Introduction
Epstein-Barr virus (EBV) is a ubiquitous human
γ-herpesvirus and was one of the first human virus linked to cancer
(1). EBV can infect, transform and
immortalize B-lymphocytes in vitro, giving rise to
lymphoblastoid cell lines (LCLs), which display elevated levels of
several cellular activation antigens (2). Consistent with this feature,
persistent, latent EBV infection is present in several lymphoid
malignancies including Burkitt’s lymphoma (BL) (3).
EBV latent membrane protein-1 (LMP-1) is a key
effector in EBV-mediated transformation of B cells (4). It consists of 386 amino acids and its
carboxy-terminal cytoplasmic domain contains two carboxy-terminal
activation regions (CTARs), CTAR-1 and CTAR-2. CTAR-1 and CTAR-2
are known to activate the nuclear factor-κB (NF-κB) signaling
pathway (5–8). NF-κB is inactive in the cytosol
because it is bound to IκBα and IκBβ, and becomes active after
phosphorylation and subsequent degradation of IκBα and IκBβ
(9). The released NF-κB from the
complex translocates to the nucleus where it activates a variety of
genes (9). The high-molecular
weight complex, IκB kinase (IKK), which is composed of two
catalytic subunits, IKKα and IKKβ, and a regulatory subunit, IKKγ,
phosphorylates IκBs (9). Members
of the p38 mitogen-activated protein kinase (MAPK) kinase kinase
protein kinase family mediate the physiological activation of IKK
(10). These kinases include
NF-κB-inducing kinase (NIK) (11)
and MAPK/extracellular signal-regulated kinase kinase 1 (12). CTAR-1 binds to tumor necrosis
factor receptor-associated factors (TRAFs) (5), whereas CTAR-2 binds to the tumor
necrosis factor receptor-associated death domain (TRADD) (6). NF-κB activation by the CTAR-1 and
CTAR-2 domains of LMP-1 is likely to be mediated by the binding of
TRAFs directly or indirectly to both the CTAR-1 and CTAR-2 domains
(5–8). NF-κB activation by LMP-1 is mediated
by NIK or a related MAPK kinase kinase, and subsequent activation
of the IKK complex (7,13). NIK is activated by aggregated TRAF2
(13). Thus, binding of LMP-1 to
TRAFs and TRADD initiates the formation of a signaling complex that
leads to activation of NF-κB and MAPK (5–8),
resulting in upregulation of several genes expression.
The human activation inducer molecule CD69 is a
disulfide-linked transmembrane homodimeric glycoprotein (14). CD69 is not detected in peripheral
blood lymphocytes, but it is expressed by small subsets of T and B
cells in peripheral lymphoid tissues (15,16).
CD69 acts as a signal transducer in inflammatory processes. Recent
studies have established the role of CD69 as an intrinsic negative
modulator of T cell responses (17). In the present report, we show that
CD69 is a transcriptional target of LMP-1 activation of NF-κB via
CTAR-1 and CTAR-2.
Materials and methods
Cell lines
Raji is a human EBV-positive BL cell line, and
LCL-Ao, LCL-Ka and LCL-Ku are EBV-immortalized human B-cell lines
generated from peripheral blood mononuclear cells of healthy
volunteers. These lymphoid cell lines were cultured in Roswell Park
Memorial Institute (RPMI)-1640 medium supplemented with 10 or 20%
heat-inactivated fetal bovine serum, 50 U/ml penicillin and 50
μg/ml streptomycin. Human embryonic kidney 293T cells were
maintained in Dulbecco’s modified Eagle’s medium supplemented with
10% heat-inactivated fetal bovine serum, 50 U/ml penicillin and 50
μg/ml streptomycin.
Flow cytometry
The expression of CD69 was analyzed by flow
cytometry with phycoerythrin (PE)-labeled mouse monoclonal antibody
against CD69 (clone TP1.55.3; Beckman Coulter, Fullerton, CA).
Analyses were carried out with isotype-matched control antibody.
The cells were incubated with the antibody for 30 min, washed with
cell WASH (Becton-Dickinson Immunocytometry Systems, San Jose, CA)
and then subjected to analysis in the cytometer.
RNA detection
Total RNA was prepared from various cell cultures by
TRIzol (Invitrogen, Carlsbad, CA) according to the protocol
provided by the manufacturer. First-strand cDNA was synthesized
from 1 μg total cellular RNA using a PrimeScript RT-PCR kit
(Takara Bio Inc., Otsu, Japan) with random primers. The primers
used were 5′-CATAGCTCTCATTGCCTTATCAGT-3′ (forward) and
5′-CCTCTCTACCTGCGTATCGTTT-3′ (reverse) for CD69,
5′-GTGACTGGACTGGAGGAGCC-3′ (forward) and
5′-GAGGGAGTCATCGTGGTGGTG-3′ (reverse) for LMP-1, and
5′-GTGGGGCGCCCCAGGCACCA-3′ (forward) and
5′-CTCCTTAATGTCACGCACGATTTC-3′ (reverse) for β-actin. The length of
the semiquantitative reverse transcription-PCR (RT-PCR) for each
gene was: 30 cycles for CD69 and LMP-1, and 28 cycles for β-actin.
The PCR products were fractionated on 2% agarose gels and
visualized by ethidium bromide staining.
Plasmids and transfections
Luciferase assay was performed to confirm that LMP-1
induces the CD69 promoter activation. Approximately
3×105 293T cells per plate were transfected by the
calcium phosphate DNA co-precipitation method. All transfections
included appropriate reporter and effector plasmids. The expression
plasmids pSG5-LMP-1, pSG5-LMP-1Δ187-351, pSG5-LMP-1Δ 349 and
pSG5-LMP-1Δ194-386 were kindly provided by Dr Martin Rowe
(University of Wales College of Medicine, Cardiff, UK) (18,19).
The CD69 promoter-luciferase gene constructs have already been
described (20,21). The single and combined internal
deletion mutants of NF-κB sites were constructed by deletion of the
NF-κB sites of the plasmid pAIM 255-LUC. The IκBαΔN- and
IκBβΔN-dominant negative mutants are IκBα and IκBβ deletion mutants
lacking the amino-terminal 36 and 23 amino acids, respectively
(22,23). The dominant negative mutants of
IKKα, IKKα (K44M), IKKβ, IKKβ (K44A), IKKγ, IKKγ (1–305) and NIK,
NIK (KK429/430AA) have been described previously (24,25).
Plasmids for truncated TRAF2 and TRAF5 proteins retaining only the
TRAF domain, ΔTRAF2 and ΔTRAF5, have been described previously
(26,27). In all cases, the reference plasmid
phRL-TK, which contains the Renilla luciferase gene under
the control of the herpes simplex virus thymidine kinase promoter,
was cotransfected to correct for transfection efficiency. After 24
h, the transfected cells were collected by centrifugation, washed
with phosphate-buffered saline and lysed in reporter lysis buffer
(Promega, Madison, WI). Luciferase assays were conducted using the
dual luciferase reporter system (Promega), in which the relative
luciferase activity was calculated by normalizing transfection
efficiency according to the Renilla luciferase
activities.
Preparation of nuclear extracts and
electrophoretic mobility shift assay (EMSA)
Nuclear proteins were extracted as described by
Antalis and Godbolt (28) with
some modifications, and NF-κB binding activity to the NF-κB element
was examined by EMSA. Briefly, 5 μg of nuclear extracts were
preincubated in a binding buffer containing 1 μg
polydeoxyinosinic-deoxycytidylic acid (GE Healthcare Biosciences,
Buckinghamshire, UK), followed by the addition of
32P-labeled oligonucleotide probes containing the NF-κB
element. The mixtures were incubated for 15 min at room
temperature. The DNA protein complexes were separated on 4%
polyacrylamide gels and visualized by autoradiography. The probes
and competitors used were prepared by annealing the sense and
antisense synthetic oligonucleotides as follows: the NF-κB element
(κB1) of the CD69 gene (5′-GATCCAGACAACAGGGAAAACCCATACTTC-3′); the
NF-κB element (κB2) of the CD69 gene (5′-GATCCAGAGTCTGGGAAAATCCCACTTTCC-3′); a
typical NF-κB element from the IL-2 receptor α chain (IL-2Rα) gene
(5′-GATCCGGCAGGGGAATCTCCCTCTC-3′) and an AP-1
element of the IL-8 gene (5′-GATCGTGATGACTCAGGTT-3′). The above
underlined sequences represent the NF-κB and AP-1 binding sites,
respectively.
Results
Upregulated CD69 expression in
EBV-immortalized human B-cell lines and EBV-positive BL cell
line
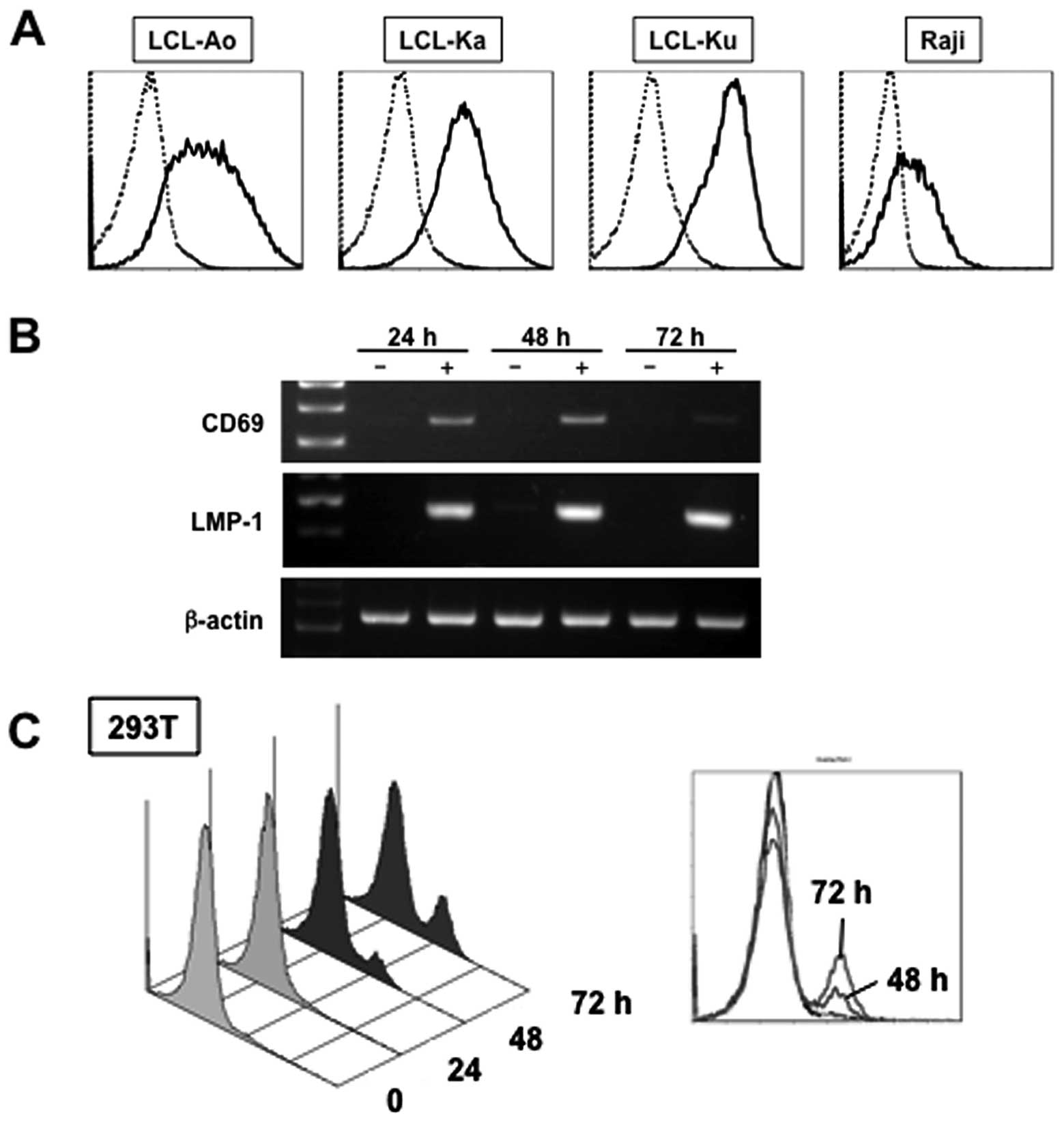
We examined first whether CD69 upregulation is a
general feature of EBV-infected B cells. All EBV-immortalized
B-cell lines, LCL-Ao, LCL-Ka and LCL-Ku, and EBV-positive BL cell
line, Raji, constitutively expressed CD69 on the cell surface
(Fig. 1A). In contrast, CD69 was
hardly expressed on normal peripheral blood mononuclear cells (data
not shown).
LMP-1 upregulates CD69 mRNA and protein
expression
EBV-immortalized B-cell lines and Raji cells
constitutively express LMP-1 (29,30).
Therefore, we analyzed the induction of CD69 expression by LMP-1.
Transient expression assays using a mammalian expression vector for
LMP-1 were performed in 293T cells. In 293T cells transfected with
empty vector (pSG5), CD69 mRNA was not detectable as determined by
RT-PCR analysis (Fig. 1B). In
contrast, ectopic expression of LMP-1 induced the expression of
CD69 mRNA in 293T cells. Next, we investigated whether LMP-1 also
transcribed the endogenous CD69 gene. The expression of CD69
antigen on the cell surface of 293T cells increased after ectopic
expression of LMP-1 in 293T cells, and such increase was
time-dependent (Fig. 1C).
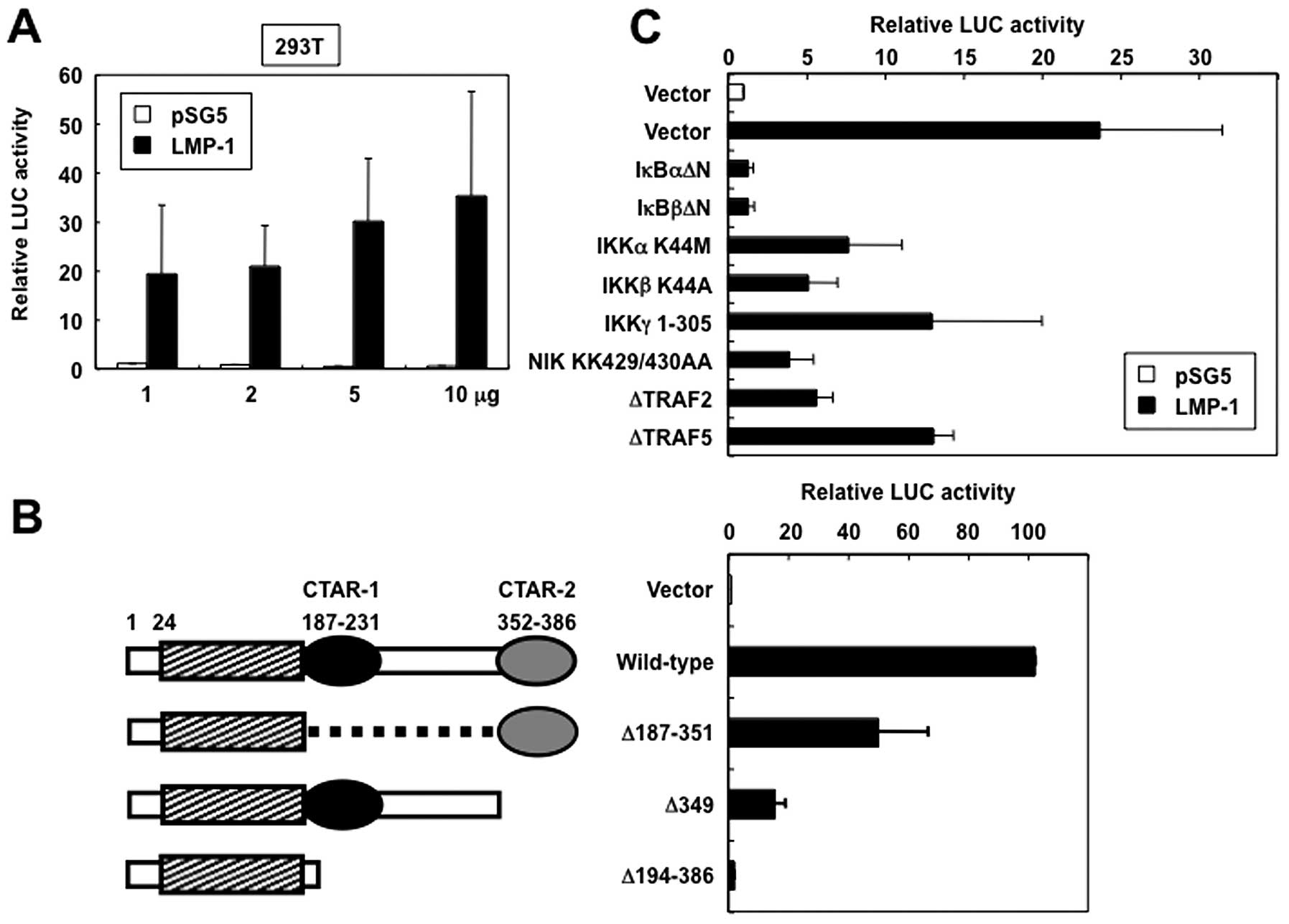
LMP-1 activates CD69 promoter
To determine whether LMP-1 regulates CD69 promoter
activity, transient expression assays using the reporter plasmid
pAIM 1.4-LUC and either an expression vector for LMP-1 or empty
vector (pSG5) were performed in 293T cells. LMP-1 induced relative
levels of CD69 promoter-directed luciferase expression in a
dose-dependent manner, suggesting that LMP-1 functionally activates
CD69 promoter (Fig. 2A). To map
the region in the LMP-1 protein that mediates activation of CD69
promoter, LMP-1 mutants were expressed in 293T cells and their
effect on CD69 promoter activity was investigated. The LMP-1
mutants used included LMP-1Δ187-351 (which contains only CTAR-2 in
the carboxy-terminus), LMP-1Δ349 (which lacks CTAR-2), and
LMP-1Δ194-386 (in which the entire carboxy-terminal cytoplasmic
region is deleted) (Fig. 2B, left
panel). In cells that expressed LMP-1Δ194-386, CD69 promoter
activity was not significantly increased (Fig. 2B, right panel). In contrast,
LMP-1Δ187-351 induced 50% of wild-type LMP-1 CD69 promoter
activation. Furthermore, LMP-1Δ349 showed substantial impairment of
CD69 promoter activation. These results suggest that LMP-1
activates CD69 expression via the cooperative activity of CTAR-1
and CTAR-2 signaling motifs.
LMP-1 activates the CD69 promoter via the
NF-κB signaling pathway
CTAR-1 and CTAR-2 are known to activate the NF-κB
signaling pathway (5–8). Based on the discussed background, we
tested the ability of the dominant interfering mutants of IκBα,
IκBβ, IKKγ, TRF2 and TRAF5, and kinase-deficient mutants of IKKα,
IKKβ and NIK to inhibit LMP-1-mediated transactivation of
CD69-driven reporter gene activity. Expression of each of these
inhibitory mutants inhibited LMP-1-induced activation of CD69
promoter (Fig. 2C). These results
demonstrated that the signaling components, TRAFs, NIK and IKKs,
which are involved in the activation of NF-κB, are also necessary
for LMP-1 transactivation of the CD69 promoter.
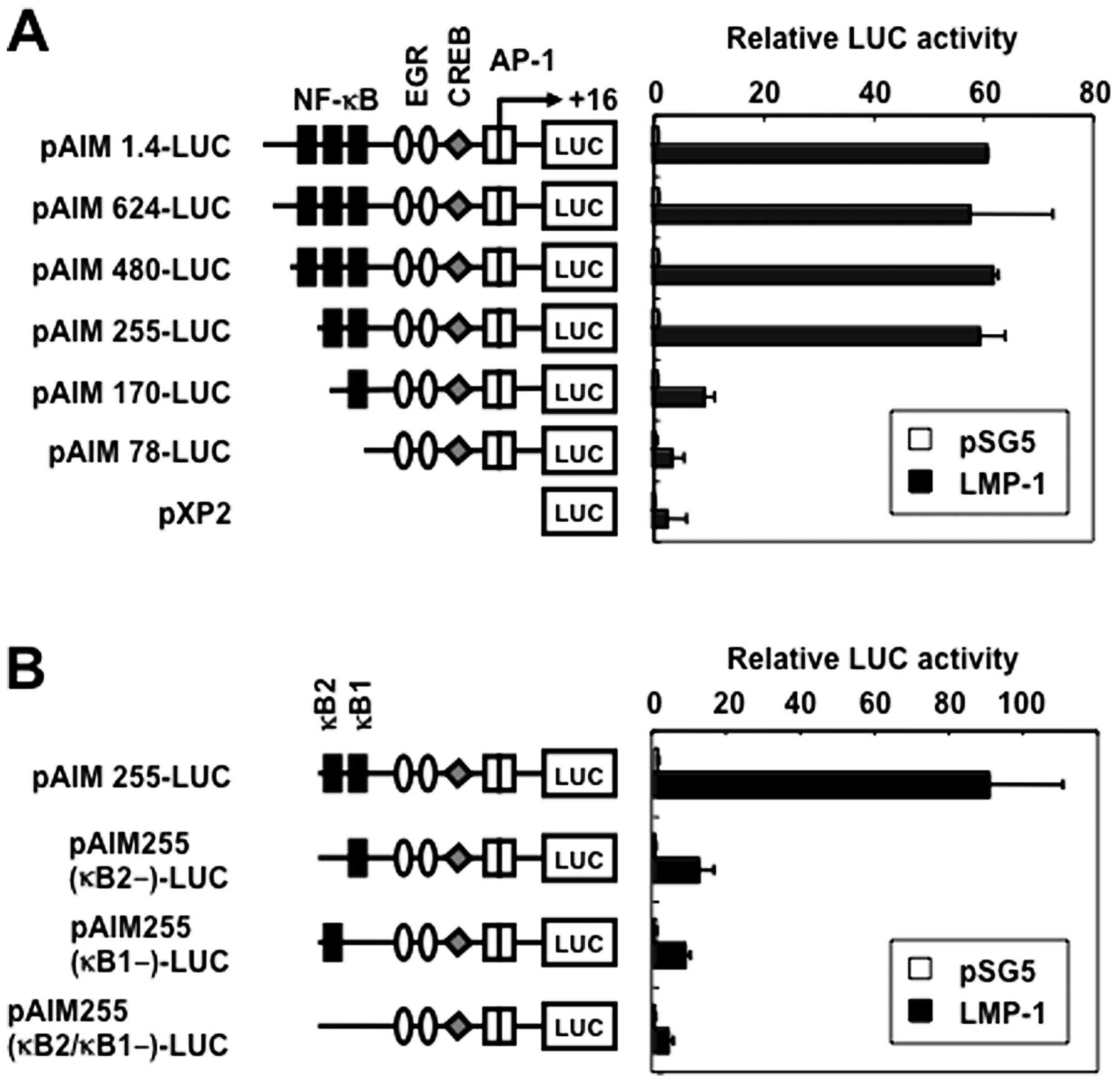
LMP-1 activates the CD69 promoter through
two NF-κB sites
The CD69 promoter contains three putative binding
sites for NF-κB [positions −373 (κB3), −223 (κB2) and −160 (κB1)],
a putative binding site for early growth response (EGR) at −69, a
composite binding site for Sp1 and EGR at −56, a cyclic adenosine
3′,5′-monophosphate response element-binding protein (CREB) binding
site at −46, and two putative AP-1 binding sites at −16 and +1
(Fig. 3A) (20). To study the role of NF-κB sites in
the transcriptional induction of CD69 gene by LMP-1, we transfected
293T cells with several 5′-deletion fragments extending from
position −1.4 k to −78 (Fig. 3A),
and an expression vector for LMP-1. Progressive removal of
5′-sequence up to position −255 did not significantly inhibit the
induced promoter activity, suggesting that the 271-bp fragment,
spanning positions −255 to +16, contained the LMP-1-responsive
elements (Fig. 3A). Further
deletion of upstream sequences up to position −170 resulted in
significant loss of responsiveness to LMP-1. Additional removal of
the 5′-sequences up to position −78 further affected the inducible
promoter activity.
Two potential NF-κB-binding sequences were
identified at positions −160 (κB1) and −223 (κB2) spanning
positions −255 to +16 bp. κB1 and κB2 were identical to those found
in the gene promoters of c-myc and IL-6, respectively (31). To test the relative contribution of
the NF-κB binding sites to the LMP-1-mediated activation of CD69,
plasmids with internal deletion mutants of these sites in the CD69
promoter were transfected (Fig.
3B). Single deletion of the κB1 or κB2 site resulted in
reduction of the inducible activity. Double deletions of the κB1
and κB2 sites further reduced Tax-mediated activation of this
reporter construct. These results indicate that the two NF-κB
binding sites in the CD69 promoter regulate LMP-1-induced
upregulation of CD69.
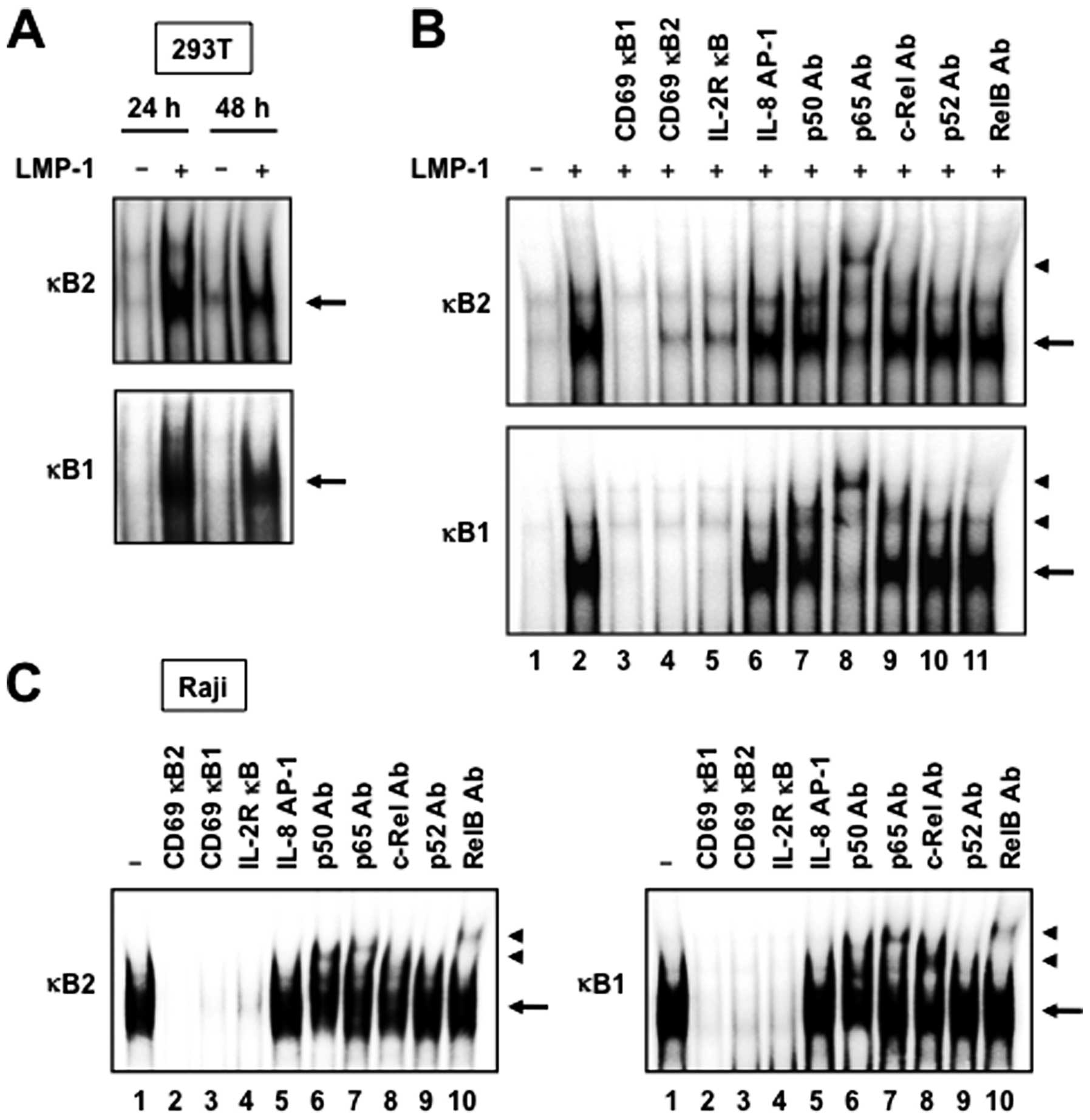
LMP-1 induces NF-κB binding to DNA
Since both NF-κB binding sites of the CD69 promoter
are required for LMP-1-mediated CD69 gene transcription, we studied
the ability of LMP-1 to activate NF-κB binding to DNA. For this
purpose, 293T cells were transfected with control plasmid (empty
vector) or LMP-1 expression plasmid for 24 or 48 h, followed by
extraction of nuclear proteins. EMSA demonstrated that LMP-1
increased the binding of NF-κB to 32P-labeled
oligonucleotides representing the κB1 and κB2 sites (Fig. 4A). The specificity of DNA-protein
complex formation was determined by competition studies with
unlabeled competitors. As expected, excess of cold CD69 κB1 or κB2
double-stranded oligonucleotide, or consensus NF-κB site from the
IL-2Rα promoter, effectively competed with the labeled probes and
eliminated the binding of nuclear extracts from 293T cells
transfected with LMP-1 expression plasmid (Fig. 4B, lanes 2–5). In contrast, the
unlabeled IL-8 AP-1 element did not compete with labeled probes
(Fig. 4B, lanes 2 and 6). The
nucleoproteins binding to both NF-κB sites were composed of p50,
p65 and c-Rel subunits of the NF-κB family, as demonstrated by
supershift experiments with specific antibodies (Fig. 4B, lanes 7–9).
Finally, to determine the role of LMP-1 on
endogenous NF-κB binding to DNA, we measured NF-κB binding to
respective NF-κB elements in the CD69 promoter in LMP-1- and
CD69-expressing Raji cells. As expected, protein complexes bound to
both κB1 and κB2 sites were detected in nuclear extracts from Raji
cells (Fig. 4C, lane 1). The
specificity of DNA-protein complexes in these extracts was
determined by competition studies using unlabeled competitors. As
observed in nuclear extracts from 293T cells transfected with LMP-1
expression plasmid, cold κB1 and κB2 oligonucleotides, and
consensus NF-κB site from the IL-2Rα promoter, but not the IL-8
AP-1 element, efficiently competed with labeled probes (Fig. 4C, lanes 1–5). Antibodies against
p50, p65, c-Rel and RelB induced a supershift of the DNA-protein
complexes (Fig. 4C, lanes 6–8 and
10). Taken together, the results indicate that NF-κB proteins bind
to both κB elements of the CD69 promoter in LMP-1- and
CD69-expressing Raji cells.
Discussion
The main issue addressed in this study was whether
LMP-1 induces CD69 gene transcription and the possible mechanisms
underlying this activity. In this study, we showed that EBV LMP-1
activates CD69 gene transcription through an NF-κB-dependent
pathway. The activation of CD69 promoter was mediated cooperatively
by CTAR-1 and CTAR-2 of LMP-1, as demonstrated by ectopic
expression of LMP-1 mutants. Interestingly, previous studies have
demonstrated that CTAR-1 and CTAR-2 cooperatively activate the
NF-κB signaling pathway (5–8), and
that CD69 promoter harbors three κB consensus sites (κB1-κB3)
(20). The results also showed
that both κB1 and κB2 domains of the CD69 promoter were absolutely
necessary for LMP-1-induced transcription.
The present results indicate that LMP-1 induces CD69
promoter activation through NIK-, IKKα- and IKKβ kinase-dependent
pathways. Dominant negative forms of NIK, IKKα, IKKβ- and IKKγ
inhibited LMP-1-induced CD69 promoter activation. These results are
consistent with previous evidence that NIK and IKKs are implicated
in the pathways through which LMP-1 induces NF-κB activation
(7,13). LMP-1 CTAR-1 directly recruits
TRAF1, 2, 3 and 5 whereas CTAR-2 indirectly recruits TRAF2 and 6
(5–8). CTAR-1 activates TRAF2-, NIK-and
IKKα-mediated NF-κB pathways, and CTAR-2 activates TRAF6-, IKKα-,
IKKβ- and IKKγ-mediated NF-κB pathways (7). Overexpression of dominant negative
forms of TRAF2 and TRAF5 inhibited LMP-1-mediated CD69 promoter
activation, indicating that NF-κB is an important component of
LMP-1-mediated CD69 gene induction from TRAFs-interacting
sites.
CD69 has been found to be rapidly upregulated on all
the leukocyte lineages studied, upon activation with the
corresponding stimuli (14). CD69
expression has also been reported in infections (32–35).
Earlier studies showed that CD69 regulates the immune response by
modulating the expression of various cytokines. CD69-deficient mice
show increased anti-tumor and autoimmune responses, which are
caused at least in part by increased production of proinflammatory
cytokines and chemokines (36,37).
More recently, tumor-derived CD69+ T cells have been
found to induce immune tolerance in the tumor environment (38). CD69 is reported to be a critical
negative regulator of immune activation during intracellular
bacterial infection (35).
LMP-1-mediated CD69 may have an important role in the immune
surveillance evasion by EBV-infected cells.
In summary, our experiments indicate that LMP-1
CTAR-1 and -2 can regulate CD69 gene at the transcriptional level
via two NF-κB-binding sites in its promoter, and TRAFs, NIK and
IKKs are effectors of CD69 promoter activation from LMP-1. Further
studies are necessary to resolve the question of the role of CD69
in EBV-associated diseases.
Acknowledgements
We thank Dr Martin Rowe, Dr Francisco
Sánchez-Madrid, Dr Dean W. Ballard, Dr Romas Geleziunas, Dr
Kuan-Teh Jeang, Dr Marta Muzio and Dr Toshiki Watanabe, for
providing the expression vectors for LMP-1 and its mutants; CD69
promoter pXP2 luciferase reporter plasmids; expression vectors for
IκBα- and IκBβ-dominant negative mutants; for NIK-, IKKα- and
IKKβ-dominant negative mutants; for IKKγ-dominant negative mutant;
and for plasmids for truncated TRAF2 and TRAF5. We also thank Dr
Zahidunnabi Dewan for providing LCL-Ao, LCL-Ka and LCL-Ku.
References
|
1.
|
Epstein MA, Achong BG and Barr YM: Virus
particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet.
1:702–703. 1964.PubMed/NCBI
|
|
2.
|
Kieff ED and Rickinson AB: Epstein-Barr
virus and its replication. Fields’ virology. Knipe DM and Howley
PM: Lippincott Williams & Wilkins; Philadelphia: pp. 2603–2654.
2006
|
|
3.
|
Taylor GS and Blackbourn DJ: Infectious
agents in human cancers: lessons in immunity and immunomodulation
from gammaherpesviruses EBV and KSHV. Cancer Lett. 305:263–278.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Kaye KM, Izumi KM and Kieff E:
Epstein-Barr virus latent membrane protein 1 is essential for
B-lymphocyte growth transformation. Proc Natl Acad Sci USA.
90:9150–9154. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Devergne O, Cahir McFarland ED, Mosialos
G, Izumi KM, Ware CF and Kieff E: Role of the TRAF binding site and
NF-κB activation in Epstein-Barr virus latent membrane protein
1-induced cell gene expression. J Virol. 72:7900–7908. 1998.
|
|
6.
|
Izumi KM and Kieff ED: The Epstein-Barr
virus oncogene product latent membrane protein 1 engages the tumor
necrosis factor receptor-associated death domain protein to mediate
B lymphocyte growth transformation and activate NF-κB. Proc Natl
Acad Sci USA. 94:12592–12597. 1997.PubMed/NCBI
|
|
7.
|
Soni V, Cahir-McFarland E and Kieff E:
LMP1 TRAFficking activates growth and survival pathways. Adv Exp
Med Biol. 597:173–187. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Schultheiss U, Püschner S, Kremmer E, Mak
TW, Engelmann H, Hammerschmidt W and Kieser A: TRAF6 is a critical
mediator of signal transduction by the viral oncogene latent
membrane protein 1. EMBO J. 20:5678–5691. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Hayden MS and Ghosh S: Shared principles
in NF-κB signaling. Cell. 132:344–362. 2008.
|
|
10.
|
Zandi E and Karin M: Bridging the gap:
composition, regulation, and physiological function of the IκB
kinase complex. Mol Cell Biol. 19:4547–4551. 1999.PubMed/NCBI
|
|
11.
|
Woronicz JD, Gao X, Cao Z, Rothe M and
Goeddel DV: IκB kinase-β: NF-κB activation and complex formation
with IκB kinase-α and NIK. Science. 278:866–869. 1997.
|
|
12.
|
Lee FS, Peters RT, Dang LC and Maniatis T:
MEKK1 activates both IκB kinase α and IκB kinase β. Proc Natl Acad
Sci USA. 95:9319–9324. 1998.
|
|
13.
|
Sylla BS, Hung SC, Davidson DM,
Hatzivassiliou E, Malinin NL, Wallach D, Gilmore TD, Kieff E and
Mosialos G: Epstein-Barr virus-transforming protein latent
infection membrane protein 1 activates transcription factor NF-κB
through a pathway that includes the NF-κB-inducing kinase and the
IκB kinases IKKα and IKKβ. Proc Natl Acad Sci USA. 95:10106–10111.
1998.PubMed/NCBI
|
|
14.
|
Sánchez-Mateos P and Sánchez-Madrid F:
Structure-function relationship and immunochemical mapping of
external and intracellular antigenic sites on the lymphocyte
activation inducer molecule, AIM/CD69. Eur J Immunol. 21:2317–2325.
1991.
|
|
15.
|
Sant’Angelo DB, Lucas B, Waterbury PG,
Cohen B, Brabb T, Goverman J, Germain RN and Janeway CA Jr: A
molecular map of T cell development. Immunity. 9:179–186. 1998.
|
|
16.
|
Testi R, D’Ambrosio D, De Maria R and
Santoni A: The CD69 receptor: a multipurpose cell-surface trigger
for hematopoietic cells. Immunol Today. 15:479–483. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
De la Fuente H, Cibrián D and
Sánchez-Madrid F: Immunoregulatory molecules are master regulators
of inflammation during the immune response. FEBS Lett.
586:2897–2905. 2012.PubMed/NCBI
|
|
18.
|
Huen DS, Henderson SA, Croom-Carter D and
Rowe M: The Epstein-Barr virus latent membrane protein-1 (LMP1)
mediates activation of NF-κB and cell surface phenotype via two
effector regions in its carboxy-terminal cytoplasmic domain.
Oncogene. 10:549–560. 1995.
|
|
19.
|
Floettmann JE and Rowe M: Epstein-Barr
virus latent membrane protein-1 (LMP1) C-terminus activation region
2 (CTAR2) maps to the far C-terminus and requires oligomerisation
for NF-κB activation. Oncogene. 15:1851–1858. 1997.PubMed/NCBI
|
|
20.
|
López-Cabrera M, Muñoz E, Blázquez MV,
Ursa MA, Santis AG and Sánchez-Madrid F: Transcriptional regulation
of the gene encoding the human C-type lectin leukocyte receptor
AIM/CD69 and functional characterization of its tumor necrosis
factor-α-responsive elements. J Biol Chem. 270:21545–21551.
1995.PubMed/NCBI
|
|
21.
|
Castellanos Mdel C, López-Giral S,
López-Cabrera M and de Landázuri MO: Multiple cis-acting elements
regulate the expression of the early T cell activation antigen
CD69. Eur J Immunol. 32:3108–3117. 2002.PubMed/NCBI
|
|
22.
|
Brockman JA, Scherer DC, McKinsey TA, Hall
SM, Qi X, Lee WY and Ballard DW: Coupling of a signal response
domain in IκBα to multiple pathways for NF-κB activation. Mol Cell
Biol. 15:2809–2818. 1995.
|
|
23.
|
McKinsey TA, Brockman JA, Scherer DC,
Al-Murrani SW, Green PL and Ballard DW: Inactivation of IκBβ by the
Tax protein of human T-cell leukemia virus type 1: a potential
mechanism for constitutive induction of NF-κB. Mol Cell Biol.
16:2083–2090. 1996.
|
|
24.
|
Geleziunas R, Ferrell S, Lin X, Mu Y,
Cunningham ET Jr, Grant M, Connelly MA, Hambor JE, Marcu KB and
Greene WC: Human T-cell leukemia virus type 1 Tax induction of
NF-κB involves activation of the IκB kinase α (IKKα) and IKKβ
cellular kinases. Mol Cell Biol. 18:5157–5165. 1998.
|
|
25.
|
Iha H, Kibler KV, Yedavalli VRK,
Peloponese JM, Haller K, Miyazato A, Kasai T and Jeang K-T:
Segregation of NF-κB activation through NEMO/IKKγ by Tax and TNFα:
implications for stimulus-specific interruption of oncogenic
signaling. Oncogene. 22:8912–8923. 2003.
|
|
26.
|
Muzio M, Ni J, Feng P and Dixit VM: IRAK
(Pelle) family member IRAK-2 and MyD88 as proximal mediators of
IL-1 signaling. Science. 278:1612–1615. 1997. View Article : Google Scholar
|
|
27.
|
Aizawa S, Nakano H, Ishida T, Horie R,
Nagai M, Ito K, Yagita H, Okumura K, Inoue J and Watanabe T: Tumor
necrosis factor receptor associated factor (TRAF) 5 and TRAF2 are
involved in CD30-mediated NFκB activation. J Biol Chem.
272:2042–2045. 1997.PubMed/NCBI
|
|
28.
|
Antalis TM and Godbolt D: Isolation of
intact nuclei from hematopoietic cell types. Nucl Acids Res.
19:43011991. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Dewan MZ, Tomita M, Katano H and Yamamoto
N, Ahmed S, Yamamoto M, Sata T, Mori N and Yamamoto N: An HIV
protease inhibitor, ritonavir targets the nuclear factor-kappaB and
inhibits the tumor growth and infiltration of EBV-positive
lymphoblastoid B cells. Int J Cancer. 124:622–629. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Xu Z-G, Iwatsuki K, Oyama N, Ohtsuka M,
Satoh M, Kikuchi S, Akiba H and Kaneko F: The latency pattern of
Epstein-Barr virus infection and viral IL-10 expression in
cutaneous natural killer/T-cell lymphomas. Br J Cancer. 84:920–925.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Baeuerle PA: The inducible transcription
activator NF-κB: regulation by distinct protein subunits. Biochim
Biophys Acta. 1072:63–80. 1991.
|
|
32.
|
Hodge G, Hodge S, Han P and Haslam R:
Multiple leucocyte activation markers to detect neonatal infection.
Clin Exp Immunol. 135:125–129. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Böhler T, Walcher J, Hölzl-Wenig G,
Schnitzler P, Geiss M, Buchholz B, Linde R, Rütschle H and Debatin
K-M: Expression of CD69 on T-cells from HIV-1-infected children and
adolescents increases with increasing viral load. Eur J Pediatr.
158:638–644. 1999.PubMed/NCBI
|
|
34.
|
Iwashiro M, Messer RJ, Peterson KE,
Stromnes IM, Sugie T and Hasenkrug KJ: Immunosuppression by
CD4+ regulatory T cells induced by chronic retroviral
infection. Proc Natl Acad Sci USA. 98:9226–9230. 2001.
|
|
35.
|
Vega-Ramos J, Alari-Pahissa E, Valle JD,
Carrasco-Marín E, Esplugues E, Borràs M, Martínez-A C and Lauzurica
P: CD69 limits early inflammatory diseases associated with immune
response to Listeria monocytogenes infection. Immunol Cell
Biol. 88:707–715. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Esplugues E, Sancho D, Vega-Ramos J,
Martínez C, Syrbe U, Hamann A, Engel P, Sánchez-Madrid F and
Lauzurica P: Enhanced antitumor immunity in mice deficient in CD69.
J Exp Med. 197:1093–1106. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Sancho D, Gómez M, Viedma F, Esplugues E,
Gordón-Alonso M, García-López MA, de la Fuente H, Martínez-A C,
Lauzurica P and Sánchez-Madrid F: CD69 downregulates autoimmune
reactivity through active transforming growth factor-β production
in collagen-induced arthritis. J Clin Invest. 112:872–882.
2003.PubMed/NCBI
|
|
38.
|
Zhao Q, Kuang D-M, Wu Y, Xiao X, Li X-F,
Li T-J and Zheng L: Activated CD69+ T cells foster
immune privilege by regulating IDO expression in tumor-associated
macrophages. J Immunol. 188:1117–1124. 2012.
|