Introduction
Irinotecan (CPT-11) is a camptothecin derivative
(CPT) selectively targeting the nuclear enzyme DNA topoisomerase I.
Like other CPTs, irinotecan-mediated cytotoxicity is mediated by a
two-step process. This includes drug-mediated accumulation of
reversible DNA-topoisomerase I complexes (called cleavable
complexes) that are associated with transient DNA single strand
breaks. Subsequently, these complexes are converted into permanent
DNA single and double strand breaks by the replication fork during
the S phase of the cell cycle (1–4). As
a result, CPTs are preferentially toxic toward S phase cells
(5–7).
Irinotecan is a major drug for treatment of patients
with metastatic colorectal cancer (CRC) (8,9) and
a promising agent for other applications such as gastric cancer.
However, its clinical activity is limited by both intrinsic
(natural) and acquired drug resistance. Patients with CRC can be
divided into two major groups according to the type of genetic
instability displayed by the tumor (10,11).
The major group (>80% of patients) is characterized by numerical
and/or structural chromosome instability (CIN) associated with loss
of heterozygosity (LOH) and aneuploidy. The other group (∼15% of
patients) is characterized by microsatellite instability (MSI/MIN).
MSI is linked to dysfunction of the DNA mismatch repair (MMR)
system, which is involved in the correction of base/base mismatches
and insertion/deletion loops during replication (12). Interestingly, it has been reported
that irinotecan is selectively active toward MMR-deficient tumor
cells in vitro as in patients (13,14)
although this needs further confirmation.
Acquired CPT resistance has been associated with
reduced drug uptake, decreased topoisomerase I expression and/or
TOP1 mutations (14–21).
In comparison, surprisingly little is known about the influence of
prolonged CPT exposure on the cell cycle machinery. This issue is
likely to gain importance in the years to come considering the
advent of novel liposomal and pegylated preparations of irinotecan
and its major metabolite SN-38 (22,23).
We have developed two independent CPT-resistant
human CRC cell lines, one derived from HT-29 cells (CIN) the other
from HCT-116 cells (MSI) by prolonged exposure to increasing
concentrations of SN-38 in the growth media. Here, we report that
in addition to classical resistance mechanisms, CPT resistance is
accompanied by increased generation doubling time, a decreased S
fraction and an increased G2 fraction in vitro as in
vivo. As a consequence, SN-38-resistant cells show
cross-resistance to S-phase selective agents such as
5-fluorouracil. SN-38 resistance is accompanied by increased basal
levels of γ-H2AX and phospho-Chk2 without notable changes in the
levels of phospho-Chk1. Taken together, our results indicate that
prolonged exposure to CPTs is accompanied by stable modifications
of cell cycle dynamics. This could have profound impact on tumor
sensitivity to a wide range of antitumor agents and may influence
tumor progression in patients.
Materials and methods
Drugs and chemicals
Irinotecan was purchased from Pfizer whereas SN-38
(7-Ethyl-10-Hydroxy-20(S)-Camptothecin) was obtained from Abatra
Technology (Sophia Ho, China). 5-Fluorouracil was purchased from
Teva-Pharma. MTT
(3-[4,5-dimethylthiazol-2-yl])-2,5-diphenyltetrazolium bromide),
propidium iodide, 4′,6-diamidino-2-phenylindole (DAPI) and RNase A
were obtained from Sigma.
Cells and culture medium
HCT-116 and HT-29 colorectal carcinoma (CRC) cells
were generously provided by Bert Vogelstein (Baltimore, MD) and
Richard Camalier (Division of Cancer Treatment and Diagnosis Tumor
Repository, NCI), respectively. The cells were maintained in Mc
Coy’s 5A (HCT-116) or DMEM medium (HT-29) from PAA supplemented
with 5% fetal calf serum (FCS, Eurobio), 100 U/ml penicillin and
100 μg/ml streptomycin.
To obtain SN-38-resistant cells, HT-29 and HCT-116
cells were exposed to increasing concentrations of SN-38 over 6–9
months as previously described (25). Specifically, cells in log phase
were first exposed to the IC50 dose of SN-38. Once
surviving cells reached 80% confluence, they were passaged twice
per week at the same concentration of SN-38. The process was
repeated with increasing doses of SN-38 until a resistant cell
population was obtained. Cells were routinely maintained in
drug-free media except for a single three day drug exposure every
three weeks. All experiments were carried out with cells grown
without drug for at least one week. The resistant phenotype was
stable over a least 20 passages in the absence of drug.
Cytotoxicity assay
The MTT (tetrazolium dye
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide],
Sigma) assay was used to determine the growth inhibitory effects of
SN-38 after 120 h continuous drug exposure as previously described
(26–28). The IC50 value
corresponds to the drug concentration inhibiting cell growth by 50%
compared with the growth of untreated control cells. All values are
averages of at least three independent experiments performed in
duplicate.
Clonogenic assay
The colony formation assay was carried out as
previously described (29).
Briefly, cells (500–2,000) were plated in 60-mm Petri dishes and
incubated overnight. Exponentially growing cells were exposed to
different drug concentrations for ten days and the colonies were
fixed with ethanol, stained with 0.5% crystal violet and examined
under a stereomicroscope. Colonies of 50 or more cells were
considered to originate from viable cells. The IC50
value corresponds to the drug concentration inhibiting colony
formation by 50% compared to the number of colonies for untreated
control cells. All values are averages of at least three
independent experiments, each performed in duplicate.
Single cell gel electrophoresis (comet
assay)
Cells were exposed to the indicated drug
concentrations for 20 min at 37°C in the dark and subjected to
single cell electrophoresis (comet assay) under alkaline conditions
as described previously (30,31).
Image analysis was performed using Komet 5.5 software (Kinetic
Imaging, Nottingham, UK) to determine the percentage of nuclear DNA
present in the comet tail. A minimum of 100 cells were analyzed per
sample. Values represent the average of at least two independent
experiments.
Relative quantitative RT-PCR
mRNA expression of TOP1 was evaluated by
relative quantitative real-time PCR (qRT-PCR) as previously
described (32,33). Validated QuantiTect®
Primer Assays (Qiagen) were used for amplification. All
quantifications were done in duplicate for three independent
experiments and normalized with respect to the endogenous β-actin
(ACTB) mRNA levels for each reaction. Target cDNA expression
was quantified using the comparative Ct method and
expressed as the fold change in samples from SN-38-resistant cells
vs samples from the corresponding parental cell lines.
TOP1 sequencing
Total RNA was extracted using RNeasy Plus mini kit
(Qiagen) and reverse-transcribed using the RevertAid Premium
reverse transcriptase (Fermentas). The resulting cDNA was used to
amplify the entire TOP1 open reading frame by polymerase
chain reaction (PCR) and PCR products were sequenced
bidirectionally (Eurofins MWG Operon, Ebersberg, Germany).
Drug uptake
HPLC was performed as previously described (34) with minor modifications. For each
cell line, three independent samples of five millions cells were
incubated with 150 nM SN-38 for 20 min at 37°C, rapidly washed in
ice-cold PBS and scraped in 2 ml of ice-cold methanol. Samples were
centrifuged at 4°C (800 g for 7 min), the cell pellets were
suspended in 1 ml methanol and samples (50 μl each)
subjected to HPLC analysis.
The chromatographic detection was achieved by using
an Atlantis-C18 (5 μm, 250×4.6 mm) analytical column
(Waters) maintained at room temperature and protected by a C18
guard cartridge (Waters). The mobile phase was a mixture of 75% 20
mM ammonium acetate, pH 3.4 and 25% acetonitrile with a flow rate
of 1.5 ml/min. The fluorescence detector excitation wavelength was
368 nm and the emission wavelength 515 nm. Run time for each
analysis was 13 min. The retention time for SN-38 was 9.8 min and
the detection limit was 0.5 ng/ml. Data collection and processing
were performed using Millenium software (Waters). Each sample was
analyzed at least twice.
Western blot analysis
Western blot analysis was carried out as described
previously (35,36). The following primary antibodies
were used: rabbit anti-ABCG2 (#4477), rabbit anti-pSer317 Chk1
(#2344), rabbit anti-pThr68 Chk2 (#2661), mouse anti-Chk1 (#2345)
and mouse anti-Chk2 (#2662), all from Cell Signaling. Rabbit
anti-Topo I (sc-10783) was from Santa Cruz, mouse anti-Pgp
(#517312) was from Calbiochem, while mouse anti-β-actin (#A5441)
was purchased from Sigma. The secondary antibodies include
horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit
secondary antibodies (Jackson ImmunoResearch Laboratories) while
the enhanced chemiluminescence reagent was obtained from
Amersham.
Flow cytometry analysis
The cell cycle distribution was measured by flow
cytometry using a FACSCalibur flow cytometer (Becton-Dickinson) as
previously described (37,38). The expression of phosphorylated
histones was determined with help of the following antibodies:
mouse anti-pSer139 histone H2AX antibody (#05-636, Millipore) and
mouse anti-pSer10 histone H3 (Cell Signaling, 9706).
Immunocytochemistry and image
acquisition
Cells were grown on coverslips and prepared for
immunocytochemistry as described previously (39,40).
Epitopes were detected with the following primary antibodies: mouse
anti-pSer139 histone H2AX, rabbit anti-Cyclin B1 (#4138, Cell
Signaling) or mouse anti-pSer1981 ATM (#4526, Cell Signaling)
followed by anti-rabbit or anti-mouse CyTM3-conjugated secondary
antibodies from Jackson ImmunoResearch Laboratories. Cells were
washed, counterstained with DAPI and mounted with Vectashield
(Vector Laboratories) before being subjected to microscopy.
Fluorescent images were captured using an inverted microscope
(Olympus CKX41) and digital compact camera (Olympus Camedia C4000)
and the fluorescence intensities were determined using the
MetaMorph software (Universal Imaging Corporation) for quantitative
analysis.
Immunohistochemistry
To measure in vivo DNA synthesis, the
thymidine analog 5-ethynyl-2′-deoxyuridine (EdU, Life Technologies)
was administered 48 h before sacrifice as previously described
(41). Incorporated EdU was
revealed by a fluorescent-azide coupling reaction (Click-iT, Life
Technologies) of paraffin-embedded tumor samples and counterstained
by DAPI to reveal the nuclei of individual cells. For quantitative
analysis of in vivo DNA synthesis (EdU incorporation), the
data represent the ratios between EdU-positive cells and the total
number of viable cells and are the averages of five fields/tumor
(each field representing approximately 1,700 cells) from three
different tumors. All images were captured with a fluorescence
microscope, and the fluorescence intensities were determined by the
MetaMorph software for quantitative analysis.
Xenograft models
The antitumor activity of irinotecan was evaluated
in athymic mice (female NMRI-Foxn1, 6 weeks old) from Taconic
(Skensved, Denmark) bearing HT-29, HT-29/SN-38, HCT-116 or
HCT-116/SN-38 CRC xenografts as described previously (41). Briefly, two to six million cells
were injected into the right flank and the treatments were started
when the tumors were palpable (median tumor volume ∼100
mm3). Animals were weighed daily and tumor sizes were
determined three times per week. Measurements (in millimeter) were
made in two dimensions (width and length) and tumor volumes were
calculated as A×B2/2, where A is tumor length and B is
tumor width as described previously (41).
For determination of drug sensitivity, mice were
treated with 35 mg/kg irinotecan i.p. every four day or with 25
mg/kg 5-FU i.p. on days 1, 2, 15 and 16 and the tumor growth
inhibition was determined by comparison with the tumor growth of
untreated controls.
Statistical analysis
The statistical analysis of experimental data was
performed using a Student’s paired t-test, and results are
presented as mean ± standard deviation (SD).
Results
Establishment of SN-38-resistant cell
lines
SN-38-resistant cells were established by continuous
exposure to increasing concentrations of SN-38 in the growth media
for 6–9 months as previously described (25). The cytotoxicity of SN-38 toward
parental and resistant CRC cells was determined by the MTT
viability assay after 120 h continued drug exposure. The parental
HCT-116 cells (MSI) were more sensitive than the parental HT-29
cells (CIN) with IC50 values of 5 and 12 nM,
respectively. In contrast, the resistance to SN-38 was more
pronounced for HCT-116/SN-38 cells than for HT-29/SN-38 cells with
IC50 values of 90 nM (18-fold resistance) and 60 nM
(5-fold resistance), respectively.
The formation of drug-induced
single-stranded DNA breaks is reduced in SN-38-resistant cells
The CPT-induced cleavable complexes are associated
with the formation of transient DNA single-strand breaks and can
thus be determined by the alkaline comet assay (single cell
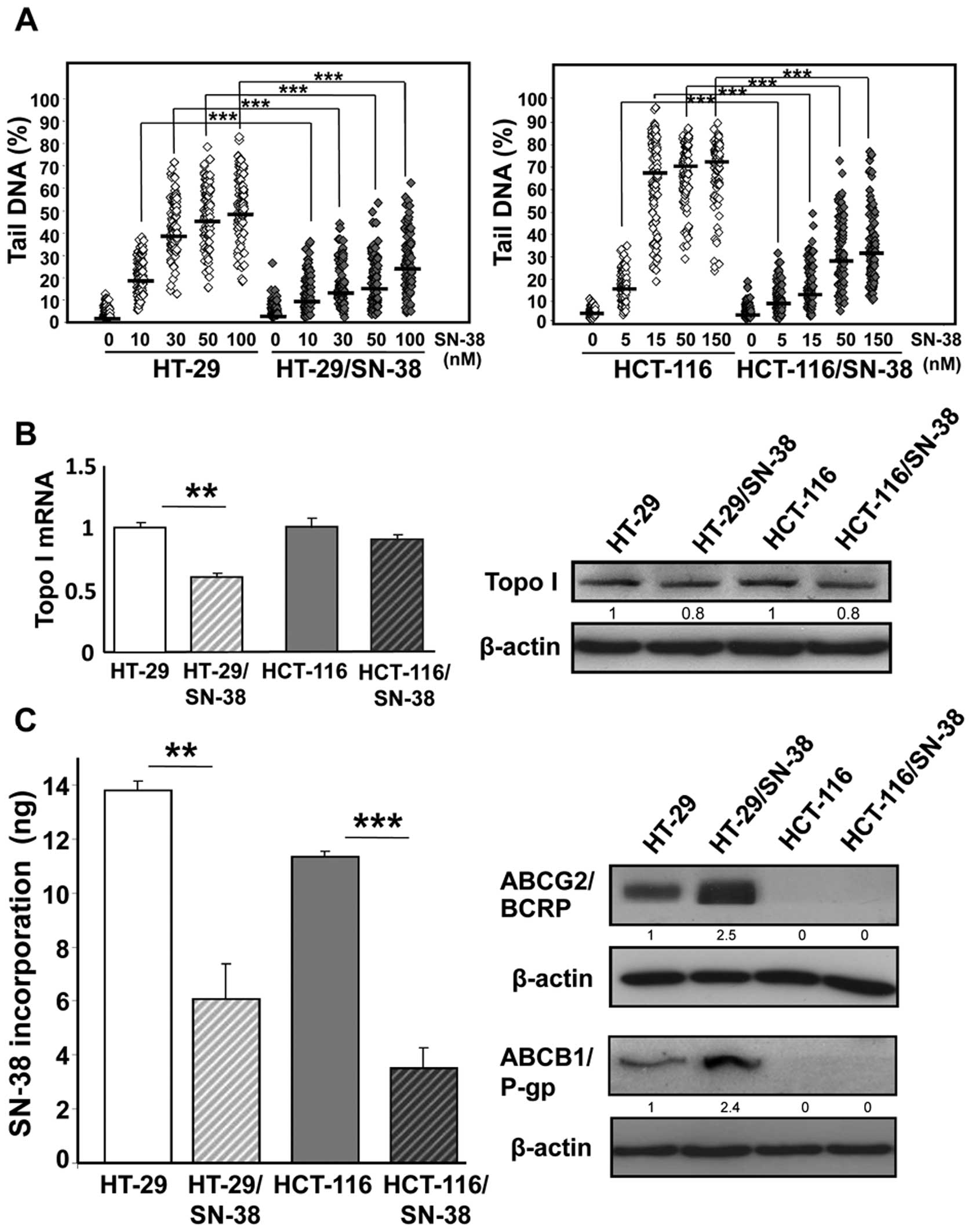
electrophoresis) after brief drug exposure (42). The results (Fig. 1A) show that the development of
SN-38 resistance was accompanied by a significant reduction in the
levels of DNA single stranded breaks (that is cleavable complexes)
at all doses examined for both HT-29/SN-38 and HCT-116/SN-38 cells,
compared with the respective parental cells (p<0.001).
Expression of topoisomerase I in parental
and SN-38-resistant cells
The attenuation of drug-induced DNA single-strand
breaks can result from decreased levels of topoisomerase I. qRT-PCR
analysis showed that the mRNA levels of topoisomerase I were
significantly decreased (p<0.01) in HT-29/SN-38 cells whereas no
notable changes were observed for HCT-116/SN-38 cells (Fig. 1B, left panel). Western blot
analysis of topoisomerase I indicated a modest decrease (∼20%) in
topoisomerase I protein levels for both resistant cells lines,
compared with the respective parental cells (Fig. 1B, right panel). These findings
indicate that the important differences between parental and
SN-38-resistant cells in the formation of DNA single-strand breaks
(Fig. 1A) can not be explained by
attenuation of topoisomerase I protein levels alone.
Topoisomerase I sequencing of parental
and SN-38 resistant CRC cells
To determine whether the decreased formation of
cleavable complexes could result from TOP1 mutations, we
sequenced the full open reading frame of TOP1 in parental
and SN-38-resistant HCT-116 and HT-29 cells. No mutations were
found in any of the four cell lines, demonstrating that resistance
to SN-38 is not linked to topoisomerase I mutations (data not
shown).
Drug accumulation is reduced in
SN-38-resistant cells
Next, the accumulation of SN-38 in parental and
resistant cells was determined by HPLC analysis after brief
exposure to SN-38 (150 nM). The results (Fig. 1C, left panel) show that SN-38
resistance was accompanied by 2- to 3-fold reduction in SN-38
accumulation for both HT-29/SN-38 cells (p<0.01) and
HCT-116/SN-38 cells (p<0.001).
Irinotecan resistance has been associated with
increased expression of drug efflux pumps belonging to the ABC
transporter superfamily including ABCG2/BCRP and
ABCB1/p-glycoprotein (21,22). In agreement, both proteins were
upregulated in HT-29/SN-38 cells, compared with HT-29 parental
cells. In comparison, no expression of P-glycoprotein or BCRP was
detected in either parental or SN-38 resistant HCT-116 cells
(Fig. 1C, right panel).
Growth and cell cycle dynamics are
altered in SN-38-resistant cells
Interestingly, both SN-38-resistant cells grow
slower than the corresponding parental cells with doubling times of
24 vs 30 h for HT-29 and HT-29/SN-38 cells, respectively, and 22 vs
29 h for HCT-116 and HCT-116/SN-38 cells, respectively (data not
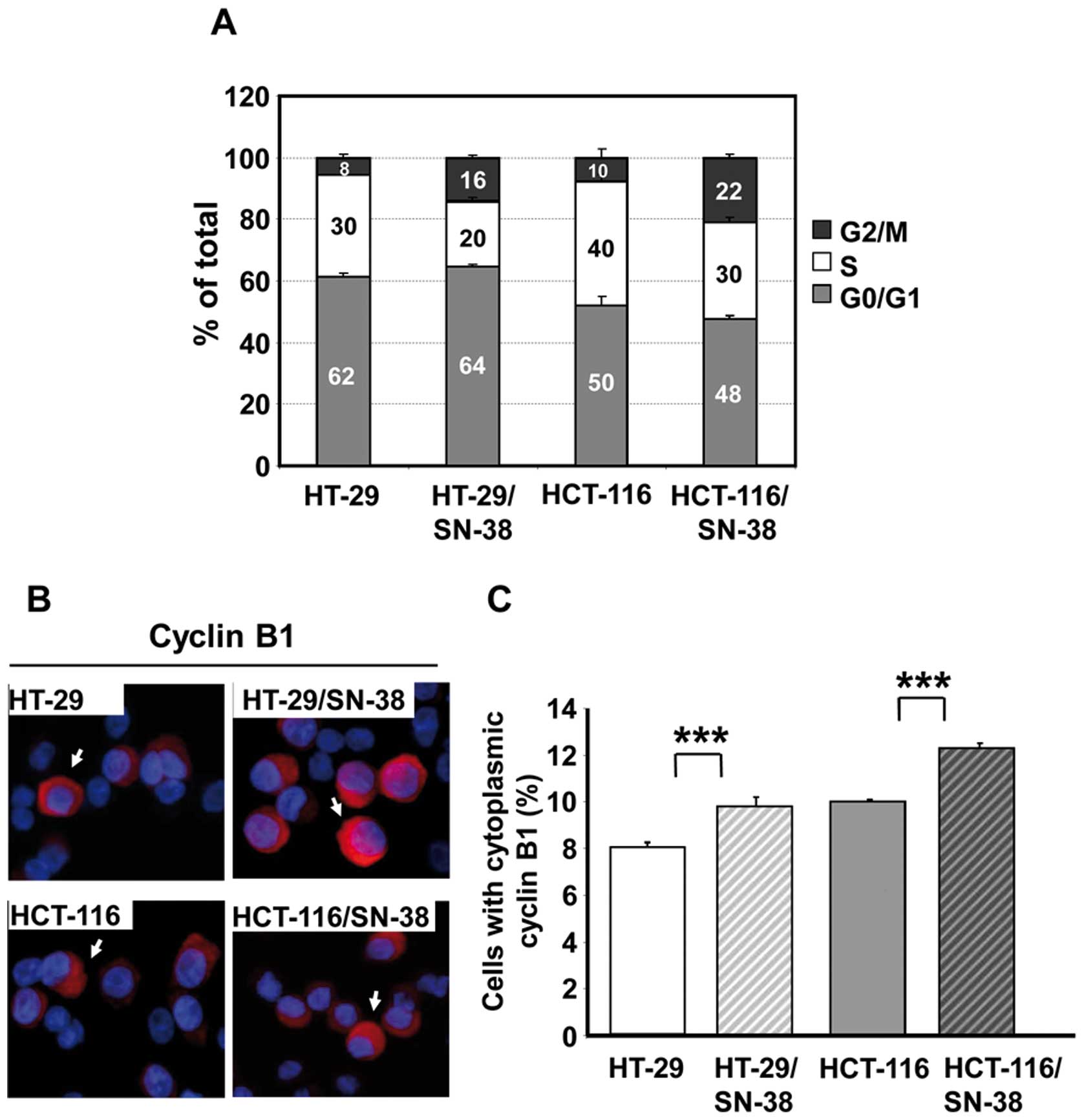
shown). Cell cycle analysis revealed that the S phase fraction was
reduced by one-fourth to one-third for both SN-38-resistant cell
lines, whereas the G2/M fraction had at least doubled (Fig. 2A). Additional analysis for
Ser10-phosphorylated histone H3, a mitotic marker (43), revealed no important differences in
the fraction of phospho-H3 positive cells which varied between 3.8
and 4.5% for all four cell lines (data not shown). Therefore, the
increased G2/M fraction is due to an important increase of cells in
the G2 phase of the cell cycle.
For further validation, the cellular distribution of
cyclin B1 was determined by immunocytochemistry. Cyclin B1
accumulates in the cytoplasm during the G2 phase and is then
translocated to the nucleus during prophase before the breakdown of
the nuclear membrane (44–46). Quantitative analysis of cells with
cytoplasmic cyclin B1 (arrows, Fig.
2B, left panel) revealed a significant (p<0.001) increase in
the fraction of such cells for both SN-38 resistant cell lines
(Fig. 2B, right panel), in
agreement with the cell cycle data.
SN-38-resistant cells show altered growth
in vivo
For in vivo characterization, tumor
xenografts were established from parental and SN-38-resistant cell
lines. The growth of HT-29/SN-38 tumors was significantly
(p<0.01) slower than that of HT-29 tumors with average tumor
volumes of 721 mm3 vs 1,362 mm3 by 28 days,
corresponding to a 47% reduction in the average tumor size
(Fig. 3A). The growth of
HCT-116/SN-38 tumors was also significantly different from that of
HCT-116 tumors (p<0.05) with average tumor volumes of 1,009
mm3 for HCT-116/SN-38 compared with 1,733 mm3
for the parental HCT-116 tumors, corresponding to a 42% reduction
in the average tumor size (Fig.
3B). Therefore, in both cases, tumor growth of the
SN-38-resistant xenografts was approximately half of that observed
for the corresponding parental tumors.
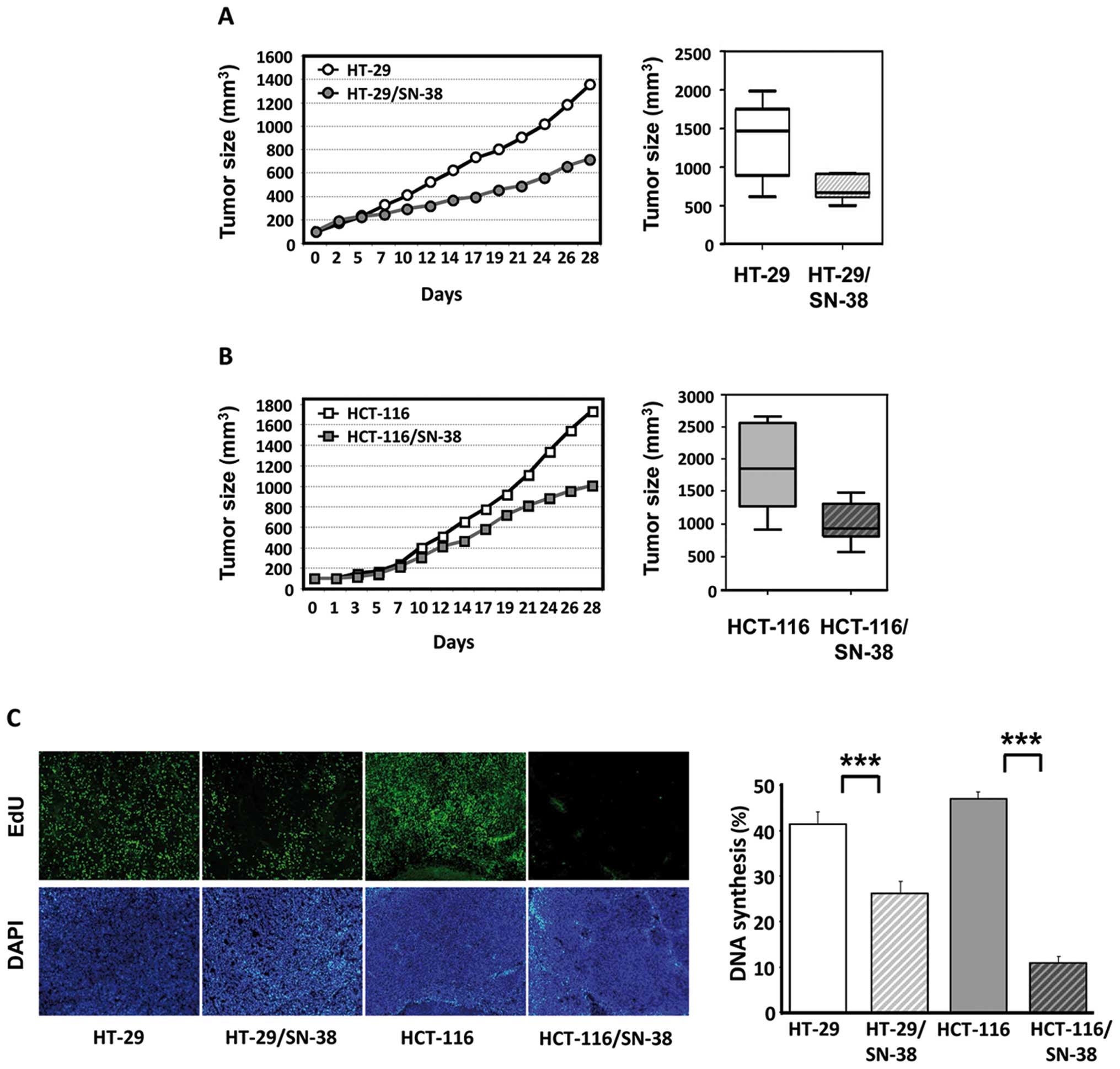 | Figure 3Growth and DNA synthesis of tumor
xenografts from parental and SN-38-resistant colorectal cancer
cells in nude mice. (A) Left, tumor growth of HT-29 (○) and
HT-29/SN-38 (•) tumors. The curves represent the average tumor
growth of at least seven animals per group. Right, box and whisker
plot of tumor volumes in mice with HT-29 or HT-29/SN-38 xenografts
by day 28. Lines, medians; boxes, 25th to 75th percentile
interquartile ranges; whiskers, the highest and lowest value for
the group. (B) Left, tumor growth of HCT-116 (□) and HCT-116/SN-38
(▪) tumors. The curves represent the average tumor growth of at
least 7 animals per group. Right, box and whisker plot of tumor
volumes in mice with HCT-116 or HCT-116/SN-38 xenografts by day 28.
Lines, medians; boxes, 25th to 75th percentile interquartile
ranges; whiskers, the highest and lowest value for the group. (C)
In vivo DNA synthesis of parental and SN-38-resistant tumor
xenografts as measured by EdU incorporation. Left, representative
images of tumors from HT-29, HT-29/SN-38, HCT-116 and HCT-116/SN-38
xenografts. Tumor cell nuclei (DAPI) appear in white in the lower
panels, whereas nuclei with active DNA synthesis (EdU) appear in
white in the upper panels. Right, the columns represent the ratio
between EdU-positive cells and the total number of viable cells and
are the averages of five fields/tumor (each field representing
approximately 1,700 cells) from three different tumors.
***p<0.001. |
Treatment of the mice with irinotecan (35 mg/kg i.p.
every four days) revealed that the HT-29/SN-38 and HCT-116/SN-38
tumors retained the resistance to irinotecan in vivo
throughout the four-week treatment. Specifically, irinotecan
treatment reduced the average tumor size by 75% for HT-29 tumors
compared with 55% for HT-29/SN-38 tumors. Irinotecan was highly
efficient toward HCT-116 tumors with complete tumor regression in
one-third of the animals and approximately 95% reduction of the
tumor size for the remaining tumors. In comparison, irinotecan only
reduced the tumor size of HCT-116/SN-38 tumors by 37% (data not
shown).
SN-38-resistant cells show reduced levels
of DNA synthesis in vivo
Next, we wished to compare the levels of in
vivo DNA synthesis in parental and SN-38-resistant cells. For
this purpose, tumor-bearing mice were injected with EdU, a
thymidine analog, 48 h before sacrifice and EdU incorporation was
subsequently determined by fluorescence histochemistry (Fig. 3C, left panel). Quantitative image
analysis showed that DNA incorporation was significantly
(p<0.001) lower in both SN-38-resistant tumors compared with
tumors derived from the respective parental cells, which was
particularly striking for the HCT-116 tumors (Fig. 3C, right panel).
SN-38-resistant cells show increased
levels of phosphorylated H2AX and Chk2
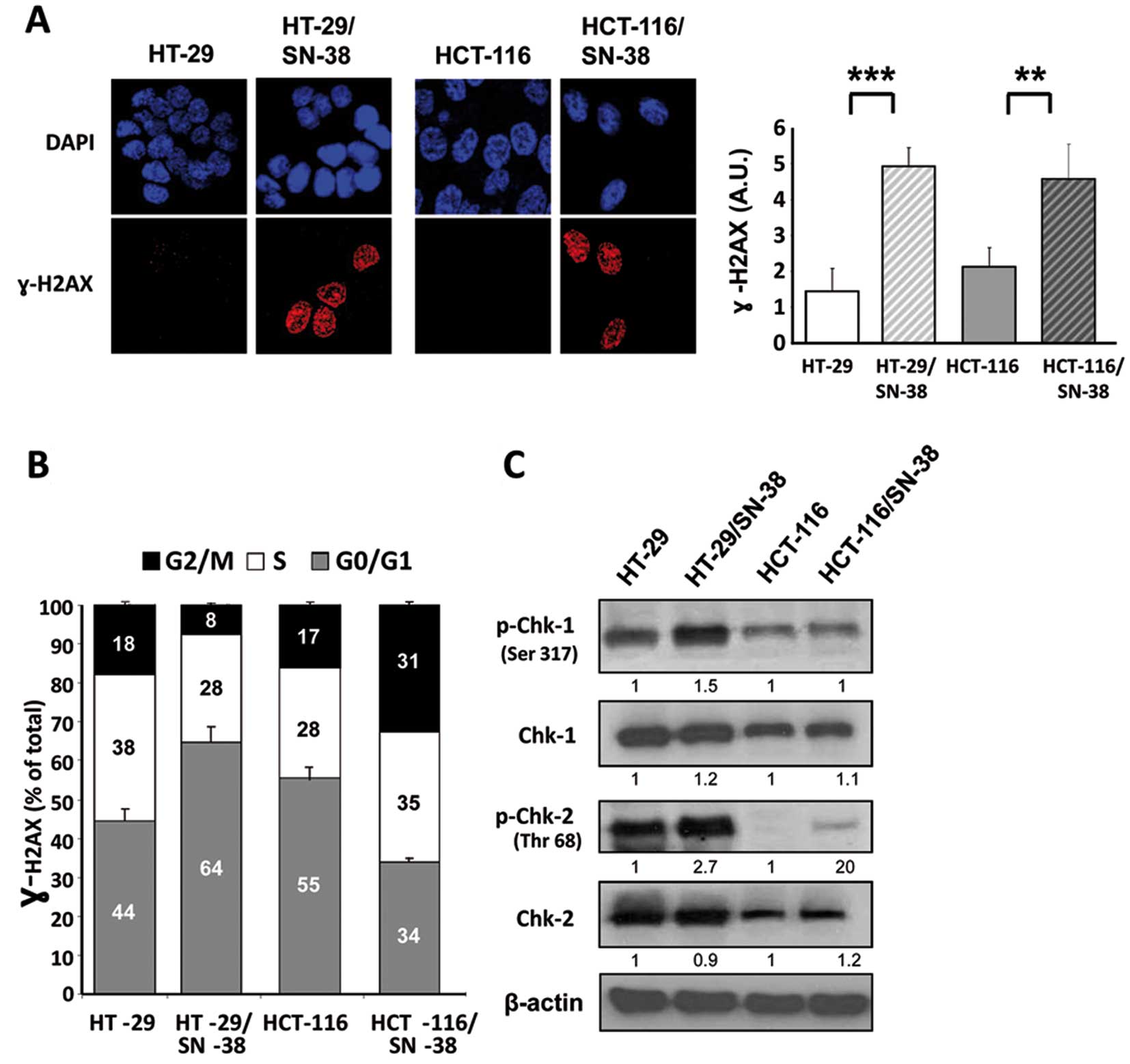
Phosphorylation of histone H2AX (γ-H2AX) is an early
response to the formation of DNA double-strand breaks and has also
been associated with increased levels of ‘oncogenic stress’, likely
due to replicative errors (47,48).
Immunocytochemistry of parental and SN-38-resistant cells showed a
significant increase in the basal levels of γ-H2AX for both
SN-38-resistant cell lines (Fig.
4A) suggesting that the development of CPT resistance is
accompanied by increased activation of the DNA damage response.
Biparametric flow cytometry analysis was used to determine whether
γ-H2AX is preferentially associated with a specific phase of the
cell cycle (Fig. 4B). The results
show that for HT-29 cells, SN-38 resistance is accompanied by an
increased fraction of γ-H2AX positive cells in the G1 phase of the
cell cycle (from 44 to 64%) whereas the fraction of γ-H2AX positive
cells in both S and G2/M is decreased. In contrast, for HCT-116
cells, SN-38 resistance is accompanied by an increase of γ-H2AX
positive cells in both the S and G2/M phase of the cell cycle
(Fig. 4B). Although apparently
contradictory, these results are coherent with the genotype of the
investigated cell lines, since MSI cells like HCT-116 are typically
diploid due to an efficient mitotic checkpoint that prevents cells
with damaged DNA from undergoing mitosis. In comparison, the
chromosome abnormalities of CIN cells like HT-29 are, at least in
part, due to defects in the mitotic checkpoint allowing mitotic
slippage of cells with damaged DNA (11).
The major regulators of the DNA-damage response are
the two PIKKs [PI3K (phosphoinoitide 3-kinase)-related kinases],
ATM [A-T (ataxia-telangiectasia) mutated] and ATR (ATM- and
Rad3-related). ATM and ATR have common phosphorylation substrates
such as H2AX, but also distinct substrates such as the checkpoint
kinase Chk2 for ATM and Chk1 for ATR (36,49,50).
Western blot analysis of the total and phosphorylated forms of Chk1
showed no consistent differences between parental and
SN-38-resistant cells (Fig. 4C).
In contrast, phospho-Chk2 was increased in both SN-38 resistant
cell lines without changes in Chk2 protein levels. Although the
selective activation of Chk2 suggests the involvement of the DNA
damage sensor ATM, we found no detectable evidence for increased
ATM activation in the SN-38-resistant cells, as indicated by the
presence of phospho-Ser1981 ATM (data not shown). Taken together,
these findings demonstrate that prolonged exposure to SN-38 is
accompanied by an upregulation of the DNA damage response, which is
coherent with the increased fraction of cells in the G2 phase of
the cell cycle.
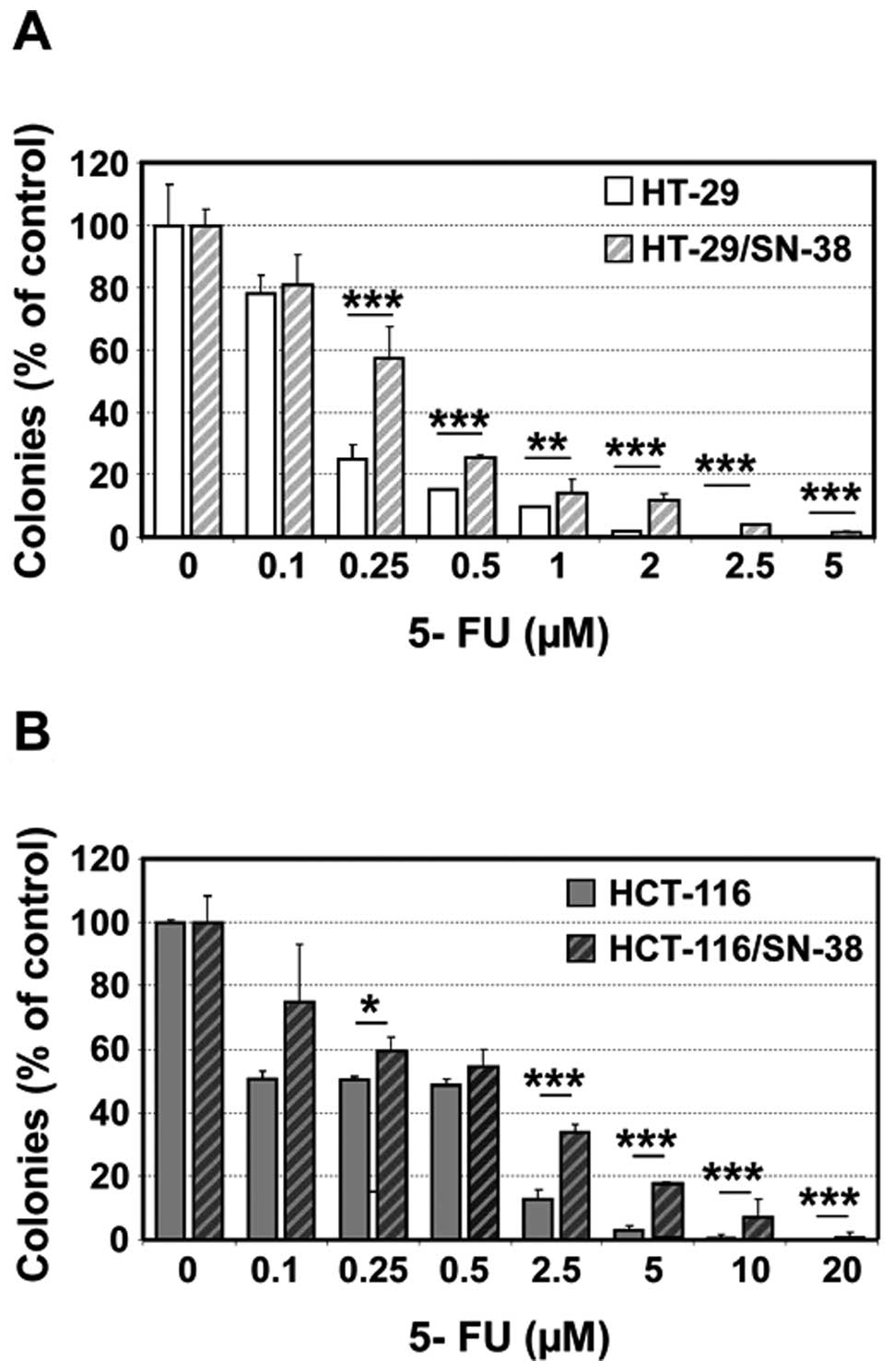
SN-38-resistant cells are cross-resistant
to 5-fluorouracil
The reduced S-phase fraction suggests that the
SN-38-resistant cells might show cross-resistance to S phase
selective anticancer agents. This is particularly relevant for
5-fluorouracil which is frequently administered in combination with
irinotecan. Both SN-38 resistant cell lines showed cross-resistance
to 5-fluorouracil as determined by colony formation, which was
particularly pronounced at high drug concentrations (Fig. 5).
We next compared the sensitivities of HCT-116 and
HCT-116/SN-38 xenografts to 5-fluorouracil (25 mg/kg i.p. on days
1, 2, 15 and 16). Treatment with 5-fluorouracil was accompanied by
a 75% decrease in the average tumor volume of HCT-116 tumors
compared with a 24% decrease for HCT-116/SN-38 tumors by day 28
(data not shown). Therefore, acquired resistance to SN-38 was
accompanied by cross-resistance to 5-FU both in vitro and
in vivo.
Discussion
Irinotecan is approved for treatment of patients
with metastatic colorectal cancer (8,9) and
shows promising activity in other applications such as gastric
cancer. However, even in patients that respond well initially, the
activity of irinotecan is eventually limited by acquisition of drug
resistance. Although irinotecan is known to be S-phase selective,
surprisingly little is known about the influence of prolonged
irinotecan exposure on the cell cycle dynamics. Microsatellite
instability (MSI) is linked to dysfunction of the DNA mismatch
repair (MMR) system, which is involved in the correction of
base/base mismatches and insertion/deletion loops during the S
phase of the cell cycle (12).
Interestingly, it has been reported that irinotecan is selectively
active toward MMR-deficient tumors cells in vitro as well as
in patients, compared to tumors with structural and numerical
chromosome instability (CIN) (13,14).
We now report the development and characterization
of two CRC cell lines, HCT-116/SN-38 (MSI) and HT-29/SN-38 (CIN)
with acquired resistance to SN-38, the active metabolite of
irinotecan. The parental MSI cells were initially more sensitive to
SN-38, compared with the parental CIN cells, in vitro as
in vivo, in agreement with previous reports (13,14).
However, the MSI cells rapidly developed high levels of SN-38
resistance coherent with the hypermutator phenotype of these cells
(11). Overall, we did not observe
any notable differences between MSI and CIN cells with regard to
the biological modifications observed following prolonged SN-38
exposure.
In agreement with the current model for CPT action,
SN-38 resistance was accompanied by decreased levels of
SN-38-induced DNA topoisomerase I cleavable complexes. This was
principally attributed to an important decrease in drug
accumulation which likely is associated with overexpression of one
or several ABC transporters. HT-29/SN-38 cells overexpressed both
ABCB1 (P-glycoprotein/mdr1) and ABCG2 (breast cancer related
protein/mitoxantrone transporter/BCRP) whereas neither parental nor
resistant HCT-116 cells expressed these two proteins. However, the
decreased drug uptake in HCT-116/SN-38 cells might involve members
of the MDR/ABCC family (21).
Strikingly, both SN-38 resistant cell lines
displayed permanent modifications of the cell cycle dynamics under
basal conditions (that is under drug-free conditions).
Specifically, SN-38 resistant cell lines showed a prolonged
generation doubling time which was accompanied by a lower
proportion of S phase cells and a doubling of the fraction of cells
in G2/M. Subsequent characterization of cells with cytoplasmic
cyclin B1, a marker for cells in G2, in combination with
quantitative analysis of cells expressing phospho-H3, a mitotic
marker, unambiguously showed that the increased G2/M fraction was
due to an increase of cells in the G2 phase of the cell cycle.
The altered growth dynamics was confirmed in
vivo for tumor xenografts established from parental and
SN-38-resistant cells. In particular, by 28 days the average tumor
size of the CPT-resistant xenografts was approximately half of that
observed for the corresponding parental tumors. In addition, in
vivo DNA synthesis (as determined by EdU incorporation) was
significantly reduced for both tumor models.
The altered growth dynamics might influence the
sensitivity to other anticancer agents. 5-Fluorouracil is of
particular interest since it is an S-phase selective agent which is
often administered in combination with irinotecan. Indeed, both
SN-38-resistant cell lines showed cross-resistance to
5-fluorouracil as determined by colony formation assays which was
subsequently confirmed in vivo for the SN-38 resistant
HCT-116 cells.
Increased activation of proteins involved in the DNA
damage response, including the phosphorylated forms of histone H2AX
(γ-H2AX) and the checkpoint kinases Chk1 and Chk2 has been
associated with ‘oncogenic stress’, probably linked to
replication-induced DNA damage. This type of DNA damage response is
quantitatively and temporally different from the classical DNA
damage response observed after acute genotoxic stress like ionizing
radiation. Specifically, replicative stress is associated with a
modest, but permanent activation of the DNA damage response
compared to the strong but transient response following ionizing
irradiation (47,48). We show here that both
SN-38-resistant cell lines had increased basal levels of
phosphorylated H2AX and Chk2, but not Chk1. This is in agreement
with previous studies reporting that certain types of
replication-dependent DNA damage may activate the ATM/Chk2 pathway
rather than the canonical S phase-specific ATR/Chk1 checkpoint
pathway (51). Although the
selective activation of Chk2 points toward the involvement of the
DNA damage sensor ATM, we found no evidence for ATM activation, as
indicated by the presence of phospho-Ser1981ATM. However, it is
possible that the putative ATM activation in the SN-38-resistant
cells might be below the detection limit. An alternative
explanation is that Chk2 might have been activated by the
DNA-dependent protein kinase, which targets the same residue on
Chk2 as ATM (52).
Our data are in line with the increasing
experimental and clinical evidence indicating that exposure to
cancer therapeutics may not only promote classical resistance
mechanisms but also change the biology of the tumor. Identification
of these mechanisms represents a major challenge in the years to
come considering that such changes may be accompanied either by
cross-resistance or by increased sensitivity to other classes of
therapeutic agents.
In summary, we present evidence that prolonged
exposure to SN-38/irinotecan is accompanied by permanent
modifications of cell cycle dynamics in vitro as in
vivo. This could have a profound impact on tumor sensitivity to
a wide range of structurally unrelated antitumor agents and may
influence tumor progression in patients.
Acknowledgments
This study was supported in part by the Association
pour la Recherche sur le Cancer (ARC), Villejuif, France and
Institut National du Cancer (INCa, CETIRICOL, PL06.008). A.P. was
supported by a fellowship from the Fondation pour la Recherche
Médicale (FRM), France. The authors thank Delphine Muller, Tatiana
Ledent and Olivier Bernadini from the animal facilities at
Saint-Antoine Research Center (PHEA); Sylvie Dumont and Fatiha
Merabtene from the pathology service and Anne-Marie Faussat from
the flow cytometry service, both IFR65.
References
|
1.
|
Hsiang YH, Lihou MG and Liu LF: Arrest of
replication forks by drug-stabilized topoisomerase I-DNA cleavable
complexes as a mechanism of cell killing by camptothecin. Cancer
Res. 18:5077–5082. 1989.PubMed/NCBI
|
|
2.
|
Larsen AK, Gilbert C, Chyzak G, Plisov SY,
Naguibneva I, Lavergne O, Lesueur-Ginot L and Bigg DC: Unusual
potency of BN 80915, a novel fluorinated E-ring modified
camptothecin, toward human colon carcinoma cells. Cancer Res.
7:2961–2967. 2001.PubMed/NCBI
|
|
3.
|
Wu J, Yin MB, Hapke G, Tóth K and Rustum
YM: Induction of biphasic DNA double-strand breaks and activation
of multiple repair protein complexes by DNA topoisomerase I drug
7-ethyl-10-hydroxy-camptothecin. Mol Pharmacol. 4:742–748. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Regairaz M, Zhang YW, Fu H, Agama KK, Tata
N, Agrawal S, Aladjem MI and Pommier Y: Mus81-mediated DNA cleavage
resolves replication forks stalled by topoisomerase I-DNA
complexes. J Cell Biol. 195:739–749. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Horwitz SB and Horwitz MS: Effects of
camptothecin on the breakage and repair of DNA during the cell
cycle. Cancer Res. 33:2834–2836. 1973.PubMed/NCBI
|
|
6.
|
Cheng MF, Chatterjee S and Berger NA:
Schedule-dependent cytotoxicity of topotecan alone and in
combination chemotherapy regimens. Oncol Res. 6:269–279.
1994.PubMed/NCBI
|
|
7.
|
Huang X, Traganos F and Darzynkiewicz Z:
DNA damage induced by DNA topoisomerase I- and topoisomerase
II-inhibitors detected by histone H2AX phosphorylation in relation
to the cell cycle phase and apoptosis. Cell Cycle. 6:614–619.
2003.PubMed/NCBI
|
|
8.
|
Saltz LB, Cox JV, Blanke C, Rosen LS,
Fehrenbacher L, Moore MJ, Maroun JA, Ackland SP, Locker PK, Pirotta
N, Elfring GL and Miller LL: Irinotecan plus fluorouracil and
leucovorin for metastatic colorectal cancer. Irinotecan Study Group
N Engl J Med. 13:905–914. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Douillard JY, Cunningham D, Roth AD,
Navarro M, James RD, Karasek P, Jandik P, Iveson T, Carmichael J,
Alakl M, Gruia G, Awad L and Rougier P: Irinotecan combined with
fluorouracil compared with fluorouracil alone as first-line
treatment for metastatic colorectal cancer: a multicentre
randomised trial. Lancet. 9209:1041–1047. 2000. View Article : Google Scholar
|
|
10.
|
Lengauer C, Kinzler KW and Vogelstein B:
Genetic instabilities in human cancers. Nature. 6712:643–649. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Cancer Genome Atlas Network: Comprehensive
molecular characterization of human colon and rectal cancer.
Nature. 487:330–337. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Jiricny J: The multifaceted
mismatch-repair system. Nat Rev Mol Cell Biol. 5:335–346. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Jacob S, Aguado M, Fallik D and Praz F:
The role of the DNA mismatch repair system in the cytotoxicity of
the topoisomerase inhibitors camptothecin and etoposide to human
colorectal cancer cells. Cancer Res. 17:6555–6562. 2001.PubMed/NCBI
|
|
14.
|
Fallik D, Borrini F, Boige V, Viguier J,
Jacob S, Miquel C, Sabourin JC, Ducreux M and Praz F:
Microsatellite instability is a predictive factor of the tumor
response to irinotecan in patients with advanced colorectal cancer.
Cancer Res. 18:5738–5744. 2003.PubMed/NCBI
|
|
15.
|
Kanzawa F, Sugimoto Y, Minato K, Kasahara
K, Bungo M, Nakagawa K, Fujiwara Y, Liu LF and Saijo N:
Establishment of a camptothecin analogue (CPT-11)-resistant cell
line of human non-small cell lung cancer: characterization and
mechanism of resistance. Cancer Res. 18:5919–5924. 1990.PubMed/NCBI
|
|
16.
|
Maliepaard M, van Gastelen MA, de Jong LA,
Pluim D, van Waardenburg RC, Ruevekamp-Helmers MC, Floot BG and
Schellens JH: Overexpression of the BCRP/MXR/ABCP gene in a
topotecan-selected ovarian tumor cell line. Cancer Res.
18:4559–4563. 1999.PubMed/NCBI
|
|
17.
|
Kawabata S, Oka M, Shiozawa K, Tsukamoto
K, Nakatomi K, Soda H, Fukuda M, Ikegami Y, Sugahara K, Yamada Y,
Kamihira S, Doyle LA, Ross DD and Kohno S: Breast cancer resistance
protein directly confers SN-38 resistance of lung cancer cells.
Biochem Biophys Res Commun. 5:1216–1223. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Rasheed ZA and Rubin EH: Mechanisms of
resistance to topoisomerase I-targeting drugs. Oncogene.
22:7296–7304. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Boyer J, McLean EG, Aroori S, Wilson P,
McCulla A, Carey PD, Longley DB and Johnston PG: Characterization
of p53 wild-type and null isogenic colorectal cancer cell lines
resistant to 5-fluorouracil, oxaliplatin, and irinotecan. Clin
Cancer Res. 6:2158–2167. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Arakawa Y, Suzuki H, Saito S and Yamada H:
Novel missense mutation of the DNA topoisomerase I gene in
SN-38-resistant DLD-1 cells. Mol Cancer Ther. 5:502–508. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Takara K, Kitada N, Yoshikawa E, Yamamoto
K, Horibe S, Sakaeda T, Nishiguchi K, Ohnishi N and Yokoyama T:
Molecular changes to HeLa cells on continuous exposure to SN-38, an
active metabolite of irinotecan hydrochloride. Cancer Lett.
1:88–96. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Tagen M, Zhuang Y, Zhang F, Harstead KE,
Shen J, Schaiquevich P, Fraga CH, Panetta JC, Waters CM and Stewart
CF: P-glycoprotein, but not multidrug resistance protein 4, plays a
role in the systemic clearance of irinotecan and SN-38 in mice.
Drug Metab Lett. 4:195–201. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Pastorino F, Loi M, Sapra P, Becherini P,
Cilli M, Emionite L, Ribatti D, Greenberger LM, Horak ID and
Ponzoni M: Tumor regression and curability of preclinical
neuroblastoma models by PEGylated SN38 (EZN-2208), a novel
topoisomerase I inhibitor. Clin Cancer Res. 19:4809–4821. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Waterhouse DN, Yapp D, Verreault M,
Anantha M, Sutherland B and Bally MB: Lipid-based nanoformulation
of irinotecan: dual mechanism of action allows for combination
chemo/angiogenic therapy. Nanomedicine (Lond). 9:1645–1654. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Petitprez A and Larsen AK: Irinotecan
resistance is accompanied by upregulation of EGFR and Src signaling
in human cancer models. Curr Pharm Des. Sep 7–2012.(Epub ahead of
print).
|
|
26.
|
Poindessous V, Koeppel F, Raymond E,
Comisso M, Waters S and Larsen AK: Marked activity of irofulven
(MGI-114) toward human carcinoma cells: comparison with cisplatin
and ecteinascidin (ET-743). Clin Cancer Res. 9:2817–2825.
2003.PubMed/NCBI
|
|
27.
|
Lemke K, Poindessous V, Skladanowski A and
Larsen AK: The antitumor triazoloacridone C-1305 is a topoisomerase
II poison with unusual properties. Mol Pharmacol. 4:1035–1042.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Poindessous V, Koeppel F, Raymond E,
Cvitkovic E, Waters SJ and Larsen AK: Enhanced antitumor activity
of irofulven in combination with 5-fluorouracil and cisplatin in
human colon and ovarian carcinoma cells. Int J Oncol. 23:1347–1355.
2003.PubMed/NCBI
|
|
29.
|
Larsen AK, Paoletti J, Belehradek J Jr and
Paoletti C: Uptake, cytofluorescence, and cytotoxicity of
oxazolopyridocarbazoles (amino acid-ellipticine conjugates) in
murine sarcoma cells. Cancer Res. 46:5236–5240. 1986.PubMed/NCBI
|
|
30.
|
Léonce S, Kraus-Berthier L, Golsteyn RM,
David-Cordonnier MH, Tardy C, Lansiaux A, Poindessous V, Larsen AK
and Pierré A: Generation of replication-dependent double-strand
breaks by the novel N2-G-alkylator S23906-1. Cancer Res.
14:7203–7210. 2006.PubMed/NCBI
|
|
31.
|
Rocca CJ, Poindessous V, Soares DG,
Ouadrani KE, Sarasin A, Guérin E, de Gramont A, Henriques JA,
Escargueil AE and Larsen AK: The NER proteins XPC and CSB, but not
ERCC1, regulate the sensitivity to the novel DNA binder S23906:
implications for recognition and repair of antitumor alkylators.
Biochem Pharmacol. 3:335–343. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Pencreach E, Guérin E, Nicolet C,
Lelong-Rebel I, Voegeli AC, Oudet P, Larsen AK, Gaub MP and Guenot
D: Marked activity of irinotecan and rapamycin combination toward
colon cancer cells in vivo and in vitro is mediated through
cooperative modulation of the mammalian target of
rapamycin/hypoxiainducible factor-1alpha axis. Clin Cancer Res.
15:1297–1307. 2009. View Article : Google Scholar
|
|
33.
|
Guérin E, Raffelsberger W, Pencreach E,
Maier A, Neuville A, Schneider A, Bachellier P, Rohr S, Petitprez
A, Poch O, Moras D, Oudet P, Larsen AK, Gaub MP and Guenot D: In
vivo topoisomerase I inhibition attenuates the expression of
hypoxia-inducible factor 1α target genes and decreases tumor
angiogenesis. Mol Med. 18:83–94. 2012.PubMed/NCBI
|
|
34.
|
Gil-Delgado MA, Bastian G, Guinet F, Spano
JP, Taillibert S, Rocher MA, Castaing D, Adam R, Urien S, Bismuth H
and Khayat D: Oxaliplatin plus irinotecan and FU-FOL combination
and pharmacokinetic analysis in advanced colorectal cancer
patients. Am J Clin Oncol. 27:294–298. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Escargueil AE, Poindessous V, Soares DG,
Sarasin A, Cook PR and Larsen AK: Influence of irofulven, a
transcription-coupled repair-specific antitumor agent, on RNA
polymerase activity, stability and dynamics in living mammalian
cells. J Cell Sci. 121:1275–1283. 2008. View Article : Google Scholar
|
|
36.
|
Soares DG, Battistella A, Rocca CJ, Matuo
R, Henriques JA, Larsen AK and Escargueil AE: Ataxia telangiectasia
mutated-and Rad3-related kinase drives both the early and the late
DNA-damage response to the monofunctional antitumour alkylator
S23906. Biochem J. 1:63–73. 2011. View Article : Google Scholar
|
|
37.
|
Skladanowski A, Côme MG, Sabisz M,
Escargueil AE and Larsen AK: Down-regulation of DNA topoisomerase
II alpha leads to prolonged cell cycle transit in G2 and early M
phases and increased survival to microtubule-interacting agents.
Mol Pharmacol. 68:625–634. 2005.PubMed/NCBI
|
|
38.
|
Skladanowski A, Bozko P, Sabisz M and
Larsen AK: Dual inhibition of PI3K/Akt signaling and the DNA damage
checkpoint in p53-deficient cells with strong survival signaling:
implications for cancer therapy. Cell Cycle. 18:2268–2275. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Soares DG, Escargueil AE, Poindessous V,
Sarasin A, de Gramont A, Bonatto D, Henriques JA and Larsen AK:
Replication and homologous recombination repair regulate DNA
double-strand break formation by the antitumor alkylator
ecteinascidin 743. Proc Natl Acad Sci USA. 32:13062–13067. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Soares DG, Machado MS, Rocca CJ,
Poindessous V, Ouaret D, Sarasin A, Galmarini CM, Henriques JA,
Escargueil AE and Larsen AK: Trabectedin and its C subunit modified
analogue PM01183 attenuate nucleotide excision repair and show
activity toward platinum-resistant cells. Mol Cancer Ther.
8:1481–1489. 2011. View Article : Google Scholar
|
|
41.
|
Poindessous V, Ouaret D, El Ouadrani K,
Battistella A, Mégalophonos VF, Kamsu-Kom N, Petitprez A,
Escargueil AE, Boudou P, Dumont S, Cervera P, Fléjou JF, André T,
Tournigand C, Chibaudel B, de Gramont A and Larsen AK: EGFR- and
VEGF(R)-targeted small molecules show synergistic activity in
colorectal cancer models refractory to combinations of monoclonal
antibodies. Clin Cancer Res. 20:6522–6530. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Godard T, Deslandes E, Sichel F, Poul JM
and Gauduchon P: Detection of topoisomerase inhibitor-induced DNA
strand breaks and apoptosis by the alkaline comet assay. Mutat
Resgg. 1–2:47–56. 2002.PubMed/NCBI
|
|
43.
|
Hans F and Dimitrov S: Histone H3
phosphorylation and cell division. Oncogene. 20:3021–3027. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Pines J and Hunter T: Cyclin-dependent
kinases: a new cell cycle motif? Trends Cell Biol. 5:117–121. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Ookata K, Hisanaga S, Okano T, Tachibana K
and Kishimoto T: Relocation and distinct subcellular localization
of p34cdc2-cyclin B complex at meiosis reinitiation in starfish
oocytes. EMBO J. 5:1763–1772. 1992.PubMed/NCBI
|
|
46.
|
Escargueil AE, Plisov SY, Skladanowski A,
Borgne A, Meijer L, Gorbsky GJ and Larsen AK: Recruitment of cdc2
kinase by DNA topoisomerase II is coupled to chromatin remodeling.
FASEB J. 12:2288–2290. 2001.PubMed/NCBI
|
|
47.
|
Bartkova J, Horejsí Z, Koed K, Krämer A,
Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C,
Ørntoft T, Lukas J and Bartek J: DNA damage response as a candidate
anti-cancer barrier in early human tumorigenesis. Nature.
7035:864–870. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Gorgoulis VG, Vassiliou LV, Karakaidos P,
Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr,
Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C and
Halazonetis TD: Activation of the DNA damage checkpoint and genomic
instability in human precancerous lesions. Nature. 434:907–913.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Jazayeri A, Falck J, Lukas C, Bartek J,
Smith GC, Lukas J and Jackson SP: ATM- and cell cycle-dependent
regulation of ATR in response to DNA double-strand breaks. Nat Cell
Biol. 8:37–45. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Reinhardt HC and Yaffe MB: Kinases that
control the cell cycle in response to DNA damage: Chk1, Chk2 and
MK2. Curr Opin Cell Biol. 21:245–255. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Soza S, Leva V, Vago R, Ferrari G, Mazzini
G, Biamonti G and Montecucco A: DNA ligase I deficiency leads to
replication-dependent DNA damage and impacts cell morphology
without blocking cell cycle progression. Mol Cell Biol.
29:2032–2041. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Li J and Stern DF: Regulation of CHK2 by
DNA-dependent protein kinase. J Biol Chem. 280:12041–12050. 2005.
View Article : Google Scholar : PubMed/NCBI
|