Introduction
A growing body of evidence indicates the existence
of an intimate link between the metabolic status and epigenetic
regulation of cells (1,2). This is exemplified by the fact that a
variety of small molecules involved in intercellular metabolism,
including adenosine triphosphate, S-adenosylmethionine,
nicotinamide adenine dinucleotide, flavin adenine dinucleotide,
folate, acetyl coenzyme A, α-ketoglutarate and iron, are essential
for the proper maintenance of the cellular epigenome.
Iron is an essential trace element for normal
cellular function. In addition to its significance in controlling a
variety of cellular processes, including proliferation, DNA
synthesis and repair, and mitochondrial electron transport, which
are essential for the accurate maintenance of normal cellular
homeostasis, iron plays a key regulatory role in the functioning of
DNA and histone-modifying proteins (3,4).
Specifically, the Jumonji domain-containing histone demethylase
(JHDM) family, which catalyzes the demethylation of tri- and
dimethylated lysine 9 and lysine 36 residues in histone H3
(5,6) and ten-eleven translocation 1-3
(TET1-3; TET methylcytosine dioxygenase 1–3), proteins that
catalyze the hydroxylation of 5-methylcytosine to form
5-hydroxymethylcytosine (7), are
members of the superfamily of α-ketoglutarate-non-heme
Fe+2-dependent oxygenases (8). This provides a direct link between
the regulation of epigenetic mechanisms and the status of cellular
iron metabolism. Both of these processes are well-balanced and
tightly controlled in normal cells; however, in cancer cells these
processes are profoundly disturbed (9,10).
For instance, it is well-established that changes in cellular iron
metabolism play a crucial role in the progression of many types of
cancer (9), including breast
cancer (11), suggesting that
deregulated iron metabolism in malignant cells may be a promising
molecular target for cancer therapy. Similarly, the dysregulation
of epigenetic mechanisms is regarded as one of the hallmarks of
cancer (12,13) and correcting the enzymatic
processes that control the epigenome has emerged as a novel
epigenetic approach for the treatment of cancer (13).
It has previously been reported that agents that
modulate iron metabolism in cancer cells, particularly those that
restrict iron availability, including iron chelating agents
(14,15) and transferrin receptor-targeted
treatments (16), exhibit potent
and broad anti-tumor activity. Several studies have demonstrated
that the mechanism of the anti-tumor action of iron chelators is
linked to the inhibition of DNA synthesis, the induction of DNA
damage, G1-S phase cell cycle arrest, and the activation
of apoptosis (15,17); however, research on targeting iron
metabolism in cancer cells for anti-tumor therapy is still in its
infancy (15).
Based on these considerations, we hypothesized that,
in addition to the well-studied molecular mechanisms for the action
of iron chelators, their anti-tumor activity may be associated with
modulating the function of chromatin-modifying proteins and their
corresponding metabolically sensitive epigenetic modifications in
cancer cells. The results of the present study demonstrate that the
treatment of wild-type TP53 MCF-7 and mutant TP53
MDA-MB-231 human breast cancer cells with desferrioxamine (DFO), a
model iron chelator, causes significant epigenetic alterations at
the global and gene-specific levels and that these alterations
activate cellular apoptotic programs and enhance the sensitivity of
cancer cells to the chemotherapeutic agents, doxorubicin and
cisplatin.
Materials and methods
Cell lines, cell culture and
treatment
MCF-7 (wild-type TP53) and MDA-MB-231 (mutant
TP53) human breast cancer cell lines were obtained from the
American Type Culture Collection (ATCC, Manassas, VA) and
maintained according to the manufacturer’s recommendations. DFO
mesylate (DFO) was purchased from Sigma-Aldrich (St. Louis, MO). In
the experiments with DFO, cells were seeded at a density of
0.5×106 viable cells per 100-mm plate. Twenty-four hours
after seeding, the medium was changed and fresh medium containing
100 μM of DFO and supplemented with 10% fetal bovine serum
was added. At 6, 24 and 48 h after the addition of DFO, the cells
were scraped on ice, washed in phosphate-buffered saline, and
immediately frozen at −80°C for subsequent analyses.
Western blot analysis of proteins
Whole cell lysates were prepared by homogenization
in 200 μl of lysis buffer (50 mM Tris-HCl, pH 7.4; 1% NP-40;
0.25% sodium deoxycholate; 150 mM NaCl; 1 mM EDTA; 1 mM PMSF; 1
μg/ml each of aprotinin, leupeptin and pepstatin; 1 mM
Na3VO4, and 1 mM NaF), sonication and
incubation at 4°C for 30 min, followed by centrifugation at 12,000
× g at 4°C for 10 min. Extracts containing equal quantities of
proteins were separated by SDS-PAGE on 8%, 10%, or 15%
polyacrylamide gels and transferred onto PVDF membranes. The
membranes were probed with primary antibodies against Jumonji
domain-containing protein 2A (JMJD2A; 1:1,000; Sigma-Aldrich), TET2
(1:1,000; Abcam, Cambridge, MA), histone demethylase SWIRM1
(lysine-specific demethylase LSD1; 1:1,000; Sigma-Aldrich),
cyclin-dependent kinase inhibitor 1A (p21; Cip1; Cell Signaling
Technology, Danvers, MA), p53 (1:1,000; Cell Signaling Technology)
and hypoxia-inducible factor 1α (HIF1α; 1:200; Santa Cruz
Biotechnology, Santa Cruz, CA). Alkaline phosphatase-conjugated
secondary antibodies (EMD Millipore, Billerica, MA) were used for
visualization. Equal protein loading was confirmed by
immunostaining against β-actin (1:4,000; Sigma-Aldrich). The signal
intensity was analyzed using ImageQuant software (Molecular
Dynamics, Sunnyvale, CA) and norma lized to β-actin.
Jumonji-type and LSD-type histone
demethylase activity assay
The activity of Jumonji-type and LSD-type histone
demethylases was determined using Demethylase Activity Assay kits
(Cayman Chemical Co., Ann Arbor, MI) according to the
manufacturer’s instructions.
Western blot analysis of histone
modifications
The histone modification status in the untreated
MCF-7 and MDA-MB-231 cells and cells treated with DFO was
determined by western blot analysis using the following primary
antibodies: anti-trimethyl histone H3 lysine 9 (H3K9me3; 1:1,000),
anti-dimethyl histone H3 lysine 9 (H3K9me2; 1:1,000), anti-acetyl
histone H3 lysine 9 (H3K9ac; 1:1,000), anti-trimethyl histone H3
lysine 27 (H3K27me3; 1:1,000), anti-trimethyl histone H3 lysine 36
(H3K36me3; 1:1,000), anti-trimethyl histone H3lysine 4 (H3K4me3;
1:1,000), anti-dimethyl histone H3 lysine 4 (H3K4me2; 1:1,000) and
anti-trimethyl histone H4 lysine 20 (H4K20me3; 1:1,000) as
previously described (18). All
antibodies were obtained from EMD Millipore.
Quantitative reverse transcription
real-time PCR (qRT-PCR)
Total RNA was extracted from the breast cancer cell
lines using TRI Reagent (Ambion, Austin, TX) according to the
manufacturer’s instructions. Reverse transcription was performed
using High Capacity cDNA Reverse Transcription kits (Applied
Biosystems, Foster City, CA) and cDNA was analyzed in a 96-well
plate assay format using a 7900HT Fast Real-Time PCR System
(Applied Biosystems). Each plate contained one experimental gene
and one housekeeping gene. All primers were obtained from Applied
Biosystems. The relative mRNA level for each gene was determined
using the 2−ΔΔCt method (19). The results are presented as a fold
change for each mRNA in the DFO-treated cells relative to the
untreated control cells.
Determination of global DNA
methylation
The extent of global DNA methylation was determined
by slight modification of the liquid chromatography-tandem mass
spectrometry (LC-MS/MS) method (20). The analyses were conducted in a
system comprising a Waters Acquity UltraPerformance Liquid
Chromatograph (UPLC; Waters Corporation, Milford, MA), coupled with
a Waters Quattro Premier XE triple quadrupole mass spectrometer
operating in positive ion electrospray mode. DNA samples (2
μg) dissolved in 20 μl of Tris-EDTA buffer, pH 8.0,
were added to 180 μl of formic acid (98%, Sigma-Aldrich) and
to each sample were added 275 ng of
cytosine-2,4-13C2,15N3
(Sigma-Aldrich) and 20 ng of
5-methyl-d3-cytosine-6-d1
(5-MeC-d4; C/D/N Isotopes, Pointe-Claire, QC,
Canada). The mixtures were sealed in 2-ml auto sampler vials and
incubated in a thermostated heating block at 140°C for 90 min.
After cooling to the room temperature the vials were opened, dried
in a centrifugal evaporator, and the contents were dissolved in 1
ml of 95% acetonitrile/5% water in a sonicator bath for 15 min.
Each sample (5 μl) was injected in the UPLC system and
eluted in a Waters UPLC BEH HILIC column (1.7 μm, 2.1 mm ×
100 mm) using an isocratic elution with 93% of acetonitrile and 7%
of 2.5 mM ammonium formate at 200 μl/min. Under these
chromatographic conditions, cytosine eluted at ca. 4.75 min
and 5-methylcytosine (5-mC) eluted at ca. 5.49 min. The mass
spectral acquisition was conducted using multiple reaction
monitoring as follows: cytosine, m/z 112.1→95.1;
13C15N-cytosine, m/z 117.1→72.2; 5-mC, m/z
126.1→109.1; and 5-mC-d4, m/z 130.2→113.2. A plot
of the response ratio for labeled versus unlabeled cytosine was
linear (r2>0.999) over a concentration range of 0–700
ng/ml cytosine with 275 ng/ml
13C15N-cytosine. A plot of the response ratio
for labeled versus unlabeled 5-mC was linear
(r2>0.999) over a concentration range of 0–80 ng/ml
5-mC with 20 ng/ml 5-mC-d4. The percentage of
methylation was calculated as the quotient between the number of
moles of 5-mC and the sum of the number of moles of 5-mC and
cytosine in each sample. The methodology was validated as regards
its accuracy and precision by analyzing, on 2 consecutive days, 5
samples of salmon testes DNA (Sigma-Aldrich) and 5 samples of
salmon testes DNA spiked with 5-mC. The intra- and inter-day
imprecision of the method, assessed by the relative standard
deviation of the replicate analyses conducted on each day was under
2.1%. The intra- and inter-day inaccuracy of the method assessed in
the analysis of the spiked versus unspiked samples ranged from
0.4–2.0%.
Determination of CpG island methylation
status by cytosine extension assay
The status of CpG island methylation evaluated with
a radiolabeled [3H]dCTP extension assay (21) following digestion of DNA with
methylation-sensitive TspMI restriction endonuclease, whose
CCCGGG hexanucleotide recognition sequences occur predominantly
within CpG islands.
Chromatin immunoprecipitation assay
Formaldehyde cross-linking and chromatin
immunoprecipitation (ChIP) assays were performed with primary
antibodies against histone H3K9me2 (EMD Millipore) and H3K4me2 (EMD
Millipore) using a MAGnify Chromatin Immunoprecipitation System
(Invitrogen, Carlsbad, CA). Purified DNA from the
immuno-precipitates and from input DNA was analyzed by quantitative
real-time PCR (qPCR) on an Applied Biosystems 7900HT Fast Real-Time
PCR System using the following primer set:
5′-GTGGCTCTGATTGGCTTTCTG-3′ (forward) and 5′-CCA
GCCCTGTCGCAAGGATC-3′ (reverse) for the promoter of the human
p21 gene (22). The results
were norma lized to the amount of input DNA and presented as the
fold change in the amount of immunoprecipitated DNA isolated from
the DFO-treated cells relative to the untreated control cells.
Drug sensitivity assay
To assay drug sensitivity, MCF-7 and MDA-MB-231
cells were plated at a density of 5×103 cells per well
in 96-well plates. Cells were cultured in medium containing 100
μM of DFO. After 48 h of incubation, the medium was changed,
and the cells were treated with doxorubicin hydrochloride (DOX) or
cis-diammineplatinum(II) dichloride (CDDP) purchased from
Sigma-Aldrich. Cell survival was analyzed with a
CellTiter-Blue® Cell Viability assay (Promega, Madison,
WI). The IC50 (inhibitory concentration to produce 50%
cell death) values were determined using the resulting
dose-response curves. The experiments were repeated twice, and each
cell line examined in triplicate.
Statistical analyses
The results are presented as the means ± SD.
Statistical analyses were conducted by one-way analysis of
variance, with pair-wise comparisons conducted using the
Student-Newman-Keuls test.
Results
DFO affects the protein level of histone
demethylases
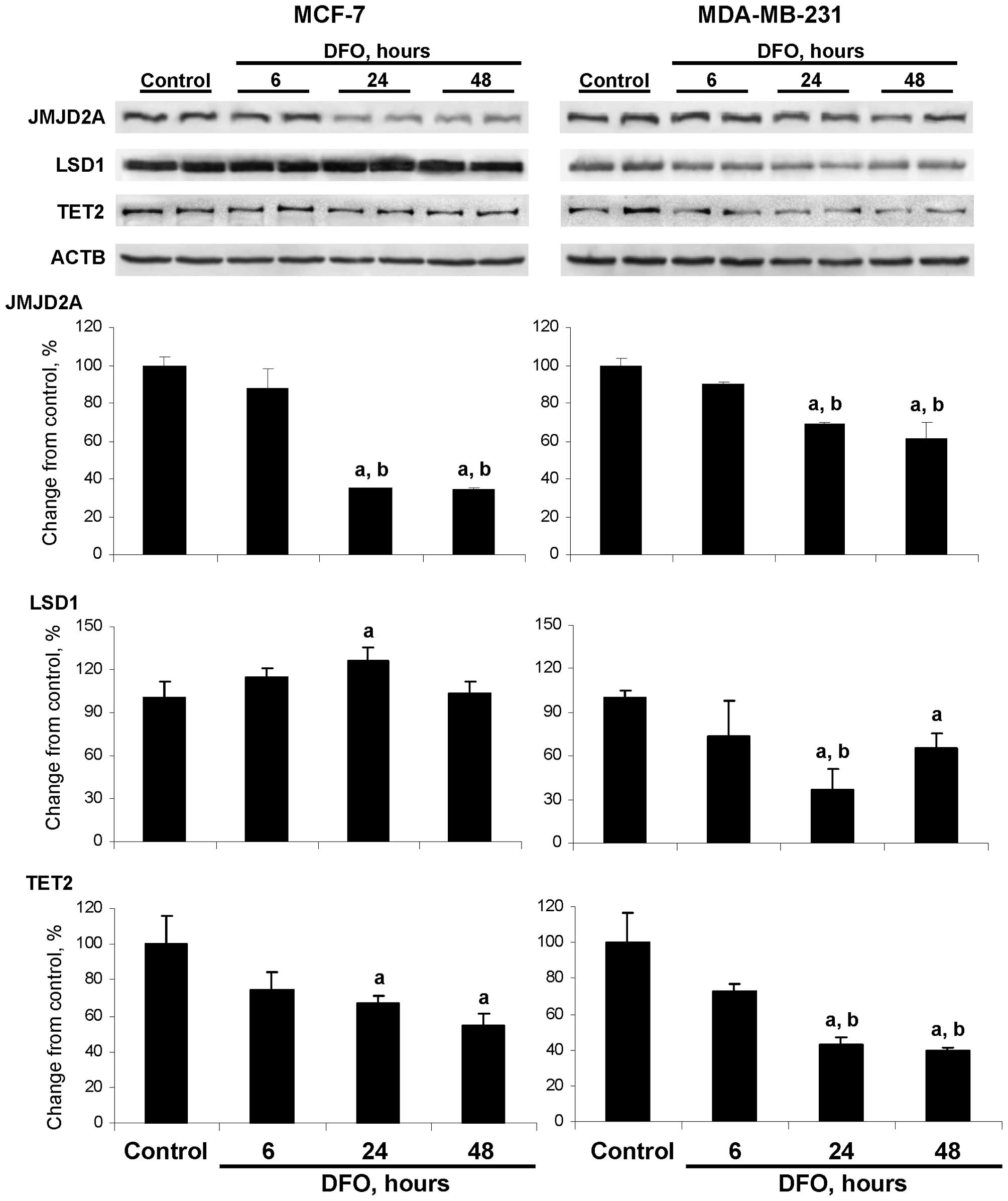
Fig. 1 demonstrates
that the protein level of JMJD2A was significantly decreased in the
MCF-7 and MDA-MB-231 cells cultured for 24 and 48 h in medium
containing DFO, with the changes being more pronounced in the
DFO-treated MCF-7 cells. This was evident by the fact that the
level of JMJD2A protein in the MCF-7 cells was decreased by 65 and
66% at 24 and 48 h, respectively after the initiation of DFO
treatment, whereas the protein level of JMJD2A in the DFO-treated
MDA-MB-231 cells was reduced only by 31 and 39%, respectively.
Culturing MDA-MB-231 cells in the DFO-containing
medium for 24 and 48 h resulted in a significant reduction in LSD1
protein levels by 63 and 40%, respectively. By contrast, the
protein level of LSD1 in the DFO-treated MCF-7 cells did not differ
from the values in the MCF-7 untreated control cells, apart from a
slight transient increase observed after 24 h of DFO treatment. The
respective changes in the protein levels of JMJD2A and LSD1 in the
DFO-treated MCF-7 and MDA-MB-231 cells were accompanied by
decreases in their enzymatic activity (data not shown).
The levels of TET2 protein in the MCF-7 and
MDA-MB-231 cells cultured in DFO-containing medium decreased, with
the values being significant at 24 and 48 h; however, the reduction
in TET2 protein levels in the DFO-treated MDA-MB-231 cells was more
pronounced compared to the MCF-7 cells at the same time points
(Fig. 1).
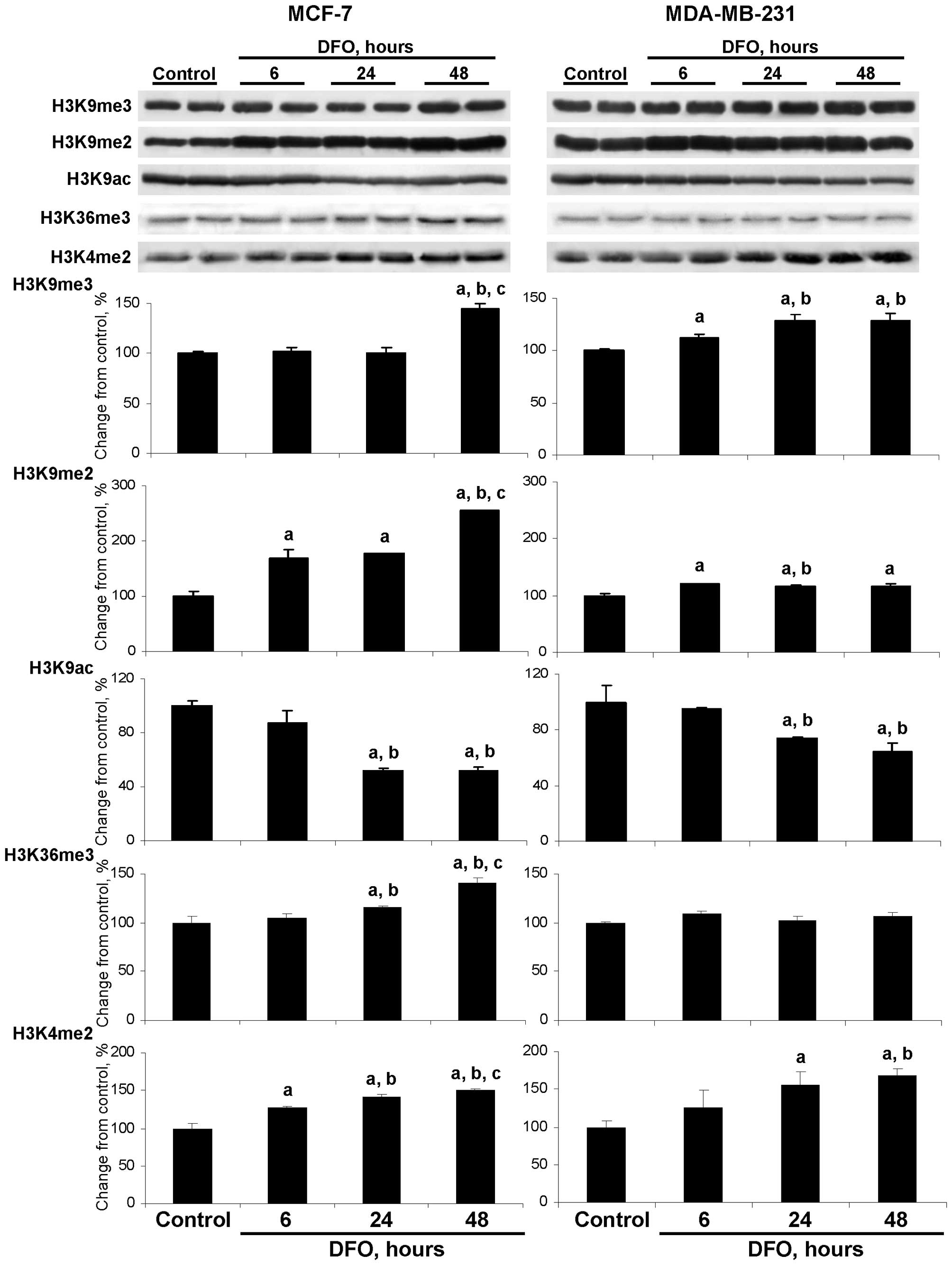
DFO affects the extent of global histone
modification pattern
The down-regulation of JMJD2A in the DFO-treated
MCF-7 cells and JMJD2A and LSD1 in the DFO-treated MDA-MB-231 cells
was accompanied by changes in the triand dimethylation of histone
H3K9, the acetylation of histone H3K9, the trimethylation of
histone H3K36 and the dimethylation of histone H3K4 (Fig. 2). Specifically, the level of
histone H3K9me3 was slightly although significantly increased in
the DFO-treated MDA-MB-231 cells at 6, 24 and 48 h, while in the
MCF-7 cells only at 48 h.
Culturing the MCF-7 and MDA-MB-231 cells in
DFO-containing medium resulted in an increase in histone H3K9me2
levels, particularly in MCF-7 cells. This was evident by a marked
time-dependent elevation of histone H3K9me2 in the DFO-treated
MCF-7 cells, with the highest values observed at 48 h. At this time
point, the level of histone H3K9me2 was 254% greater than in the
untreated MCF-7 control cells and significantly greater than the
values at 6 and 24 h in the DFO-treated MCF-7 cells (Fig. 2). In the DFO-treated MDA-MB-231
cells, the levels of histone H3K4me2 were only slightly increased
compared to the untreated MDA-MB-231 control cells and were
relatively constant at each time point.
The levels of histone H3K9ac were decreased in the
DFO-treated MCF-7 and MDA-MB-231 cells at 24 and 48 h; however, the
extent of histone H3K9ac reduction was greater in the MCF-7 cells.
This corresponds to the well-established inverse association
between histone H3K9 methylation and histone H3K9 acetylation
(23).
The pattern of changes in the levels of histone
H3K36me3 in the DFO-treated MCF-7 cells was similar to the
alterations in the levels of histone H3K9me2 in these cells, which
may be attributed to the fact that JMJD2A is responsible for the
demethylation of both of these histone modification markers
(5,6). By contrast, the level of histone
H3K36me3 in the DFO-treated MDA-MB-231 cells did not differ from
the values in the untreated cells.
Culturing the MCF-7 and MDA-MB-231 cells in
DFO-containing medium resulted in an increase in histone H3K4me2
levels in both cell lines. By contrast, the levels of histone
H3K4me3 did not change (data not shown), which may be explained by
the fact that LSD1 demethylates only dimethylated histone H3 lysine
4 residues (24).
Additionally, the levels of histone H3K27me3 and
histone H4K20me3 in the DFO-treated MCF-7 and MDA-MB-231 cells did
not differ from the values in the respective untreated cells (data
not shown).
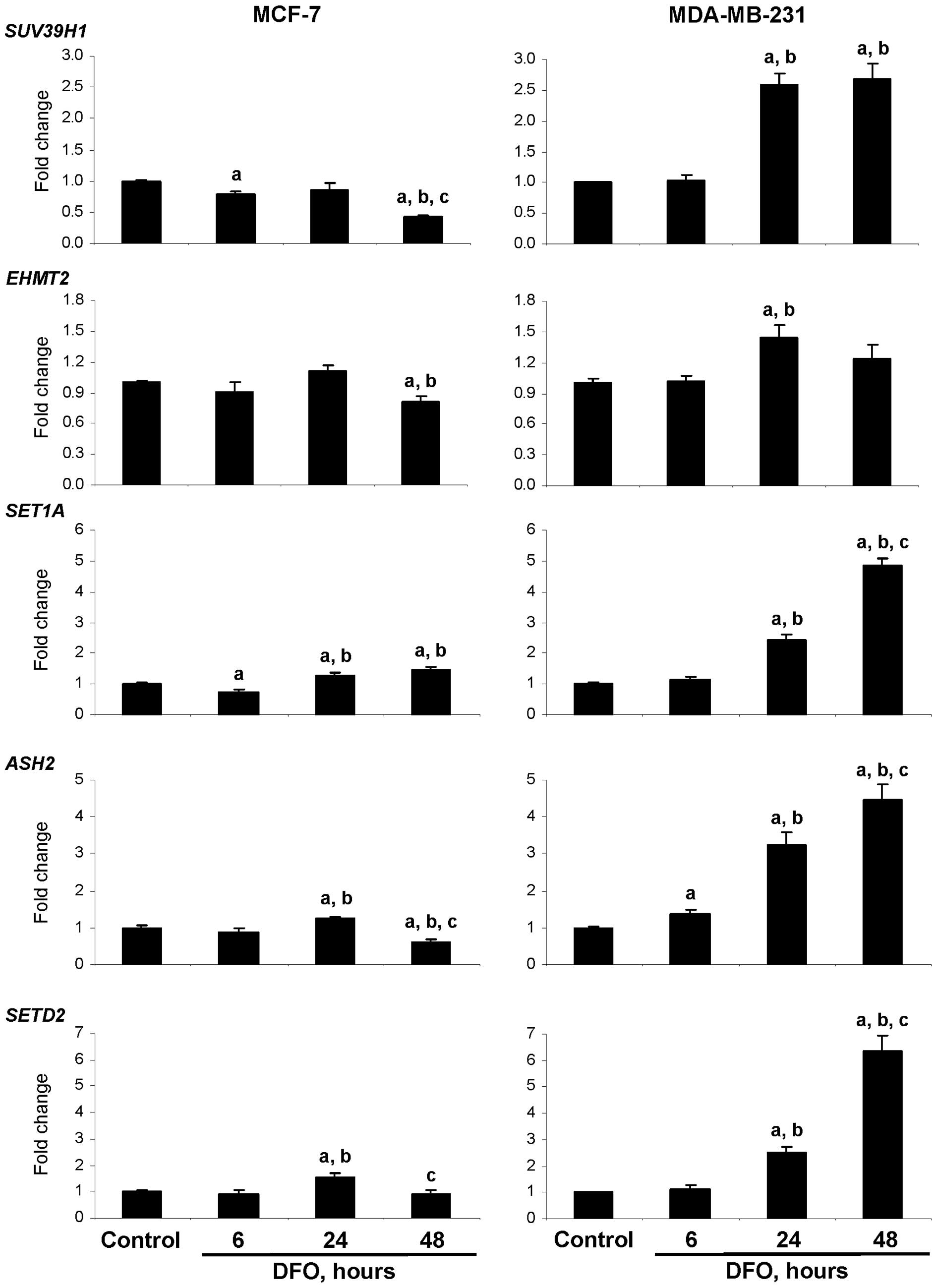
Effect of DFO on DNA and the expression
of histone-modifying genes
To further evaluate the effect of DFO treatment on
the functioning of DNA and histone methylation machinery, qRT-PCR
was conducted to examine the expression of histone
methyltransferases (HMTs), including the histone H3K9
methyltransferases, SUV39H1 and EHMT2, the histone
H3K4 methyltransferases, SET1 and ASH2, and the
histone H3K36 methyltransferase SETD2, as well as that of
the DNA methyltransferases (DNMTs), DNMT1, DNMT3A and
DNMT3B.
Fig. 3 demonstrates
that culturing the MCF-7 and MDA-MB-231 cells in DFO-containing
medium resulted in cell type-dependent alterations in the
expression of HMT genes. This was evident by a difference in the
trends and magnitude of the expression changes in the DFO-treated
MCF-7 and MDA-MB-231 cells. The most noticeable changes were a
distinct up-regulation in the expression of the SUV39H1,
SET1A, ASH2 and SETD2 HMTs in the DFO-treated
MDA-MB-231 cells at 24 h and, specifically, at 48 h. By contrast,
the expression of the SUV39H1, EHMT2, ASH2 and
SETD2 HMTs in the DFO-treated MCF-7 cells was significantly
lower at 48 h. Similar to the HMTs, the expression of DNMT1,
DNMT3A and DNMT3B in the MDA-MB-231 cells cultured in
the presence of DFO was markedly increased at 24 and 48 h with the
magnitude being greater at 48 h (Fig.
4A). By contrast, the expression of DNMTs in the DFO-treated
MCF-7 cells was decreased, reaching the lowest level 48 h after the
initiation of DFO treatment. At this time, the expression of
DNMT1, DNMT3A and DNMT3B in the DFO-treated
MCF-7 cells was 75, 65 and 72% lower, respectively, as compared to
similar values in the untreated MCF-7 control cells.
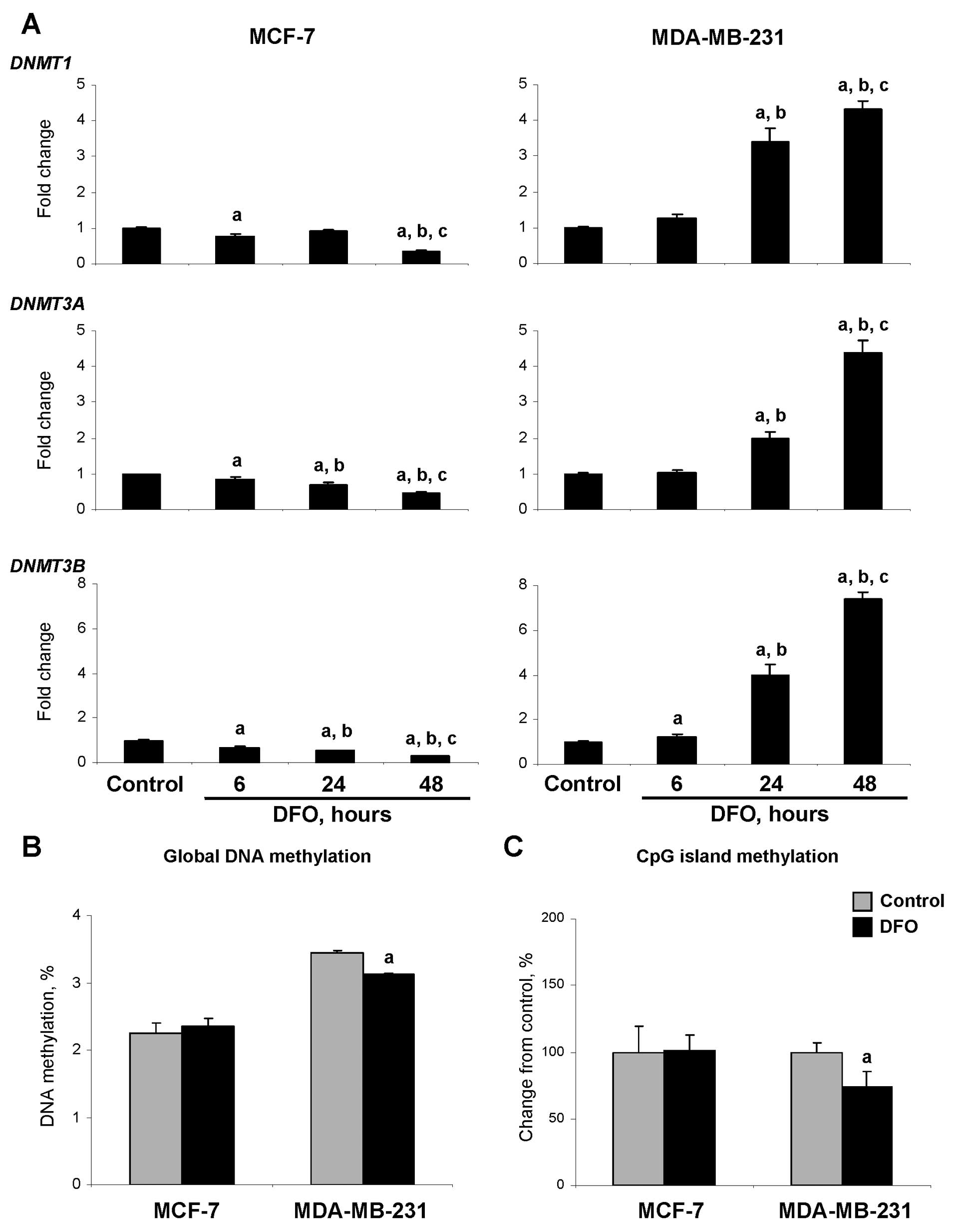 | Figure 4Effects of DFO treatment on the
expression of the DNA methyltransferases, DNMT1, DNMT3A and DNMT3B,
and DNA methylation in the MCF-7 and MDA-MB-231 human breast cancer
cells. (A) The expression of DNMT1, DNMT3A and
DNMT3B genes was determined by qRT-PCR as detailed in
‘Materials and methods’. Data are presented as an average fold
change in the expression of each gene in the DFO-treated cells
relative to that in the corresponding untreated control cells,
which were assigned the value 1. The letters ‘a’, ‘b’ and ‘c’
denote a significant (p<0.05) difference (n=3) compared to the
corresponding untreated control cells (a), or the cells treated
with DFO for 6 (b) or 24 h (c). (B) Global DNA methylation in the
untreated control and DFO-treated cells was measured using the
LC-MS/MS method as detailed in ‘Materials and methods’. Data are
presented as a percentage of DNA methylation. The letter ‘a’
denotes a significant (p<0.05) difference (n=3) compared to the
corresponding untreated control cells. (C) CpG island methylation
in the untreated control and DFO-treated cells was measured by
[3H]dCTP extension assay after digestion of DNA with
methylation-sensitive TspMI restriction endonuclease. The
TspMI hexanucleotide recognition sequences, CCCGGG, occur
predominantly within CpG islands. Data are presented as a
percentage change compared to the corresponding untreated control
cells, which were assigned a value of 100%. The letter ‘a’ denotes
a significant (p<0.05) difference (n=3) compared to the
corresponding untreated control cells. |
Effect of DFO on DNA methylation
Fig. 4B
demonstrates that culturing the MCF-7 cells in DFO-containing
medium did not affect the level of 5-mC in the DNA. By contrast,
the level of 5-mC in the DFO-treated MDA-MB-231 cells was reduced
by 10%. The level of 5-mC in the CpG islands in the DFO-treated
MCF-7 cells did not differ from that in the untreated MCF-7 control
cells, whereas in the DFO-treated MDA-MB-231 cells, it was
significantly decreased (Fig.
4C).
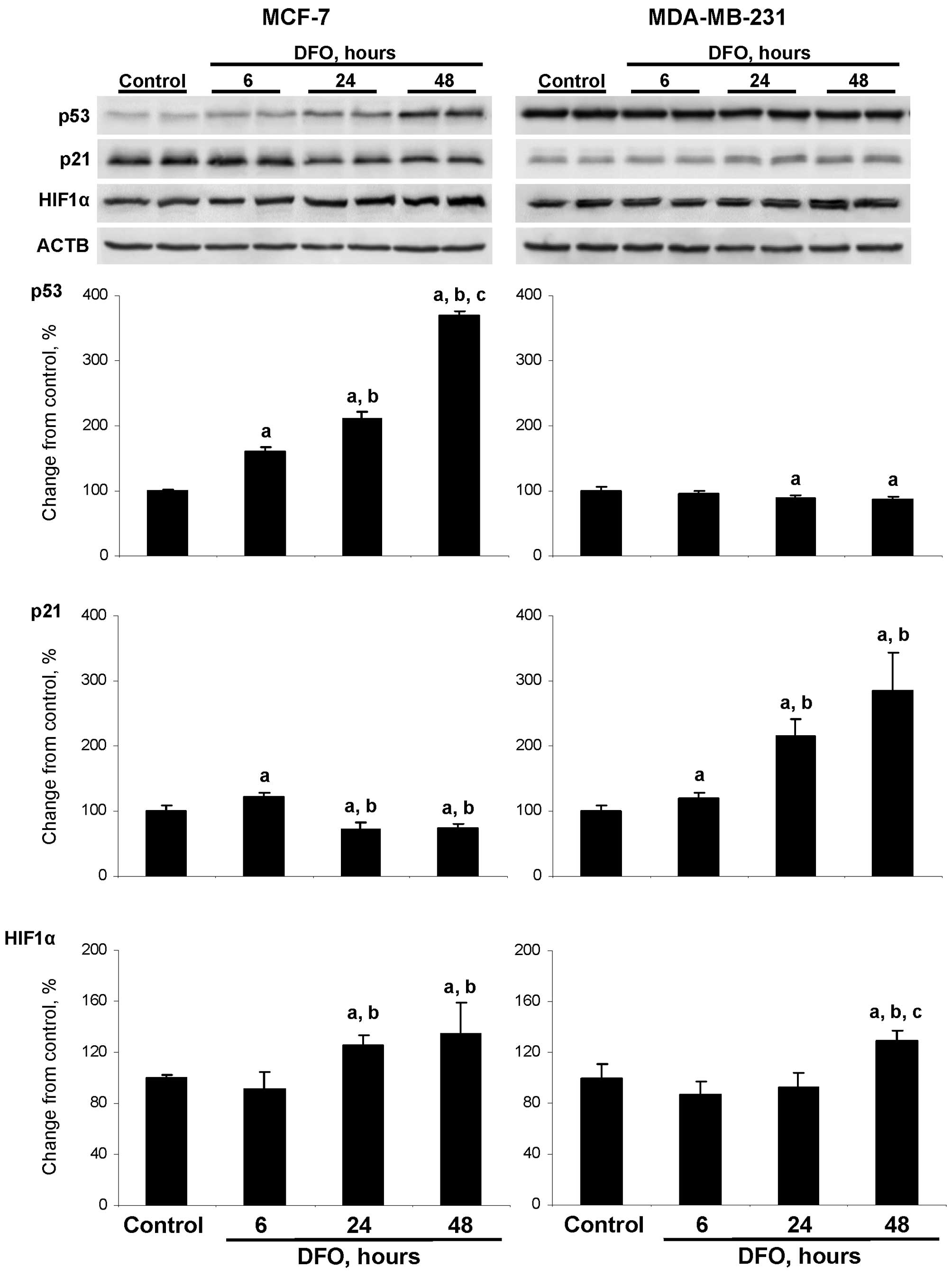
DFO affects the level of p53 and p21
proteins
Previous studies have indicated that the anti-tumor
action of DFO and other iron chelators is associated with the
activation of apoptosis (25–28);
however, the mechanism underlying the induction of apoptosis has
remained unexplored. Fig. 5
demonstrates that the treatment of wild-type TP53 MCF-7
cells with DFO resulted in an increase in the level of p53 protein
in a time-dependent manner. The level of p53 protein in the
DFO-treated cells at 6, 24 and 48 h was 160, 211 and 370% greater,
respectively, compared to the untreated MCF-7 cells. By contrast,
an opposite trend in p53 alterations was observed in the
DFO-treated mutant TP53 MDA-MB-231 cells. The treatment of
MDA-MB-231 cells with DFO caused a progressive increase in p21
protein levels, with the highest values being detected at 24 and 48
h, while the levels of p21 protein in the DFO-treated MCF-7 cells
decreased. The treatment of MCF-7 and MDA-MBA-231 cells with DFO
caused a moderate elevation in HIF1α protein levels with changes
being significant at 24 an 48 h in the MCF-7 cells and at 48 h in
the MDA-MB-231 cells.
DFO affects the histone methylation
pattern at the p21 gene promoter
Previous reports have suggested a link between the
induction of apoptosis and an epigenetic mechanism for the
transcriptional activation of the p21 gene (22,29).
In view of this, and considering the substantial global epigenetic
changes induced by DFO, we used ChIP assay to examine the levels of
histones H3K9me2 and H3K4me2, 2 epigenetic markers associated with
the alteration of gene expression, at the promoter of the
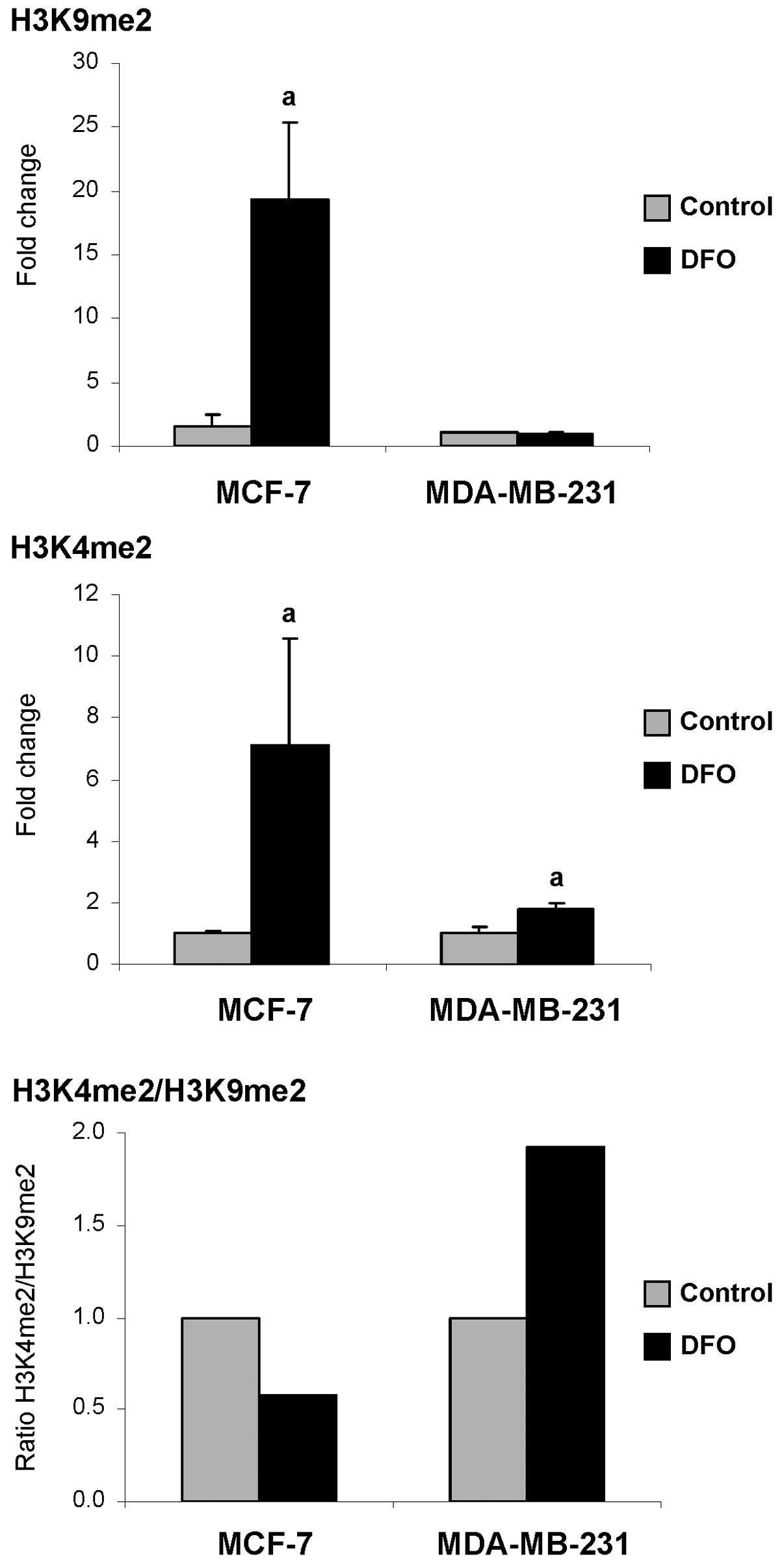
p21 gene. Fig. 6
demonstrates that at 48 h after treatment, the DFO-treated MCF-7
cells were characterized by a prominent increase in the levels of
histones H3K9me2 and H3K4me2. At this time point, the levels of
histones H3K9me2 and H3K4me2 were 12.3- and 7.1-fold greater,
respectively, compared to the untreated control cells. By
comparison, the DFO-treated MDA-MB-231 cells exhibited a 1.8-fold
increase in the levels of histone H3K4me2, while the levels of
histone H3K9me2 did not differ from the control values. More
importantly, the H3K4me2/H3K9me2 ratio in the DFO-treated
MDA-MB-231 cells was 1.9-fold greater compared to the control
cells, indicating an enrichment of the p21 promoter by
histone H3K4me2. By contrast, in the DFO-treated MCF-7 cells, the
H3K4me2/H3K9me2 ratio was decreased, indicating an enrichment of
the p21 gene promoter by the transcriptional silencing of
histone H3K9me2.
DFO enhances the sensitivity of MCF-7 and
MDA-MB-231 cancer cells to chemotherapeutic drugs
Given that DFO treatment induced pro-apoptotic
pathways in the MCF-7 and MDA-MB-231 cells, we investigated whether
the activation of these pathways results in increased cancer cell
sensitivity to chemotherapeutic drugs. Table I demonstrates that in the
DFO-treated MCF-7 and MDA-MB-231 cells, the IC50 values
for DOX and CDDP were lower than the IC50 values in the
untreated cells. The most dramatic increase in drug sensitivity
occurred with the MDA-MB-231 cells cultured in DFO-containing
medium and treated with DOX.
 | Table IDrug sensitivity of MCF-7 and
MDA-MB-231 cells following treatment with desferrioxamine
(DFO). |
Table I
Drug sensitivity of MCF-7 and
MDA-MB-231 cells following treatment with desferrioxamine
(DFO).
| MCF-7
| MDA-MB-231
|
|---|
| Drug | Control | DFO | Control | DFO |
|---|
| DOX | 4.4±0.2 | 3.7±0.2 | 9.2±0.2 | 1.7±0.1a |
| CDDP | 25.9±3.7 | 14±2.9a | 38.9±8.6 | 23.6±5.2a |
Discussion
Breast cancer is one of the most prevalent
malignancies in women (30).
Despite the statistically significant decline in breast cancer
incidence in recent years, breast cancer is currently the leading
cause of cancer-related mortality among women worldwide (31) and second leading cause of
cancer-related mortality among women in the US (30). The success of breast cancer
treatment relies on a better understanding of the underlying
molecular mechanisms involved in breast cancer initiation and
progression. Therefore, further the comprehensive elucidation of
breast cancer-associated molecular abnormalities is critical in
order to improve the clinical management of breast cancer.
In recent years, the role and mechanisms of breast
cancer-related abnormalities in iron metabolism have been
investigated in a variety of experimental and clinical settings
(11,32–34),
with particular emphasis on altering the dependence of cancer cells
on iron as a potential therapeutic strategy for the treatment of
tumors and/or increasing the efficacy of anti-cancer agents
(17,35,36).
This strategy is driven largely by the well-established fact that
neoplastic cells have an increased requirement for iron, as well as
recent evidence demonstrating that ‘high intracellular iron
phenotype’ is associated with poor prognosis in breast cancer
patients (33). Several potential
mechanisms have been proposed for the anti-cancer effects of iron
chelating compounds, including the inhibition of iron-dependent
ribonucleotide reductase (15), a
rate-limiting enzyme in DNA synthesis, the inhibition of cell cycle
progression by inducing G1-S phase arrest (15), and the inhibition of
epithelial-to-mesenchymal transition via the up-regulation of N-Myc
downstream-regulated gene 1 (NDRG1) (37).
Iron is a key regulator of cellular metabolic
reactions, including several DNA and histone-modifying proteins
(7,8). In the present study, we demonstrate
that the iron chelator, DFO, substantially decreases the protein
levels of the histone H3K9 demethylase, JMJD2A, in MCF-7 and
MDA-MB-231 breast cancer cells (Fig.
1). The reduction in JMJD2A levels following DFO treatment was
not a surprising discovery, since JMJD2A belongs to the superfamily
of iron-dependent oxygenases (11). This suggests that the
down-regulation of JMJD2A is associated directly with the iron
chelating properties of DFO; however, in the DFO-treated MCF-7
cells, HIF1α may also actively contribute to this effect (38), since it was moderately up-regulated
(Fig. 5).
DFO treatment also caused a down-regulation of the
histone H3K4 demethylase, LSD1, in the MDA-MB-231 cells (Fig. 1). This was unexpected since LSD1
belongs to the family of flavin-dependent amine oxidases (39); however, it has been recently
demonstrated that LSD1 exhibits folate-binding activity (40). Considering the intimate
interdependence between iron, flavin and folate metabolic pathways,
alterations in intracellular iron metabolism may indirectly affect
the level and function of LSD1.
The down-regulation of JMJD2A and LSD1 in the
DFO-treated cells was accompanied by the altered expression of a
number of histone H3K9, H3K36 and H3K4 methyltransferase genes
(Fig. 3). These down- and
up-regulation events were accompanied by marked changes in the
global levels of corresponding metabolically sensitive histone
markers. Specifically, the DFO-treated MCF-7 cells exhibited a
prominent increase in the levels of H3K9me2 and H3K36me3, while the
DFO-treated MDA-MB-231 cells were characterized by an increase in
the levels of H3K4me2 (Fig. 6).
The differential expression levels of H3K9me2 and H3K4me2 in the
DFO-treated MCF-7 and MDA-MB-231 cells were associated with the
differential expression patterns of histone demethylases and HMTs
in these cells.
Previous reports have shown that the treatment of
various cancer cells with DFO induces apoptosis through the
activation of various pro-apoptotic pathways (25–28).
Likewise, the results of the present study demonstrated that the
exposure of MCF-7 and MDA-MB-231 human breast cancer cells to DFO
induced the pro-apoptotic program; however, the underlying
mechanisms of this activation differed in these cell lines. The
treatment of MCF-7 cells that possess wild-type TP53 with
DFO activated a p53-dependent pro-apoptotic pathway, which was
evident by the profound increase in p53 protein levels in the
MCF-7-treated cells (Fig. 5). It
has been suggested that the JMJD2A histone demethylase plays a
major role in the regulation of apoptosis. Specifically, several
studies have demonstrated that the depletion of JMJD2A increases
p53-dependent apoptosis (41–43).
The findings of the present study showing a substantial reduction
in JMJD2A protein expression concomitant with an increase in p53
protein levels in the DFO-treated MCF-7 cells are in accord with
this suggestion.
By contrast, the treatment of mutant TP53
MDA-MB-231 cells with DFO also activated a pro-apoptotic program,
although in a p53-independent manner, as evident by the substantial
increase in p21 protein levels. Several possible explanations exist
for the mecha nism of p21-up-regulation in the DFO-treated
MDA-MB-231 cells. First, it may be attributed to the enrichment of
the promoter region of the p21 gene by H3K4me2, a histone
modification marker that is strongly associated with active
transcription. This was evident by the more extensive increase in
histone H3K4me2 levels compared to histone H3K9me2 levels at the
promoter region of the p21 gene, which corresponds to
previous findings showing that the methylation of histone H3K4
impairs the methylation of histone H3K9 (44). By contrast, in the DFO-treated
MCF-7 cells with down-regulated p21, the promoter region of
the p21 gene was enriched by the transcriptional silencing of
H3K9me2. Second, it has been demonstrated that the histone
demethylase, JMJD2A, can bind to the promoter of the p21
gene and repress its expression (43). Therefore, the DFO-induced decrease
in JMJD2A protein expression may block its recruitment to the
p21 gene with a resultant enhancement in the expression of
p21. Third, recent evidence has indicated that a DFO-induced
up-regulation of the NDRG1 gene (37) may up-regulate p21 expression via
p53-independent mechanisms (45).
The results of this study also show that the
activation of apoptotic pathways in the DFO-treated breast cancer
cells enhances the anti-cancer activity of chemotherapeutic drugs.
The most pronounced changes in drug-sensitivity were the increased
sensitivity of MDA-MB-231 cells, which possess an advanced
mesenchymal and drug-resistant phenotype (46), to DOX, one of the main
chemotherapeutic agents for the treatment of breast cancer. In
addition to the up-regulation of the p53-independent pro-apoptotic
pathway, this may be attributed to the activation of other
epigenetically silenced tumor suppressor genes in the MDA-MB-231
cells, which was evident by a decrease in the 5-mC levels.
In conclusion, to our knowledge, the results of the
present study provide evidence for the first time that the iron
chelator, DFO, activates apoptotic programs in human breast cancer
cells and enhances their sensitivity to chemotherapeutic agents via
epigenetic mechanisms by affecting the functioning of the chromatin
remodeling machinery. The activation of the pro-apoptotic program
in wild-type TP53 MCF-7 cells was p53-dependent, triggered
mainly by the down-regulation of the JMJD2A histone demethylase. In
mutant TP53 MDA-MB-231 cells, DFO caused similar effects via
the activation of the p53-independent apoptotic program driven
predominantly by the epigenetic up-regulation of p21. Importantly,
the results of the present study provide experimental proof of the
interdependence between iron and epigenetic regulatory mechanisms
and suggest that the modification of intracelluar iron metabolism
may enhance the efficacy of epigenetic therapy.
Abbreviations:
|
DFO
|
desferrioxamine;
|
|
JHDM
|
Jumonji domain-containing histone
demethylase;
|
|
TET
|
TET methylcytosine dioxygenase;
|
|
p21
|
cyclin-dependent kinase inhibitor
1A;
|
|
HIF1α
|
hypoxiainducible factor 1α;
|
|
H3K9me3
|
trimethyl histone H3 lysine 9;
|
|
H3K9me2
|
dimethyl histone H3 lysine 9;
|
|
H3K9ac
|
acetyl histone H3 lysine 9;
|
|
H3K27me3;
|
trimethyl histone H3 lysine 27;
|
|
H3K36me3
|
trimethyl histone H3 lysine 36;
|
|
H3K4me3
|
trimethyl histone H3 lysine 4;
|
|
H3K4me2
|
dimethyl histone H3 lysine 4;
|
|
H4K20me3
|
trimethyl histone H4 lysine 20;
|
|
LC-MS/MS
|
liquid chromatography-tandem mass
spectrometry;
|
|
ChIP
|
chromatin immunoprecipitation;
|
|
DOX
|
doxorubicin hydrochloride;
|
|
CDDP
|
cis-diammineplatinum(II)
dichloride;
|
|
HMT
|
histone methyltransferase
|
Acknowledgements
The views expressed in this study do
not necessarily represent those of the US Food and Drug
Administration.
References
|
1.
|
Teperino R, Schoonjans K and Auwerx J:
Histone methyltransferases and demethylases; can they link
metabolism and transcription? Cell Metab. 12:321–327. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Burgio G, Onorati MC and Corona DFV:
Chromatin remodeling regulation by small molecules and metabolites.
Biochim Biophys Acta. 1799:671–680. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Hou H and Yu H: Structural insights into
histone lysine demethylation. Curr Opin Struct Biol. 20:739–748.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Iyer LM, Abhiman S and Aravind L: Natural
history of eukaryotic DNA methylation systems. Prog Mol Biol Transl
Sci. 101:25–104. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Tsukada Y-i, Fang J, Erdjument-Bromage H,
Warren ME, Borchers CH, Tempst P and Zhang Y: Histone demethylation
by a family of JmjC domain-containing proteins. Nature.
439:811–816. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Yamane K, Toumazou C, Tsukada Y-i,
Erdjument-Bromage H, Tempst P, Wong J and Zhang Y: JHDM2A, a
JmjC-containing H3K9 demethylase, facilitates transcription
activation by androgen receptor. Cell. 125:483–495. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Wu H and Zhang Y: Mechanisms and functions
of Tet protein–mediated 5-methylcytosine oxidation. Genes Dev.
25:2436–2452. 2011.
|
|
8.
|
McDonough MA, Loenarz C, Chowdhury R,
Clifton IJ and Schofield CJ: Structural studies of human
2-oxoglutarate dependent oxygenases. Curr Opin Struct Biol.
20:659–672. 2010. View Article : Google Scholar
|
|
9.
|
Torti SV and Torti FM: Ironing out cancer.
Cancer Res. 71:1511–1514. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Jones PA and Baylin SB: The epigenomics of
cancer. Cell. 128:683–692. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Miller LD, Coffman LG, Chou JW, Black MA,
Berg J, D’Agostino R Jr, Torti SV and Torti FM: An iron regulatory
gene signature predicts outcome in breast cancer. Cancer Res.
71:6728–6737. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Baylin SB and Jones PA: A decade of
exploring the cancer epigenome - biological and translational
implications. Nat Rev Cancer. 11:726–734. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Buss JL, Torti FM and Torti SV: The role
of iron chelation in cancer therapy. Curr Med Chem. 10:1021–1034.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Yu Y, Gutierrez E, Kovacevic Z, Saletta F,
Obeidy P, Suryo Rahmanto Y and Richardson DR: Iron chelators for
the treatment of cancer. Curr Med Chem. 19:2689–2702. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Kawamoto M, Horibe T, Kohno M and Kawakami
K: A novel transferring receptor-targeted hybrid peptide
disintegrates cancer cell membrane to induce rapid killing of
cancer cells. BMC Cancer. 11:3592011. View Article : Google Scholar
|
|
17.
|
Rao VA, Klein SR, Agama KK, Toyoda E,
Adachi N, Pommier Y and Shacter EB: The iron chelator Dp44mT causes
DNA damage and selective inhibition of topoisomerase IIα in breast
cancer cells. Cancer Res. 69:948–957. 2009.PubMed/NCBI
|
|
18.
|
Chekhun VF, Lukyanova NY, Kovalchuk O,
Tryndyak VP and Pogribny IP: Epigenetic profiling of
multidrug-resistant MCF-7 breast cancer adenocarcinoma cells
reveals novel hyper- and hypomethylated targets. Mol Cancer Ther.
6:1089–1098. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative CT method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Zhang JJ, Zhang L, Zhou K, Ye X, Liu C,
Zhang L, Kang J and Cai C: Analysis of global DNA methylation by
hydrophilic interaction ultra high-pressure liquid chromatography
tandem mass spectrometry. Anal Biochem. 413:164–170. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Pogribny I, Yi P and James SJ: A sensitive
new method for rapid detection of abnormal methylation patterns in
global DNA and within CpG islands. Biochem Biophys Res Commun.
262:624–628. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Kim TD, Shin S, Berry WL, Oh S and
Janknecht R: The JMJDA demethylase regulates apoptosis and
proliferation in colon cancer cells. J Cell Biochem. 113:1368–1376.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Stewart MD, Li J and Wong J: Relationship
between histone H3 lysine 9 methylation, transcription repression,
and heterochromatin protein 1 recruitment. Mol Cell Biol.
25:2525–2538. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Shi Y, Lan F, Matson C, Mulligan P,
Whetstine JR, Cole PA, Casero RA and Shi Y: Histone demethylation
mediated by the nuclear amine oxidase homolog LSD1. Cell.
119:941–953. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Hileti D, Panayiotidis P and Hoffbrand AV:
Iron chelators induce apoptosis in proliferating cells. Br J
Haematol. 89:181–187. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Pan YJ, Hopkins RG and Loo G: Increased
GADD153 gene expression during iron chelation-induced apoptosis in
Jurkat T-lymphocytes. Biochim Biophys Acta. 1691:41–50. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
So EY, Ausman M, Saeki T and Ouchi T:
Phosphorylation of SMC1 by ATR is required for desferrioxamine
(DFO)-induced apoptosis. Cell Death Dis. 2:e1282011. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Saletta F, Suryo Rahmanto Y, Siafakas AR
and Richardson DR: Cellular iron depletion and the mechanisms
involved in the iron-dependent regulation of the growth arrest and
DNA damage family of genes. J Biol Chem. 286:35396–35406. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Escoubet-Lozach L, Lin IL, Jensen-Pergakes
K, Brady HA, Gandhi AK, Schafer PH, Muller GW, Worland PJ, Chan KW
and Verhelle D: Pomalidomide and lenalidomide induce p21WAF-1
expression in both lymphoma and multiple myeloma through a
LSD1-mediated epigenetic mechanism. Cancer Res. 69:7347–7356. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
31.
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
32.
|
Huang X: Does iron have a role in breast
cancer? Lancet Oncol. 9:803–807. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Pinnix ZK, Miller LD, Wang W, D’Agostino R
Jr, Kute T, Willngham MC, Hatcher H, Tesfay L, Sui G, Di X, Torti
SV and Torti FM: Ferroportin and iron regulation in breast cancer
progression and prognosis. Sci Transl Med. 2:43ra562010. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Shpyleva SI, Tryndyak VP, Kovalchuk O,
Starlard-Davenport A, Chekhun VF, Beland FA and Pogribny IP: Role
of ferritin alterations in human breast cancer cells. Breast Cancer
Res Treat. 126:63–71. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Hoke EM, Maylock CA and Shacter E:
Desferal inhibits breast tumor growth and does not interfere with
the tumoricidal activity of doxorubicin. Free Radic Biol Med.
39:403–411. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Whitnall M, Howard J, Ponka P and
Richardson DR: A class of iron chelators with a wide spectrum of
potent antitumor activity that overcomes resistance to
chemotherapeutics. Proc Natl Acad Sci USA. 103:14901–14906. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Chen Z, Zhang D, Yue F, Zheng M, Kovacevic
Z and Richardson DR: The iron chelators Dp44mT and DFO inhibit
TGF-β-induced epithelial-mesenchymal transition via up-regulation
of N-Myc downstream-regulated gene-1 (NDRG1). J Biol Chem.
287:17016–17028. 2012.PubMed/NCBI
|
|
38.
|
Pollard PJ, Loenarz C, Mole DR, McDonough
MA, Gleadle JM, Schofield CJ and Ratcliffe PJ: Regulation of
Jumonji-domain-containing histone demethylases by hypoxia-inducible
factor (HIF)-1α. Biochem J. 416:387–394. 2008.
|
|
39.
|
Culhane JC and Cole PA: LSD1 and the
chemistry of histone demethylation. Curr Opin Chem Biol.
11:561–568. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Luka Z, Moss F, Loukachevitch LV, Bornhop
DJ and Wagner C: Histone demethylase LSD1 is a folate-binding
protein. Biochemistry. 50:4750–4756. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Black JC, Allen A, Van Rechem C, Forbes E,
Longworth M, Tschöp K, Rinehart C, Quiton J, Walsh R, Smallwood A,
Dyson NJ and Whetstine JR: Conserved antagonism between
JMJD2A/KDM4A and HP1γ during cell cycle progression. Mol Cell.
40:736–748. 2010.PubMed/NCBI
|
|
42.
|
Mallette FA, Mattiroli F, Cui G, Young LC,
Hendzel MJ, Mer G, Sixma TK and Richard S: RNF8- and
RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1
recruitment to DNA damage sites. EMBO J. 31:1865–1878. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Kim TD, Oh S, Shin S and Janknecht R:
Regulation of tumor suppressor p53 and HCT116 cell physiology by
histone demethylase JMJD2D/KDM4D. PLoS One. 7:e346182012.
View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Binda O, LeRoy G, Bua DJ, Garcia BA,
Gozani O and Richard S: Trimethylation of histone H3 lysine 4
impairs methylation of histone H3 lysine 9. Regulation of lysine
methyltransferases by physical interaction with their substrates.
Epigenetics. 5:767–775. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Kovacevic Z, Sivagurunathan S, Mangs H,
Chikhani S, Zhang D and Richardson DR: The metastasis suppressor,
N-myc downstream regulated gene 1 (NDRG1), upregulates p21 via
p53-independent mechanisms. Carcinogenesis. 32:732–740. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Tryndyak VP, Beland FA and Pogribny IP:
E-cadherin transcriptional down-regulation by epigenetic and
microRNA-200 family alterations is related to mesenchymal and
drug-resistant phenotypes in human breast cancer cells. Int J
Cancer. 126:2575–2583. 2010.PubMed/NCBI
|