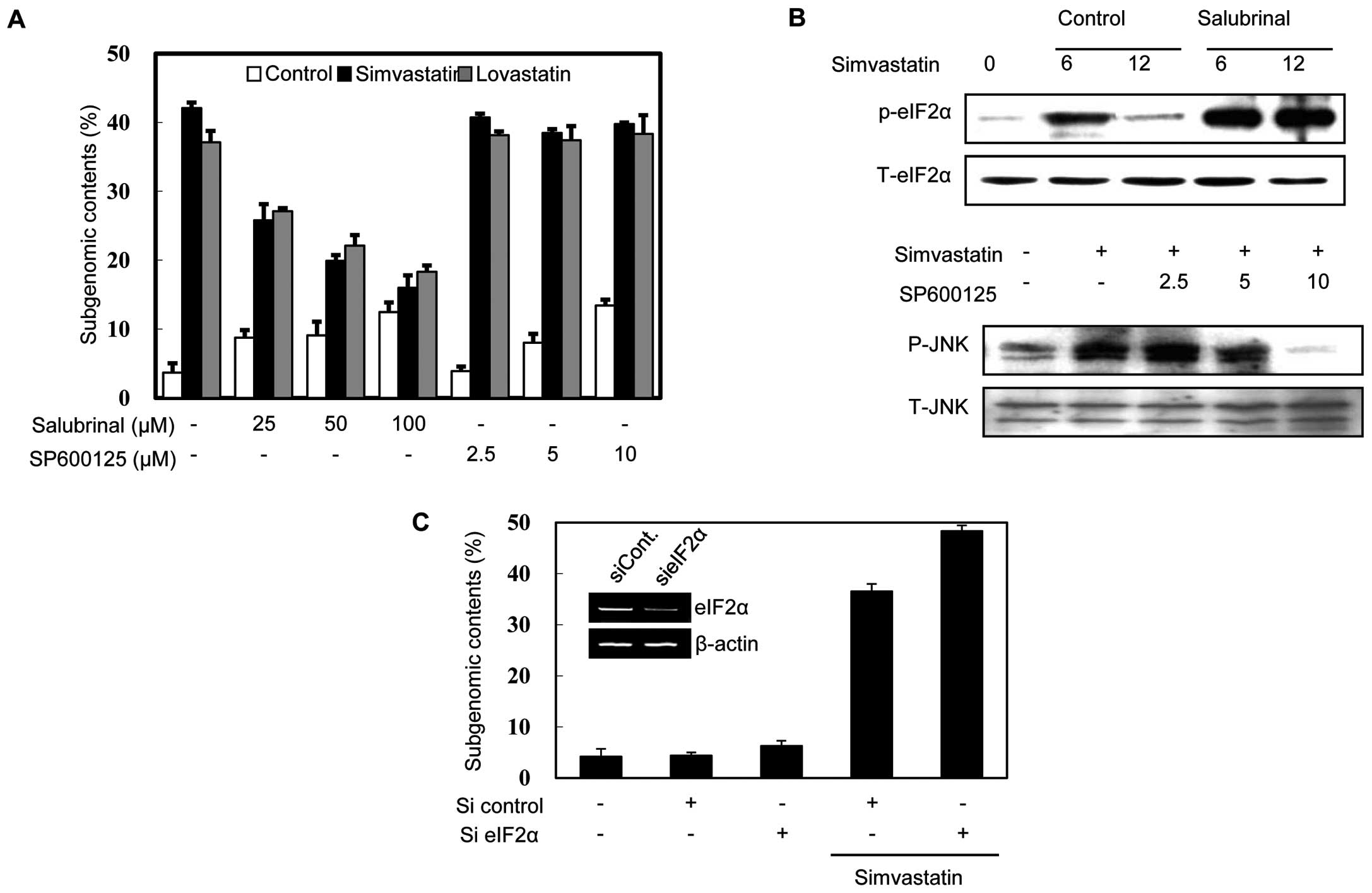
To investigate whether the phosphorylation of eIF2α
and JNK induced by statins is involved in apoptosis, we assessed
the effects of salubrinal (a selective inhibitor of eIF2α
dephosphorylation) and SP600125 (a specific inhibitor of JNK
kinase). Notably, salubrinal treatment increased the
phosphorylation of eIF2α and reduced the DNA fragmentation induced
by statins (Fig. 2A and B),
whereas SP600125 treatment had no effect, although it clearly
inhibited JNK phosphorylation (Fig. 2A
and B). This result suggests that the phosphorylation of eIF2α,
but not that of JNK, plays a role in statin-induced apoptosis. To
confirm this finding, we investigated the effect of the eIF2α
knockdown on DNA fragmentation. As shown Fig. 2C, the MethA cells in which eIF2α
had been knocked down were more sensitive to simvastatin-induced
apoptosis than the mock-transfected cells. Taken together, these
results support the notion that the phosphorylation of eIF2α
negatively modulates statin-induced apoptosis in MethA cells.
Inhibition of eIF2α dephosphorylation
decreases the stabilization of p53 and its translocation to the
mitochondria in statin-induced apoptosis
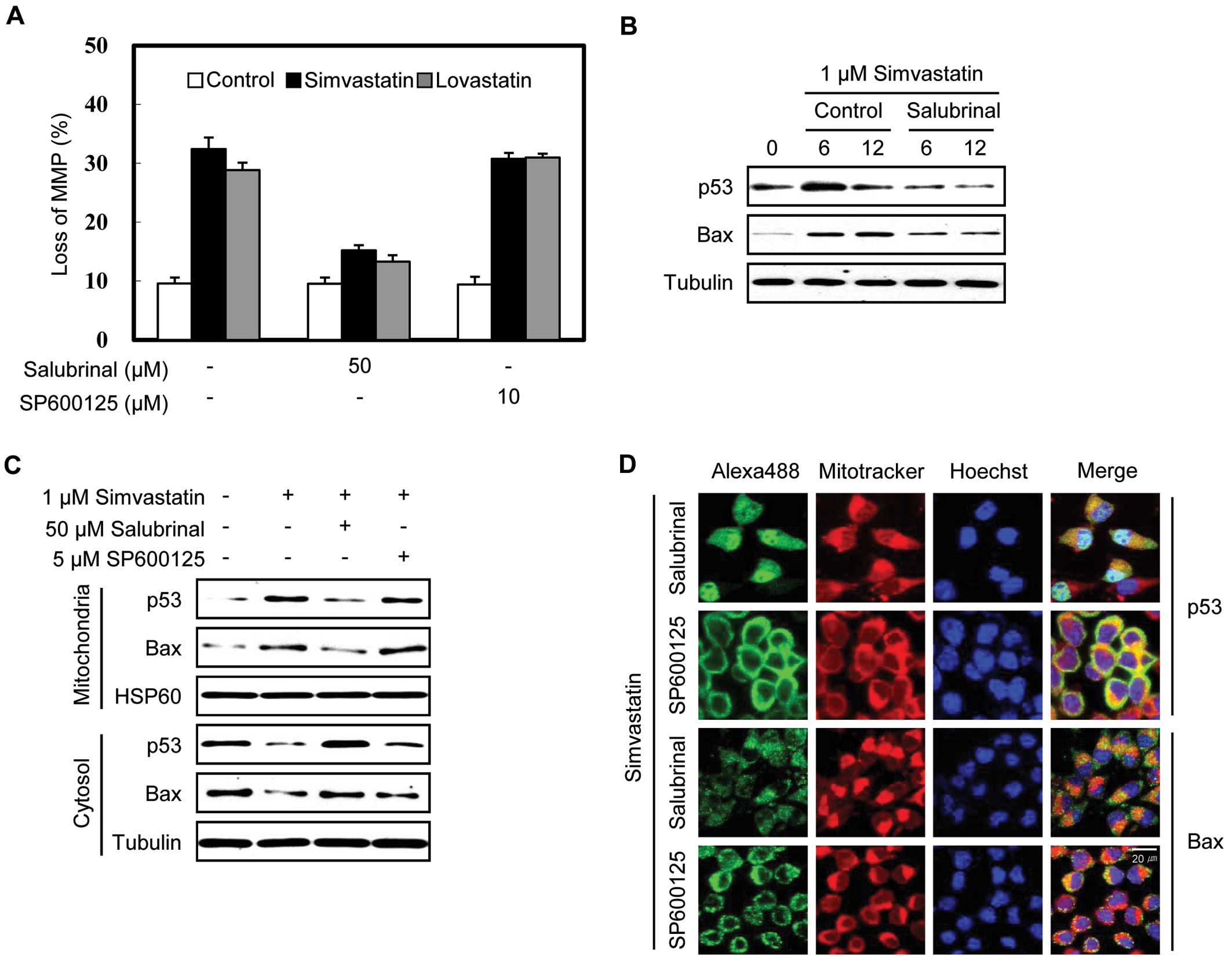
We previously reported that simvastatin activates
the mitochondrial apoptotic pathway in MethA cells accompanied by
the stabilization of the p53 protein and its translocation with Bax
to the mitochondria and that the knockdown of p53 expression
decreases apoptosis (9). To
determine whether the phosphorylation of eIF2α and JNK contributes
to the simvastatin-induced MMP loss and translocation of p53 to the
mitochondria, we treated the cells with salubrinal (an inhibitor of
the dephosphorylation of eIF2α) and SP600125 (a JNK kinase
inhibitor). Salubrinal treatment significantly reduced
statin-induced MMP loss (Fig. 3A)
and the stabilization of p53 and Bax by simvastatin in the MethA
cells (Fig. 3B), as well as the
simvastatin-induced translocation of p53 and Bax to the
mitochondria (Fig. 3C). By
contrast, SP600125 had no discernible effect. The inhibitory effect
of salubrinal on the simvastatin-induced translocation of p53 and
Bax to the mitochondria was also evident via immunofluorescence
staining (Fig. 3D). In the
presence of salubrinal, the p53 and Bax mitochondrial
co-localization was less apparent than in the presence of SP600125.
These results demonstrate that the phosphorylation of eIF2α
counteracts the p53-mediated mitochondrial apoptotic pathway in
simvastatin-induced apoptosis.
Phosphorylation of eIF2α on Ser51 is
responsible for the stabilization of p53 and its translocation to
the mitochondria
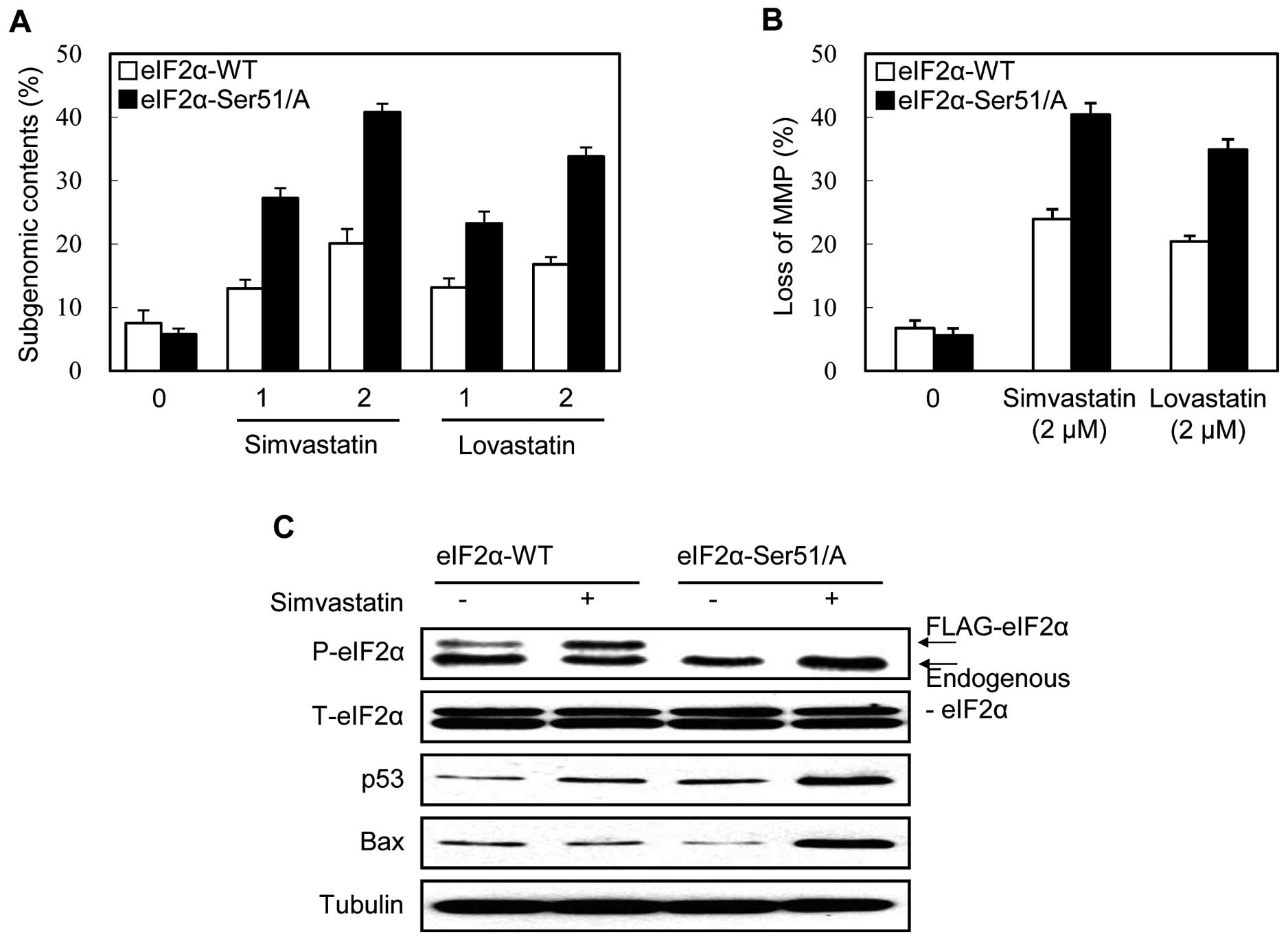
Several kinases, including PERK, phosphorylate eIF2α
on Ser51 in response to various ER stress inducers. To investigate
whether the phosphorylation of eIF2α on Ser51 is involved in the
statin-induced ER stress response and apoptosis, we examined the
effect of the overexpression of the non-phosphorylatable mutant
form of eIF2α (Ser51/Ala) on simvastatin-induced apoptosis. The
overexpression of the mutant eIF2α (Ser51/Ala) attenuates the eIF2α
phosphorylation pathway (10). The
flag-tagged wild-type eIF2α-transfected clone was used as the
control. Both the DNA fragmentation and MMP loss induced by
simvastatin were enhanced in the eIF2α mutant-expressing clone
compared to the wild-type-expressing clone (Fig. 4A and B). The effect of the
overexpression of the eIF2α mutant on p53 and Bax protein in
response to simvastatin was also analyzed by immunoblot analysis.
As shown in Fig. 4C, simvastatin
treatment induced the phosphorylation of the flag-tagged eIF2α
wild-type protein, but not that of the flag-tagged mutant protein.
At the same time, simvastatin effectively stabilized p53 and Bax
proteins in the flag-tagged eIF2α mutant clone compared with the
flag-tagged eIF2α wild-type clone (Fig. 4C). These results indicate that the
phosphorylation of eIF2α on Ser51 is responsible for the cell
survival effect in the p53-mediated mitochondrial apoptosis of
statin-treated MethA cells.
Discussion
Under stress conditions, p53 is stabilized and acts
as a transcription factor for the expression of pro-apoptotic
target genes, such as Puma, Noxa, Bax and Bid (11). In addition, cytoplasmic p53
directly activates the mitochondrial apoptotic pathway in a
transcription-independent manner; i.e., p53 interacts with the
Bcl-2 family members, Bcl-2 or Bcl-xL, leading to the translocation
of Bax and Bid to the mitochondrial outer membrane (12). We previously reported that in
response to statin, p53 itself is stabilized and translocated along
with Bax to the mitochondria, thus activating the mitochondrial
apoptotic pathway (9). In the
present study, we demonstrate that statins induce the ER stress
response in MethA cells by depleting the isoprenyl products of the
mevalonate pathway and that the phosphorylation of eIF2α on Ser51
plays a role in promoting cell survival. To our knowledge, this is
the first study to demonstrate that the phosphorylation of eIF2α
counteracts the stabilization of p53 and its translocation to the
mitochondria in statin-induced apoptosis.
Statins exert stress on cancer cells and display
various cardinal features of ER stress response, which are pro- or
anti-apoptotic. Lovastatin has been shown to induce apoptosis
accompanied by reduced global protein translation by inducing eIF2α
phosphorylation. It has also been shown to induce general control
non-repressed 2 (GCN2)-mediated activating transcription factor
(ATF)4 production followed by the increased expression of ATF3 and
CHOP in a head and neck squamous cell carcinomas cell line
(6). Lovastatin-induced apoptosis
was attenuated in CHOP−/− and GCN2−/− murine
embryonic fibroblasts (MEFs), thus demonstrating the involvement of
ATF4, CHOP and ATF3 in apoptosis. In multiple myeloma cells,
lovastatin has been shown to induce BiP/Grp78 and CHOP protein
expression, the phosphorylation of eIF2α and apoptosis, as well as
the cleavage of poly(ADP-ribose) polymerase (PARP) (13). In contrast to these apoptotic
effects of statin-induced ER stress, numerous studies have
demonstrated that the ER stress response also attenuates the
apoptotic response. Fluvastatin has been shown to induce BiP/Grp78
expression and to activate ATF6 and X-box binding protein-1
(XBP-1), but not CHOP and ATF4 in RAW264.7 cells (5). Among the induced proteins, the
induction of BiP/ Grp78 is responsible for the cytoprotective
effect of fluvastatin pre-treatment against hypoxia-induced cell
death. Simvastatin pre-treatment also exerted a neuroprotective
effect by attenuating the ER stress response, with concomitant
increases in ATF6 and XBP-1 protein expression during acute
ischemia and reperfusion in rats (14). Recently, a novel protective effect
of statins against atherosclerosis was proposed based on the
finding that stearic acid-induced ER stress in macrophages was
attenuated by statins (15). In
sum, statins seem to exert differential ER stress responses
depending on the strength, nature and duration of the stress.
However, we observed that statins induce the majority of indicators
of the ER stress response in MethA cells: the early phosphorylation
of PERK, eIF2α and JNK and the induction of CHOP and BiP/Grp78
protein expression.
The induction of CHOP and JNK phosphorylation
suggests that, under our experimental conditions, ER stress in
response to statins triggers apoptotic signals. The MethA clone in
which CHOP was knocked down by siRNA exhibited increased resistance
to statin-induced apoptosis, pointing to a pro-apoptotic role of
CHOP (data not shown). However, the JNK inhibitor, SP600125 did not
exert any effect on statin-induced apoptosis in our study (Figs. 2 and 3). The JNK kinase pathway is activated
during lovastatin-induced apoptosis in the NB4 acute promyelocytic
leukemia cell line (16) and human
breast cancer cells (17). By
contrast, the JNK pathway is downregulated (18) or not affected (19) in statin-induced apoptosis. Further
study is required to elucidate the role of JNK phosphorylation in
statin-induced ER stress.
Salubrinal, an inhibitor of eIF2α dephosphorylation,
clearly attenuated statin-induced apoptosis, suggesting that the
phosphorylation of eIF2α plays a role in triggering cell survival
signals. Previous studies have also demonstrated that
phosphorylation of eIF2α protected cells under glucose deprivation
stress (20,21), and other ER stress (22). When global translation is inhibited
by the phosphorylation of eIF2α, the translation of certain mRNAs,
such as GCN2, ATF4 and X-linked inhibitor of apoptosis protein
(XIAP), is increased and this promotes tumor cell survival and
chemoresistance (23,24). In renal medullary cells exposed to
urea stress, the phosphorylation of eIF2α by activated GCN2 also
exerts cytoprotective effects (25). On the other hand, there have been
several reports demonstrating that the phosphorylation of eIF2α is
involved in apoptosis under various stress conditions, such as
proteasome inhibition (26,27),
hypoxia (28) and tunicamycin
(10). Moreover, salubrinal
enhances cisplatin-induced nephrotoxicity (29). Depending on the nature and strength
of the stress, the phosphorylation of eIF2α seems to shift from its
primary role of cytoprotection to apoptosis induction (4). More importantly, further studies are
required to elucidate the signaling flows from the phosphorylation
of eIF2α to the reduced stability of p53 in response to statins.
Unveiling the missing link between p53 stabilization and eIF2α
phosphorylation may contribute to the expansion of therapeutic
approaches against hypercholesterolemia and cancer.
Acknowledgements
This study was supported by a grant
from the Korea Healthcare Technology R&D project, Ministry and
Welfare, Republic of Korea (A084773).
References
|
1
|
Demierre MF, Higgins PD, Gruber SB, Hawk E
and Lippman SM: Statins and cancer prevention. Nat Rev Cancer.
5:930–942. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Harding HP, Zhang Y and Ron D: Protein
translation and folding are coupled by an
endoplasmic-reticulum-resident kinase. Nature. 397:271–274. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hampton RY: ER-associated degradation in
protein quality control and cellular regulation. Curr Opin Cell
Biol. 14:476–482. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Holcik M and Sonenberg N: Translational
control in stress and apoptosis. Nat Rev Mol Cell Biol. 6:318–327.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen JC, Wu ML, Huang KC and Lin WW:
HMG-CoA reductase inhibitors activate the unfolded protein response
and induce cytoprotective GRP78 expression. Cardiovasc Res.
80:138–150. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Niknejad N, Morley M and Dimitroulakos J:
Activation of the integrated stress response regulates
lovastatin-induced apoptosis. J Biol Chem. 282:29748–29756. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li J, Lee B and Lee AS: Endoplasmic
reticulum stress-induced apoptosis: multiple pathways and
activation of p53-up-regulated modulator of apoptosis (PUMA) and
NOXA by p53. J Biol Chem. 281:7260–7270. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vaseva AV and Moll UM: The mitochondrial
p53 pathway. Biochim Biophys Acta. 1787:414–420. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee SK, Kim YC, Song SB and Kim YS:
Stabilization and translocation of p53 to mitochondria is linked to
Bax translocation to mitochondria in simvastatin-induced apoptosis.
Biochem Biophys Res Commun. 391:1592–1597. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fritsch RM, Schneider G, Saur D, Scheibel
M and Schmid RM: Translational repression of MCL-1 couples
stress-induced eIF2 alpha phosphorylation to mitochondrial
apoptosis initiation. J Biol Chem. 282:22551–22562. 2007.
View Article : Google Scholar
|
|
11
|
Vousden KH and Lu X: Live or let die: the
cell’s response to p53. Nat Rev Cancer. 2:594–604. 2002.
|
|
12
|
Chipuk JE and Green DR: Dissecting
p53-dependent apoptosis. Cell Death Differ. 13:994–1002. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Holstein SA and Hohl RJ: Isoprenoid
biosynthetic pathway inhibition disrupts monoclonal protein
secretion and induces the unfolded protein response pathway in
multiple myeloma cells. Leuk Res. 35:551–559. 2011. View Article : Google Scholar
|
|
14
|
Urban P, Pavliková M, Sivonová M, et al:
Molecular analysis of endoplasmic reticulum stress response after
global forebrain ischemia/reperfusion in rats: effect of
neuroprotectant simvastatin. Cell Mol Neurobiol. 29:181–192. 2009.
View Article : Google Scholar
|
|
15
|
Breder I, Coope A, Arruda AP, et al:
Reduction of endoplasmic reticulum stress - a novel mechanism of
action of statins in the protection against atherosclerosis.
Atherosclerosis. 212:30–31. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sassano A, Katsoulidis E, Antico G, et al:
Suppressive effects of statins on acute promyelocytic leukemia
cells. Cancer Res. 67:4524–4532. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Koyuturk M, Ersoz M and Altiok N:
Simvastatin induces apoptosis in human breast cancer cells: p53 and
estrogen receptor independent pathway requiring signalling through
JNK. Cancer Lett. 250:220–228. 2007. View Article : Google Scholar
|
|
18
|
Campbell MJ, Esserman LJ, Zhou Y, et al:
Breast cancer growth prevention by statins. Cancer Res.
66:8707–8714. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu J, Wong WW, Khosravi F, Minden MD and
Penn LZ: Blocking the Raf/MEK/ERK pathway sensitizes acute
myelogenous leukemia cells to lovastatin-induced apoptosis. Cancer
Res. 64:6461–6468. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Scheuner D, Song B, McEwen E, et al:
Translational control is required for the unfolded protein response
and in vivo glucose homeostasis. Mol Cell. 7:1165–1176. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Muaddi H, Majumder M, Peidis P, et al:
Phosphorylation of eIF2α at serine 51 is an important determinant
of cell survival and adaptation to glucose deficiency. Mol Biol
Cell. 21:3220–3231. 2010.
|
|
22
|
Boyce M, Bryant KF, Jousse C, et al: A
selective inhibitor of eIF2alpha dephosphorylation protects cells
from ER stress. Science. 307:935–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Thakor N and Holcik M: IRES-mediated
translation of cellular messenger RNA operates in eIF2α-independent
manner during stress. Nucleic Acids Res. 40:541–552.
2011.PubMed/NCBI
|
|
24
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cai Q and Brooks HL: Phosphorylation of
eIF2α via the general control kinase, GCN2, modulates the ability
of renal medullary cells to survive high urea stress. Am J Physiol
Renal Physiol. 301:F1202–F1207. 2011.
|
|
26
|
Jiang HY and Wek RC: Phosphorylation of
the alpha-subunit of the eukaryotic initiation factor-2 (eIF2alpha)
reduces protein synthesis and enhances apoptosis in response to
proteasome inhibition. J Biol Chem. 280:14189–14202. 2005.
View Article : Google Scholar
|
|
27
|
Schewe DM and Aguirre-Ghiso JA: Inhibition
of eIF2alpha dephosphorylation maximizes bortezomib efficiency and
eliminates quiescent multiple myeloma cells surviving proteasome
inhibitor therapy. Cancer Res. 69:1545–1552. 2009. View Article : Google Scholar
|
|
28
|
Liu Y, László C, Liu W, Chen X, Evans SC
and Wu S: Regulation of G(1) arrest and apoptosis in hypoxia by
PERK and GCN2-mediated eIF2alpha phosphorylation. Neoplasia.
12:61–68. 2010.PubMed/NCBI
|
|
29
|
Sayan BS, Sayan AE, Knight RA, Melino G
and Cohen GM: p53 is cleaved by caspases generating fragments
localizing to mitochondria. J Biol Chem. 281:13566–13573. 2006.
View Article : Google Scholar : PubMed/NCBI
|