Introduction
Programmed cell death is a crucial cellular process
under both physiological and pathological conditions. Emerging
evidence provides deep insights into the molecular pathways
regulating and executing programmed cell death. Basing on a range
of measurable biochemical features and morphological criteria,
programmed cell death is further sub-classified as extrinsic
apoptosis, caspase-dependent or -independent intrinsic apoptosis,
programmed necrosis, autophagic cell death and others (1). Resembling apoptosis, necrosis can
also proceed under a regulated way. This regulated necrosis is
frequently termed as ‘programmed necrosis’, which is highly
regulated by receptor-interacting protein kinases 1 (RIP1) and 3
(RIP3) (2). In classical
programmed necrosis (or ‘necroptosis’) triggered by TNF, assembly
of RIP1and RIP3 forming a necrotic complex (termed as ‘necrosome’)
is critical in initiation of programmed necrotic process. However,
current studies on programmed necrosis triggered by other stimuli
demonstrate programmed necrosis can also be initiated in a
RIP1-independent but RIP3-dependent manner. Accumulating evidence
indicates RIP3 is indispensable for programmed necrosis and
participates in constitution of all demonstrated necrotic complex
triggered by various stimuli, whereas RIP1 participates in certain
stimuli induced programmed necrosis (3). Apoptotic and programmed necrotic
signaling pathways share some vital corporate molecules.
Consequently, it is comprehensible that certain chemicals trigger
different cell death modes in dose-dependent manner (autophagy or
apoptosis after lower-dose exposure and necrosis at higher-dose
exposure).
Honokiol (HNK) is a pharmacologically active small
molecule with multifunctional antitumor effects (4). Apoptosis is the best-demonstrated
mechanism through which HNK accomplish its antitumor effects in
various cancer cell lines. HNK induces apoptosis through both death
receptor (extrinsic) pathway (5–8) and
mitochondrial (intrinsic) pathway (9,10).
Besides apoptosis, we previously reported HNK induced programmed
necrotic cell death in HL60, MCF-7 and HEK293 cells at certain
doses through mitochondrial permeability transition pore (11). This programmed necrosis is
highly-regulated by cyclophilin D (CypD) (11). Although emerging evidence concerns
programmed cell death triggered by HNK, little attention has been
paid to the potential existence of death modes transition from
apoptosis to programmed necrosis and the possible mechanism. We
investigated honokiol-induced cell death mode transition, and
potential regulation mechanism in the current study.
Materials and methods
Reagents and chemicals
Honokiol was purchased from the National Institute
for the Control of Pharmaceutical and Biological Products (Beijing,
China) with >99% purity. Honokiol power was dissolved in DMSO at
20 mg/ml (75 mM). z-VAD-fmk was from Beyotime Institute of
Biotechnology (Shanghai, China). PrimeScript® RT reagent
Kit (Perfect Real-Time) and SYBR® Premix Ex Taq™ II (Tli
RNase H Plus) were purchased from Takara Biotechnology Co. Ltd
(Dalian, China). DNA Ladder Detection Kit was from KeyGEN Biotech
(Nanjing, China). M-PER® Mammalian Protein Extraction
Reagent was from Thermo Scientific (Pierce Biotechnology, Rockford,
IL, USA). RIP1 antibody and Annexin V-FITC/PI kit were from R&D
Systems (Minneapolis, MN, USA). RIP3 and Bcl-2 antibodies were
purchased from Epitomics (Burlingame, CA, USA), PTEN, Bcl-xl and
GAPDH antibodies were from Cell Signaling Technology (Danvers, MA,
USA).
Cell culture
Human breast carcinoma cell lines were obtained from
Cancer Institution of Zhejiang University (Zhejiang, China). MCF-7
was maintained in RPMI-1640 medium. MDA-MB-231 was cultured in
L-15. Bcap-37, T47D and SKBR-3 were maintained in DMEM. All media
were supplemented with 10% FBS. The multidrug resistant human
breast carcinoma cell line MCF-7/ADR and Bcap-37/ADR was developed
as reported previously (12) and
maintained in RPMI-1640 (supplemented with 10% FBS) with 1 and 0.5
μg/ml doxorubicin.
Cell toxicity assay
Breast cancer cells of cultured cell lines were
seeded in 96-well culture plates at density of 6,000–8,000
cells/well and cultured for 24 h pretreatment. Then, cells were
incubated with drug-free medium, honokiol of different
concentrations for different time durations. Cell toxicity assays
were preformed using MTT method as described previously (12).
Flow cytometric analysis for cell
apoptosis and necrosis
MCF-7 cells were cultured in 6-well culture plates
at 8–10×105/well in 2 ml medium for 24 h. Cells were
treated with 0.1% DMSO (vehicle control) and honokiol at different
concentrations (liquid volume: 1.5 ml/well) for different time
durations. After treatment, cells were stained with Annexin
V-FITC/PI and assayed by flow cytometry as previously described
(12).
Assessment of chromatin condensation
using Hoechst 33342 staining by confocal imaging
Cells were seeded and treated as in flow cytometric
analysis for cell apoptosis and necrosis. Cells were fixed in 4%
phosphate-buffered paraformaldehyde for 10 min and were washed
twice with PBS subsequently. Then cells were incubated with 10
μg/ml Hoechst 33342 for 10 min in dark at room temperature.
Nuclei stains of Hoechst 33342 were examined under a confocal
microscope (Leica TCS SP5). Hoechst reagent was taken up by the
cell nuclei, and apoptotic cells exhibited a bright blue
fluorescence.
DNA ladder detection
Cells were seeded and treated as in flow cytometric
analysis for cell apoptosis and necrosis. After treatment, cells
were collected and washed twice with chilled PBS. DNA gel
electrophoresis was done after DNA extraction according to the
manufacturer’s instructions of DNA Ladder Detection Kit. HL60 cells
treated with 20 μg/ml VP-16 for 6 h were used as positive
control.
Real-time RT-PCR quantification of
mRNA
Cells were seeded and treated as in flow cytometric
analysis for cell apoptosis and necrosis. Total RNA was extracted
using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Concentration
and quality of RNA were measured by Nanodrop 1000 spectrophotometer
(Thermo Scientific). Total RNA were reverse transcribed using
PrimeScript® RT reagent Kit (Takara Biotechnology Co.
Ltd). Subsequently, real-time PCR reactions were performed using
SYBR® Premix Ex Taq™ II (Tli RNaseH Plus) Kit (Takara
Biotechnology Co. Ltd) on Stepone Plus System (Applied Biosystems,
Foster City, CA, USA). Detailed primer sequences are summarized in
Table I. PCR reaction conditions
were: 95°C for 30 sec, 40 cycles with 95°C for 5 sec and 60°C for
30 sec. β-actin was applied as internal reference for
normalization. All the above operation procedures were performed
according to the manufacturer’s protocols. ΔΔCt method was
introduced to determine the relative mRNA expression level.
 | Table IPrimer used for quantitative
real-time PCR analyses of gene expression. |
Table I
Primer used for quantitative
real-time PCR analyses of gene expression.
| Targeted gene | Primer sequences
(5′ to 3′) |
|---|
| CypD | Forward:
AACCTGCTAAATTGTGCGTTATTG |
| Reverse:
TAATAGCCATCTCCCAGTTCTG |
| β-actin | Forward:
TTCCAGCCTTCCTTCCTGGG |
| Reverse:
TTGCGCTCAGGAGGAGCAAT |
Western blot analysis
Cells were treated and harvested as in flow
cytometric analysis for cell apoptosis and necrosis, protein was
extracted using M-PER® Mammalian Protein Extraction
Reagent. Protein concentrations were determined with BCA method. A
total of 20 μg of each sample was loaded on the gel and
procedure of electrophoresis and western blot analysis were
performed as previously described (11). After PVDF membranes were ready for
antibody staining, they were incubated with appropriate primary
antibodies at 4°C overnight and HRP-conjugated second antibodies at
room temperature for 1 h in sequence. ECL detection kit was
introduced for signal development. Images were acquired using
chemiluminesence. Jurkat cells were used as positive control in
analysis RIP3 protein expression.
Statistical analysis
All the experiments were done in triplicate, and
similar results were obtained in three different experimental
setups. The data represent means ± standard deviation. One-way
analysis of variance (ANOVA) was used for evaluations of time- and
dose-response curves. Student’s t-test was used in single group
comparisons. P<0.05 was considered statistically
significant.
Results
Cytotoxic effect of HNK on breast cancer
cell lines
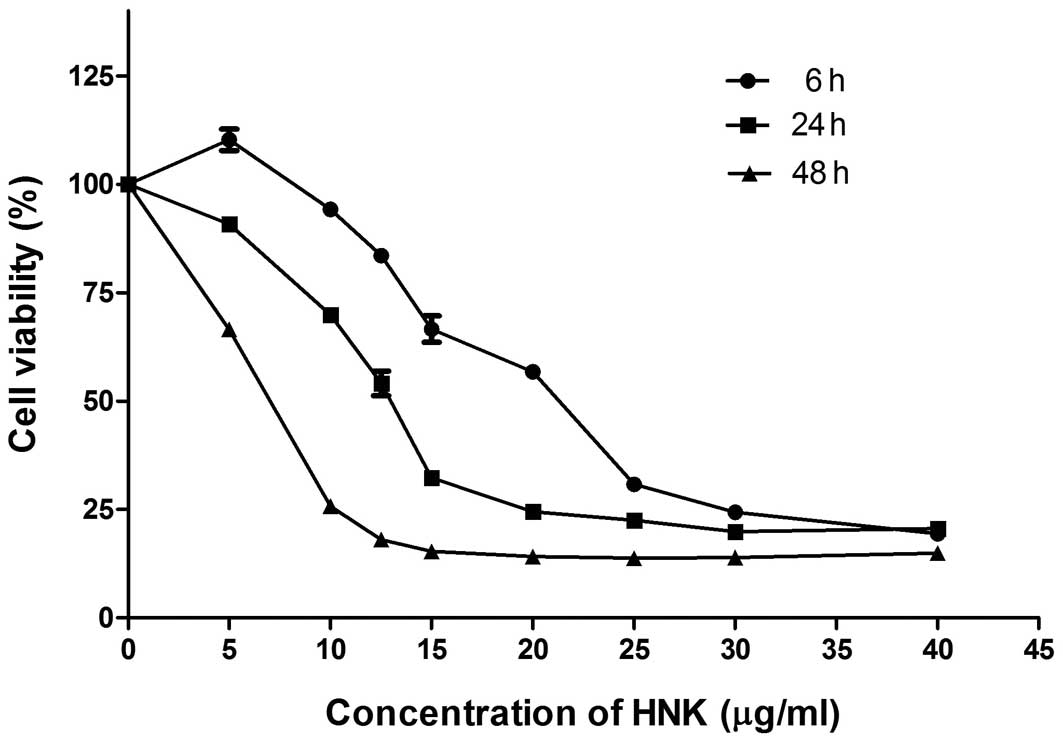
Our data revealed that HNK repressed cell viability
of all tested breast cancer cell lines (MCF-7, MDA-MB-231, Bcap-37,
T47D, SKBR-3, MCF-7/ADR and Bcap-37/ADR) after 24-h treatment with
IC50 ranged from 18.389 to 21.940 μg/ml (Table II). HNK showed parallel cytotoxical
effect on multidrug resistant cells and their corresponding
sensitive cells (Table II).
Further, we demonstrated that HNK had time- and dose-dependent
cytotoxic effects on MCF-7 cells (Fig.
1).
| Table IIThe effects of HNK on breast cancer
cell viability. |
Table II
The effects of HNK on breast cancer
cell viability.
| Cell line | IC50
(μg/ml) |
|---|
| MDA-MB-231 | 19.708 |
| SKBR-3 | 20.712 |
| T47D | 18.670 |
| Bcap-37 | 18.389 |
| MCF-7 | 21.940 |
| Bcap-37/ADR | 21.037 |
| MCF-7/ADR | 19.381 |
HNK induces cell death mode transition in
a time- and dose-dependent manner
A range of chemicals trigger different cell death
modes in a dose-dependent manner. Frequently, lower-dose exposure
induces autophagy or apoptosis, whereas higher-dose induces
necrosis (13). Our group
previously reported that HNK at a concentration of 40 μg/ml
triggered programmed necrosis in MCF-7 cells (11). Accordingly, we hypothesized HNK may
trigger apoptosis to programmed necrosis transition in a time- and
dose-dependent manner.
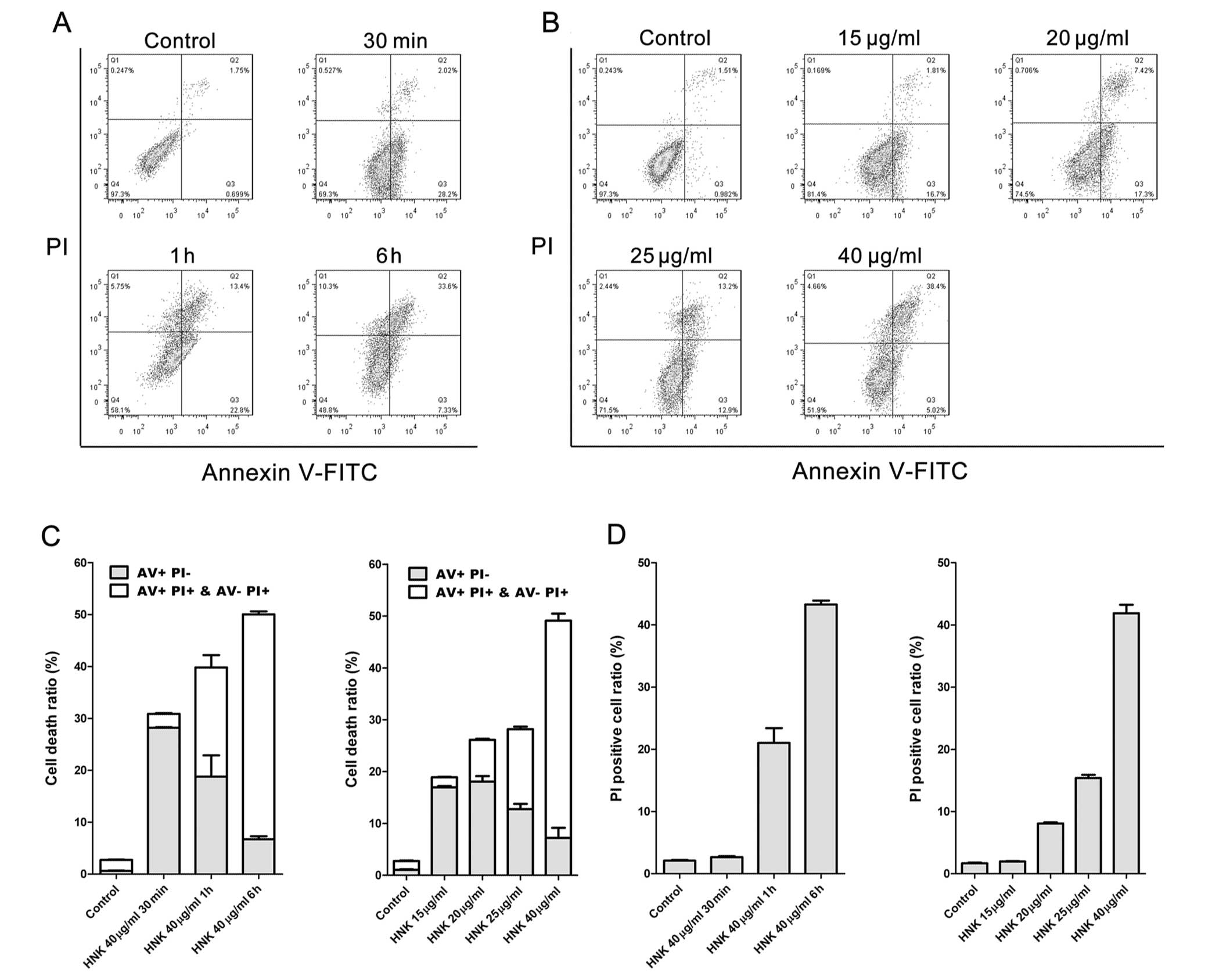
Cell death modes triggered by HNK at a fixed
concentration (40 μg/ml) of different exposure durations
were characterized using Hoechst 33342 staining, DNA laddering and
flow cytometric analysis of Annexin V-FITC/PI staining. MCF-7 cells
were incubated with 40 μg/ml HNK for 30 min, 1 and 6 h,
respectively, and then labeled with Annexin V-FITC (AV) and PI
(Fig. 2). Even for the shortest
incubating time (30 min), cell viability was significantly reduced
by 40 μg/ml HNK (P<0.001) (Fig. 2A and C). Cell death was accelerated
with the extension of incubating time (Fig. 2A and C). Necrotic cell death
(PI+) triggered by 40 μg/ml HNK increased
(P<0.001) with the extension of incubating time, whereas early
apoptotic cell death (AV staining positive and PI negative)
manifested contrary tendency (Fig. 2C
and D). Internucleosomal DNA fragmentation is one of the
hallmarks of apoptosis at later stage (or degradation phase), which
are simply detectable as a ladder pattern in electrophoresis of
isolated DNA. Nuclear chromatin condensation and apoptotic bodies
are both morphological characteristic manifestations of apoptosis
and are detectable using nuclei staining by Hoechst 33342. No
obvious ladder pattern was observed in any of the treatment groups
(40 μg/ml HNK incubation for 30 min, 1, 2, 4 and 6 h,
respectively) (Fig. 3A). Confocal
imaging of Hoechst 33342-stained nuclei revealed no remarkable
chromatin condensation or apoptotic bodies in any of the treatment
groups, but anomalous diffuse staining were detected in groups with
longer treatment durations (4 and 6 h) (Fig. 3B). Both DNA ladder and Hoechst
33342 staining assays demonstrated later-stage apoptosis were not
the predominant cell death mode of 40 μg/ml HNK triggered
cell death. Taken together, 40 μg/ml HNK triggered a cell
death mode transition from early-stage apoptosis to programmed
necrosis in a time-dependent manner.
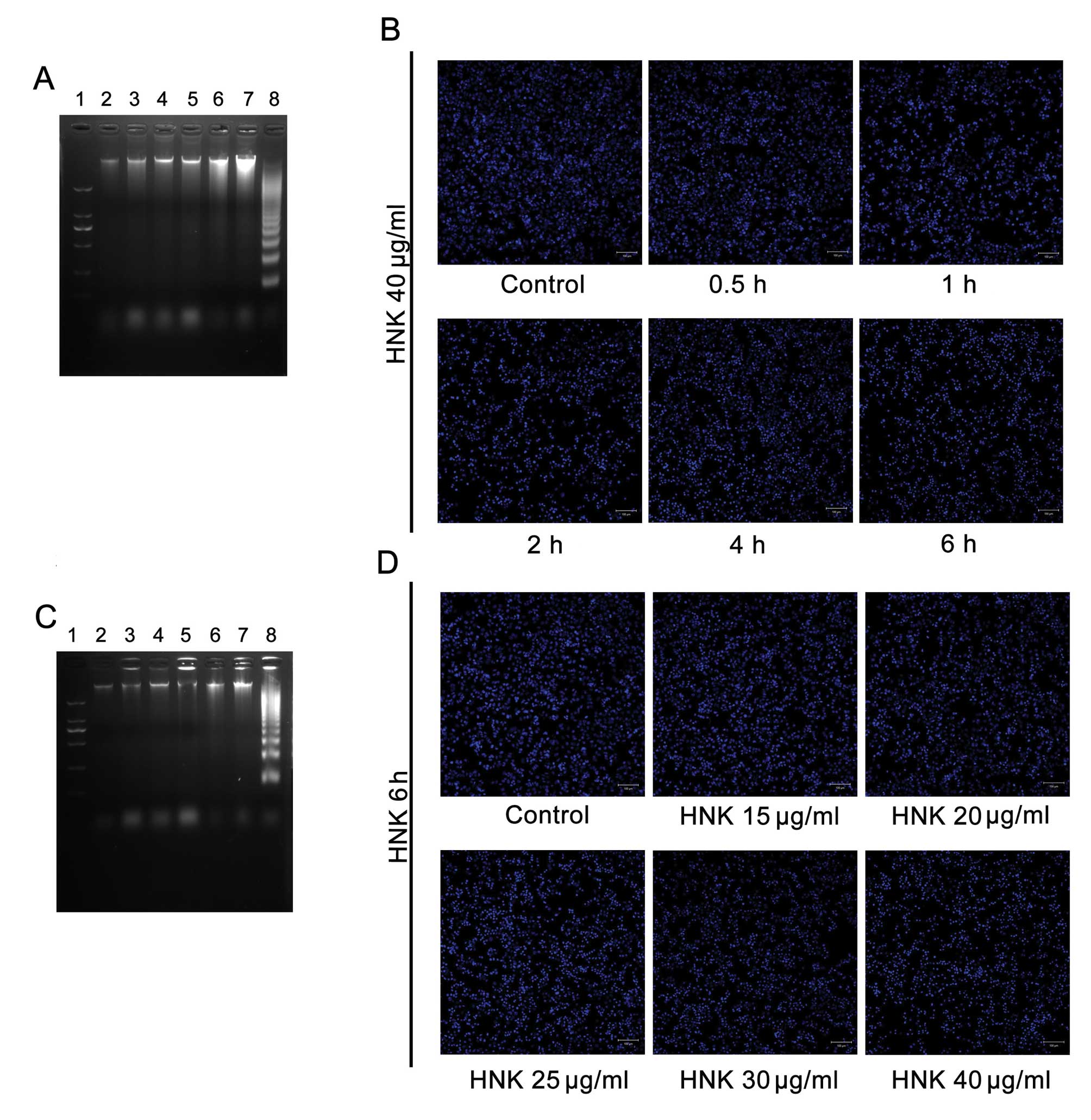 | Figure 3No marked manifestation of late-stage
apoptosis was detected by Hoechst 33342 staining and DNA laddering.
There were no obvious DNA ladder patterns in all treatment groups
after both (A) time-gradient and (C) dose-gradient treatments with
HNK. Simultaneously, no chromatin condensation (apoptotic bodies)
was detected in any treatment groups after (B) time-gradient or (D)
dose-gradient treatments with HNK. Nos. 1 to 8 marked in (A) stands
for DNA marker; control; HNK 40 μg/ml treatments for 0.5, 1,
2, 4 and 6 h in MCF-7 cells; and HL-60 cells treated with VP-16 for
6 h, respectively. Nos. 1 to 8 marked in (B) stands for DNA marker;
control; HNK 15, 20, 25, 30 and 40 μg/ml treatments for 6 h
in MCF-7 cells; and HL-60 cells treated with VP-16 for 6 h,
respectively. In DNA ladder detection, HL-60 cells treated with
VP-16 for 6 h were introduced as a positive control. |
Death modes triggered by different concentrations
(20, 25, 30 and 40 μg/ml) of HNK for 6 h were further
analyzed. Cell fraction of early-stage apoptosis decreased,
fraction of necrotic cell death conversely increased (P<0.001)
with the extension of doses (Fig.
2B–D), which demonstrated similar tendency to that of
time-dependent manner. However, no obvious DNA ladders or apoptotic
bodies were detected using DNA laddering or Hoechst 33342-staining
in any of the treatment groups (Fig.
3C and D). Such results also demonstrated HNK triggered cell
death mode transition from early-stage apoptosis to programmed
necrosis in a dose-dependent manner.
Collectively, these experimental results support the
original hypothesis that HNK triggered cell death mode transition
from apoptosis to programmed necrosis in a time- and dose-dependent
manner.
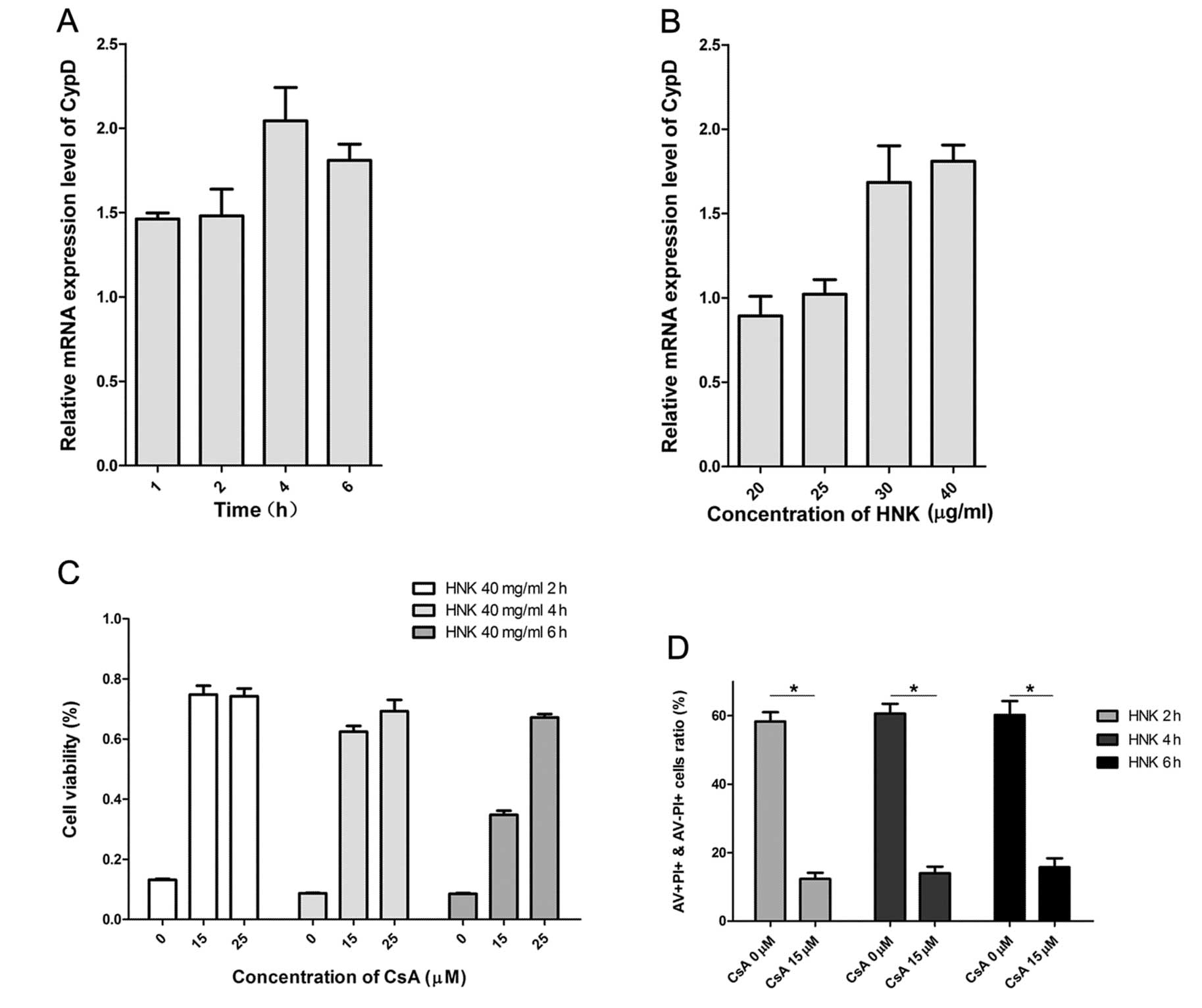
Cyclophilin D modulates HNK-induced
apoptosis to programmed necrotic cell death transition
Cyclophilin D (CypD) is a vital protein involved in
mitochondrial permeability transition (mPT) and critical regulator
in HNK-induced programmed necrotic cell death (11). Recent reports demonstrated
cyclophilin D-dependent mPT regulates some necrotic but not
apoptotic cell death (14), and
comports as apoptotic repressor (15,16).
In consequence, we explored whether HNK-induced cell death mode
transition was specifically regulated by CypD. As we previously
demonstrated, cellular CypD expression levels of mRNA and protein
coincided with each other perfectly (11). We then measured the expression
levels of CypD mRNA after HNK exposure in MCF-7 cells using RT-PCR.
CypD mRNA levels in MCF-7 cells after HNK exposure was evaluated in
all groups with different incubation durations (Fig. 4A and B). CypD mRNA levels increased
with the extension of incubating time in the early death phase and
declined in the late-phase (6 h). A possible explanation for the
declined CypD mRNA levels in late-phase is that cells are under
cellular collapse. Fig. 4B
provides details of CypD mRNA levels between groups at different
doses of incubation. CypD mRNA levels manifested a gradually
increasing tendency with the extension of doses (Fig. 4B) (P<0.001). CypD mRNA level was
suppressed in apoptosis-predominant group (HNK 20 μg/ml),
was parallel in apoptosis and necrosis-balanced group (HNK 25
μg/ml), whereas elevated in necrosis-predominant group (HNK
30 and 40 μg/ml) (Fig. 4B). These
results indicated CypD was a potential modulator of HNK-triggered
cell death mode transition time- and dose-dependently.
Cyclosporin A (CsA) targets and binds to the
mitochondria CypD, which subsequently inhibites modulated-function
of CyPD in cell death (17). We
found CsA dramatically inhibited HNK induced cell death using MTT
assay (Fig. 4C) at doses of 15 and
25 μM. Further, we used AV/PI staining methods to determine
whether HNK induced time-dependent necrotic cell death was
inhibited by CsA. We increased total dose (through increasing
volume of drug, 2 ml/well) in each well with a similar
concentration of HNK (40 μg/ml) in order to amplify its
necrotic death induction effects. Pretreatment with 15 μM
CsA for 2 h significantly inhibited HNK-induced necrotic cell death
(PI staining-positive cells) with HNK incubation of 2, 4 and 6 h,
respectively (P<0.001) (Fig.
4D). The evidence supports CypD was a potential modulator of
HNK-triggered cell death mode transition.
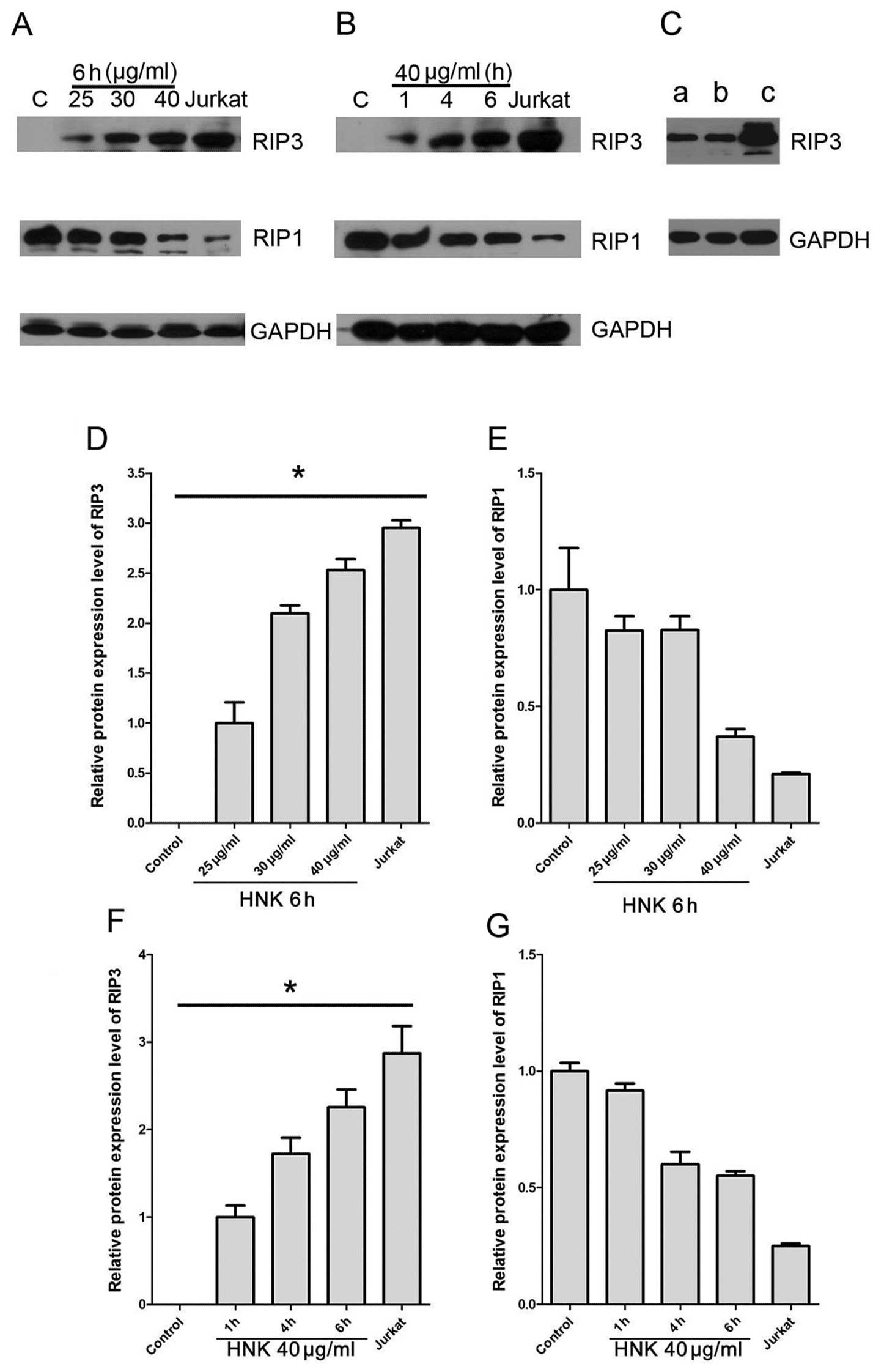
RIP3 expression correlates with
HNK-induced cell death switch from apoptosis to programmed
necrosis
Programmed necrosis is defined as necrosis highly
regulated by RIP1 and RIP3 (2).
RIP3 is essential and crucial component in the initiation of
programmed necrosis (18,19), but has no role in apoptosis
(20,21). Enhanced expression of RIP3 is
strongly associated with increased programmed necrosis (2) and enable the switch of TNF-induced
cell death from apoptosis to necrosis (20). We further assessed RIP3 expression
during HNK-triggered cell death mode transition using western blot
analysis. Untreated MCF-7 did not express any detectable RIP3,
which accords with a previous report (21). Expressions of RIP3 increased
significantly after HNK treatment in a dose- and time-dependent
manner (P<0.0001) (Fig. 5A, B, D
and F). Furthermore, in programmed necrosis predominant groups,
RIP3 expressions were approximate or more than 2 times compared
with early-apoptosis predominant groups (HNK 25 μg/ml for 6
h or HNK 40 μg/ml for 1 h) (Fig. 5A, B, D and F).
RIP1 is a critical switch in regulation cell fate,
including NF-κB activation associated cell survival and
proliferation, apoptosis and programmed necrosis (22–24).
Under caspase-inhibited condition, RIP1 and RIP3 form a so-called
‘necrosome’ complex to initiate programmed necrosis signaling
pathway (20). We further assessed
RIP1 expression during HNK treatment using western blot analysis.
Results indicated MCF-7 cells expressed reduced-RIP1 in all
treatment groups compared with baseline level of normal MCF-7 cells
(Figs. 5A, 6B). The RIP1 expression inhibition after
HNK treatments was dose- and time-dependent (Fig. 5E and G).
We also detected, despite pretreatment with 15
μM CsA for 2 h significantly inhibited HNK-induced necrotic
cell death, it did not affect enhanced RIP3 expression triggered by
HNK (Fig. 5C). Overall, these
results specifically revealed RIP3 was a potential regulator in
HNK-induced programmed necrosis and cell mode transition from
apoptosis to programmed necrosis.
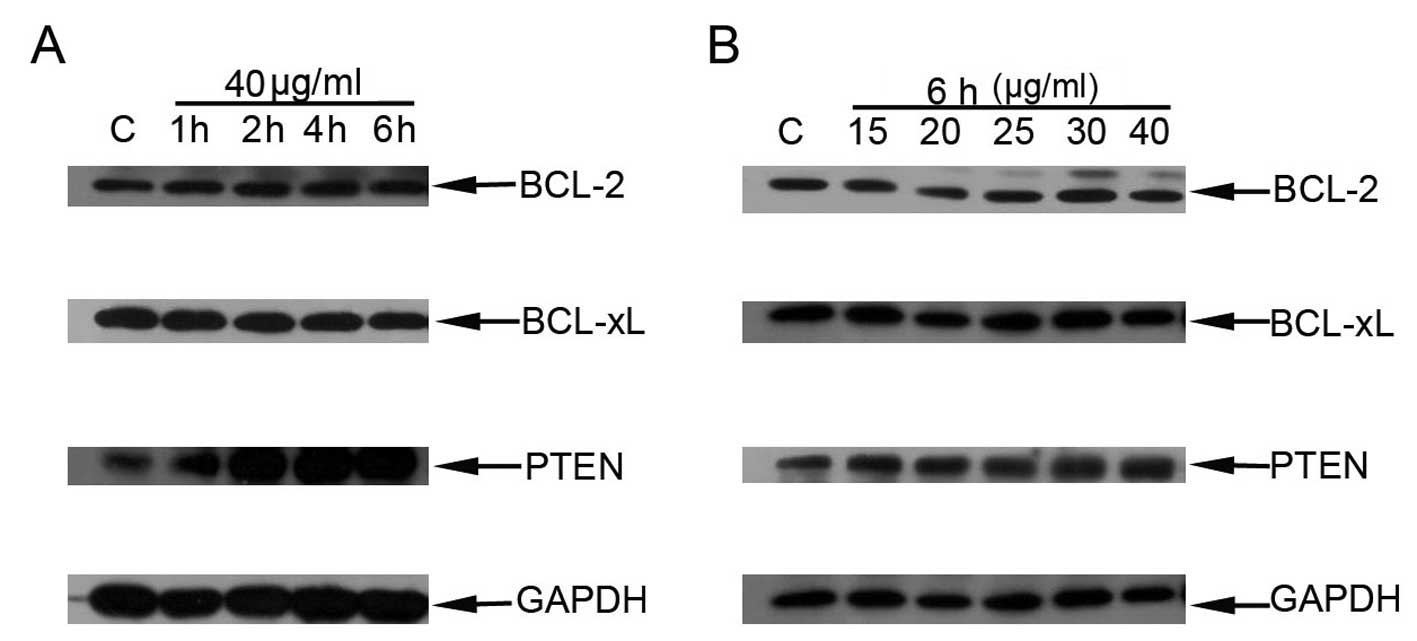
HNK induces PTEN overexpression
paralleling with cell death mode transition
Bcl-2 and Bcl-xl are pro-survival proteins of the
Bcl-2 family. They excert anti-apoptotic effect through
sequestering and inhibiting pro-apoptotic Bcl-2 proteins, which may
trigger mitochondrial outer membrane permeabilization (25–27).
Overexpression of these anti-apoptotic Bcl-2 proteins protects
cells from apoptosis induction from a broad range of apoptotic
stimuli (26). We aimed to detect
the relevance between HNK-triggered apoptosis to programmed
necrotic cell death transition and expression of Bcl-2 and Bcl-xl.
No obvious changes in expressions of Bcl-2 and Bcl-xl proteins were
detected paralleling with increased doses or treatment durations
using western blot analysis (Fig. 6A
and B).
Loss of expression or inactivation of PTEN amplifies
the PI3K-Akt-mTOR pathway, which promotes cell survival and
proliferation (28–30). PTEN protein expression during cell
death mode transition triggered by increased doses or duration of
incubation was also determined. Western blot analysis results
revealed that PTEN protein expressions increased paralleling with
HNK incubated-duration (Fig. 6A)
and apparently enhanced in necrosis-predominant group (HNK 40
μg/ml for 2, 4 and 6 h). Similarly, intensive expression of
PTEN was also detected with increased doses of HNK, especially in
necrosis-predominant group (HNK 30 and 40 μg/ml for 6 h)
(Fig. 6B).
Discussion
Cell death is classified as extrinsic apoptosis,
caspase-dependent or -independent intrinsic apoptosis, regulated
necrosis, autophagic cell death and others by measurable
biochemical features (1).
Different from conventional concept, recent studies demonstrate
necrosis can also proceed in a well-regulated manner, which is
frequently defined as programmed necrosis. Current evidence
indicates chemicals often trigger different cell death modes in a
dose-dependent manner, including autophagy or apoptosis after
lower-dose and necrosis at higher-dose exposure (13). We previously reported honokiol at
higher doses induced programmed necrosis highly-regulated by
cyclophilin D (CypD) in HL-60, MCF-7 and HEK293 cell lines
(11), whereas other groups
revealed HNK-induced apoptosis in such cell lines (31,32).
From this perspective, we supposed HNK could trigger programmed
cell death switching from apoptosis to programmed necrosis in a
dose- and time-dependent manner. Data from flow cytometric analysis
indicated that a proportion of early-stage apoptosis
(AV+ PI−) decreased and that of necrosis
(PI+) increased with extension of treatment time at a
fixed dose of HNK (40 μg/ml) in MCF-7 cells.
Internucleosomal DNA fragmentation is one of the hallmarks of
apoptosis at later stage (or degradation phase), which are simply
detectable as a ladder pattern in electrophoresis of isolated DNA.
Nuclear chromatin condensation and apoptotic bodies are both
morphological characteristic manifestations of apoptosis and are
detectable using nuclei staining by Hoechst 33342. Hoechst 33342
staining, DNA laddering demonstrated that the increased
PI+ cells were not late-stage apoptosis as no obvious
DNA ladder pattern or chromatin condensation (apoptotic bodies)
were detected. These results reveal that with the extension of
treatment time, HNK 40 μg/ml triggered early-stage apoptosis
which did not switch to late-stage apoptosis but to programmed
necrosis. Similar results were obtained with dose extension of HNK
with a fixed treatment time (6 h). Such experimental results in the
current study provide substantial evidence for the original
assumptions and confirm HNK triggered cell death mode transition
from early-stage apoptosis to programmed necrosis.
The most well-characterized form of programmed
necrosis is ‘necroptosis’, which is triggered by TNF and dependent
on the assembly of RIP homotypic interaction motif (RHIM)-dependent
necrotic signaling complex of RIP1 and RIP3 (3,21,22,33,34).
Expression of RIP1 or RIP3 is frequently-used to determine the
existence of programmed necrosis. In necroptosis, RIP1 is critical
component in formation of necrotic complex ‘necrosome’. RIP1
protein is frequently overexpressed in cells that have undergone
necroptosis and couples with RIP3 forming a functional amyloid
signaling complex required for necroptosis (22). Our results demonstrated RIP1
protein expression was downregulated in a dose- and time-dependent
manner coupling with HNK triggered cell death mode transition.
Consequently, these results indicate HNK-induced programmed
necrosis and cell death mode transition are potentially
RIP1-independent. Although distinct from necroptosis, it is still
explainable that programmed necrosis can also proceed in the
absence of RIP1 in some cell context triggered by certain stimuli,
such as double-stranded DNA virus (murine cytomegalovirus, MCMV)
and toxic stress (35–37). Different from RIP1, current
evidence indicates RIP3 is indispensable for programmed necrosis
and participates in constitution of all demonstrated necrotic
complexes triggered by various stimuli (3). Data in the current study revealed
protein expression of RIP3 was upregulated in a dose- and
time-dependent manner parallelling with HNK-triggered cell death
mode transition. These data provide additional evidence that HNK
triggered programmed necrosis and cell death modes transition may
initiate and be highly-regulated by RIP3. RIP3 can trigger
programmed necrosis without RIP1. Besides RIP1, RIP3 may also
either form a homotypic complex or interact with other cellular
RHIM-containing proteins (such as DNA-dependent activator of
interferon regulatory factors, DAI) to form a necrotic complex and
trigger a RIP1-independent necrosis (3,33,38).
We speculate HNK-triggered programmed necrosis and cell death modes
transition maybe highly-regulated by RIP3 through assembly of a
necrotic complex with some other proteins besides RIP1 and initiate
programmed necrosis process. The definite signal pathway initiating
the HNK-induced programmed necrosis still needs further
evaluation.
Although current evidence associated with execution
process of programmed necrosis is still unclear, a series of
cellular events are revealed downstream of RIP1/RIP3-containing
complex activation. These cellular events generally take place in
mitochondria and include reactive oxygen species (ROS)
over-production, cellular ATP depletion, enhance glycogenolysis,
glycolysis and glutaminolysis. Mitochondrial permeability
transition (mPT) is a crucial event in signaling network of cell
death, which participates in regulation of both apoptosis and
programmed necrosis. Recent evidence indicates cyclophilin D (CypD)
is a vital protein involved in the mPT and acts as ‘gatekeeper’,
which regulates some necrotic but not apoptotic cell death
(14), and comports as apoptotic
repressor (15,16). We previously reported CypD is a
critical regulator in HNK induced programmed necrotic cell death,
downregulated CypD by Cyps inhibitor (CsA) and siRNAs can
significantly suppress HNK-induced programmed necrosis (11). In the current study we revealed
CypD modulated HNK-triggered cell death mode transition. CypD
expression gradually increased in parallel with HNK-triggered cell
death switch from early-stage apoptosis to programmed necrosis.
Pretreatment with CsA observably increased cell viabilities and
inhibited HNK-induced necrosis. These results indicate upregulated
CypD expression may block the apoptosis process between early and
late stage and trigger the switch from early-stage apoptosis to
programmed necrosis in MCF-7 cells after HNK treatment.
Our results indicate CypD may act downtream of RIP3
as a pivotal modulator in execution process of programmed necrosis
for the following reasons: i) RIP3 is essential and crucial
component in the initiation of programmed necrosis, while
mitochondrial events act downstream of the RIP1/RIP3-contained
complex activation and take responsibility for execution of
programmed necrosis; ii) cyclophilin D (CypD) is a vital protein
involved in the mPT and regulate mitochondrial events; iii) our
results indicated CsA blocked HNK triggered programmed necrosis but
without influence on RIP3 expression.
Bcl-2 and Bcl-xl are anti-apoptotic proteins of
Bcl-2 family. They inhibit pro-apoptotic Bcl-2 proteins to suppress
their triggered mitochondrial outer membrane permeabilization and
perform anti-apoptotic effects (25–27).
Proteins of Bcl-2 family regulate programmed necrosis (3), which is evidenced as TNF-induced
programmed necrosis may be restrained by Bcl-xl (39). We also assessed their expressions
after time gradient and concentration gradient treatments of HNK.
Results indicate HNK does not conspicuously affect Bcl-2 and Bcl-xl
expressions. We have previously demonstrated overexpression of
Bcl-2 and Bcl-xl did not apparently suppress HNK triggered necrosis
(11). Based on this evidence,
Bcl-2 and Bcl-xl do not participate in regulation of HNK triggered
necrosis. On the other hand, these results are in variance with
previous reports, which demonstrate Bcl-2 and Bcl-xl are frequently
downregulated during HNK-induced apoptosis in a series of tumor
cell lines (6,40–44).
A possible explanation for this difference is that Bcl-2 takes
different roles in the process of apoptosis and necrosis. Besides,
previously evidence demonstrates CypD may exert anti-apoptotic
effect through interacting with Bcl-2 (16). Existence of interation between CypD
and Bcl-2 (and other anti-apoptotic proteins) in HNK-triggered cell
death mode transition still needs further evaluation.
This study revealed that HNK has dramatic antitumor
effects in breast cancer cell lines. It triggers cell death mode
transition from early-stage apoptosis to programmed necrosis time-
and dose-dependently. This programmed necrosis and death mode
transition may be potentially RIP3-dependent. The death mode
transition and execution process of programmed necrosis is highly
regulated by cyclophilin D.
Acknowledgments
The authors are thankful for the research grants
from National Natural Science Foundation of China (no. 30901741)
and the Department of Education of Zhejiang Province of China (no.
Y201225802).
References
|
1.
|
Galluzzi L, Vitale I, Abrams JM, et al:
Molecular definitions of cell death subroutines: recommendations of
the Nomenclature Committee on Cell Death 2012. Cell Death Differ.
19:107–120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Vanlangenakker N, Vanden Berghe T and
Vandenabeele P: Many stimuli pull the necrotic trigger, an
overview. Cell Death Differ. 19:75–86. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Han J, Zhong CQ and Zhang DW: Programmed
necrosis: backup to and competitor with apoptosis in the immune
system. Nat Immunol. 12:1143–1149. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Tian W, Xu D and Deng YC: Honokiol, a
multifunctional tumor cell death inducer. Pharmazie. 67:811–816.
2012.PubMed/NCBI
|
|
5.
|
Battle TE, Arbiser J and Frank DA: The
natural product honokiol induces caspase-dependent apoptosis in
B-cell chronic lymphocytic leukemia (B-CLL) cells. Blood.
106:690–697. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Park EJ, Min HY, Chung HJ, et al:
Down-regulation of c-Src/EGFR-mediated signaling activation is
involved in the honokiol-induced cell cycle arrest and apoptosis in
MDA-MB-231 human breast cancer cells. Cancer Lett. 277:133–140.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Raja SM, Chen S, Yue P, et al: The natural
product honokiol preferentially inhibits cellular FLICE-inhibitory
protein and augments death receptor-induced apoptosis. Mol Cancer
Ther. 7:2212–2223. 2008. View Article : Google Scholar
|
|
8.
|
Shigemura K, Arbiser JL, Sun SY, et al:
Honokiol, a natural plant product, inhibits the bone metastatic
growth of human prostate cancer cells. Cancer. 109:1279–1289. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Chen YJ, Wu CL, Liu JF, et al: Honokiol
induces cell apoptosis in human chondrosarcoma cells through
mitochondrial dysfunction and endoplasmic reticulum stress. Cancer
Lett. 291:20–30. 2010. View Article : Google Scholar
|
|
10.
|
Mannal PW, Schneider J, Tangada A,
McDonald D and McFadden DW: Honokiol produces anti-neoplastic
effects on melanoma cells in vitro. J Surg Oncol. 104:260–264.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Li L, Han W, Gu Y, et al: Honokiol induces
a necrotic cell death through the mitochondrial permeability
transition pore. Cancer Res. 67:4894–4903. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Xu D, Lu Q and Hu X: Down-regulation of
P-glycoprotein expression in MDR breast cancer cell MCF-7/ADR by
honokiol. Cancer Lett. 243:274–280. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Orrenius S, Nicotera P and Zhivotovsky B:
Cell death mechanisms and their implications in toxicology. Toxicol
Sci. 119:3–19. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Nakagawa T, Shimizu S, Watanabe T, et al:
Cyclophilin D dependent mitochondrial permeability transition
regulates some necrotic but not apoptotic cell death. Nature.
434:652–658. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Schubert A and Grimm S: Cyclophilin D, a
component of the permeability transition-pore, is an apoptosis
repressor. Cancer Res. 64:85–93. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Eliseev RA, Malecki J, Lester T, Zhang Y,
Humphrey J and Gunter TE: Cyclophilin D interacts with Bcl2 and
exerts an anti-apoptotic effect. J Biol Chem. 284:9692–9699. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Rasola A and Bernardi P: The mitochondrial
permeability transition pore and its involvement in cell death and
in disease pathogenesis. Apoptosis. 12:815–833. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Zhang DW, Zheng M, Zhao J, et al: Multiple
death pathways in TNF-treated fibroblasts: RIP3- and RIP1-dependent
and independent routes. Cell Res. 21:368–371. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Kreuzaler P and Watson CJ: Killing a
cancer: what are the alternatives? Nat Rev Cancer. 12:411–424.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Zhang DW, Shao J, Lin J, et al: RIP3, an
energy metabolism regulator that switches TNF-induced cell death
from apoptosis to necrosis. Science. 325:332–336. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
He S, Wang L, Miao L, et al: Receptor
interacting protein kinase-3 determines cellular necrotic response
to TNF-alpha. Cell. 137:1100–1111. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Li J, McQuade T, Siemer AB, et al: The
RIP1/RIP3 necrosome forms a functional amyloid signaling complex
required for programmed necrosis. Cell. 150:339–350. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Moquin D and Chan FK: The molecular
regulation of programmed necrotic cell injury. Trends Biochem Sci.
35:434–441. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Declercq W, Vanden Berghe T and
Vandenabeele P: RIP kinases at the crossroads of cell death and
survival. Cell. 138:229–232. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Kurokawa M and Kornbluth S: Caspases and
kinases in a death grip. Cell. 138:838–854. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Kelly PN and Strasser A: The role of Bcl-2
and its pro-survival relatives in tumourigenesis and cancer
therapy. Cell Death Differ. 18:1414–1424. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Youle RJ and Strasser A: The BCL-2 protein
family: opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Song MS, Salmena L and Pandolfi PP: The
functions and regulation of the PTEN tumour suppressor. Nat Rev Mol
Cell Biol. 13:283–296. 2012.PubMed/NCBI
|
|
29.
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Martelli AM, Evangelisti C, Chappell W, et
al: Targeting the translational apparatus to improve leukemia
therapy: roles of the PI3K/PTEN/Akt/mTOR pathway. Leukemia.
25:1064–1079. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Wolf I, O’Kelly J, Wakimoto N, et al:
Honokiol, a natural biphenyl, inhibits in vitro and in
vivo growth of breast cancer through induction of apoptosis and
cell cycle arrest. Int J Oncol. 30:1529–1537. 2007.
|
|
32.
|
Liu H, Zang C, Emde A, et al: Anti-tumor
effect of honokiol alone and in combination with other anti-cancer
agents in breast cancer. Eur J Pharmacol. 591:43–51. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Mocarski ES, Upton JW and Kaiser WJ: Viral
infection and the evolution of caspase 8-regulated apoptotic and
necrotic death pathways. Nat Rev Immunol. 12:79–88. 2012.PubMed/NCBI
|
|
34.
|
Sun L, Wang H, Wang Z, et al: Mixed
lineage kinase domain-like protein mediates necrosis signaling
downstream of RIP3 kinase. Cell. 148:213–227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Motani K, Kushiyama H, Imamura R,
Kinoshita T, Nishiuchi T and Suda T: Caspase-1 protein induces
apoptosis-associated speck-like protein containing a caspase
recruitment domain (ASC)-mediated necrosis independently of its
catalytic activity. J Biol Chem. 286:33963–33972. 2011. View Article : Google Scholar
|
|
36.
|
Cho YS, Challa S, Moquin D, et al:
Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates
programmed necrosis and virus-induced inflammation. Cell.
137:1112–1123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Upton JW, Kaiser WJ and Mocarski ES: Virus
inhibition of RIP3-dependent necrosis. Cell Host Microbe.
7:302–313. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Rebsamen M, Heinz LX, Meylan E, et al:
DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction
motifs to activate NF-kappaB. EMBO Rep. 10:916–922. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Ono K, Wang X, Kim SO, Armstrong LC,
Bornstein P and Han J: Metaxin deficiency alters mitochondrial
membrane permeability and leads to resistance to TNF-induced cell
killing. Protein Cell. 1:161–173. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Jeong JJ, Lee JH, Chang KC and Kim HJ:
Honokiol exerts an anticancer effect in T98G human glioblastoma
cells through the induction of apoptosis and the regulation of
adhesion molecules. Int J Oncol. 41:1358–1364. 2012.
|
|
41.
|
Arora S, Bhardwaj A, Srivastava SK, et al:
Honokiol arrests cell cycle, induces apoptosis, and potentiates the
cytotoxic effect of gemcitabine in human pancreatic cancer cells.
PLoS One. 6:e215732011. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Han LL, Xie LP, Li LH, Zhang XW, Zhang RQ
and Wang HZ: Reactive oxygen species production and Bax/Bcl-2
regulation in honokiol-induced apoptosis in human hepatocellular
carcinoma SMMC-7721 cells. Environ Toxicol Pharmacol. 28:97–103.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Deng J, Qian Y, Geng L, et al: Involvement
of p38 mitogen-activated protein kinase pathway in honokiol-induced
apoptosis in a human hepatoma cell line (hepG2). Liver Int.
28:1458–1464. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Hahm ER, Arlotti JA, Marynowski SW and
Singh SV: Honokiol, a constituent of oriental medicinal herb
Magnolia officinalis, inhibits growth of PC-3 xenografts in
vivo in association with apoptosis induction. Clin Cancer Res.
14:1248–1257. 2008.PubMed/NCBI
|