Introduction
Esophageal cancer is one of the most aggressive
types of cancer and the eighth most common cause of cancer-related
death in the world (1,2). In spite of recent advances in the
development of anticancer and molecular target drugs, the
combination regimen of 5-fluorouracil (5-FU) and cisplatin (CDDP)
still plays an important role in the treatment of esophageal
cancer, as the gold standard chemotherapy.
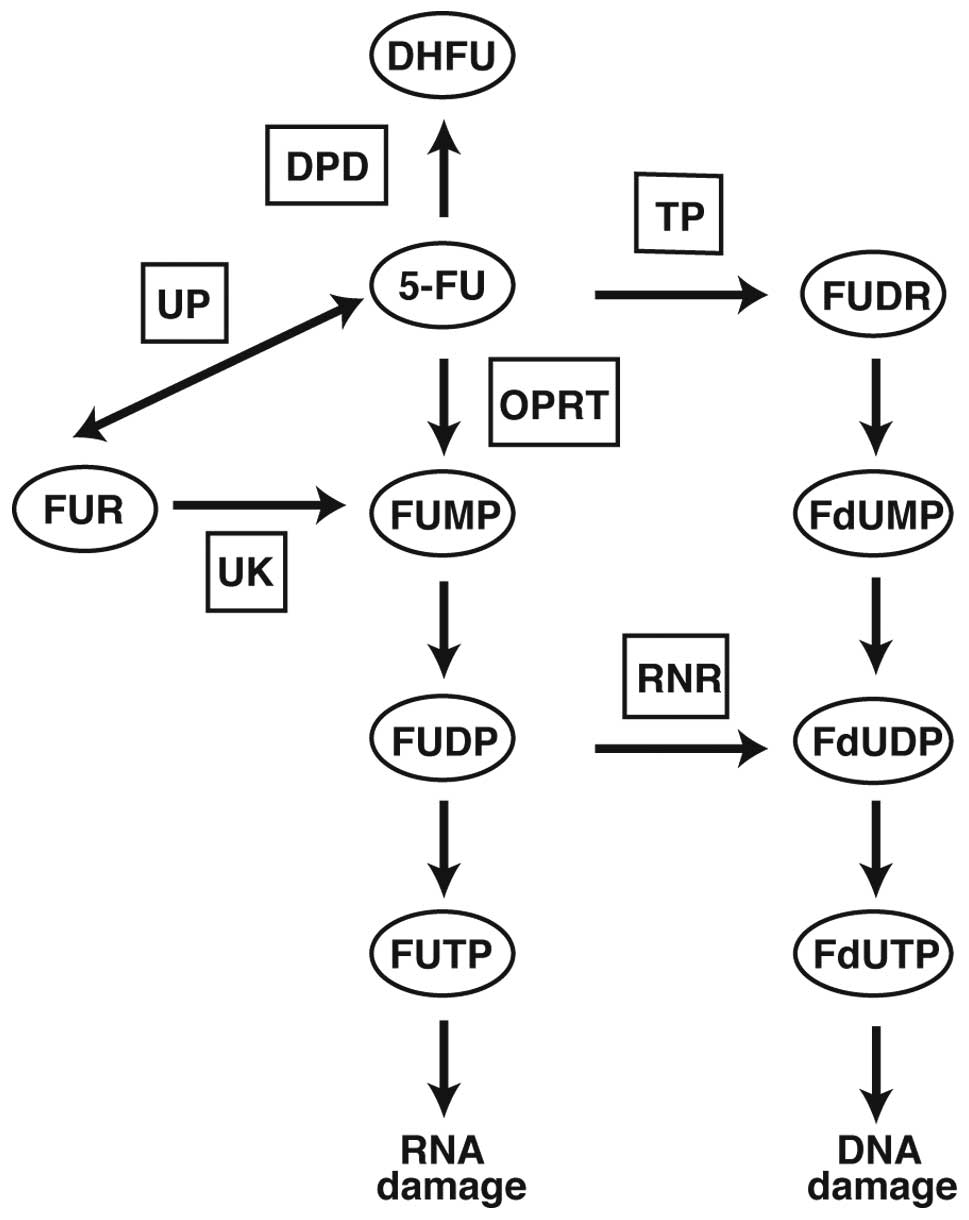
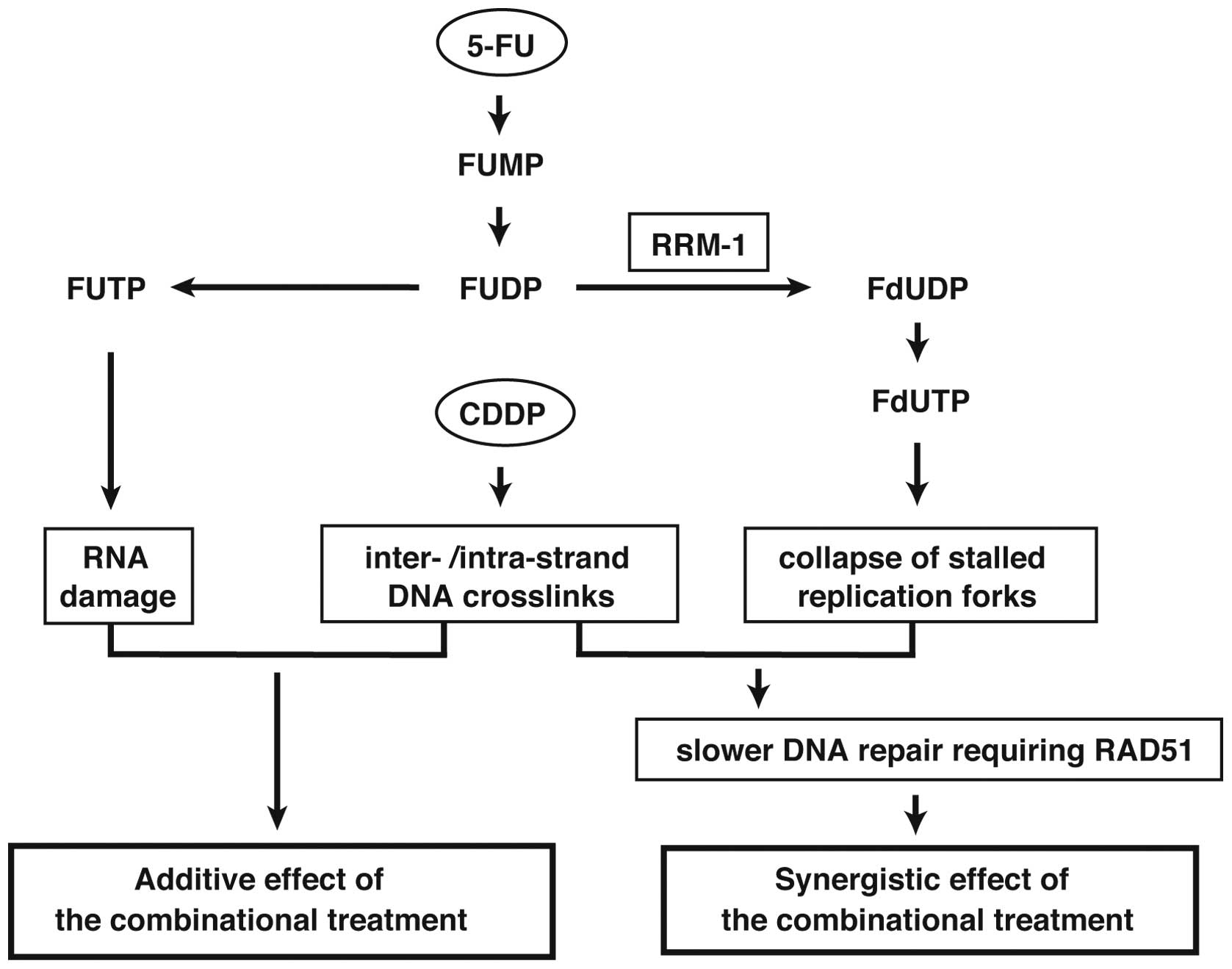
The nucleoside analogue 5-FU is converted to several
active metabolites, including fluorouridine triphosphate (FUTP),
fluorodeoxyuridine triphosphate (FdUTP) and fluorodeoxyuridine
monophosphate (FdUMP), to exert its cytotoxic activity (Fig. 1). FUTP is largely incorporated into
the RNA strand, preventing the processing and function of various
types of RNA including mRNA, rRNA, tRNA and snRNA to exert
anticancer effect of 5-FU (3).
Recently, several lines of evidence suggest that these metabolites
of 5-FU also interrupt DNA metabolism by misincorporation or enzyme
inhibition (3). For example, FdUMP
disturbs DNA replication by inhibiting the activity of thymidylate
synthetase (TS), through the depletion of dTMP. 5-FU may also
generate DNA damage, such as DNA single-strand breaks and/or DNA
double strand breaks (DSBs) via the collapse of stalled replication
forks. Although the induction of DNA damage has been shown to play
a role in the anticancer effect of 5-FU (4–6), the
means by which the metabolites of 5-FU exert their anticancer
effects through the disturbance of DNA or RNA metabolism remain
obscure (7).
Several 5-FU related enzymes, such as
dihydropyrimidine dehydrogenase (DPD), orotate phosphoribosyl
transferase (OPRT), thymidine phosphorylase (TP) and ribonucleotide
reductase (RNR), are considered as major determinants of the
outcome of 5-FU treatment (8–10)
(Fig. 1). Among these enzymes,
ribonucleotide reductase, composed of large subunit RRM-1 and small
subunit RRM-2, is the rate-limiting enzyme in de novo DNA
synthesis (11). RNR converts
fluorouridine diphosphate (FUDP) to fluorodeoxyuridine diphosphate
(FdUDP), which preferentially affects DNA metabolism. Recently, RNR
has also been shown to play an important role in DNA repair
(12). However, how RNR is
involved in the regulation of the anticancer effect of 5-FU remains
to be clarified.
DNA damage, such as stalled replication forks and
DSBs, induces the phosphorylation of histone H2AX on Ser-139
(γ-H2AX), and the focus formation of γ-H2AX (γ-H2AX foci) (13). γ-H2AX is required to retain DNA
repair proteins and checkpoint proteins at damaged sites, for the
efficient processing of damaged DNA (14,15).
Homologous recombination (HR) is a versatile DNA repair mechanism,
because it can promote the elimination of a variety of lesions,
including DSBs, single-strand gaps and stalled DNA replication
forks (16,17). RAD51 is one of the key proteins for
DNA repair by HR, since it mediates homologous pairing and strand
exchange between DNA duplexes (18). RAD51 has also been shown to form
nuclear foci at DSB sites (19).
Since many anticancer drugs induce DNA damage, analyses of DNA
repair proteins, such as γ-H2AX and RAD51, could be useful to
predict the outcome of anticancer drugs (20–22).
Recently, a dose-dependent increase in the number of γ-H2AX foci
after 5-FU treatment was reported (23). CDDP also induces the γ-H2AX and
RAD51 foci by the formation of interstrand and intrastrand DNA
crosslinks, which lead to the formation of DSBs when they meet
replication forks (24–26). Although the combination regimen of
5-FU and CDDP shows synergistic effects both in vitro and
in vivo, the involvement of DNA damage in this effect
remained to be clarified (27,28).
In this study, we investigated the mechanism of the
anticancer effect of 5-FU, by analyzing the kinetics of γ-H2AX and
RAD51 foci formation in esophageal cancer cells. RRM-1 was required
for the γ-H2AX focus formation after 5-FU treatment. Moreover,
RRM-1 enhanced the formation of RAD51 foci, but not γ-H2AX foci,
when cells were treated with 5-FU and CDDP in combination. These
findings clearly suggest that RRM-1 is involved in the anticancer
effect of 5-FU by modulating DNA repair in human esophageal cancer
cells.
Materials and methods
Cell lines, culture medium and
chemicals
Two esophageal carcinoma cell lines (TE1 and TE11)
were obtained from the Cell Resource Center for the Biomedical
Research Institute of Development, Aging and Cancer (Tohoku
University, Sendai, Japan). TE1 is a well differentiated squamous
cell carcinoma, and TE11 is a moderately differentiated squamous
cell carcinoma. These cell lines were routinely grown in RPMI-1640
(Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine
serum, and incubated at 37°C in a humidified atmosphere of 5%
CO2 in air. 5-FU (Kyowa Hakko Co., Japan) and CDDP
(Nihonkayaku Co., Japan) were dissolved in PBS at 1 mM.
Clonogenic assay for cell survival
The drug sensitivity of the cells was assessed by
their colony forming ability. In brief, approximately
2.0×103 cells were seeded in a 60-mm tissue culture dish
and incubated overnight. To assess the synergistic effect of 5-FU
and CDDP, the concomitant treatment regimen involved the
simultaneous treatment of cells with 5-FU (1–3 μM) for 24 h
and CDDP (1–5 μM) for 1 h, alongside suitable controls of
cells treated with appropriate doses of CDDP (0–10 μM) alone
for 1 h, or 5-FU (0–10 μM) alone for 24 h. The cells were
washed in PBS and cultured for 7–10 days in a 5% CO2
atmosphere at 37°C in air. The colonies were fixed with 100%
methanol for 10 min, and stained with Giemsa in phosphate buffer
(pH 6.4). Colonies composed of 50 or more cells were scored as
survivors, and the surviving fraction for a given treatment dose
was calculated as the relative plating efficiency of treated versus
untreated (control) cultures. All experiments were performed three
times and yielded similar results.
Dose-response curves were analyzed using the
CalcuSyn Software (Biosoft), which is based on the median effect
model of Chou and Talalay. A combination index (CI) of 1 indicated
an additive drug interaction, whereas a CI of >1 was
antagonistic and a score lower than 1 was synergistic.
Immunofluorescence analysis
The cells were seeded in 6-well plates containing
coverslips, and were treated with 3 μM 5-FU for 24 h, 5
μM CDDP for 1 h, or the concomitant treatment, and were
allowed to recover for the indicated periods. The coverslips were
then fixed with 4% paraformadehyde at room temperature for 10 min.
The cells were washed twice with 0.1% Triton X-100 in PBS (PBS-T),
permeablized with 0.5% Triton X-100 and 0.1% SDS in PBS for 10 min,
and washed with 0.05% glycine and 0.1% BSA in PBS (PBS+). The
coverslips were incubated with the primary antibody at 37°C for 90
min. The primary antibodies were used at the following ratios:
anti-RAD51 (Calbiochem, La Jolla, CA, USA), 1:500; anti-γ-H2AX
(Upstate Biotechnology, Lake Plasid, NY, USA) 1:40,000. The
coverslips were incubated at 37°C for 1 h with the secondary
antibody, either sheep-anti-mouse-Cy3 (BioSource, USA) at 1:400 or
goat-anti-rabbit-FITC (Jackson ImmunoResearch Laboratories Inc.,
West Grove, PA, USA) at 1:500. DNA counter staining was
accomplished with DAPI (4′, 6-diamidino-2-phenylindole). After
staining, the coverslips were sealed with VectaShield (Vector
Laboratories Inc., Burlingame, CA, USA) and stored in the dark.
Images were obtained with a Zeiss Axiovert 200 M
fluorescence microscope, using the AxioVision Rel 4.4 software
(Carl Zeiss, Germany). Cells were scored as positive for
damage-induced foci if there were ≥15 spots per cell for γ-H2AX and
≥5 for RAD51. About 200 cells were observed for each regimen, and
the averaged data are from three independent experiments.
siRNA treatment
TE11 cells were transfected with either the
non-targeting control (NT) siRNA (Dharmacon, Inc., USA) or the
siRNA for human RRM-1 (GGAUCGCUGUCUCUAA CUUtt). The siRNA for human
RRM-1 was kindly provided by Dr H. Niida (Department of Molecular
Biology, Hamamatsu University School of Medicine, Shizuoka, Japan)
(12). Transfection was performed
24 h before 5-FU and/or CDDP treatment using Lipofectamine 2000
(Invitrogen) according to the manufacturer’s instructions, and the
final concentrations of both the NT siRNA and RRM-1 siRNA were 1 nM
in OptiMEM (Invitrogen) medium per well.
Immunoblot analysis
Cells were harvested and lysed in Laemmli’s buffer.
Total cell protein extracts were fractionated by SDS-PAGE using a
multigel II (Cosmo Bio, Japan), and were electrophoretically
transferred onto PVDF membranes. The membranes were blocked with 5%
non-fat dried milk in TBS containing 0.1% Tween-20 (TBS-T) for 1 h.
The membranes were then incubated with primary antibodies against
β-actin (Sigma, St. Louis, MO, USA), RRM-1 or RRM-2 (Santa Cruz
Biotechnology Inc., Santa Cruz, CA, USA) for 1 h. The membranes
were then washed three times with TBS-T and incubated with the
appropriate secondary antibodies. After three washes with TBS-T,
the immunoreactive proteins were visualized by enhanced
chemiluminescence (Amersham Biosciences, Uppsala, Sweden).
Flow cytometry for γ-H2AX analysis
The cells were harvested with 2% trypsin and washed
three times in cold PBS. After permeation, the cells were incubated
with the anti-γ-H2AX antibody (1: 40,000 dilution) for 1 h. The
cells were washed and resuspended in the secondary antibody, sheep
anti-mouse-Cy3 (1:400 dilution), for 30 min. After fixation in 0.5%
paraformadehyde, the fluorescence emitted by Cy3 was measured by
flow cytometry.
Statistical analysis
The differences in the cell viability and the
percentage of focus formation of γ-H2AX and RAD51 between samples
were evaluated with the Student’s unpaired t-test. P<0.05 was
regarded as statistically significant.
Results
Induction of DNA damage with 5-FU in
esophageal cancer cell lines
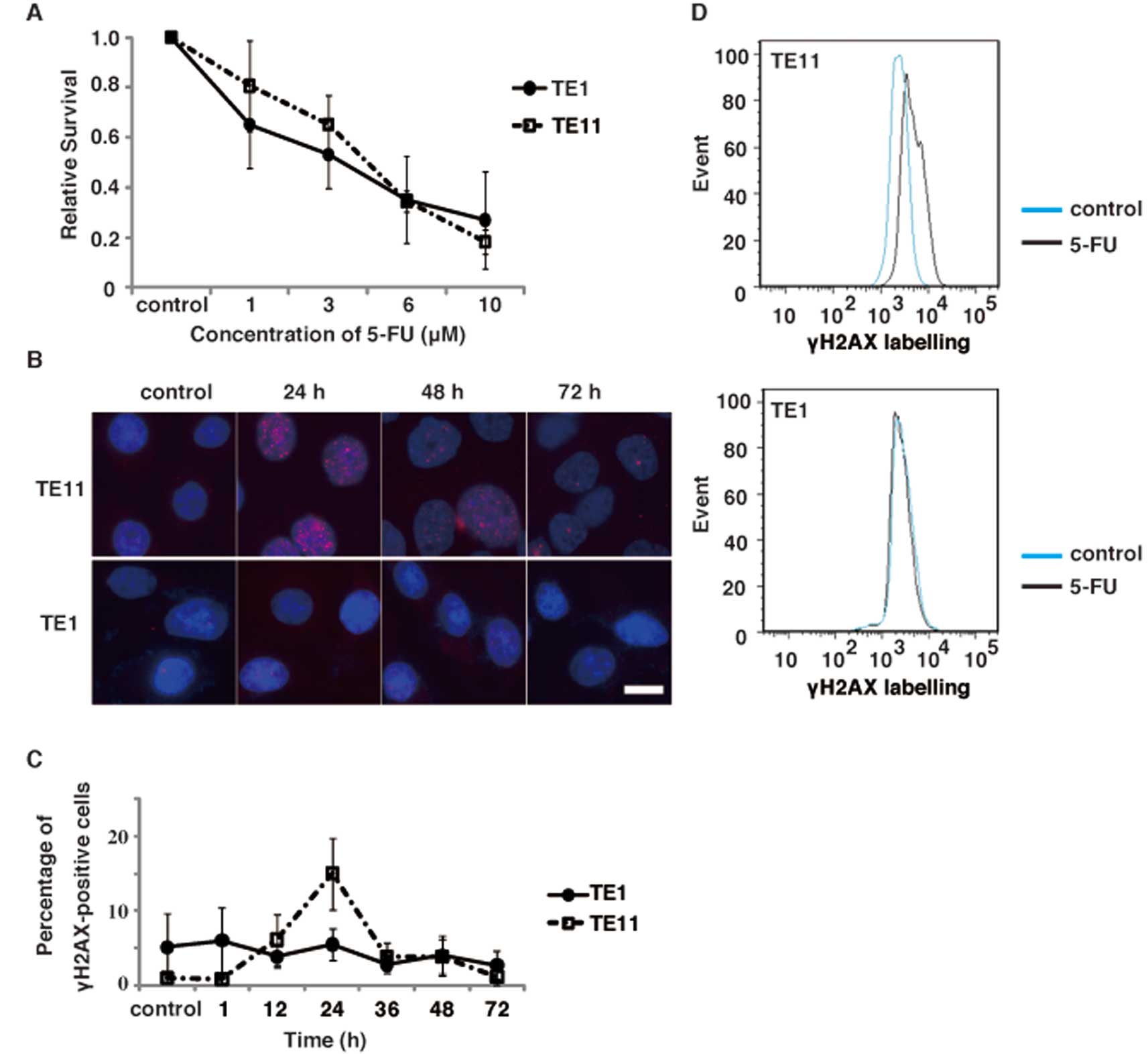
To study the mechanisms of the anticancer activity
of 5-FU, we investigated the survival of two esophageal cell lines,
TE1 and TE11, after 5-FU treatment by a clonogenic assay.
Representative dose-survival responses of 5-FU are depicted in
Fig. 2A. The Dm values for 5-FU
were estimated as 3.6 and 2.7 μM in TE11 and TE1,
respectively. This indicates that there are no significant
differences in the 5-FU sensitivities between these two cell
lines.
Next, we examined the role of the induction of DNA
damage in the anticancer effect of 5-FU (4,5). The
formation of γ-H2AX foci at damaged sites is one of the highly
sensitive markers of DNA damage induced by ionizing radiation
and/or chemotherapeutic agents, such as CDDP (21). Therefore, we performed
immunofluorescence staining of 5-FU-treated TE11 cells, using
anti-γ-H2AX antibodies (Fig. 2B).
The percentage of cells with γ-H2AX foci increased in a
time-dependent manner. The peak was observed at 24 h after 5-FU
exposure in TE11 cells (Fig. 2C).
The flow cytometry analysis of γ-H2AX confirmed the increase in
γ-H2AX positive TE11 cells after 5-FU treatment (Fig. 2D). In contrast to TE11, TE1 cells
failed to show an increase in γ-H2AX foci positive cells after 5-FU
treatment (Fig. 2B–D). These
findings suggest that 5-FU induces more DNA damage in TE11 cells,
as compared to TE1 cells. Since the sensitivity of TE11 cells to
5-FU is similar to that of TE1 (Fig.
2A), the mechanism by which 5FU exerts its anticancer effect
could be different in these esophageal cell lines.
Metabolism of 5-FU in the two esophageal
cancer cell lines
The difference in the induction of DNA damage by
5-FU in TE1 and TE11 cells led us to examine the enzymes involved
in the metabolism of the drug (3).
The pharmacogenetic variability of 5-FU-related enzymes, such as
DPD, OPRT, TS and RNR, is associated with the anticancer activity
of 5-FU (8–10). Among these enzymes, RNR is required
to supply ribonucleotides for both DNA replication and repair
(12). Thus, we examined the
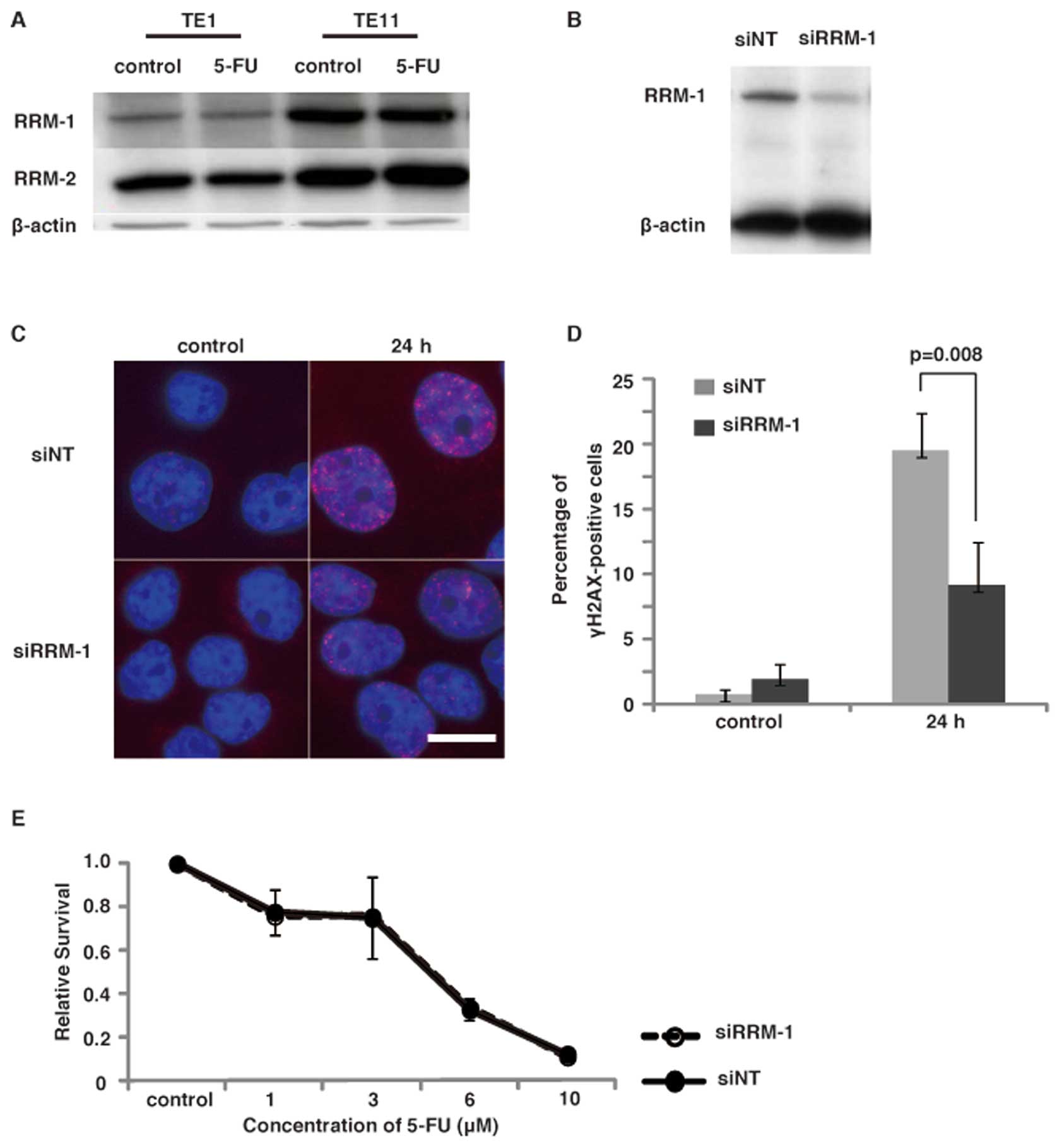
protein levels of the RNR subunits, RRM-1 and RRM-2, in the two
cell lines, TE1 and TE11. The immunoblot analyses using the
anti-RRM-1 antibody revealed that TE11 expressed a significantly
higher level of RRM-1, as compared to TE1 cells. The expression of
RRM-2 was slightly higher in TE11 cells than TE1 cells (Fig. 3A). The treatment of cells with 5-FU
did not alter the expression of these enzymes. These results
suggest that the high expression of RNR, especially RRM-1, in TE11
facilitates the induction of DNA damage, by converting 5-FU to
5-FdU.
To confirm the involvement of RRM-1 in the induction
of DNA damage by 5-FU, we next examined the effect of RRM-1
depletion in TE11 cells. RRM-1 depletion by the siRNA was confirmed
by an immunoblot analysis of TE11 cells (Fig. 3B). To investigate the effect of
RRM-1 depletion on DNA damage in TE11 cells, we first performed
immunofluorescence staining of γ-H2AX after 5-FU treatment
(Fig. 3C). The RRM-1 depletion by
siRNA significantly repressed the γ-H2AX focus formation after 5-FU
treatment (p<0.01) (Fig. 3D).
This finding strongly supports the notion that RRM-1 is involved in
the induction of DNA damage in TE11 cells after 5-FU treatment.
Next we investigated the survival of these cells
after 5-FU treatment by a clonogenic assay, to determine whether
RRM-1 depletion is involved in 5-FU sensitivity. Representative
dose-survival responses of 5-FU are depicted in Fig. 3E. The Dm values for 5-FU were
estimated as 3.34 μM in RRM-1 siRNA treated cells and 3.44
μM in control cells. Therefore, a significant change in 5-FU
sensitivity by the depletion of RRM-1 was not observed. The reduced
anticancer activity of 5-FU, through the lower induction of DNA
damage, might be compensated by the increased disturbance of RNA
metabolism by the depletion of RRM-1.
Induction of DNA damage and damage
response with the combination of 5-FU and CDDP in esophageal cancer
cell lines
Having established that 5-FU induces DNA damage in
TE11 cells, but not in TE1 cells, we elucidated the mechanism of
the synergistic effect of the combination regimen of 5-FU and CDDP.
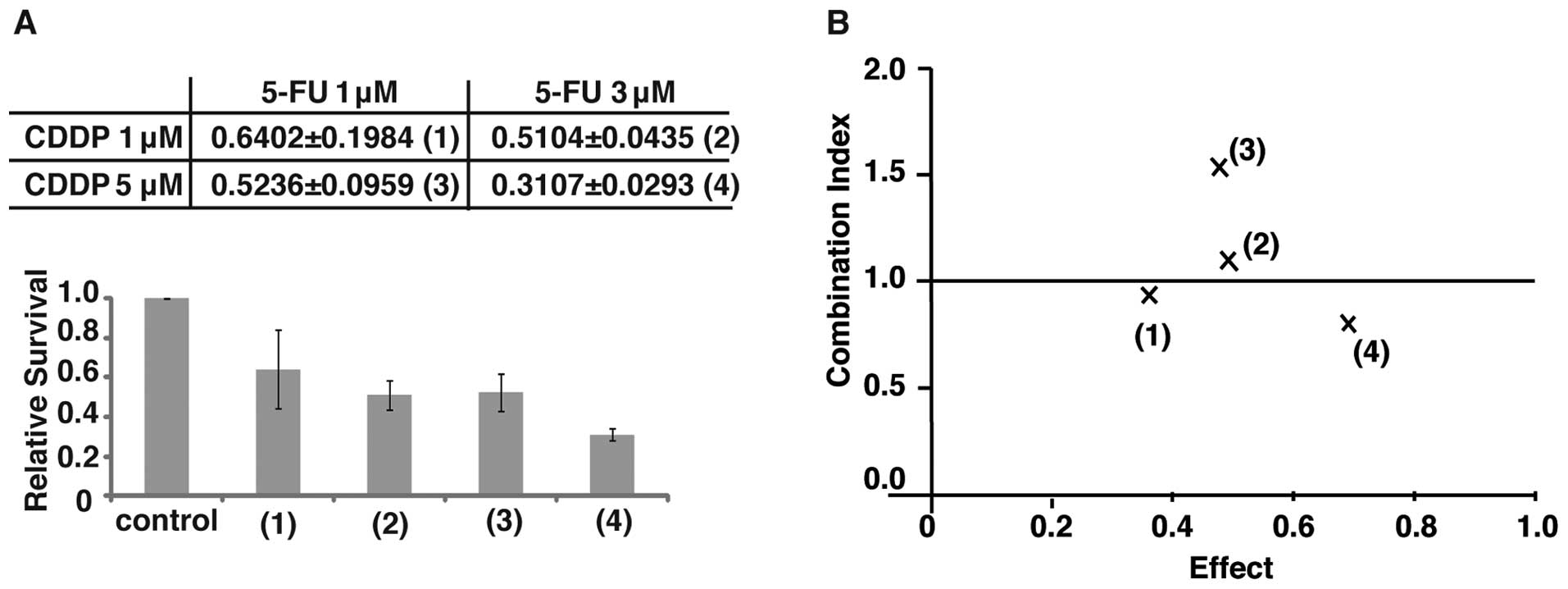
First we performed clonogenic assays to determine if the
synergistic effect could be observed in the TE11 cell line. As a
result, the co-incubation of TE11 cells with 5 μM CDDP and 3
μM 5-FU showed the most significant synergistic inhibition
of cell growth (Fig. 4A and
B).
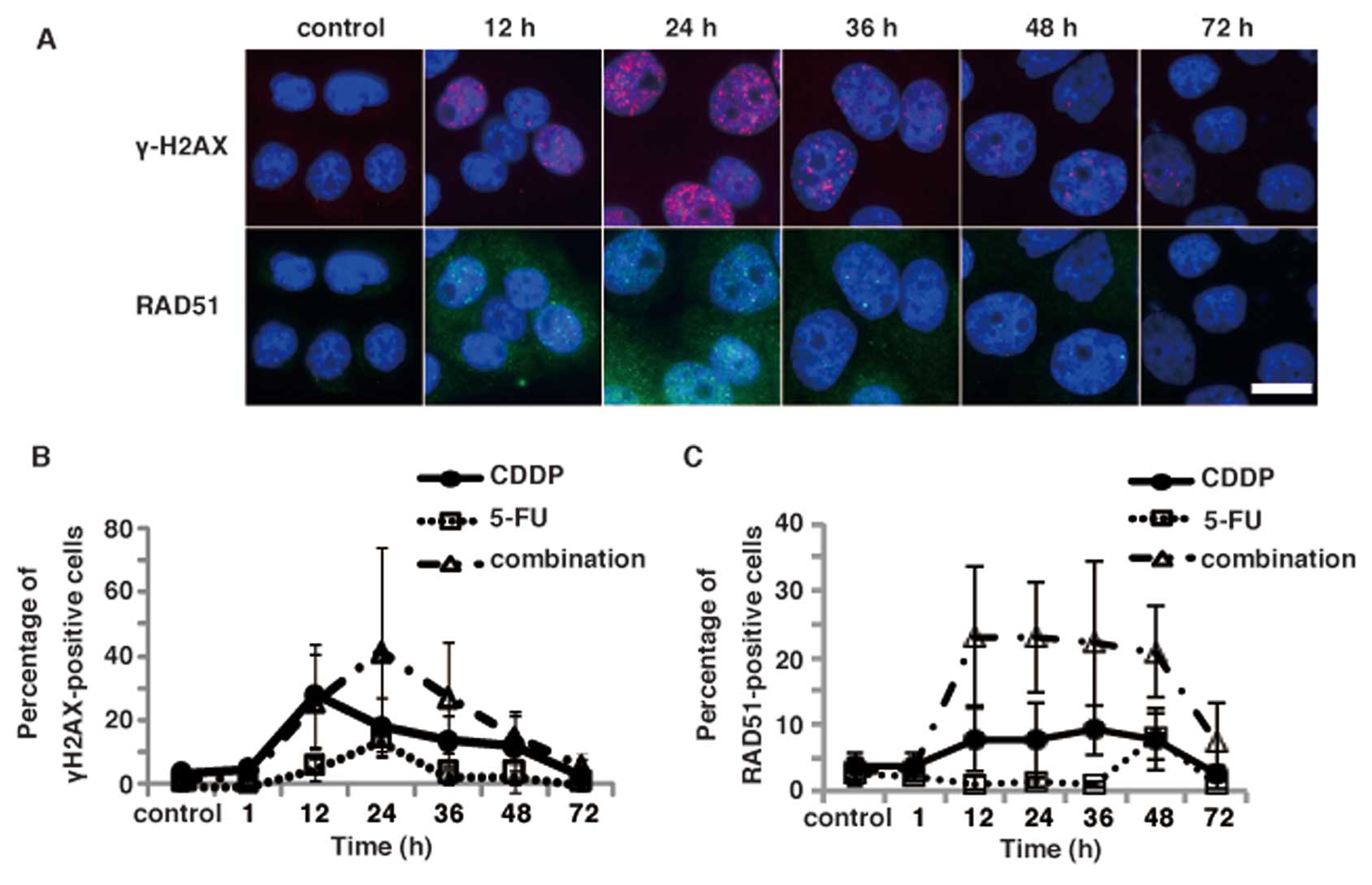
Since CDDP is a widely used platinum-containing
anticancer drug with an activity mechanism that involves the
formation of DNA damage, we next examined the induction of DNA
damage in TE11 cells after treatments with either CDDP alone or 3
μM 5-FU and 5 μM CDDP. Immunofluorescence staining of
TE11 cells with the anti-γ-H2AX antibody revealed that CDDP alone
and in the combination treatment induced γ-H2AX focus formation in
a time-dependent manner (Fig. 5A and
B). The percentage of γ-H2AX foci was slightly higher in the
cells treated with the combination of 5-FU and CDDP than in those
treated with CDDP or 5-FU alone. The peak was observed at 24 h
after the combination regimen, and was similar to 5-FU alone
(Fig. 5B). In contrast, the peak
of γ-H2AX focus formation was observed at 12 h after CDDP
treatment. Moreover, the increase in the percentage of γ-H2AX focus
positive cells by the combinational treatment was additive.
Therefore, the synergic effect may be due to a modulation of DNA
repair, rather than an increase in the induction of DNA damage by
the combinational treatment.
To further investigate the involvement of DNA damage
induced by the combinational treatment in the synergic effect, we
examined the kinetics of RAD51 focus formation, another DNA damage
marker formed by a recombinational repair protein (Fig. 5A). Immunofluorescence analyses
using an anti-RAD51 antibody revealed that the percentage of RAD51
foci in TE11 cells was not significantly increased after a
treatment with 5-FU alone. While CDDP treatment slightly increased
the percentage of RAD51 foci positive cells, the combinational
treatment with 5-FU and CDDP drastically induced the RAD51 focus
formation in TE11 cells (Fig. 5C).
Interestingly, the increased focus formation of RAD51 persisted
longer than that of γ-H2AX after the combinational treatment.
Moreover, increase of the focus formation of RAD51 is more
significant than that of γ-H2AX by the combinational treatment
compared to either CDDP or 5-FU alone (Fig. 5B and C). Therefore, the modulation
of DNA repair, rather than the increased induction of DNA damage,
could contribute more to the synergic effect of the combinational
treatment observed in TE11 cells.
RRM-1 is involved in the synergic effect
of the combination treatment
We next examined if RRM-1 is involved in the
synergic effect of the combinational treatment, since RRM-1 plays a
role in the 5-FU-induced DNA damage in TE11 cells (Fig. 3). We performed a clonogenic assay
of RRM-1-depleted cells after the 5-FU/CDDP treatment. As expected,
the depletion of RRM-1 with siRNA significantly increased the
survival of TE11 cells after the combinational treatment
(p<0.05) (Fig. 6A). Since RRM-1
depletion did not affect the 5-FU or CDDP sensitivity of TE11
(Figs. 3E and 7C, p=0.90), this finding suggests the
involvement of RRM-1 in the synergic effect of the combination
regimen with 5-FU and CDDP in TE11 cells.
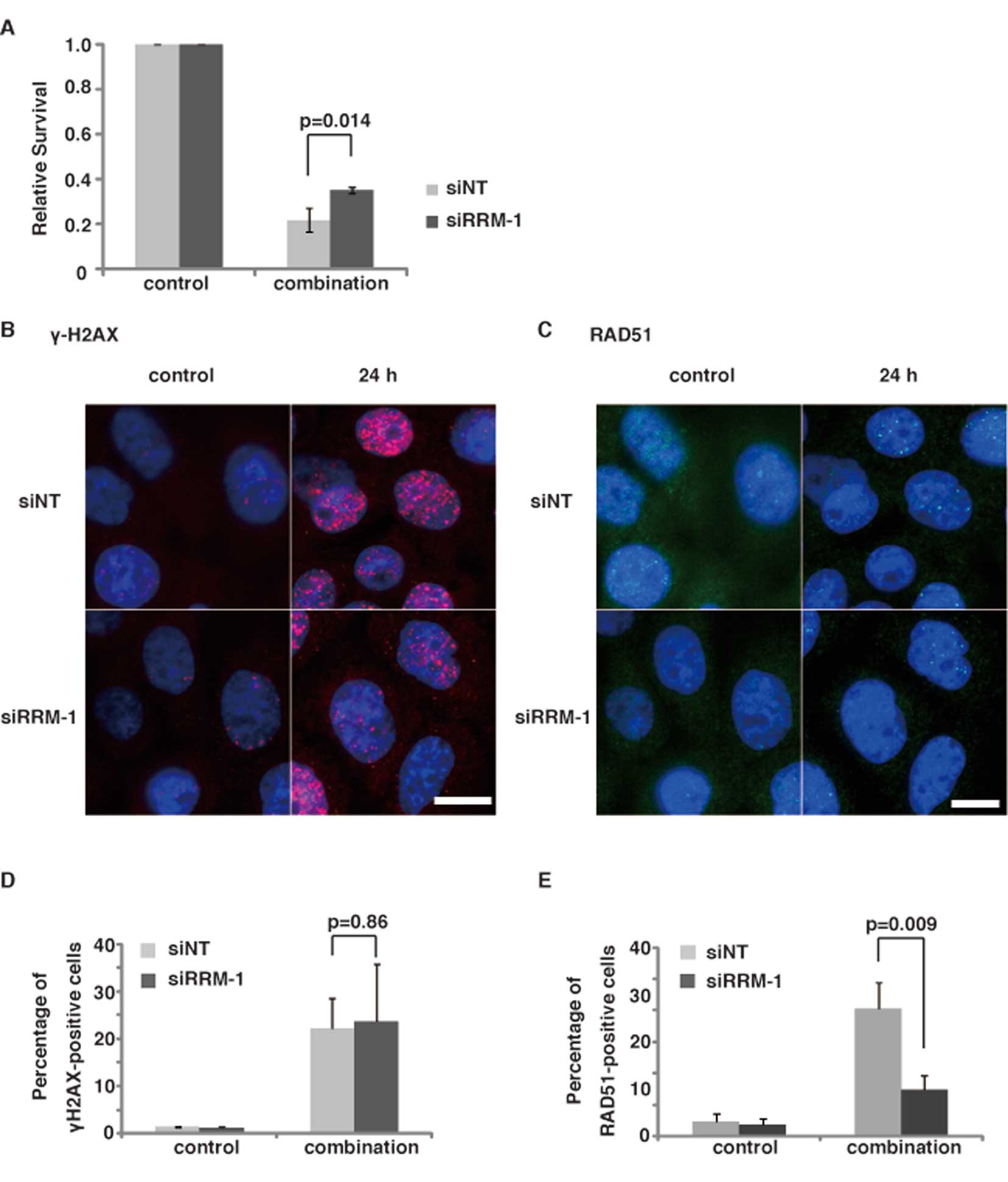 | Figure 6Effect of RRM-1 depletion on the
induction of γ-H2AX and RAD51 focus formation in TE11 cells treated
with the combination regimen. (A) The cytotoxicity of cells
expressing either the NT siRNA (siNT) or RRM-1 siRNA (siRRM-1) was
determined, using the clonogenic assay. Results are the means of at
least three independent experiments, and error bars show the
standard deviation of the mean. (B and C) Immunostaining of TE11
cells transfected with RRM-1 siRNA and NT siRNA, using anti-γ-H2AX
and RAD51 antibodies. Cells were treated with 3 μM 5-FU for
24 h and 5 μM CDDP for 1 h. γ-H2AX, RAD51 and DNA are shown
in red, green and blue, respectively. Scale bars, 20 μm. (D)
Percentage of γ-H2AX foci-positive RRM-1 depleted cells, before and
after the combinational treatment. All results are the means of
three independent experiments, and error bars show the standard
deviation of the mean. (E) Percentage of RAD51 foci-positive cells.
All results are the means of three independent experiments, and
error bars show the standard deviation of the mean. |
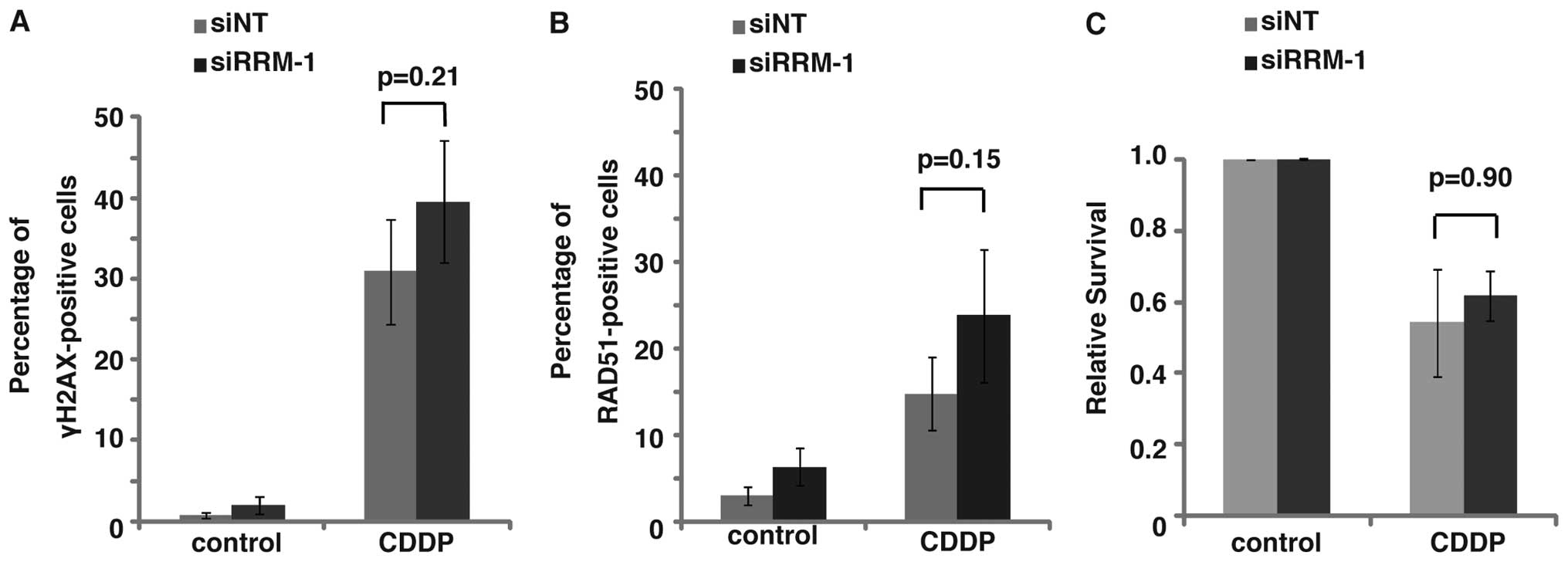
To study the role of RRM-1 in the synergic effect,
we examined the effect of RRM-1 depletion on the induction of DNA
damage after the combinational treatment of TE11 cells.
Immunofluorescence staining with the anti-γ-H2AX antibody revealed
that the depletion of RRM-1 by siRNA did not significantly change
the percentage of TE11 cells with γ-H2AX foci after the
combinational treatment (p=0.86, Fig.
6B and D). The γ-H2AX focus formation was not significantly
increased by the combinational treatment, as compared to the
treatments with 5-FU or CDDP alone (Fig. 5B). Moreover, the γ-H2AX focus
formation after CDDP treatment was not affected by the RRM-1
depletion (Fig. 7A, p=0.21). Taken
together, these findings strongly suggest that the facilitation of
DNA damage induction by RRM-1 is not involved in the synergic
effect of the combinational treatment.
If the synergic effect of the combinational
treatment was not due to increased DNA damage by the combinational
treatment, then the modulation of the repair system could be
involved in this effect. To test this hypothesis, we next examined
the effect of the depletion of RRM-1 on the RAD51 focus formation
(Fig. 6C). Immunfluorescence
analyses using an anti-RAD51 antibody revealed that, in contrast to
γ-H2AX, RAD51 focus formation was significantly repressed by the
depletion of RRM-1 in TE11 cells, after the combinational treatment
with 5-FU and CDDP (p<0.01) (Fig.
6E). In contrast, the RAD51 focus formation in TE11 cells
treated with CDDP alone was not interrupted by the depletion of
RRM-1 (Fig. 7B, p=0.15). Taken
together, these findings suggest that RRM-1 is involved in the
repair of the DNA damage induced by the combinational treatment,
but not by the CDDP treatment alone. Therefore, the modulation of
DNA repair, rather than the increased induction of DNA damage, by
the combinational treatment through RRM-1 activity could be
responsible for the synergic effect of the combinational treatment
with CDDP and 5-FU in TE11 cells.
Discussion
In the present study, we showed that RRM-1, large
subunit of RNR involved in the metabolism of 5-FU to 5-FdU, is
required for the induction of DNA damage by 5-FU. Interestingly,
5-FU treatment significantly increased the CDDP-induced RAD51 focus
formation in an RRM-1-dependent manner. Taken together, these
findings indicate that RRM-1 plays an important role in the
anticancer effect of combinational therapy of 5-FU with CDDP
through either the induction of DNA damage or modulation of DNA
repair.
A significant synergistic inhibition of cell growth
was observed after the combinational treatment with CDDP and 5-FU.
A recent report showed that the retention of DNA damage-induced
γ-H2AX foci and RAD51 foci is apparently indicative of lethal DNA
damage (29). The percentage of
γ-H2AX-positive cells treated with the combination regimen was
slightly higher than that treated with CDDP or 5-FU alone. Since
both CDDP and 5-FU can induce DNA damage, this could be due to
either the increased induction of DNA damage or the slowed DNA
repair by the combinational treatment. The decline in the
percentage of γ-H2AX-positive cells occurred earlier in the cells
treated with CDDP, as compared to those subjected to the
combinational treatment (Fig. 5B).
Moreover, in contrast to the slight increase in γ-H2AX foci
positive cells, the percentage of TE11 cells with RAD51 foci was
significantly increased after the combinational treatment (Fig. 5C). Although we could not exclude
the possible role of the increased induction of DNA damage, these
findings strongly support the notion that slower DNA repair, due to
the combinational treatment, plays a role in the synergic effect in
TE11 cells treated with 5-FU and CDDP.
We found that RRM-1 plays important roles in the
synergic effect of the combinational treatment with 5-FU and CDDP
(Fig. 6A). However, the formation
of γ-H2AX foci in cells treated with the combinational regimen was
not affected by the depletion of RRM-1 (Fig. 6D). In contrast to the γ-H2AX focus
formation, the depletion of RRM-1 significantly reduced the focus
formation by RAD51, a recombinational repair protein, after the
combinational treatment (Fig. 6E).
Since RRM-1 is required for the production of FdUDP and FdUTP which
could disturb DNA metabolism including repair, these findings also
support the notion that the synergic effect of the combinational
treatment is due to slower DNA repair requiring RAD51 (Fig. 8).
In spite of the requirement of RRM-1 for the
induction of DNA damage, the expression levels of RRM-1 did not
affect the survival of TE11 cells after 5-FU alone treatment
(Fig. 3E). Metabolites of 5-FU can
disturb either DNA or RNA metabolisms (3). RRM-1 depletion could lead to
increased conversion of 5-FU into 5-FUTP which contributes to the
anticancer effect of 5-FU at several levels in RNA metabolism
(30). The misincorporation of
FUTP into RNA may inhibit the processing of pre-rRNA into mature
rRNA (31,32). In addition, it may disrupt the
post-transcriptional modifications of tRNAs (33,34)
and the assembly and activity of snRNA/protein complexes, thus
inhibiting the splicing of pre-mRNAs (35,36).
Therefore, the decreased induction of DNA damage by the depletion
of RRM-1 after 5-FU treatment could be compensated by the increased
disturbance of RNA metabolism, through the increased level of
5-FUTP in TE11 cells. Still further investigation to clarify the
disturbance of RNA metabolism by the RRM-1 depletion is required,
RRM-1 may play a key role in the regulation of the anticancer
effect of 5-FU alone by disturbing either DNA or RNA
metabolism.
In this study, we found that RRM-1 is involved in
the regulation of the anticancer effect of 5-FU, in single and
combinational treatments with CDDP. Although further studies are
required to clarify the role of RRM-1 in the synergic effect of the
combinational therapy, the expression level of RRM-1 may be a
useful predictive marker for the treatment of patients with the
combination regimen of CDDP and 5-FU. Moreover, the modulation of
RRM-1 expression could be a novel therapeutic strategy to enhance
the effects of CDDP and 5-FU.
Acknowledgements
This study was supported by the
Grants-in-Aid Program from the Ministry of Education, Culture,
Sports, Science and Technology of Japan.
References
|
1
|
Kamangar F, Dores GM and Anderson WF:
Patterns of cancer incidence, mortality, and prevalence across five
continents: defining priorities to reduce cancer disparities in
different geographic regions of the world. J Clin Oncol.
24:2137–2150. 2006. View Article : Google Scholar
|
|
2
|
Enzinger PC and Mayer RJ: Esophageal
cancer. N Engl J Med. 349:2241–2252. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Longley DB, Harkin DP and Johnston PG:
5-fluorouracil: mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Matuo R, Sousa FG, Escargueil AE,
Grivicich I, Garcia-Santos D, Chies JA, Saffi J, Larsen AK and
Henriques JA: 5-Fluorouracil and its active metabolite FdUMP cause
DNA damage in human SW620 colon adenocarcinoma cell line. J Appl
Toxicol. 29:308–316. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Matuo R, Sousa FG, Escargueil AE, Soares
DG, Grivicich I, Saffi J, Larsen AK and Henriques JA: DNA repair
pathways involved in repair of lesions induced by 5-fluorouracil
and its active metabolite FdUMP. Biochem Pharmacol. 79:147–153.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wyatt MD and Wilson DM III: Participation
of DNA repair in the response to 5-fluorouracil. Cell Mol Life Sci.
66:788–799. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
El-Awady RA, Saleh EM and Dahm-Daphi J:
Targeting DNA double-strand break repair: is it the right way for
sensitizing cells to 5-fluorouracil? Anticancer Drugs. 21:277–287.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Diasio RB and Johnson MR:
Dihydropyrimidine dehydrogenase: its role in 5-fluorouracil
clinical toxicity and tumor resistance. Clin Cancer Res.
5:2672–2673. 1999.PubMed/NCBI
|
|
9
|
Mizutani Y, Wada H, Fukushima M, Yoshida
O, Nakanishi H, Li YN and Miki T: Prognostic significance of
orotate phosphoribosyltransferase activity in bladder carcinoma.
Cancer. 100:723–731. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Salonga D, Danenberg KD, Johnson M,
Metzger R, Groshen S, Tsao-Wei DD, Lenz HJ, Leichman CG, Leichman
L, Diasio RB and Danenberg PV: Colorectal tumors responding to
5-fluorouracil have low gene expression levels of dihydropyrimidine
dehydrogenase, thymidylate synthase, and thymidine phosphorylase.
Clin Cancer Res. 6:1322–1327. 2000.
|
|
11
|
Kolberg M, Strand KR, Graff P and
Andersson KK: Structure, function, and mechanism of ribonucleotide
reductases. Biochim Biophys Acta. 1699:1–34. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Niida H, Katsuno Y, Sengoku M, Shimada M,
Yukawa M, Ikura M, Ikura T, Kohno K, Shima H, Suzuki H, Tashiro S
and Nakanishi M: Essential role of Tip60-dependent recruitment of
ribonucleotide reductase at DNA damage sites in DNA repair during
G1 phase. Genes Dev. 24:333–338. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rogakou EP, Pilch DR, Orr AH, Ivanova VS
and Bonner WM: DNA double-stranded breaks induce histone H2AX
phosphorylation on serine 139. J Biol Chem. 273:5858–5868. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Uziel T, Lerenthal Y, Moyal L, Andegeko Y,
Mittelman L and Shiloh Y: Requirement of the MRN complex for ATM
activation by DNA damage. EMBO J. 22:5612–5621. 2003. View Article : Google Scholar
|
|
15
|
Branzei D and Foiani M: Regulation of DNA
repair throughout the cell cycle. Nat Rev Mol Cell Biol. 9:297–308.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Arnaudeau C, Lundin C and Helleday T: DNA
double-strand breaks associated with replication forks are
predominantly repaired by homologous recombination involving an
exchange mechanism in mammalian cells. J Mol Biol. 307:1235–1245.
2001. View Article : Google Scholar
|
|
17
|
Pardo B, Gomez-Gonzalez B and Aguilera A:
DNA repair in mammalian cells: DNA double-strand break repair: how
to fix a broken relationship. Cell Mol Life Sci. 66:1039–1056.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Suwaki N, Klare K and Tarsounas M: RAD51
paralogs: roles in DNA damage signalling, recombinational repair
and tumorigenesis. Semin Cell Dev Biol. 22:898–905. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tashiro S, Walter J, Shinohara A, Kamada N
and Cremer T: Rad51 accumulation at sites of DNA damage and in
postreplicative chromatin. J Cell Biol. 150:283–291. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bolderson E, Richard DJ, Zhou BB and
Khanna KK: Recent advances in cancer therapy targeting proteins
involved in DNA double-strand break repair. Clin Cancer Res.
15:6314–6320. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Clingen PH, Wu JY, Miller J, Mistry N,
Chin F, Wynne P, Prise KM and Hartley JA: Histone H2AX
phosphorylation as a molecular pharmacological marker for DNA
interstrand crosslink cancer chemotherapy. Biochem Pharmacol.
76:19–27. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Asakawa H, Koizumi H, Koike A, Takahashi
M, Wu W, Iwase H, Fukuda M and Ohta T: Prediction of breast cancer
sensitivity to neoadjuvant chemotherapy based on status of DNA
damage repair proteins. Breast Cancer Res. 12:R172010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fujinaka Y, Matsuoka K, Iimori M, Tuul M,
Sakasai R, Yoshinaga K, Saeki H, Morita M, Kakeji Y, Gillespie DA,
Yamamoto K, Takata M, Kitao H and Maehara Y: ATR-Chk1 signaling
pathway and homologous recombinational repair protect cells from
5-fluorouracil cytotoxicity. DNA Repair (Amst). 11:247–258. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Scharer OD: DNA interstrand crosslinks:
natural and drug-induced DNA adducts that induce unique cellular
responses. Chembiochem. 6:27–32. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Limoli CL, Giedzinski E, Bonner WM and
Cleaver JE: UV-induced replication arrest in the xeroderma
pigmentosum variant leads to DNA double-strand breaks, gamma-H2AX
formation, and Mre11 relocalization. Proc Natl Acad Sci USA.
99:233–238. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ewald B, Sampath D and Plunkett W: H2AX
phosphorylation marks gemcitabine-induced stalled replication forks
and their collapse upon S-phase checkpoint abrogation. Mol Cancer
Ther. 6:1239–1248. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Johnston PG, Geoffrey F, Drake J, Voeller
D, Grem JL and Allegra CJ: The cellular interaction of
5-fluorouracil and cisplatin in a human colon carcinoma cell line.
Eur J Cancer. 32A:2148–2154. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Matsusaka S, Nagareda T and Yamasaki H:
Does cisplatin (CDDP) function as a modulator of 5-fluorouracil
(5-FU) antitumor action? A study based on a clinical trial. Cancer
Chemother Pharmacol. 55:387–392. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Banath JP, Klokov D, MacPhail SH, Banuelos
CA and Olive PL: Residual gammaH2AX foci as an indication of lethal
DNA lesions. BMC Cancer. 10:42010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huehls AM, Wagner JM, Huntoon CJ, Geng L,
Erlichman C, Patel AG, Kaufmann SH and Karnitz LM: Poly
(ADP-Ribose) polymerase inhibition synergizes with
5-fluorodeoxyuridine but not 5-fluorouracil in ovarian cancer
cells. Cancer Res. 71:4944–4954. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kanamaru R, Kakuta H, Sato T, Ishioka C
and Wakui A: The inhibitory effects of 5-fluorouracil on the
metabolism of preribosomal and ribosomal RNA in L-1210 cells in
vitro. Cancer Chemother Pharmacol. 17:43–46. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ghoshal K and Jacob ST: Specific
inhibition of pre-ribosomal RNA processing in extracts from the
lymphosarcoma cells treated with 5-fluorouracil. Cancer Res.
54:632–636. 1994.PubMed/NCBI
|
|
33
|
Santi DV and Hardy LW: Catalytic mechanism
and inhibition of tRNA (uracil-5-) methyltransferase: evidence for
covalent catalysis. Biochemistry. 26:8599–8606. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Randerath K, Tseng WC, Harris JS and Lu
LJ: Specific effects of 5-fluoropyrimidines and 5-azapyrimidines on
modification of the 5 position of pyrimidines, in particular the
synthesis of 5-methyluracil and 5-methylcytosine in nucleic acids.
Recent Results Cancer Res. 84:283–297. 1983.PubMed/NCBI
|
|
35
|
Patton JR: Ribonucleoprotein particle
assembly and modification of U2 small nuclear RNA containing
5-fluorouridine. Biochemistry. 32:8939–8944. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Doong SL and Dolnick BJ: 5-Fluorouracil
substitution alters pre-mRNA splicing in vitro. J Biol Chem.
263:4467–4473. 1988.PubMed/NCBI
|