Introduction
Epstein-Barr virus (EBV) is a human
gamma-herpesvirus, infecting over 90% of the adult human population
(1). EBV, as one of the most
common viruses, is the causative factor of infectious mononucleosis
and strongly involved with the development of various human
malignant diseases, including Hodgkin’s lymphoma, Burkitt’s
lymphoma, post transplant lymphoproliferative disorders,
nasopharyngeal carcinoma, immunoblastic B lymphoma associated with
HIV, some gastric carcinomas, and autoimmune diseases such as
multiple sclerosis, Sjögren’s syndrome and rheumatoid arthritis
(2). However, the precise role of
EBV in the pathogenesis of these diseases is not yet clear.
Glucocorticoids (GCs) are known to regulate cell
proliferation, differentiation, development and inflammation.
Dexamethasone (Dex), a synthetic GCs, prevents the cell growth of
many hematologic malignant cells and solid tumor cell types,
including multiple myeloma (3),
leukemia (4), prostate cancer
(5), hepatoma (6), melanoma (7), osteosarcoma (8), lung cancer (9), breast cancer (10) and ovarian cancer cells (11). However, the biological effect and
molecular events leading from Dex treatment in EBV-transformed B
cells are still not understood completely.
XIAP-associated factor 1 (XAF1) is a nuclear protein
and a binding partner that directly interacts with endogenous XIAP
(X-linked inhibitor of apoptosis), resulting in the redistribution
of XIAP from the cytoplasm to the nucleus for sequestration,
thereby antagonizing the anti-caspase activity of XIAP (12). XAF1 was not only an
apoptosis-promoting factor, but is also involved in the cellular
stress response (13). XAF1 is
expressed ubiquitously in all normal cells, in contrast to
extremely low or undetectable levels in several cancer cells
(14). Likewise, deficiency of
XAF1 expression is strongly associated with tumor progression.
Overexpression of XAF1 enhances chemosensitivity and cell death,
and inhibits tumor growth in various cancers including gastric,
colon, pancreatic and prostate cancers (15–18).
Although XAF1 is thought to be a pro-apoptotic nuclear protein,
after the re-localization of XAF1 to mitochondria, it promotes
translocation of Bax into mitochondria and cytochrome c
release from mitochondria (19).
However, the role of XAF1 in apoptosis of EBV-transformed B cells
and its putative correlation with the reactive oxygen species (ROS)
and ERK1/2 pathway have not been studied.
In this study, we aimed to study the effect of XAF1
on cellular response to Dex in EBV-transformed B cells and the
underlying molecular mechanisms. We were interested in whether
ERK1/2 would have any regulatory role in other apoptotic pathways,
such as the XAF1 signaling pathway, upon Dex treatment. We report a
study on Dex-induced apoptosis in EBV-transformed B cells
demonstrating that caspase-9 activation and XAF1 expression are
induced by ROS production and ERK1/2 activation and mediate both in
the induction of apoptosis and in translocation of Bax into
mitochondria.
Materials and methods
Preparation of stock of EBV virions and
generation of EBV-transformed B cells
Cell-free EBV virions were prepared from culture
supernatant of B95-8 marmoset cell line. To establish EBV infection
of B cells from normal peripheral blood mononuclear cells (PBMCs),
PBMCs were isolated from peripheral blood of a healthy donor by
Ficoll-Paque (Amersham Life Science, Buckinghamshire, UK) gradient
centrifugation. PBMCs were added to EBV virions stock in a culture
flask, and after 2-h incubation at 37°C, RPMI-1640 culture medium
(HyClone) and 1 mg/ml of cyclosporine A (Sigma-Aldrich, St. Louis,
MO, USA) were added to cells (1×106 cells/ml). The
cultures were incubated for 2–4 weeks. This study was approved by
the Institutional Bioethics Review Board at the Medical College of
Inje University, and all donors gave informed consent for the
study.
Proliferation measurement by
AlamarBlue
Cells (5×104 cells/well) were cultured in
medium containing 10% FBS in 96-well plates. After 24 h, cell
proliferation was measured by AlamarBlue (Serotec Ltd, Kidlington,
UK) assay. AlamarBlue was added (10% by volume) to each well and
relative fluorescence was determined 9 h later by SpectraMax M2e
Multi-Detection Microplate Reader (Molecular Devices, Sunnyvale,
CA; excitation, 530 nm; emission, 590 nm). Relative fluorescence
unit (RFU) values were expressed as mean ± SEM of three
determinations.
Quantification of apoptotic cells by flow
cytometry
The level of Dex-induced apoptosis in human
EBV-transformed B cells (4 weeks, 5×105 cells/ml) and
normal PBMCs was measured by flow cytometry using FITC-labeled
Annexin V and 7-AAD (BD Biosciences, San Diego, CA, USA). To decide
optimal conditions, experiments were performed using variable
concentrations (0, 10, 50, 100 and 200 μM) and variable
durations of incubation (2, 4, 8, 16 and 24 h). To investigate the
effects of caspase inhibitors, cells were pretreated with
z-LEHD-fmk (z-Leu-Glu(OMe)-His-Asp-(OMe)-fluoremethylketone, 20
μM, a caspase-9 inhibitor; Calbiochem, San Diego, CA, USA)
for 2 h before Dex treatment. To inhibit generation of ROS or
ERK1/2 cascade, cells were pretreated with NAC (N-acetylcysteine,
10 mM, antioxidant; Sigma-Aldrich) or PD98059 (10 μM,
Calbiochem) for 1 h. Cells were then harvested, washed in PBS, and
incubated with Annexin V and 7-AAD in binding buffer at room
temperature for 15 min in the dark. The stained cells were analyzed
using a FACSCalibur flow cytometer (BD Biosciences) equipped with
CellQuest Pro software (BD Biosciences).
Detection of mitochondria membrane
potential (Δψm) and intracellular reactive oxygen
species (ROS) generation
The changes in mitochondrial membrane potential
(Δψm) were determined using DiOC6
(3,3’-dihexyloxacarboxyanine iodide; Molecular Probes, Eugene, OR,
USA). Cells were treated with methanol (MetOH) or Dex for 24 h,
harvested, washed twice in PBS, resuspended in PBS supplemented
with DiOC6 (20 nM), incubated at 37°C for 15 min in the
dark, and immediately analyzed by flow cytometry. The intracellular
accumulation of ROS was examined by flow cytometry after being
stained with the fluorescent probe, DCFH-DA (10 μM,
2′,7′-dichlorodihydrofluorescein diacetate; Molecular Probes).
DCFH-DA was deacetylated in cells by esterase to a non-fluorescent
compound, DCFH, which remains trapped within the cell and is
cleaved and oxidized by ROS in the presence of endogenous
peroxidase to a highly fluorescent compound, DCF
(2′,7′-dichlorofluorescein). EBV-transformed B cells were seeded in
6-well plates (5×105 cells/ml), treated with or without
Dex, and incubated with 10 μM DCFH-DA for 30 min at 37°C.
Then cells were washed, resuspended in PBS, and ROS levels were
determined using a FACSCalibur flow cytometer (BD Biosciences).
Reverse transcription polymerase chain
reaction
Total RNA was isolated using an RNeasy mini kit
(Qiagen, Hilden, Germany). RNA was transcribed into cDNA using
oligo(dT) primers (Bioneer, Daejeon, Korea) and reverse
transcriptase. To investigate apoptosis-associated molecules, PCR
amplification was performed using specific primer sets (Bioneer)
for XAF1 (upstream primer, 5′-TTCAGCTCCTGAAAGGGAAA; downstream
primer, 5′-TTCAGCAGCTTGACTTGGAA), XIAP (upstream primer,
5′-GTGCCACGCAGTCTACAAATT CTGG; downstream primer,
5′-CGTGCTTCATAATCTGCCA TGGATGG), Bax (upstream primer,
5′-CCAAGAAGCTGAG CGAGTGT; downstream primer, 5′-CAGCCCATGATGGTT
CTGAT), Noxa (upstream primer, 5′-AGGACTGTTCGTGTT CAGCTC;
downstream primer, 5′-GTGCACCTCCTGAG AAAACTC), and Puma (upstream
primer, 5′-GTGTAGAGG AGACAGGAATC; downstream primer, 5′-GCTCGTACTGT
GCGTTGAGG). A specific primer set for β-actin (upstream primer,
5′-ATCCACGAAACTACCTTCAA; downstream primer, 5′-ATCCACACGGAGTACTTGC)
was used as a control and PCR was performed using Prime Taq Premix
(GeNet Bio, Chungnam, Korea). PCR products were analyzed by agarose
gel electrophoresis and visualized with ethidium bromide under UV
light using the multiple Gel DOC system (Fujifilm, Tokyo, Japan).
Data were analyzed using ImageJ 1.38 software (National Institutes
of Health, Bethesda, MD). Experiments were performed in
triplicate.
Western blot analysis
After treatment, cells were harvested and lysed in
RIPA buffer (Elpis Biotech, Daejeon, Korea) containing a protease
inhibitor cocktail (AEBSF, aprotinin, Bestatin hydrochloride, E-64,
EDTA and leupeptin hemisulfate salt; Sigma-Aldrich). To address
phosphorylation events, an additional set of phosphatase inhibitors
(Cocktail II, sodium orthovanadate, sodium molybdata, sodium
tartrate and imidasole; Sigma-Aldrich) was added to the RIPA buffer
(Elpis Biotech, Daejeon, Korea). Protein concentration was
determined using a BCA assay kit (Pierce, Rockford, IL). Proteins
(10 μg/sample) were immediately heated for 5 min at 100°C.
Total cell lysates (5×106 cells/sample) were subjected
to SDS-PAGE on gel containing 15% (w/v) acrylamide under reducing
conditions. Separated proteins were transferred to nitrocellulose
membranes (Millipore Corp., Billerica, MA, USA), and then the
membranes were blocked with 5% skim milk and commercial western
blot analysis was performed. Chemiluminescence was detected using
and ECL kit (Advansta Corp., Menlo Park, CA, USA) and the multiple
Gel DOC system (Fujifilm). The following primary Abs were used:
caspase-8, caspase-3, caspase-9, β-actin, Bax, Puma, Noxa, XAF1,
phospho-JNK (Thr183/Tyr185), JNK, phospho-p38
MAPK (Thr180/Tyr182), p38 MAPK,
phospho-ERK1/2 (Thr202/Tyr204), and ERK1/2
(Cell Signaling Technology, Beverly, MA, USA); Ref-1 and COX-IV
(Santa Cruz Biotechnology, Santa Cruz, CA, USA); and β-tubulin (BD
Biosciences). Data were analyzed using ImageJ 1.38 software.
Measurement of XAF1, Bax and Puma
translocation
Following treatment, mitochondrial and cytosol
cellular fractions were prepared using a Cytosol/Mitochondria
Fractionation kit (Calbiochem). Cells (1×107) with or
without various treatments were harvested by centrifugation at 600
× g for 5 min at 4°C and washed twice with cold PBS. Afterward, the
cells were resuspended in 250 μl Cytosol Extraction buffer
containing a protease inhibitor cocktail and 1 mM dithiothreitol
(DTT). After incubation on ice for 10 min, cells were homogenized
on ice using a dounce tissue homogenizer. Homogenized cells were
centrifuged at 700 × g for 10 min at 4°C, and supernatants were
collected. Supernatants were then centrifuged again at 10,000 × g
for 30 min at 4°C. The resulting supernatants were harvested and
designated as cytosolic fractions and the pellets were resuspended
in 50 μl Mitochondria Extraction buffer containing a
protease inhibitor cocktail and 1 mM DTT and designated as
mitochondrial fractions. All fractions were stored at −80°C until
use.
Co-immunoprecipitation (co-IP) assay
For XAF1-Bax binding or Puma-Bax binding assay,
cells were treated with Dex for 24 h. Cells (5×106
cells/sample) were then harvested and lysed in RIPA buffer (Elpis
Biotech) containing a protease inhibitor cocktail (Sigma-Aldrich).
To reduce non-specific binding of protein, we performed
pre-clearing on equal amounts of cell lysates by incubating samples
with washed protein G PLUS-agarose beads (Santa Cruz
Biotechnology). For IP, pre-cleared lysate plus the optimal amount
of anti-XAF1 or -Puma antibody was incubated at 4°C for 2 h on a
rotator. The immunoprecipitates were harvested by protein G
PLUS-agarose beads (Santa Cruz Biotechnology) and incubated at 4°C
for 2 h under rotary agitation. When incubation time was over,
supernatant was removed and the beads were washed in lysis buffer
four times. Finally, immunoprecipitates were eluted by boiling the
beads in SDS-PAGE sample buffer for 5 min and then characterized by
western blot analysis with appropriate antibodies.
Results
Dex induces apoptosis in EBV-transformed
B cells but not in normal PBMCs
We measured the effect of Dex on the proliferation
of EBV-transformed B cells. Cells were treated with various doses
of Dex (10, 100, 200, 500 and 1,000 μM) for 24 h, and its
effect on cell proliferation was analyzed by AlamarBlue assay. In
the presence of Dex, EBV-transformed B cell proliferation was
reduced in a dose-dependent manner, suggesting potential antitumor
activity (Fig. 1A). Dex exhibited
approximately 50% proliferation inhibition at dose of 100
μM. To characterize the apoptosis response to Dex treatment
in EBV-transformed B cells, Annexin V/7-AAD staining was performed.
The cells were treated with different doses of Dex (0, 10, 50, 100
and 200 μM) for 16 h. Fig.
1B shows that different doses of Dex has apoptotic-inducing
effect on cells and that Annexin V and 7-AAD positive cells in
MetOH-treated cells was 2.27%, whereas in treated cells with 10,
50, 100 and 200 μM of Dex these were 4.97, 9.28, 31.81 and
78.87% after 16-h treatment, respectively. As shown in Fig. 1C, cells were exposed to Dex for
diverse time intervals and Annexin V and 7-AAD positive cells in
treated cells with Dex for 2, 4, 8, 16 and 24 h were 4.57, 15.10,
35.91, 55.45 and 75.50%. Moreover, Dex markedly induced
Δψm dissipation (Fig. 1B,
lower panel), especially between 8- and 16-h treatment
(Fig. 1C, lower panel; from 25.21
to 75.41%). Because the optimal dose and time of Dex treatment were
100 μM and 24 h, we chose this condition to evaluate protein
alterations in Dex-induced apoptosis. Furthermore, we investigated
whether Dex has any cytotoxic effect on normal human PBMCs and did
not observed significant cell death in normal human PBMCs after Dex
treatment (Fig. 1D). Our data
suggest that Dex more selectively induces the cytotoxic effect on
cancerous EBV-transformed B cells than normal human PBMCs.
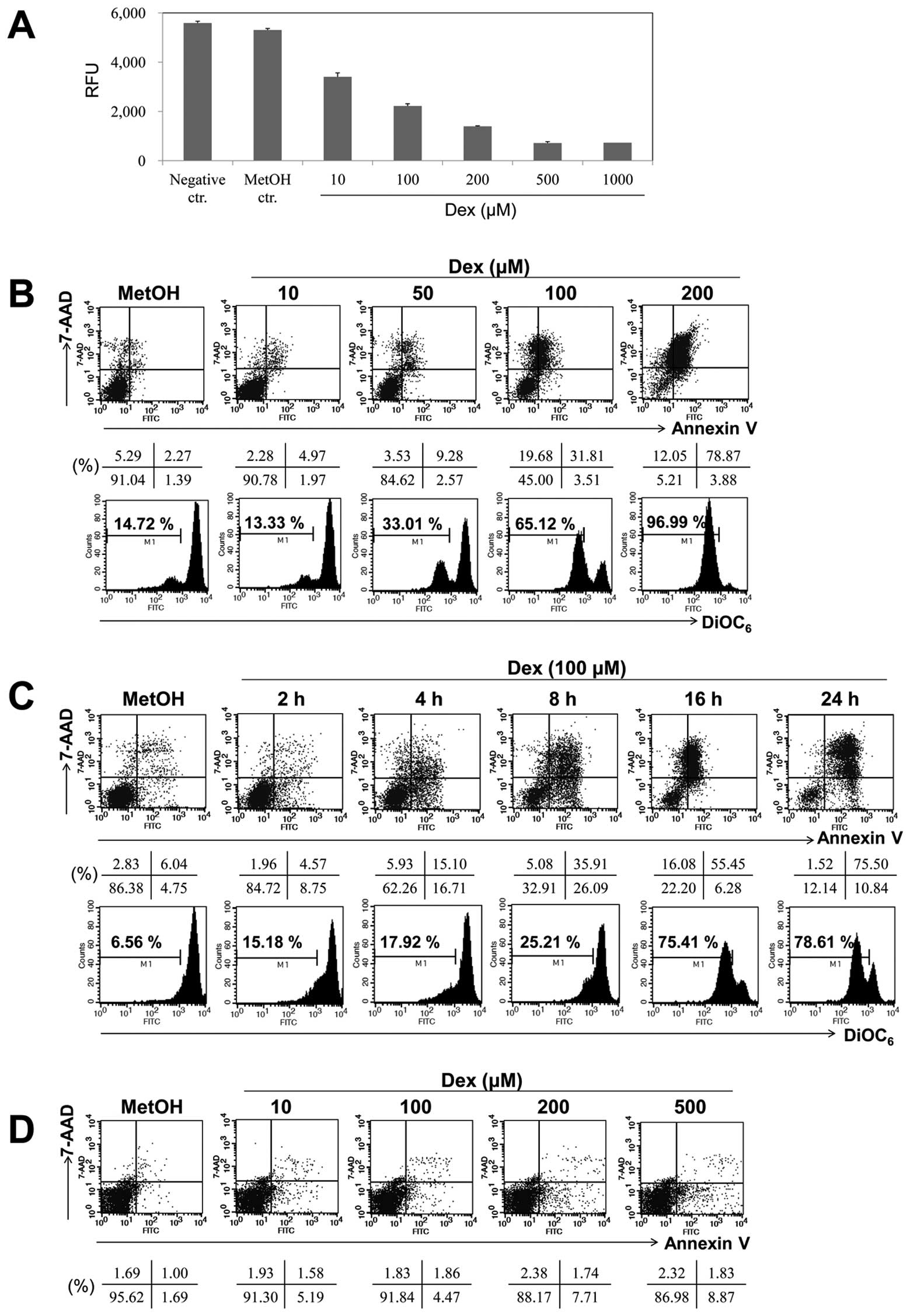 | Figure 1.Dex induced apoptosis in a dose- and
time-dependent manner in EBV-transformed B cells. (A)
EBV-transformed B cells (5×104 cells/well) were cultured
in 96-well plates. After 24 h, cell proliferation was measured by
AlamarBlue assay. RFU is the relative fluorescence unit. (B and C)
EBV-transformed B cells and (D) PBMCs were treated with 10, 50, 100
and 200 μM of Dex for 2, 4, 8, 16 and 24 h. The percentage
of apoptotic cells was estimated by Annexin V/7-AAD staining. Dot
plot graphs show percentage of viable cells (Annexin
V−/7-AAD−), early-phage apoptotic cells
(Annexin-V+/7-AAD−), late-phage apoptotic
cells (Annexin-V+/7-AAD+), and necrotic cells
(Annexin-V−/7-AAD+). To measure disruption of
Δψm, cells were stained with DiOC6.
Diminished DiOC6 fluorescence indicates Δψm
disruption and percentages indicates the cell proportion in each
bar. Results are representative of three independent
experiments. |
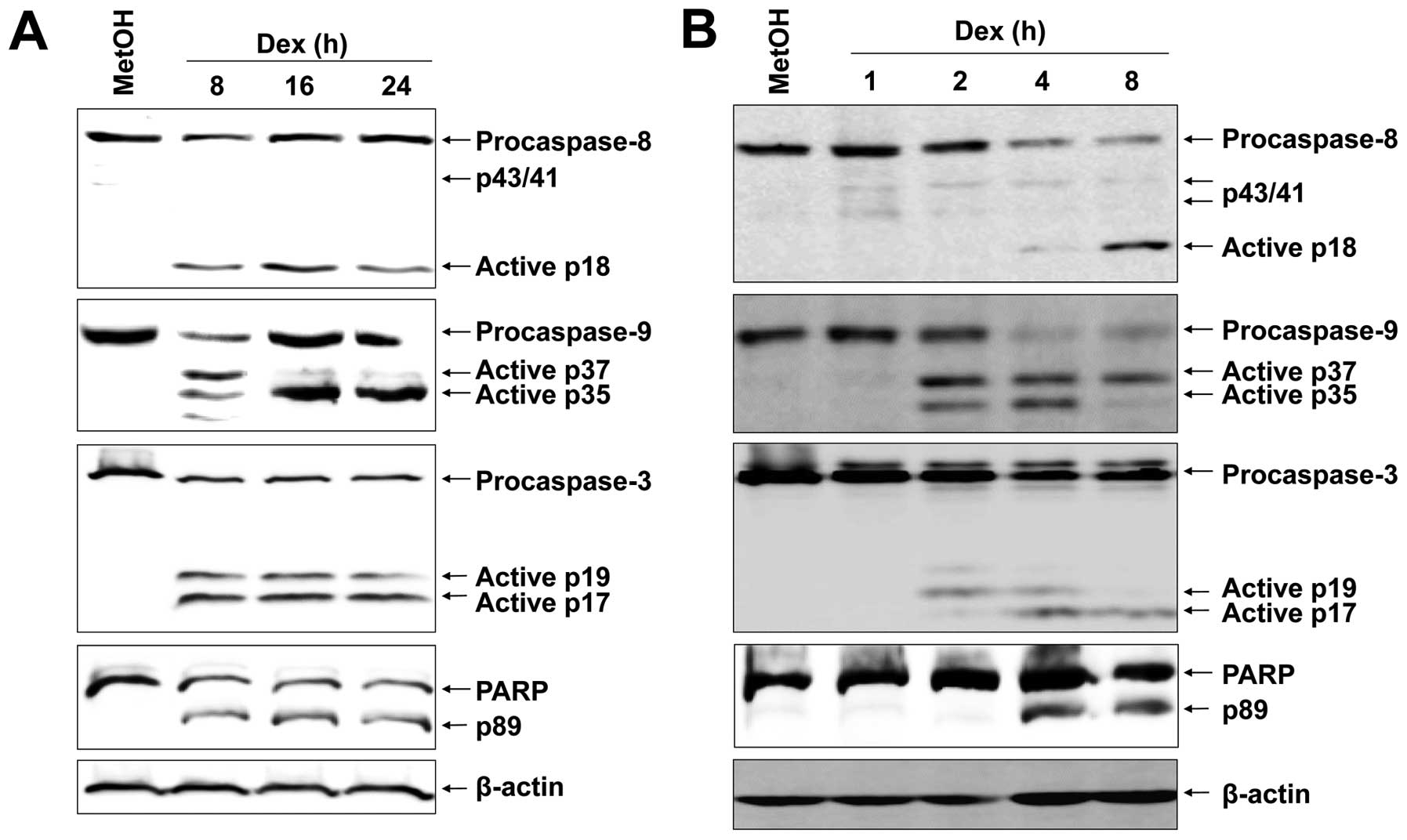
Dex induces caspase-dependent apoptosis
in EBV-transformed B cells
Based on the preliminary assays where a strong
apoptotic effect of Dex was elicited in EBV-transformed B cells,
the possible molecular mechanism underlying Dex-induced apoptosis
was scrutinized. We first examined proteolytic processing of
caspases by western blot analysis because activation of caspases
has been reported to play a role in apoptosis mediated by various
stimuli. Dex-induced apoptosis of EBV-transformed B cells were
involved with activation of caspase-8, -9, -3 and PARP, as assessed
by the appearance of the respective cleaved active caspases
(Fig. 2A). Further, to elucidate
the mechanistic order of caspases and PARP, we carried out
short-term treatment with Dex. Interestingly, cleavage of
procaspase-9 was detected as early as 2 h after Dex treatment
(Fig. 2B). Cleavage of
procaspase-8 into the characteristic 18-kDa active fragments was
apparent 8 h after Dex treatment. We then examined effector
caspase-3 and its substrate, PARP, and they were already increased
at 4 h after Dex treatment and the amount dramatically increased at
8 h (Fig. 2B).
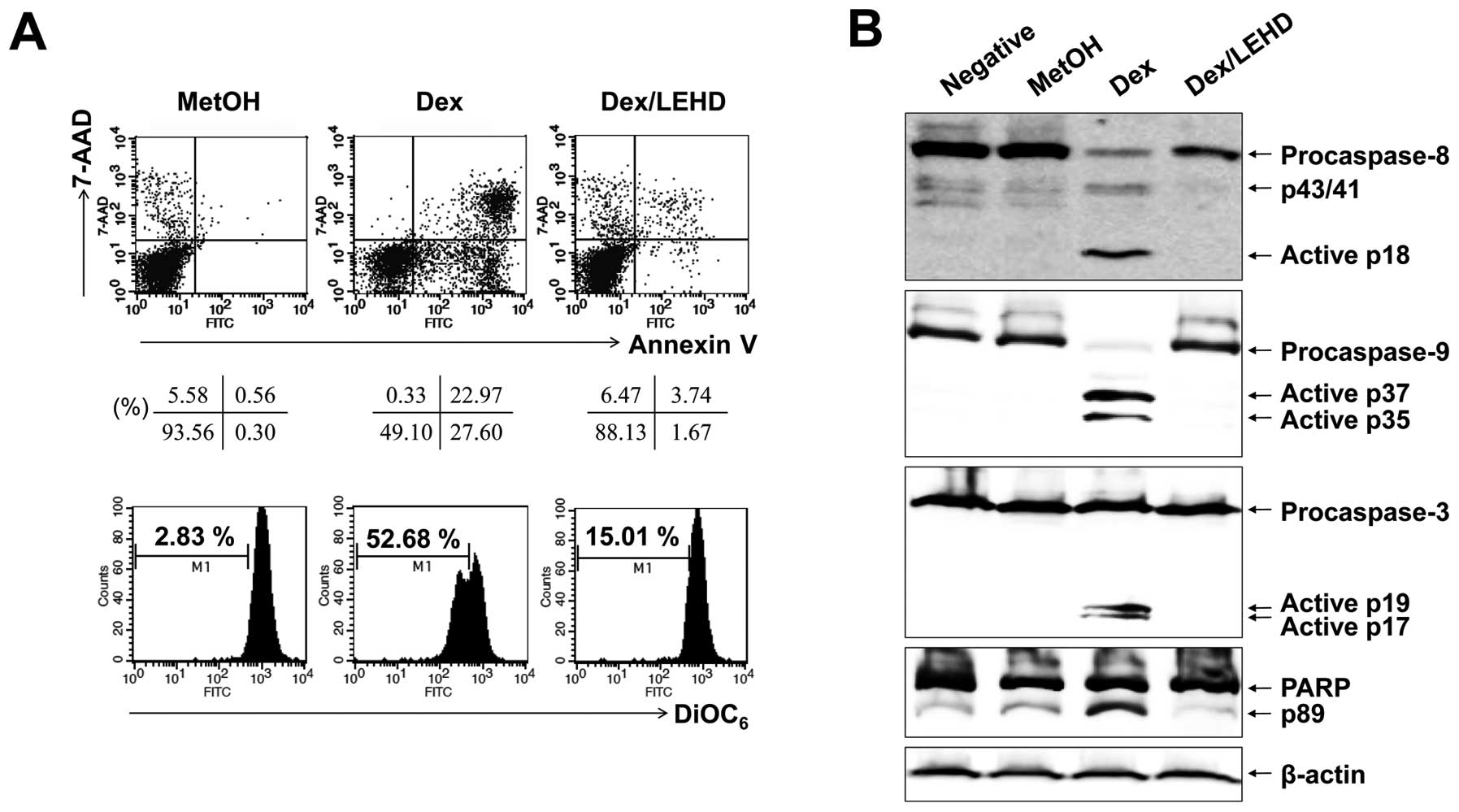
Caspase-9 inhibition blocks activation of
caspase-8
The activation of caspase-8 is an initial caspase
and could be due to direct activation of the death-receptor
pathway. However, it could also be secondary signal to
caspase-9/-3-linked cleavage, as reported in PUVA-induced apoptosis
of T leukemia cells (20). Our
experiments had shown that no FasL surface expression increased
after Dex treatment of EBV-transformed B cells (data not shown).
However, to survey this possibility further, we used selective
caspase inhibitors. As depicted in Fig. 3, when EBV-transformed B cells had
been pre-treated with a caspase-9 inhibitor (z-LEHD-fmk), this
inhibitor blocked Dex-induced apoptosis and no cleavage of
caspase-8 could be detected by western blot analysis performed on
cells 24 h after Dex treatment. These results pointed to the
likelihood that, in our system, activation of caspase-8 is
secondary to the activation of caspase-9.
Dex induces mitochondrial events related
to apoptosis in EBV-transformed B cells
Mitochondria-related proteins include anti-apoptotic
proteins (Bcl-2, Bcl-xL, XIAP and survivin) and pro-apoptotic
proteins (Bax, Bad, Bid, Puma, Noxa and XAF1). They can inhibit or
activate the release of downstream factors which lead to the
activation of caspase-3 and PARP in the execution of apoptosis. To
investigate the apoptosis-related proteins, anti-apoptotic (Bcl-2,
Bcl-xL and XIAP) and pro-apoptotic (Bax, Puma, Noxa and XAF1) were
detected by RT-PCR and western blot analysis after cells were
treated for different times with 100 μM Dex. We found that
Dex diminished the expression of XIAP, Bcl-2 and Bcl-xL mRNA, and
protein levels. In contrast, Dex significantly increased the
expression of XAF1 and slightly increased expression of Puma and
Bax but had little effect on Noxa (Fig. 4A and B). It has been reported that
Bax translocation promotes the rupture of mitochondrial outer
membrane and facilitates the disruption of Δψm(19). We separated the mitochondrial and
cytosolic fractions after 12 and 24 h of Dex exposure to determine
the Bax level by western blot analysis. As shown in Fig. 4C, there was a significant
enhancement in translocation of Bax from cytosol to mitochondria at
12 and 24 h compared with control. In general, under normal
environments, XAF1 was localized fundamentally in the nuclear
(12), whereas XAF1 relocalization
from nuclear to cytoplasm and mitochondria was observed after Dex
exposure (Fig. 4C). Bax is
required for XAF1 or Puma-mediated apoptosis and is translocated by
XAF1 or Puma activation (19). To
investigate whether XAF1 and Puma can interact with Bax directly,
the interplay between XAF1 or Puma and Bax was performed using
co-IP tool. The results of co-IP analysis indicate that the amount
of Bax binding to XAF1 or Puma enhanced strongly after Dex exposure
(Fig. 4C) and that the amount of
XAF1 binding to Puma also increased after Dex exposure, suggesting
that XAF1 and Puma have a parallel action and activate Bax. In
addition, z-LEHD-fmk impeded translocation of XAF1, Puma, and Bax
to the mitochondria (Fig. 4D),
indicating that caspase-9 activation may precede translocation of
XAF1, Puma and Bax.
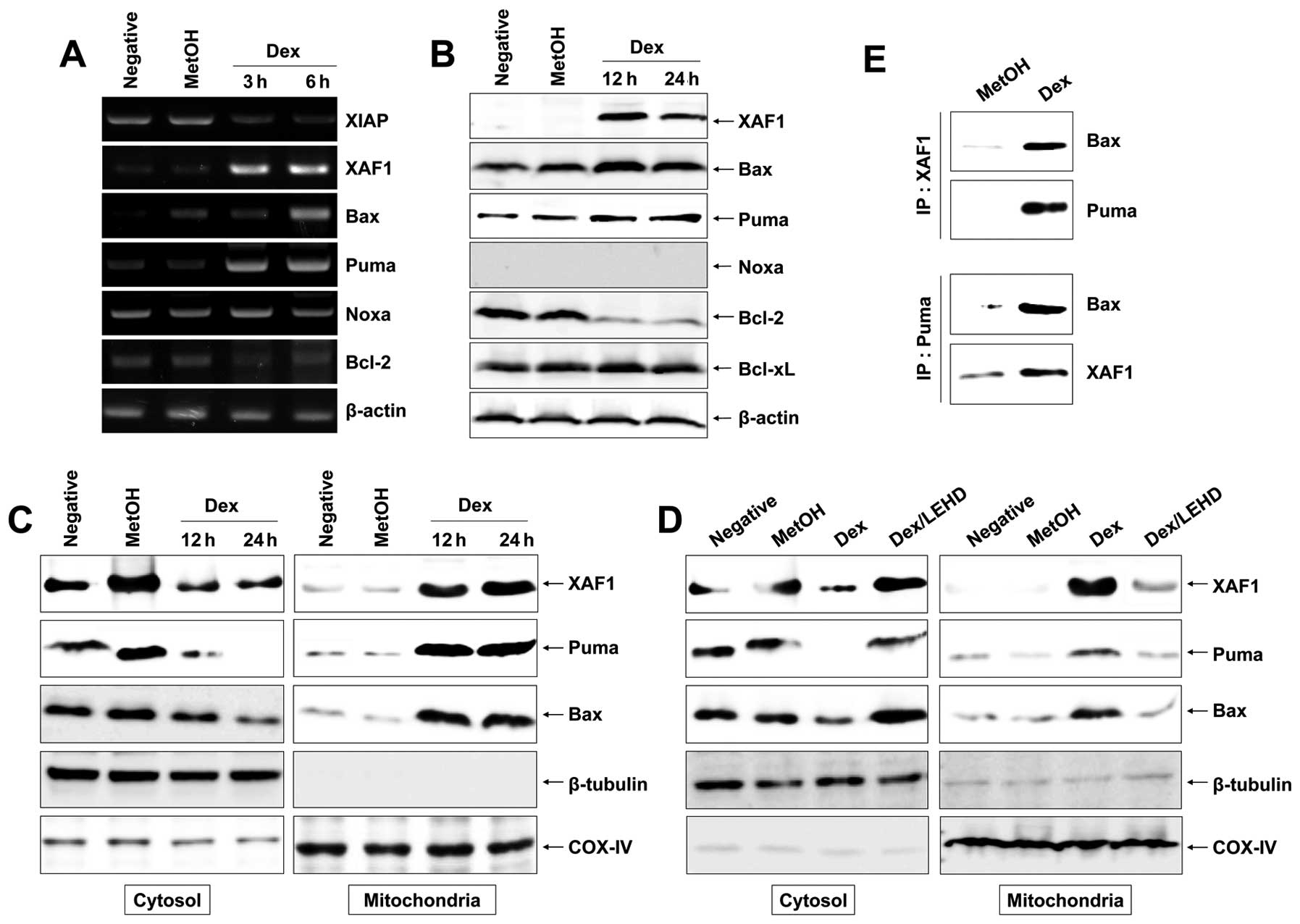 | Figure 4.Activation of mitochondrial events in
Dex-induced apoptosis in EBV-transformed B cells. Cells were
treated with 100 μM Dex for the indicated times. Total RNA
and proteins were extracted from cell lysates and (A) RT-PCR for
XIAP, XAF1, Bax, Puma, Noxa, Bcl-2 and β-actin mRNA and (B) western
blot analysis for XAF1, Bax, Puma, Noxa, Bcl-2 and Bcl-xL protein
were performed. (C and D) Cells were harvested and then the amount
of Bax, XAF1 and Puma in cytosol and mitochondria fractions was
determined. The mitochondria marker, COX-IV, and cytosol marker,
β-tubulin were used to verify the purity of each fraction performed
as described in Materials and methods. To block activation of
caspase-9, cells were pretreated with z-LEHD-fmk (20 μM) for
2 h. (E) In binding assay, XAF1 or Puma was immunoprecipitated
using specific Ab, followed by immunodetection of Bax in
immunoprecipitate as detailed in Materials and methods. Results are
representative of three independent experiments. IP,
immunoprecipitation; IB, immunoblot analysis. |
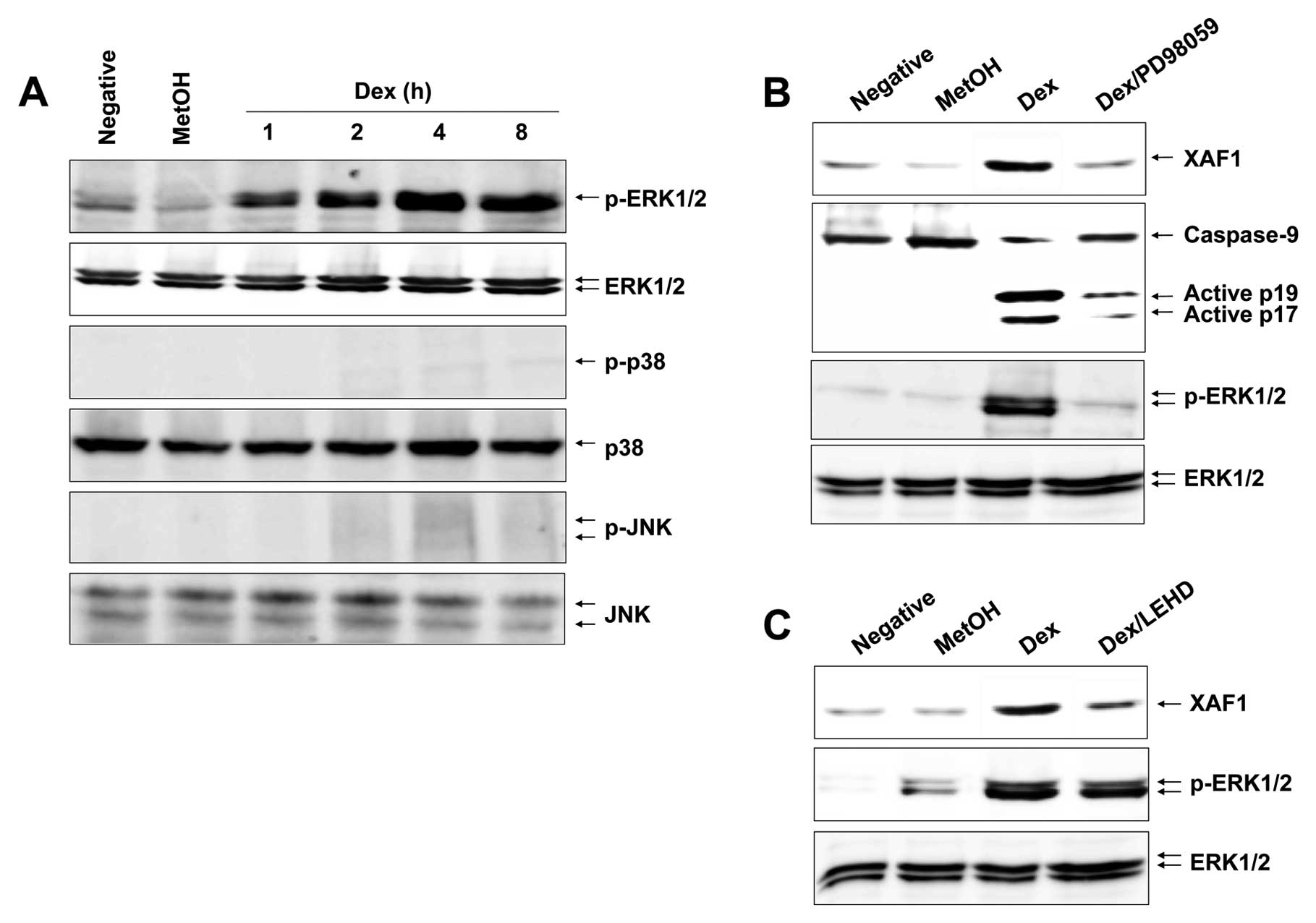
Dex leads to a persistent ERK1/2
phosphorylation in EBV-transformed B cells
MAPK signaling associated with various cellular
stresses and stimuli contributes to induction of apoptosis
(21). We therefore tested whether
certain MAPKs could induce expression and translocation of XAF1,
Puma and Bax after Dex treatment. Cells were treated with Dex and
analyzed for various MAPK activities, including ERK1/2, JNK and p38
MAPK. Fig. 5A shows that Dex
apparently induced an activation of ERK1/2 after 1 h and a
persistent phosphorylation level was observed up to 8 h in a
time-dependent manner, whereas it had no effect on JNK and p38
MAPK. These results indicate that ERK1/2 is the potential inducer
of Dex-induced XAF1 expression and trans-location. To corroborate
the role of ERK1/2 in Dex-induced apoptosis, cells were treated
with Dex in the presence or absence of the ERK1/2 inhibitor
PD98059. The results show that the PD98059 inhibited Dex-induced
XAF1 expression and attenuated caspase-9 activation (Fig. 5B). Moreover, caspase-9 inhibitor
z-LEHD-fmk impeded XAF1 expression, whereas it did not block
phosphorylation of ERK1/2 (Fig.
5C). Collectively, these observations substantiate the role of
ERK1/2 in caspase-9 activation and XAF1 expression.
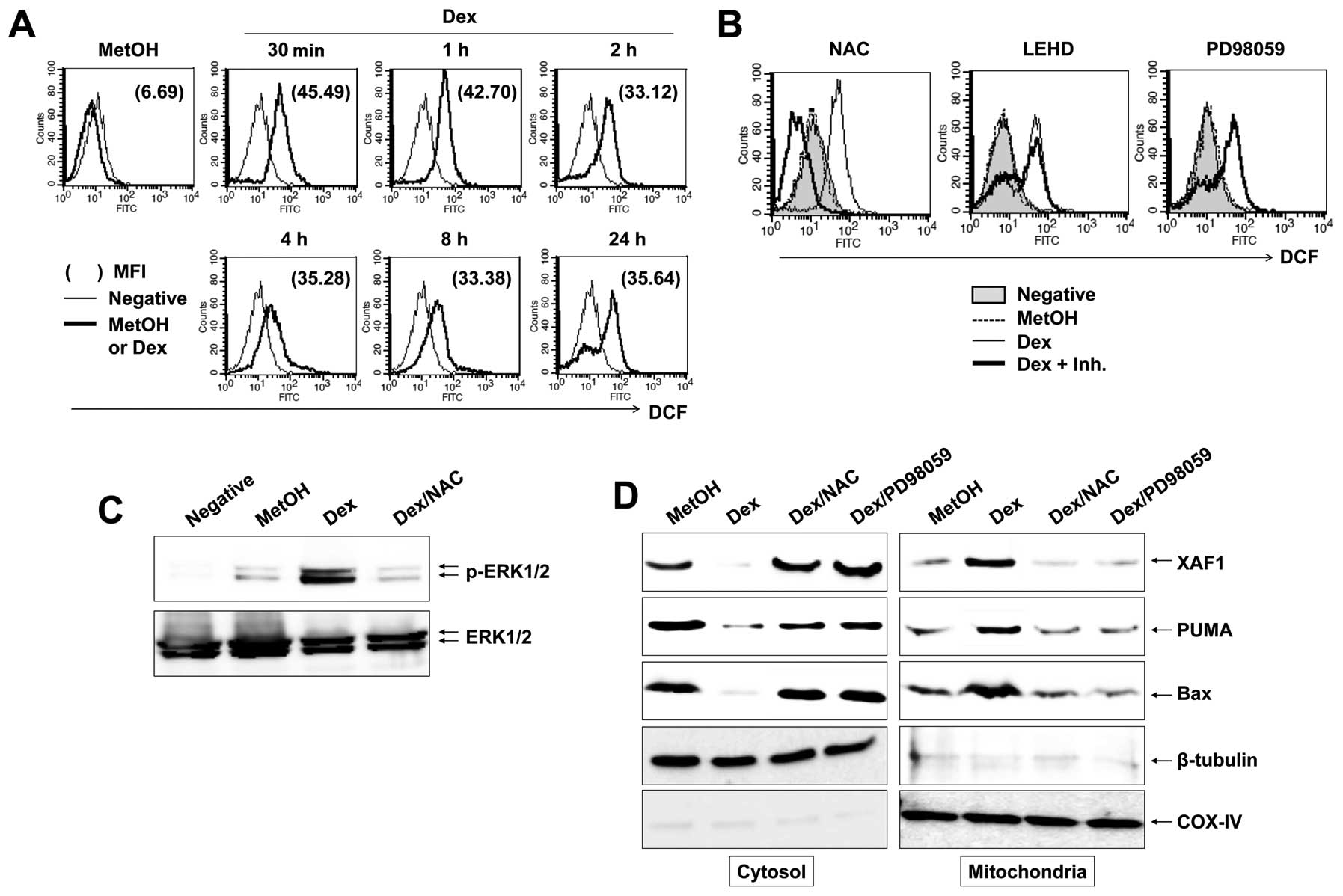
ROS is critical for ERK1/2 activation and
mitochondrial events by Dex
In apoptosis, ROS is directly associated with
activation of MAPKs and caspases (22). In addition, ROS is an early signal
that provokes apoptosis (23) and
a major source of action of many antineoplastic drugs. To establish
whether reactive oxygen species (ROS) participate in Dex-induced
apoptosis, cells were treated with 100 μM Dex for the
indicated times, followed by addition of DCFH-DA to measure
intracellular ROS level. We found that Dex induced a marked
increase in DCF fluorescence within 30 min and Dex-induced ROS
level was maintained until 24 h (Fig.
6A). To elucidate the role of Dex-induced ROS generation on a
persistent phosphorylation of ERK1/2 and caspase-9 activation
during apoptosis, we treated cells with NAC, PD98059 or z-LEHD-fmk;
inhibitors of ROS, ERK1/2 and casapse-9, respectively. Despite the
inhibitors diminishing Dex-induced apoptosis remarkably (data not
shown), as illustrated in Fig. 6B,
NAC was the only inhibitor that significantly suppressed
Dex-induced ROS production. Further, NAC inhibited phosphorylation
of ERK1/2 by Dex treatment (Fig.
6C) and prevented translocation of XAF1/Puma/Bax complex to
mitochondria (Fig. 6D). These
findings strongly support that the critical role of ROS in
Dex-induced persistent ERK1/2 phosphorylation.
Discussion
Dex induces apoptosis and suppresses the
mitogen-stimulated proliferation in various normal cells, including
lymphoid cells (24) and
fibroblasts (25). Therefore, it
also represses cell growth of multiple myeloma (3), leukemia (4), prostate cancer (5), hepatoma (6), melanoma (7), osteosarcoma (8), lung cancer (9), breast cancer (10) and ovarian cancer cells (11). In the current study, we set out to
elucidate the action mechanisms of Dex by which it induces
apoptosis on EBV-transformed B cells. However, there is no
information available concerning which proteins are the key
inducers of apoptosis. We demonstrated, for the first time to our
knowledge, that ROS generation and ERK1/2 activation induced XAF1
up-regulation in apoptosis.
Our results showed that Dex treatment of
EBV-transformed B cells induced activation of caspase-9 and -8, as
well as caspase-3. In time course experiments, our results
suggested that caspase-9 was at the top of the hierarchy of the
caspase cascade and is an initiator caspase to be evoked. This
activation occured as early as 2 h after Dex treatment in
EBV-transformed B cells, whereas caspase-3 and -8 showed activation
after 4–8 h (Fig. 2). This
postponement might agree with the formation of apoptosomes
resultant from mitochondrial disruption (26), happening prior to adequate
activation of the caspase signaling. Caspase-9 activation is
consistent with the appearance of mitochondrial dysfunction
reported by others in PUVA-treated Jurkat cells (27). The dramatic inhibition of
Dex-induced apoptosis by z-LEHD-fmk strongly suggested that
caspase-9 was the predominant upstream caspase in Dex-treated
EBV-transformed B cell apoptosis (Fig.
3).
XAF1 is an essential mediator of apoptosis and plays
a critical role in the induction of cell death (17,19).
Previous reports have shown that the activation of JNK pathway was
closely involved in XAF1-mediated apoptosis induction (28). Puma can be induced by DNA damaging
drugs and is important in apoptosis (29). XAF1 or Puma induces
mitochondria-mediated apoptosis by directly translocating Bax into
mitochondria (29). Our current
results indicate that Dex-induced apoptosis fulfills the molecular
characteristics of the intrinsic pathway of apoptosis, including
phosphatidylserine exposure, dissipation of Δψm, and
Bax, Puma and Bcl-2 conformational changes. More importantly, we
found a significant increase both at the mRNA and protein level of
the XAF1 during apoptosis. Our studies indicated that the
expression of XAF1 was up-regulated and translocated into
mitochondria in Dex-treated cells and XAF1 with Puma elicited
translocation of Bax into mitochondria (Fig. 4). This suggested that XAF1 could be
responsible for triggering the pro-apoptotic conformational changes
of Bax. Immunoprecipitation further supported the main role of XAF1
in this apoptosis (Fig. 4E),
suggesting that XAF1 is the most critical pathways through which
Dex exerts an apoptotic effect in these cells.
The MAPK family proteins are mediators of diverse
cellular reaction including proliferation, apoptosis and
differentiation (30). They are
composed of three protein kinases: ERK1/2, JNK and p38 MAPK
(31). In general, transient
activation of ERK1/2 takes part in the survival pathway (32). However, a previous report suggests
that persistent activation of ERK1/2 contributes to cellular
apoptosis in cervical cancer cells (33). Here we found that Dex treatment
caused a persistent activation of ERK1/2 rather than JNK and p38
MAPK and persistent activation of ERK1/2 was involved in
Dex-induced growth inhibition and apoptosis in EBV-transformed B
cells (Fig. 5). Intriguingly, this
was the first report that ERK1/2 functioned upstream of the XAF1 to
induce cell apoptosis (Figs. 5B
and 6D). PD98059, a specific
inhibitor of ERK1/2, effectively reversed Dex-induced apoptosis and
attenuated Dex-induced XAF1 expression, suggesting the
pro-apoptotic effects of Dex in EBV-transformed B cells are
mediated by a persistent activation of the ERK1/2 signaling
pathway. Our study indicated that the ERK1/2-induced XAF1 with Puma
promoted translocation of Bax into mitochondria in Dex-exposed
cells (Fig. 6D).
Oxidative stress can be elicited by ROS, which
refers to persistent excessive ROS production and limited
antioxidant shield, and has been involved in various biological
responses such as apoptosis (22,23).
Accumulating evidence indicates that chemical-mediated ROS
production gives rise to alteration of cellular function and
eventually results in apoptosis. Dysfunction of mitochondria,
induced by the production of excessive ROS, leads to dissipation of
Δψm and apoptosis (23). Moreover, ROS signaling appears to
be triggered by the activation of the mitochondrial-dependent cell
death pathway through activation of MAPK pathways (22). Our data indicated that a persistent
phosphorylation of ERK1/2 was caused by ROS generation after Dex
treatment. Thus, Dex was shown to induce a boost of ROS with a peak
after a 30 min exposure, and a pretreatment with the ROS scavenger
NAC significantly decreased the persistent ERK1/2 phosphorylation.
We found that ERK1/2 was involved in this apoptotic effect of Dex
in a ROS-dependent manner (Fig.
5). Inhibition of ERK1/2 reversed Dex-mediated apoptosis. Dex
induced apoptosis of EBV-transformed B cells by ROS-dependent
ERK1/2-mediated XAF1 up-regulation (Fig. 6).
In conclusion, we found that Dex inhibited cell
growth and induced apoptosis in EBV-transformed B cells. Our
results indicated that Dex-induced apoptosis was involved in the
reduction of XIAP, Bcl-xL and Bcl-2 expression and induction of
Bax, Puma and XAF1 expression, and caused the dissipation of
Δψm in EBV-transformed B cells. Our results also
demonstrated that Dex induced the activation of caspase-9 as
initiator caspase and subsequently induced the activation of
caspase-3 and -8. More importantly, ROS, ERK1/2 and XAF1
participated in Dex-induced apoptosis. Therefore, we demonstrate
that Dex mediates apoptosis of EBV-transformed B cells through a
novel ROS-dependent ERK1/2-mediated XAF1 signaling pathway.
Abbreviations:
|
EBV
|
Epstein-Barr virus;
|
|
Dex
|
dexamethasone;
|
|
ROS
|
reactive oxygen species;
|
|
NAC
|
N-acetyl-l-cysteine
|
Acknowledgements
This study was supported by the 2008
Inje University Research Grant and the National R&D Program for
Cancer Control, Ministry for Health, Welfare and Family Affairs,
Republic of Korea (grant no. 0920040).
References
|
1.
|
Young LS and Rickinson AB: Epstein-Barr
virus: 40 years on. Nat Rev Cancer. 4:757–768. 2004.PubMed/NCBI
|
|
2.
|
Kuppers R: B cells under influence:
transformation of B cells by Epstein-Barr virus. Nat Rev Immunol.
3:801–812. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Lopez-Royuela N, Balsas P, Galan-Malo P,
Anel A, Marzo I and Naval J: Bim is the key mediator of
glucocorticoid-induced apoptosis and of its potentiation by
rapamycin in human myeloma cells. Biochim Biophys Acta.
1803:311–322. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Laane E, Panaretakis T, Pokrovskaja K, et
al: Dexamethasone-induced apoptosis in acute lymphoblastic leukemia
involves differential regulation of Bcl-2 family members.
Haematologica. 92:1460–1469. 2007. View Article : Google Scholar
|
|
5.
|
Li Z, Chen Y, Cao D, Wang Y, Chen G, Zhang
S and Lu J: Glucocorticoid upregulates transforming growth factor-β
(TGF-β) type II receptor and enhances TGF-β signaling in human
prostate cancer PC-3 cells. Endocrinology. 147:5259–5267. 2006.
|
|
6.
|
Li M, Chen F, Liu CP, Li DM, Li X, Wang C
and Li JC: Dexamethasone enhances trichosanthin-induced apoptosis
in the HepG2 hepatoma cell line. Life Sci. 86:10–16. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Dobos J, Kenessey I, Timar J and Ladanyi
A: Glucocorticoid receptor expression and antiproliferative effect
of dexamethasone on human melanoma cells. Pathol Oncol Res.
17:729–734. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Yamamoto T, Nishiguchi M, Ioue N, et al:
Inhibition of murine osteosarcoma cell proliferation by
gluocorticoid. Anticancer Res. 22:4151–4156. 2002.PubMed/NCBI
|
|
9.
|
Greenberg AK, Hu J, Basu S, et al:
Glucocorticoids inhibit lung cancer cell growth through both the
extracellular signal-related kinase pathway and cell cycle
regulators. Am J Respir Cell Mol Biol. 27:320–328. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Wang H, Wang Y, Rayburn ER, Hill DL,
Rinehart JJ and Zhang R: Dexamethasone as a chemosensitizer for
breast cancer chemotherapy: potentiation of the antitumor activity
of adriamycin, modulation of cytokine expression, and
pharmacokinetics. Int J Oncol. 30:947–953. 2007.PubMed/NCBI
|
|
11.
|
Xu MJ, Fang GE, Liu YJ and Song LN:
Effects of glucocorticoid on proliferation, differentiation, and
glucocorticoid receptor expression in human ovarian carcinoma cell
line 3AO. Acta Pharmacol Sin. 23:819–823. 2002.PubMed/NCBI
|
|
12.
|
Liston P, Fong WG, Kelly NL, et al:
Identification of XAF1 as an antagonist of XIAP anti-Caspase
activity. Nat Cell Biol. 3:128–133. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Wang J, He H, Yu L, et al: HSF1
down-regulates XAF1 through transcriptional regulation. J Biol
Chem. 281:2451–2459. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Fong WG, Liston P, Rajcan-Separovic E, St
Jean M, Craig C and Korneluk RG: Expression and genetic analysis of
XIAP-associated factor 1 (XAF1) in cancer cell lines. Genomics.
70:113–122. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Byun DS, Cho K, Ryu BK, Lee MG, Kang MJ,
Kim HR and Chi SG: Hypermethylation of XIAP-associated factor 1, a
putative tumor suppressor gene from the 17p13.2 locus, in human
gastric adenocarcinomas. Cancer Res. 63:7068–7075. 2003.PubMed/NCBI
|
|
16.
|
Tu SP, Sun YW, Cui JT, et al: Tumor
suppressor XIAP-associated factor 1 (XAF1) cooperates with tumor
necrosis factor-related apoptosis-inducing ligand to suppress colon
cancer growth and trigger tumor regression. Cancer. 116:1252–1263.
2010. View Article : Google Scholar
|
|
17.
|
Huang J, Yao WY, Zhu Q, Tu SP, Yuan F,
Wang HF, Zhang YP and Yuan YZ: XAF1 as a prognostic biomarker and
therapeutic target in pancreatic cancer. Cancer Sci. 101:559–567.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Lee MG, Huh JS, Chung SK, et al: Promoter
CpG hyper-methylation and downregulation of XAF1 expression in
human urogenital malignancies: implication for attenuated p53
response to apoptotic stresses. Oncogene. 25:5807–5822. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Straszewski-Chavez SL, Visintin IP,
Karassina N, et al: XAF1 mediates tumor necrosis
factor-alpha-induced apoptosis and X-linked inhibitor of apoptosis
cleavage by acting through the mitochondrial pathway. J Biol Chem.
282:13059–13072. 2007. View Article : Google Scholar
|
|
20.
|
Martelli AM, Cappellini A, Tazzari PL, et
al: Caspase-9 is the upstream caspase activated by
8-methoxypsoralen and ultraviolet-A radiation treatment of Jurkat T
leukemia cells and normal T lymphocytes. Haematologica. 89:471–479.
2004.
|
|
21.
|
Park GB, Kim YS, Lee HK, Song H, Cho DH,
Lee WJ and Hur DY: Endoplasmic reticulum stress-mediated apoptosis
of EBV-transformed B cells by cross-linking of CD70 is dependent
upon generation of reactive oxygen species and activation of p38
MAPK and JNK pathway. J Immunol. 185:7274–7284. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Matsuzawa A and Ichijo H:
Stress-responsive protein kinases in redox-regulated apoptosis
signaling. Antioxid Redox Signal. 7:472–481. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Chiu WH, Luo SJ, Chen CL, et al: Vinca
alkaloids cause aberrant ROS-mediated JNK activation, Mcl-1
downregulation, DNA damage, mitochondrial dysfunction, and
apoptosis in lung adenocarcinoma cells. Biochem Pharmacol.
83:1159–1171. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Baghdassarian N, Catallo R, Mahly MA,
French P, Chizat F, Bryon PA and French M: Glucocorticoids induce
G1 as well as S-phase lengthening in normal human stimulated
lymphocytes: differential effects on cell cycle regulatory
proteins. Exp Cell Res. 240:263–273. 1998. View Article : Google Scholar
|
|
25.
|
Ramalingam A, Hirai A and Thompson EA:
Glucocorticoid inhibition of fibroblast proliferation and
regulation of the cyclin kinase inhibitor p21Cip1. Mol Endocrinol.
11:577–586. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Gupta S: Molecular signaling in death
receptor and mitochondrial pathways of apoptosis. Int J Oncol.
22:15–20. 2003.PubMed/NCBI
|
|
27.
|
Canton M, Caffieri S, Dall’Acqua F and Di
Lisa F: PUVA-induced apoptosis involves mitochondrial dysfunction
caused by the opening of the permeability transition pore. FEBS
Lett. 522:168–172. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Wang J, Zhang W, Zhang Y, et al: c-Jun
N-terminal kinase (JNK1) upregulates XIAP-associated factor 1
(XAF1) through interferon regulatory factor 1 (IRF-1) in
gastrointestinal cancer. Carcinogenesis. 30:222–229. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Melino G, Bernassola F, Ranalli M, et al:
p73 induces apoptosis via PUMA transactivation and Bax
mitochondrial translocation. J Biol Chem. 279:8079–8083. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Roman M, Chen W and Cobb MH: Differential
regulation and properties of MAPKs. Oncogene. 26:3100–3112. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Zhang W and Liu HT: MAPK signal pathways
in the regulation of cell proliferation in mammalian cells. Cell
Res. 12:9–18. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Desbarats J, Birge RB, Mimouni-Rongy M,
Weinstein DE, Palerme JS and Newell MK: Fas engagement induces
neurite growth through ERK activation and p35 upregulation. Nat
Cell Biol. 5:118–125. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Zhuang S and Schnellmann RG: A
death-promoting role for extracellular signal-regulated kinase. J
Pharmacol Exp Ther. 319:991–997. 2006. View Article : Google Scholar : PubMed/NCBI
|