Introduction
Invasive cervical cancer can be divided in two major
histological types: squamous cell carcinoma (SCC) and
adenocarcinoma (AC). Although the incidence of SCC of the uterine
cervix has been decreasing, that of cervical adenocarcinoma (AC)
has been increasing in recent years (1). Compared with SCC, AC of the cervix is
rare. Due to the relative rarity, only a few large studies have
addressed risk factors in adenocarcinoma (2). Previous epidemiologic studies of the
association between human papillomavirus (HPV) and cervical adeno
carcinoma have shown strong associations; this information has
suggested that there appears to be a difference in HPV types and
prevalence (3). In SCC, HPV 16 is
the dominant type, while in adenocarcinoma, HPV 18 is seen more
frequently (4). However, the
preference and capability of HPV 18 above HPV 16 to infect and
transform glandular epithelial cells is unclear (3). Some evidence indicates that cofactors
that contribute to the progression of HPV-infected cervical cells
to AC are distinct from those that contribute to the progression to
squamous cell carcinoma (5).
Smoking, for example, is associated with an increased risk for SCC
but there is no association for AC (6). Although several factors have been
established as important initial events for the tumorigenesis of
cervical carcinoma, the reports regarding the molecular biology
involved in the cellular process are rare. Only a few studies have
been published comparing global transcript expressions for both
tumor types (7). A major
limitation of these studies is the fact that no adjustment for the
molecular pathogenesis has been made.
In this study, we used oligomicroarray technology
and pathway analyses to describe the role of molecular networks in
the development of AC and SCC. Evaluations were carried out using
bioinformatic analysis of genomic data from expression patterns of
genes with emphasis on network and pathway analysis. The expression
profiles were histology-dependent and clearly differentiated.
Therefore, the expression of the genes in this study clearly
displayed evidence indicating similarities and differences between
the molecular environments in AC and SCC. Our findings also have
important implications for the diagnostic and possible therapeutic
intervention of patients with cervical cancer as well as future
research.
Materials and methods
Ethics statement
All patients involved in the study signed a
declaration of consent stating that the patients specimens may be
used for scientific intentions. Specimens were obtained from the
patients in the Department of Obstetrics and Gynecology in
concordance with procedures approved by the Institutional Review
Board of The Catholic University of Korea (06BR131).
Tissue samples
The disease status was assigned according to the
International Federation of Gynecology and Obstetrics. Briefly, all
patients were Korean. Of the 28 patients with cervical cancer, 11
patients were classified as AC and 17 patients were classified as
SCC. Normal cervix samples were obtained from 17 uterine leiomyoma
patients and stored in liquid nitrogen. Common reference RNAs from
17 different normal cervixes were used as a consistent control. All
samples were filled up to a depth of 1–2 mm using a microscope,
carefully avoiding the underlying stromal tissue. The results were
then examined with the microscope. The samples were immediately
placed in vials containing 2 ml TRIzol (Gibco-BRL, Grand Island,
NY), stored at 4°C for up to 12 h, and then frozen at −80°C.
Probe hybridization
Reverse transcription was carried out using total
RNA isolated from samples using TRIzol. Experimental procedures for
microarray assay were performed according to the Macrogen Magic
II-10K technical manual. Total RNA was converted into double
stranded cDNA using the cDNA synthesis system (Roche) using a
T7-(dT) 24 primer. The fluorescent labeled-cDNA was hybridized with
Magic II-10K microarray (Macrogen, Seoul, Korea) for 16 h at 42°C.
Arrays were then washed and scanned with a GenePix 4000B scanner
(Axon Instruments, Union City, CA). Each chip contained a total of
10,368 elements of which 10,108 were unique genes/clusters.
Western blot analysis
The proteins were electrophoresed for 2 h with
SDS-PAGE and western blotting was performed for 1 h and 30 min with
a Hybond-ECL membrane (Amersham, Uppsala, Sweden) at 100 V. Protein
bands were visualized using an ECL kit according to the
manufacturer’s protocol (Amersham, Arlington Heights, IL).
Analysis
Acquired images were processed and analyzed
statistically for interpretation of the analyzed spot intensity
results using Imagene v4.1 software (Roche). Non-biological factors
that contributed to variability of data were minimized using global
normalization/scaling with data from all probe sets; normalization
for the microarrays was also carried out. For each gene, its
relative fold change in expression was the ratio of the sum of the
median expression levels of cervical cancer tissues compared to the
common control. Genes were excluded from the analysis if their
expression was negative or if they were too smeared to read. Genes
that showed differences in their expression levels, of at least
2.0-fold, were selected for the different analyses (hierarchical
cluster analysis, functional cluster analysis and biological
pathway analysis). Supervised hierarchical clustering was performed
in clusters and a two-way average linkage clustering was applied.
To classify the observed profiles of gene expression, functional
analysis was carried out as follows. Each gene was annotated by
integrating the information (as of October, 2011) on the Gene
Ontology website (http://GenMapp.org). First, each gene
was associated with its corresponding current curated gene entry in
UniGene (http://www.ncbi.nlm.nih.gov). Next,
the Ingenuity Pathway Analysis software (IPA, Ingenuity Systems,
Mountain View, CA) was utilized to identify networks of interacting
genes and other functional groups. Semantically consistent pathway
relationships were modeled based on a continual, formal extraction
from the public domain literature and covered >10,400 human
genes (www.ingenuity.com/products/pathways_knowledge.html).
These genes were then used as a starting point for generating
biologic networks. The resulting networks are presented in a
graphic format.
Statistical analysis
Statistical analysis was done using χ2
test and ANOVA. Values from the different groups were compared. A
P-value <0.05 was considered significant.
Results
Hierarchical cluster analysis
Using unsupervised hierarchical clustering, by
average linkage analysis, we successfully grouped AC and SCC into 2
distinctive groups (Fig. 1A). Gene
expression levels of 46 transcripts separated hierarchical
clustering samples into 2 groups on the basis of the expression
patterns. These data are represented by a dendrogram with the
closest branches of the tree representing arrays with similar gene
expression patterns. The results showed that there was a
significant difference between AC and SCC. Even though the overall
signal patterns found on the AC and SCC hybridized arrays were
similar, a small subset of 46 transcripts show differential
expression signals in comparisons between the AC and SCC samples.
These genes can be characterized as new putative histology-specific
cervical cancer genes. Among genes significantly upregulated in AC,
we have found VIPR1, NIFUN, SPEN, MAD and BCL3 of particular
interest. The genes upregulated in SCC included KRT17, CALCA and
CHMP2A. KRT17 is one of the pathological situation genes that are
expressed only in pathological situations such as metaplasia and
carcinoma of the uterine cervix as well as in psoriasis vulgaris.
Western blots were performed to confirm gene expression patterns in
this study. As shown in Fig. 1B,
several up- and downregulated proteins confirmed the patterns
obtained from the microarray and showed the consistency of the
assays.
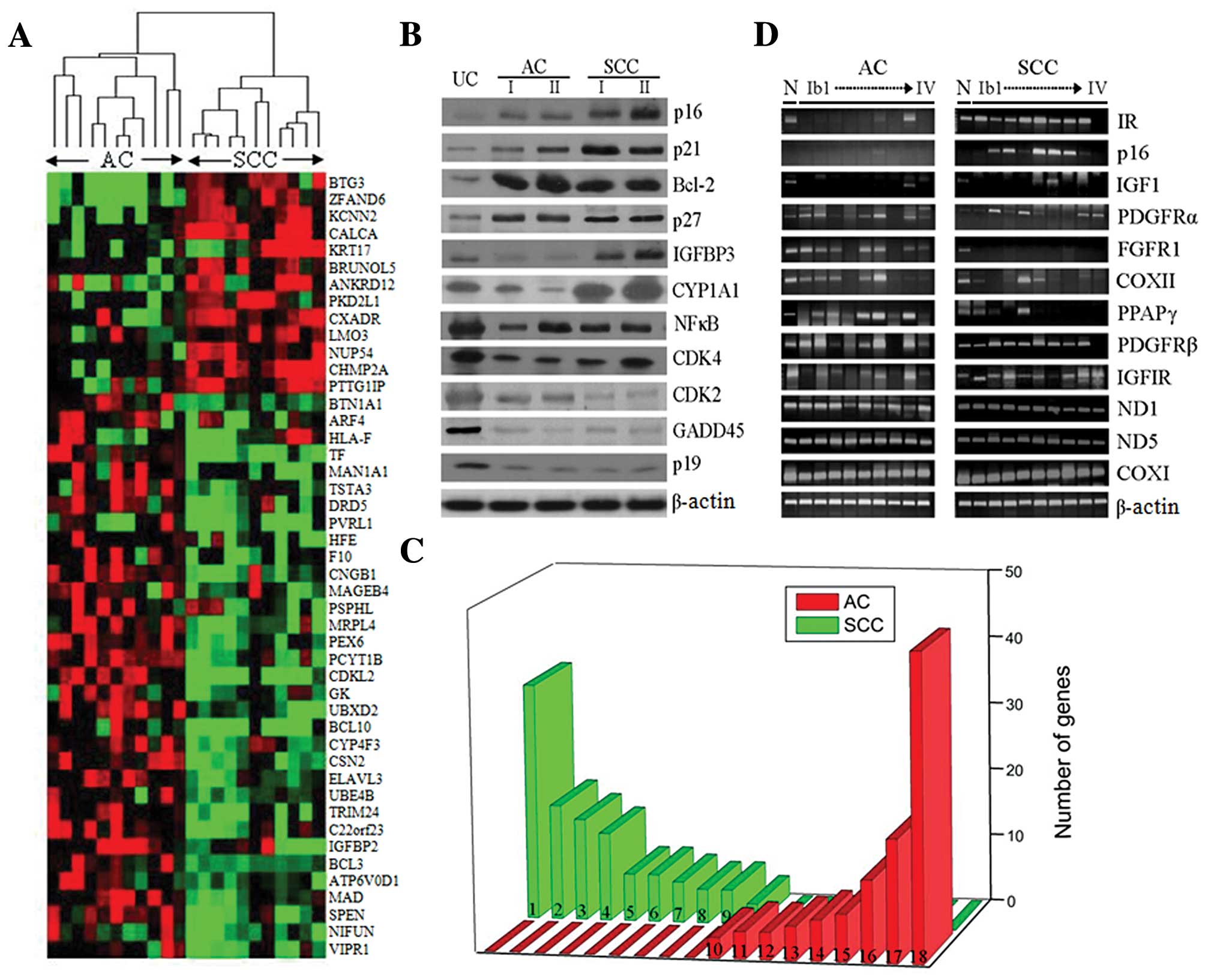 | Figure 1.Gene expression profiles measured by
microarrays. (A) Hierarchical clustering analysis in AC and SCC. A
dendrogram (tree graph) shows the grouping of the genes based on
the similarity between them. Supervised analysis of 46 transcripts
was carried out using the ‘Euclidean distance’ to determine the
similarity measure and the input rank as the ordering function
(green, underexpression; red, overexpression). Dendrogram with
tissues labels corresponding to the experiments. (B) Western blot
analysis of differentially expressed genes of the microarray.
Columns refer to common reference (N) and each cancer stage (I and
II). (C) Differential display of key functional patterns between AC
and SCC (P-value for all <0.05). Representative functional
clusters were obtained using Gene Ontology analysis based on
cluster visualization. (1) Apoptosis of neurons, (2) cell death of
pheochromocytoma cells, (3) damage to DNA, (4) binding of synthetic
promoter, (5) polyploidization, (6) proliferation of granulocytes,
(7) differentiation of erythroid progenitor cells, (8) emigration
of T lymphocytes, (9) colony formation of colony-forming erythroid
cells, (10) internalization of virus, (11) anoikis of epithelial
cells, (12) proliferation of ovarian cancer cells, (13) G1 phase of
epithelial cells, (14) stabilization of mitochondrial membrane,
(15) binding of red blood cells, (16) splenomegaly, (17) hepatic
system disorder and (18) immunological disorder. The differential
clusters include several functions of the core enzyme activity, as
well as several related families that share similar phenotypes. (D)
Carcinogenesis-related transcripts by RT-PCR analysis. Total RNAs
obtained from common reference RNAs (lane 1) and cervical cancer
stages (lanes 2–9) were subjected to RT-PCR. |
Functional cluster analysis
To show how the transcripts, identified by the gene
expression signature, were related in AC and SCC, we placed the
transcripts in the context of present interactome knowledge, using
Ingenuity Pathways Analysis tools. Using gene ontology analysis,
closer examination of the genes resulted in a variety of 372 and
523 mutually-dependent molecular functions to be identified in AC
and SCC, respectively; this evaluation revealed that the functional
profiling was not randomly distributed, as shown in Fig. 1C (P-value for all <0.05).
Significantly downregulated molecular functions for SCC were cell
death in pheochromocytoma cells (P=0.0037), apoptosis in neurons
(P=0.009) and damage to DNA (P=0.0038). The upregulated molecular
function was internalization of virus (P=0.0091). By contrast, the
upregulated molecular functions for AC were immunological disorder
(P=0.006), splenomegaly (P=0.0053) and hepatic system disorder
(P=0.006). Of note, the internalization of virus with viral
infection (BCAR1, CSF2RA, LDLR) was clearly upregulated for both
SCC (P=0.0091) and AC (P=0.0085). In Tables I and II, differenti ally up- or down-regulated
putative cervical cancer-network processes are summarized according
to biological pathways. To detect the differences in the functional
profiles between AC and SCC, we placed differentially expressed
transcripts in the context of present interactome knowledge, using
the Ingenuity Pathways Analysis tools (P-value for all <0.05).
Biologic pathway analysis revealed the antigen presenting canonical
pathway (P=0.03) as a significant molecular pathway in AC. Network
analysis, based on predetermined knowledge on individually modeled
relationships between genes, identified seven highly significant,
overlapping networks in the data set. The top-scoring network,
built around the antigen presenting pathway, displayed high-level
functions in the apoptosis signaling, GM-CSF signaling, death
receptor signaling and PI3k/AKT signaling pathways. By contrast,
the pathway analysis revealed the G2/M DNA damage checkpoint regul
ation pathway (P=0.05) as a significant molecular pathway in SCC.
Network analysis identified highly significant overlapping of the
networks. The top-scoring network displayed high-level functions in
the estrogen receptor signaling, Notch signaling, death receptor
signaling and apoptosis signaling pathways. The relative expression
of the transcripts SPEN, HLA-DRB3, CDKN1A, MDM2 and TAF5L showed a
correlation of the expression levels revealed by the
microarray.
 | Table I.Functional network analysis based on
the transcript signature in AC. |
Table I.
Functional network analysis based on
the transcript signature in AC.
| Network no. | Genes in the
network | Scorea | High-level
functions | No. of associated
genesb | Significancec |
|---|
| 1 | APAF1,
ARHGAP1, BBC3, BCL2, BCL2L1,
BCL2L10, CLCN3, DCX, DDR1,
DNAJA3, FBXO2, HAP1, IGFBP6,
ITPR1, L1CAM, MRAS, NFKBIB,
NRP1, PDCD2, PRDX1, RASA1,
RASGRP2, RIN1, RRAS, RRAS2,
SCN3A, SEMA3F, SERPINF1, SKP1A,
SNAP91, SRP72, STAT6, TBX5,
TXNIP, WISP1 | 35 | Cancer | 24 |
1.6E−8-7.6E−3 |
| Cell morphology | 17 |
1.6E−8-7.6E−3 |
| Cell death | 21 |
1.5E−7-7.6E−3 |
| 2 | ALAS2,
ARNT, CBX1, CHAF1B, CSF2RA,
DDB2, DNAJB1, FANCA, FANCC,
G1P3, IER3, IFNA6, IL12RB1,
IL12RB2, IL27RA, ING1, IRF3,
JAK2, LBP, PGK1, PML, REST,
SAP30, SF3A1, SF3A3, SH2B, SIM2,
SKI, SMARCC1, STAT1, TBK1, TIF1,
TRIM28, ZNF74, ZNF197 | 35 | Gene
expression | 22 |
8.7E−8-3.0E−2 |
| Cell death
immunological disease | 21 |
4.0E−7-3.0E−2 |
| 11 |
4.0E−7-3.3E−2 |
| 3 | B2M,
BCL3, BOK, CDH4, CDKL2, CLU,
COL1A2, COPS2, COPS5, CYR61,
EEF1A1, EIF3S6, FUS, G22P1,
GCNT2, GDF5, HCST, HNRPC, HNRPU,
ILF3, JUND, KIR3DL1, KLRC2,
KLRC3, LIG3, MAN1B1, MCL1, MIF,
MLF1, NRG1, PDE3A, RFX5, TF,
TYROBP, VAV1 | 35 | Cell death | 20 |
6.9E−6-2.5E−2 |
| Cancer | 19 |
1.8E−5-2.3E−2 |
| DNA
replication | 10 |
2.8E−5-2.2E−2 |
| 4 | ADORA2B,
ANXA4, BAIAP2, CCND2, CCNE1,
CCR5, CDT1, CUL3, EDNRA, ELL,
ERG, FOS, HOXB4, KRT8, LDHA,
LDHB, MLLT7, MXD1, NCKAP1, NMB,
PCDHGC3, PSMA2, PSMA5, PSMA6,
PSMB3, PSMC5, PSMD6, RBP1,
RPS6KA4, SFRP4, SLC19A1, SOD1,
TACR1, VIL2, WASF2 | 35 | Cell death | 18 |
1.07E−4-2.06E−2 |
| Cellular
development | 14 |
2.13E−4-3.03E−2 |
| Hematological
system | 7 |
2.13E−4-1.91E−2 |
| 5 | ACVR2B,
AUP1, BRS3, CAV1, CD9, CD47,
CD151, COL6A2, CSK, CSPG2, CTSK,
DAG1, EGFR, ELN, ERBB2, FBLN2,
GNAS, GNRHR, HTR4, ITzNF4A,
IFITM1, MUC6, NAP1L1, PHLDA2,
PPARG, PRG4, PTPRU, SPAG11,
SUFU, TAX1BP3, TDGF1, TRERF1 | 35 | Cell-to-cell
signaling and | 20 |
1.9E−7-3.8E−3 |
| 9 | Gene
expression | 20 |
6.9E−11-3.8E−3 |
| Cancer | 19 |
8.9E−10-3.8E−3 |
| Cellular growth and
proliferation | 20 |
8.9E−10-3.8E−3 |
| Table II.Functional network analysis based on
the transcript signature in SCC. |
Table II.
Functional network analysis based on
the transcript signature in SCC.
| Network no. | Genes in the
network | Scorea | High-level
functions | No. of associated
genesb |
Significancec |
|---|
| 1 | ADD1,
ANXA5, CBX4, CCRK, CDK2, CDK7,
CDKN1A, CYR61, DYRK1B, EXO1,
FHL1, FOXG1B, GTF2H2, HES1,
LOC58486, MAML1, MCM4, MCM6,
MFNG, MLLT7, MNAT1, MS4A3,
PCGF4, PCGF6, PLK2, PPP1R12A,
PPP1R12B, RBL2, RBPSUH, ROCK2,
RRM1, RRM2, SART2, SPEN,
ST8SIA1 | 36 | Cell cycle | 14 |
4.2E−6-2.2E−2 |
| Gene
expression | 13 |
5.4E−6-1.9E−2 |
| Cancer | 13 |
6.1E−6-2.2E−2 |
| 2 | APAF1,
APPL, BAX, BBC3, BCL2L1, BFAR,
BNIP1, CALB1, CASP1, CASP3,
CASP8, CASP10, CFLAR, CHST8,
DCC, EIF4A2, EIF4G3, HSPB2,
IFI16, IL18, ITPR1, MADD, MDM2,
NALP1, NOL3, NUMB, PPP1CA, PRNP,
PTGS2, RIPK1, RIPK4, SEPT4,
SNCB, TRAF1, TRIP | 36 | Cell death | 30 |
2.6E−17-3.8E−3 |
| Cancer | 22 |
9.8E−15-3.8E−3 |
| Hematological
disease | 13 |
5.5E−11-3.8E−3 |
| 3 | ADRA1D,
ATP2B4, BDP1, BRF1, BST2, CEBPZ,
CHRNA5, DDX20, EGR3, FGF9,
GEMIN4, GLDC, GSTM5, GTF3C1,
GTF3C4, HOXB4, LAMB3, MEST,
MICB, MTA2, PAX3, PKIA, PTPN14,
RB1, SMC1L1, STAG3, STX3A,
SYBL1, TM4SF2, TMPO, TOP2A,
TOP2B, TRIP11, TSPY1, YWHAE | 36 | Cellular assembly
and organization | 11 |
1.4E−5-4.8E−2 |
| Cancer | 7 |
2.1E−4-4.8E−2 |
| Cell cycle | 9 |
2.1E−4-4.8E−2 |
| 4 | ACADM,
APOA1, APOA2, ATP2B1, BCAR1,
CAPN3, CCL8, CCL27, CCRL1, CD33,
CD36, CDK5R1, CXCL12, CXCL16,
CXCR6, CYP3A5, DCX, FOXA3,
IFNAR1, IL12B, ISGF3G, ITGAV,
ITGB3, L1CAM, LTF, PCTK1, PHKA2,
PLCB1, PPAP2B, RALA, SEC10L1,
SEC8L1, SEPT7, STAT2, TNFRSF8 | 36 | Cellular
movement | 19 |
1.2E−9-7.6E−3 |
| Cancer | 16 |
2.4E−9-7.6E−3 |
| Immunological
disease | 7 |
4.7E−8-3.8E−3 |
| 5 | AXIN2,
BLM, CCNT1, EDD1, ESR2, GCK,
GYS2, ILF3, IPF1, MEIS2, NR0B1,
PFKFB2, PML, POLM, POLR2A,
POLR2H, RAD18, RAD51C, RAD51L3,
RBM17, RFC2, SKI, SMAD3, STUB1,
SUMO1, TIF1, TINF2, TITF1,
TOPBP1, WRN, XAB2, XRCC5, ZIC2,
ZNF198, ZNF221 | 36 | Gene
expression | 19 |
8.7E−8-3.4E−2 |
| DNA
replication | 10 |
4.5E−6-3.4E−2 |
| Cell cycle | 10 |
6.0E−6-3.2E−2 |
| 6 | ADCY5,
ATF7, BMP2, BRAF, CNKSR1, GAS,
GNB5, MAP3K6, MAPK7, MAPK9,
MAPK10, MEF2C, MINK1, MRAS,
MYLK, NTRK3, PLN, RASGRP2, RGS9,
SCNN1A, SGK, SLC18A2, TBX5,
TNFRSF11B, TNIK, WWP2 | 19 | Amino acid
metabolism | 9 |
2.1E−6-3.8E−3 |
| Post-translational
modification | 8 |
2.1E−6-1.2E−3 |
| Small molecule
biochemistry | 17 |
2.1E−6-3.1E−2 |
| 7 | 76P,
ACBD3, AK3, AKAP9, CD276, COIL,
IL12RB2, IL17E, IL8RA, KLRD1,
MAPK14, NOLC1, NUP155, PCNT2,
POLE2, POLE3, PRKCBP1, PTPN4,
SNCB | 10 | Cellular assembly
and organization | 12 |
3.4E−11-3.8E−3 |
| Cellular function
and maintenance | 9 |
1.8E−9-3.8E−3 |
| Cellular
development | 6 |
1.0E−6-3.8E−3 |
| 8 | ALDOB,
APOM, BZRAP1, CD1B, CYP11A1,
ESR2, GAPD, HNF4A, IFI30,
KCNJ11, NOS2A, NR0B2, NR1I2,
SMAD3, STAMBP, TM7SF2, WWP1 | 8 | Amino acid
metabolism | 23 |
4.7E−12-7.6E−3 |
| Post-translational
modification | 18 |
3.9E−8-7.6E−3 |
| Small molecule
biochemistry | 15 |
3.9E−8-7.6E−3 |
Correlation of canonical pathway and
carcinogenesis-related genes
We searched the carcinogenesis-related transcript
expression patterns for interaction of additional members of these
putative pathways. Although none existed in the microarray, the
expression patterns of additional carcinogenesis-related genes were
observed in our model. As shown in Fig. 1D, the differential transcript
expressions are shown for SCC and AC. The overexpressed transcripts
in SCC were IR, p16, IGF1, IGF1R, PDGFRα and PDGFRβ. By contrast,
the overexpressed transcripts in AC were PDGFRα, FGFR1, PPARγ and
PDGFRβ. Differential expression of the transcripts could be related
to the tumor classification. The ND1, ND5 and COX1 showed no
differential expression patterns. The antigen presenting canonical
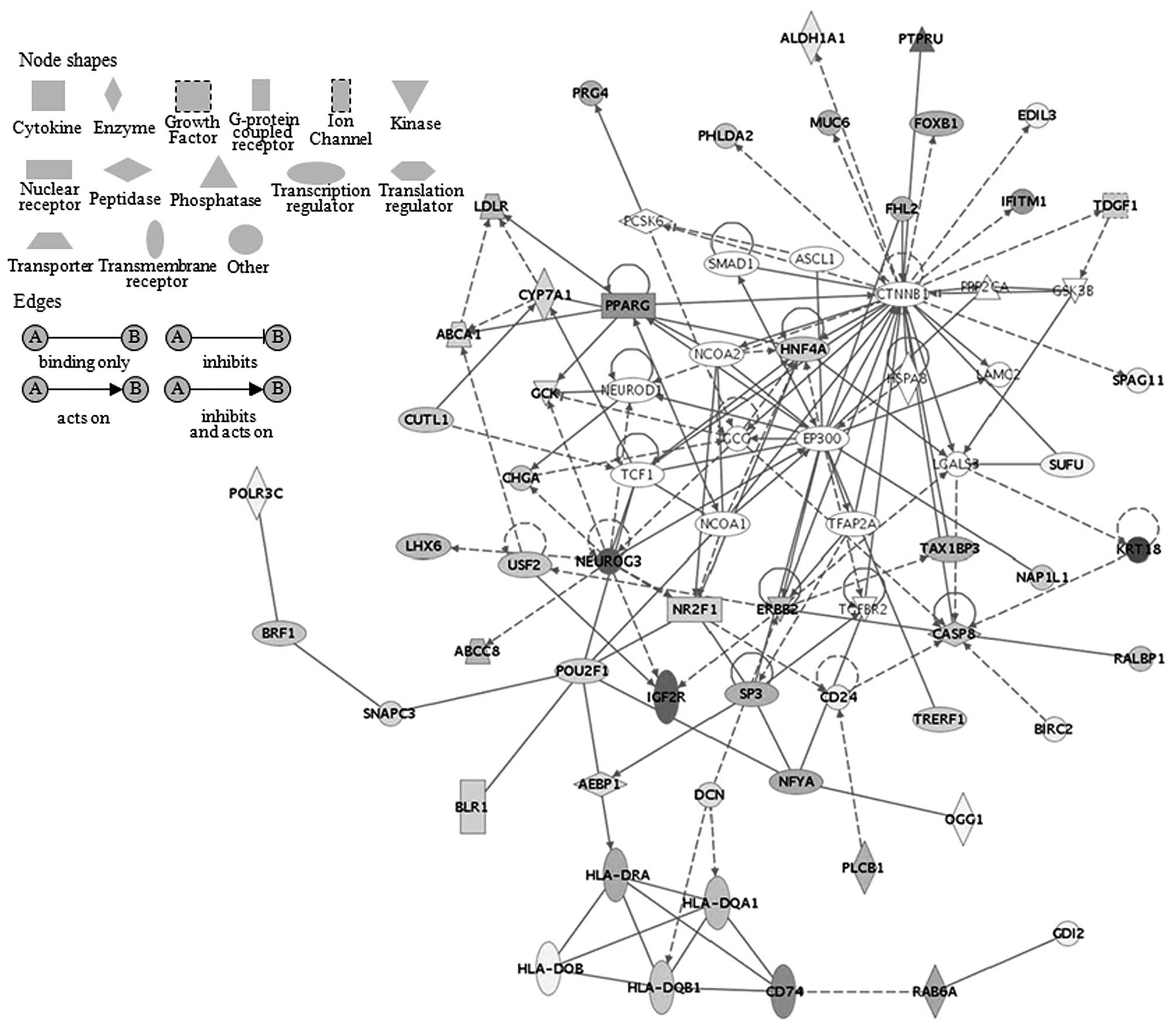
pathway was revealed to be linked to PPARγ in AC. The
overexpression of this gene was observed to be related to HLA-DRA
overexpression in the pathway as shown in Fig. 2. In addition, the overexpression of
the PPARγ and HAL-DRA was significantly associated with NEUROG3
overexpression. This analysis included complete overexpression of
the PPARγ gene in AC from stage Ib1a to IV. The G2/M DNA damage
checkpoint regulation pathway was observed to be significantly
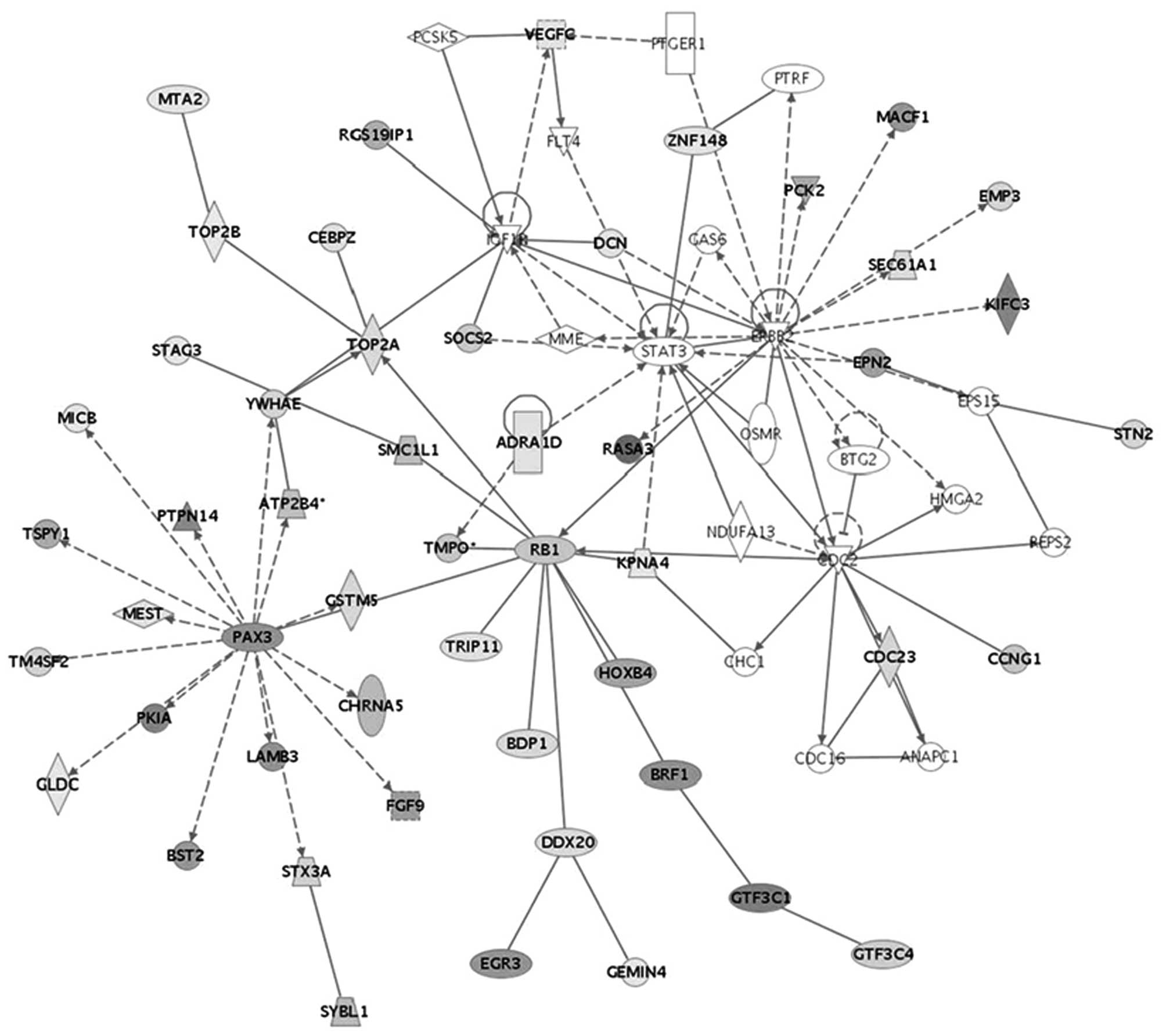
linked to IGF1R in SCC. The upregulation of this gene was noted to
be related to YWHAE and TOP2A downregulation in the pathway
(Fig. 3). In addition, the
upregulation of IGF1R was significantly associated with RB1
downregulation. This analysis included the upregulation of the
IGF1R gene in SCC from stage Ib1 to IVb. Therefore, the molecular
pathways of SCC and AC appear to be related to different
carcinogenesis-related genes resulting from cell
transformation.
Discussion
The specific details of the reference RNA, as a
control, from several cell lines or tissues have previously been
reported (8). The properties of a
cell line derived reference RNA sample has failed to address the
long-term needs and highly reproducible reference samples (9). Immortal cell lines are obviously not
normal. In this study, we show that a well maintained reproducible
reference sample can be created from a pool of RNAs derived from
normal cervixes. Comparison of two duplicate samples demonstrated
the reproducibility of this experimental approach (correlation
coefficient >0.95).
Even though the overall signal patterns found on the
AC and SCC hybridized arrays were similar, a small subset of
regions showed differential expression signals in comparisons
between the AC and SCC. Using hierarchical clustering, 46
transcripts separated the samples into two distinct groups on the
basis of the expression patterns. Therefore, these genes can be
characterized as new putative histology-specific cervical cancer
genes. KRT17 is coexpressed with KRT16 only in pathological
situations such as metaplasia and carcinoma of the uterine cervix
as well as in psoriasis vulgaris (10). It was noted that KRT17 showed
increased gene expression only in SCC specimens. This is compatible
with the previous finding that during progression of cervical
intraepithelial neoplasia a clear increase in the expression of
keratin 17 was observed (11). The
IGFBP2 transcript was significantly increased in high grade
prostate intraepithelial neoplasia (PIN) and adenocarcinoma
(12). A significant
overexpression of IGFBP2 was observed in patients with localized
prostate adenocarcinoma; this suggests that overexpression of
IGFBP2 is a powerful marker for malignant transformation in
prostate epi thelium. IGFBP2 was only downregulated in SCC in this
study. In the case of VIPR1, the normalized overexpression
distribution for tissue type (http://genome-www5.stanford.edu/cgi-bin/source/sourceSearch)
was 13.7% in the adrenal gland, and only expressed in AC in this
study. VIPR1 has been characterized and localized in the neoplastic
cells of most adenocarcinomas such as ovarian adenocarcinomas,
colonic adenocarcinomas and pancreatic adenocarcinomas (13). Future studies are needed to clarify
the regulatory mechanisms of these 46 genes and their role in AC
and SCC.
Combined application of our network analysis of
differentially regulated genes in AC and SCC revealed that most of
the genes were assigned to tens of the cellular processes
(P<0.05), revealed as key functions of a variety of cellular
activities involved in cervical carcinogenesis. Genes (BCAR1,
CSF2RA and LDLR) associated with internalization of virus functions
(P=0.0091) were commonly overexpressed in both AC and SCC. We also
observed significant upregulated functions associated with
immunological (P=0.006), hepatic system (P=0.006) and splenomegaly
(P=0.0053) disorders in AC. The genes coding for 32 out of 45
immunological disorder functions were upregulated; this finding
suggests the importance of these gene products in the developmental
pathway of cervical adenocarcinoma. In addition, the genes coding
for 9 out of 12 splenomegaly immune diseases were upregulated. It
has been reported that the hepatic changes, as well as marked
splenomegaly, may represent an altered immunophenomenon of uterine
cervical cancer (14). Numerous
genes coding for proteins involved in immunological disorders were
notably upregulated in AC. In several types of carcinoma, a number
of genes responsible for the immune response have been reported to
be upregulated (15). By contrast,
the significant downregulated functions observed in SCC were
apoptosis of neurons (P=0.009), cell death of pheochromocytoma
cells (P=0.006) and damage to DNA (P=0.0057). The genes coding for
25 out of 35 apoptosis of neuron function were downregulated. In
addition, the genes coding for 13 out of 15 damage to DNA function
were downregulated; this suggests the importance of these gene
products in the developmental pathway of squamous cell cervical
carcinoma. Disruption of apoptosis and the cell cycle progression
pathways has been implicated in abnormal cell growth and
carcinogenesis (16). We observed
that genes involved in cell death were significantly downregulated
in SCC compared to AC.
In this study our investigation focused on the main
molecular pathway of SCC and AC, and its involvement in response to
carcinogenesis and cell transformation. The functional profiling
consequence of SCC and AC was the G2/M DNA damage checkpoint
regulation pathway and antigen presenting pathway, respectively.
The DNA damage-induced cell cycle checkpoint has broad implications
for human disease, particularly cancer (17). The G2/M cell cycle checkpoint is
especially emerging as an attractive candidate for new cancer
therapies (18). The G2/M
checkpoint prevents cells from attempting to undergo mitosis in an
inappropriate state; due to defects in G1 checkpoint mechanisms,
cancer cells depend on the G2/M checkpoint far more than normal
cells and it suggested that therapeutic agents acting at this cell
cycle phase may confer tumor selectivity. p53 can regulate the G2/M
transition either through the induction of p21 and 14-3-3σ, a
protein that normally sequesters cyclin B1-Cdc2 complexes in the
cytoplasm or through the induction of apoptosis (19). The human papilloma virus E6 protein
(HPV-E6) appears to allow genetic or epigenetic events to occur
that severely impair the G2 checkpoint by binding and inactivating
of p53 that is required for G2 arrest. The different observations
made with HPV-E6 and the truncated form of p53 suggests that other
targets of HPV-E6 may contribute to the G2 checkpoint (20).
The expression patterns of the
carcinogenesis-related genes were different in the comparisons
between SCC and AC. The overexpressed transcripts in SCC, such as
p16, IGF1 and PDGFRα were reported previously (21). By contrast, the overexpressed
transcripts in AC were PDGFRα, PPARγ and PDGFRβ (22). The G2/M DNA damage checkpoint
regulation pathway in SCC was observed to be significantly linked
to IGF1R via YWHAE protein-protein interaction (23). IGF-I and IGF-IR are known to play
an important role in cell transformation induced by viral
oncogenes, such as E6 and E7 (24). It has been reported that YWHAE
(14-3-3 proteins) interacts with IGFIR in vivo and that this
interaction may play a role in a transformation pathway signaled by
the IGFIR (25). The 14-3-3
proteins are found in all eukaryotic cells and have been shown to
bind to molecules involved in cell cycle control, apoptosis and
oncogenesis (26). Overexpression
of 14-3-3 caused most cells to arrest in G2. However, binding of
14-3-3 to phosphoserine 1283, in IGFIR, may play a role in IGFIR
mediated transformation. In this study, the analysis included
upregulation of the IGF1R transcript in SCC stage. It has also been
reported that 14-3-3 proteins do not bind to the insulin receptor
(IR) and the IR does not cause transformation (27). In addition, the upregulation of
IGF1R was significantly associated with the downregulation of RB1.
Rb family members are required to downregulate Cdc2 and Cyclin B1,
which is necessary to maintain prolonged G2 arrest.
Malignant transformation is frequently associated
with escape of tumor cells from immune recognition. The present
study showed that the antigen presenting canonical pathway was
observed to be linked to peroxisome proliferator-activated receptor
gamma (PPARγ) in AC in response to cell transformation. The
overexpression of PPARγ has been related to MHC class II (HLA-DRA,
DPA1, DQA1 and DQB1 overexpression) in this pathway. It has been
shown that expression of PPARγ protein was higher in an
adenocarcinoma cell line (TE-7 cells) than in a squamous cell
carcinoma cell line (TE-1 cells) (28). PPARγ activation has been implicated
in tumor promotion, cellular differentiation and apoptosis
(29). Many of the effects of
PPARγ are mediated through the inhibition of proinflammatory
transcription pathways such as NF-κB, AP-1, NFAT, C/EBP or Smad3,
via protein-protein interaction and competition for cofactor
recruitment (30). The
downregulation of IFNγ and IL-12 by PPARγ is known to prevent the
development of inflammatory diseases (31). In addition, the effects of PPARγ on
dendritic cells can drive the local immune response by favoring the
differentiation of TH2 cells, thus orienting the immune response
toward a humoral response. In human T cells, PPARγ activators
reduce the secretion of IFNγ, TNFα and IL-2 (32). These data suggest an important role
of PPARγ in AC and the immune system with a potential profound
impact on the immune response. It has been reported that
metabolites that physically binds to PPARγ as a ligand leads to
apoptosis of lung adenocarcinoma cells, and this may be beneficial
for the therapy of such cancers (33). Understanding the role of IGF1R and
PPARγ in cervical carcinogenesis is important for designing
diagnostic and therapeutic interventions as well as future research
(34).
In this study, microarray data and knowledge-based
information analyses were used for tumor classification. Different
expression profiles of genes and their specific molecular networks
were involved in the progression associated with SCC and AC. In
addition, we identified the utilization of the G2/M DNA damage
checkpoint regulation pathway in SCC and the antigen presenting
canonical pathway in AC. Therefore, good candidate genes identified
from this study clearly displayed specific association with each
aggressive biological subset of cervical cancer. Further studies
can be carried out to identify tumor markers that may be useful for
patient diagnosis and treatment of SCC and AC.
Acknowledgements
The study was supported by the
National Research Foundation of Korea (NRF), Seoul, Republic of
Korea (Grant no. 5-2011-A0154-00120). The funders had no role in
study design, data collection and analysis, decision to publish or
preparation of the manuscript.
References
|
1.
|
Bulk S, Visser O, Rozendaal L, Verheijen
RH and Meijer CJ: Cervical cancer in the Netherlands 1989–1998:
decrease of squamous cell carcinoma in older women, increase of
adenocarcinoma in younger women. Int J Cancer. 113:1005–1009.
2005.
|
|
2.
|
International Collaboration of
Epidemiological Studies of Cervical Cancer: Comparison of risk
factors for invasive squamous cell carcinoma and adenocarcinoma of
the cervix: collaborative reanalysis of individual data on 8,097
women with squamous cell carcinoma and 1,374 women with
adenocarcinoma from 12 epidemiological studies. Int J Cancer.
120:885–891. 2007. View Article : Google Scholar
|
|
3.
|
Ciapponi A, Bardach A, Glujovsky D,
Gibbons L and Picconi MA: Type-specific HPV prevalence in cervical
cancer and high-grade lesions in Latin America and the Caribbean:
systematic review and meta-analysis. PLoS One. 6:e254932011.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Clifford GM, Smith JS, Plummer M, Munoz N
and Franceschi S: Human papillomavirus types in invasive cervical
cancer worldwide: a meta-analysis. Br J Cancer. 88:63–73. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Castellsague X, Diaz M, de Sanjose S, et
al: Worldwide human papillomavirus etiology of cervical
adenocarcinoma and its cofactors: implications for screening and
prevention. J Natl Cancer Inst. 98:303–315. 2006. View Article : Google Scholar
|
|
6.
|
Plummer M, Herrero R, Franceschi S, et al:
Smoking and cervical cancer: pooled analysis of the IARC
multi-centric case-control study. Cancer Causes Control.
14:805–814. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Wingo SN, Gallardo TD, Akbay EA, et al:
Somatic LKB1 mutations promote cervical cancer progression. PLoS
One. 4:e51372009. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Wessels JM, Edwards AK, Zettler C and
Tayade C: Selection and validation of reference genes for miRNA
expression studies during porcine pregnancy. PLoS One.
6:e289402011. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Yang IV, Chen E, Hasseman JP, et al:
Within the fold: assessing differential expression measures and
reproducibility in microarray assays. Genome Biol.
3:research00622002.PubMed/NCBI
|
|
10.
|
Bae SM, Lee CH, Cho YL, et al:
Two-dimensional gel analysis of protein expression profile in
squamous cervical cancer patients. Gynecol Oncol. 99:26–35. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Smedts F, Ramaekers F, Troyanovsky S, et
al: Basal-cell keratins in cervical reserve cells and a comparison
to their expression in cervical intraepithelial neoplasia. Am J
Pathol. 140:601–612. 1992.PubMed/NCBI
|
|
12.
|
Zumkeller W: IGFs and IGFBPs: surrogate
markers for diagnosis and surveillance of tumour growth? Mol
Pathol. 54:285–288. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Reubi JC: In vitro identification of
vasoactive intestinal peptide receptors in human tumors:
implications for tumor imaging. J Nucl Med. 36:1846–1853. 1995.
|
|
14.
|
Nakanuma Y, Kouda W, Nakano T, Uneno K,
Tachibana S and Araki I: A case report of early idiopathic portal
hypertension. Pathol Res Pract. 197:759–767. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Iizuka N, Oka M, Yamada-Okabe H, et al:
Comparison of gene expression profiles between hepatitis B virus-
and hepatitis C virus-infected hepatocellular carcinoma by
oligonucleotide microarray data on the basis of a supervised
learning method. Cancer Res. 62:3939–3944. 2002.
|
|
16.
|
Wyllie AH: Apoptosis and carcinogenesis.
Eur J Cell Biol. 73:189–197. 1997.
|
|
17.
|
Dixon BP, Henry J, Siroky BJ, Chu A, Groen
PA and Bissler JJ: Cell cycle control and DNA damage response of
conditionally immortalized urothelial cells. PLoS One.
6:e165952011. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Luk SC, Siu SW, Lai CK, Wu YJ and Pang SF:
Cell cycle arrest by a natural product via G2/M checkpoint. Int J
Med Sci. 2:64–69. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Choi HJ, Fukui M and Zhu BT: Role of
cyclin B1/Cdc2 up-regulation in the development of mitotic
prometaphase arrest in human breast cancer cells treated with
nocodazole. PLoS One. 6:e243122011. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Gao Q, Srinivasan S, Boyer SN, Wazer DE
and Band V: The E6 oncoproteins of high-risk papillomaviruses bind
to a novel putative GAP protein, E6TP1, and target it for
degradation. Mol Cell Biol. 19:733–744. 1999.PubMed/NCBI
|
|
21.
|
Arai H, Ueno T, Tangoku A, et al:
Detection of amplified oncogenes by genome DNA microarrays in human
primary esophageal squamous cell carcinoma: comparison with
conventional comparative genomic hybridization analysis. Cancer
Genet Cytogenet. 146:16–21. 2003. View Article : Google Scholar
|
|
22.
|
Schneider G, Weber A, Zechner U, et al:
GADD45alpha is highly expressed in pancreatic ductal adenocarcinoma
cells and required for tumor cell viability. Int J Cancer.
118:2405–2411. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Parvaresch S, Yesilkaya T, Baer K,
Al-Hasani H and Klein HW: 14-3-3 binding to the IGF-1 receptor is
mediated by serine auto-phosphorylation. FEBS Lett. 532:357–362.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Nakamura K, Hongo A, Kodama J, Miyagi Y,
Yoshinouchi M and Kudo T: Down-regulation of the insulin-like
growth factor I receptor by antisense RNA can reverse the
transformed phenotype of human cervical cancer cell lines. Cancer
Res. 60:760–765. 2000.
|
|
25.
|
Spence SL, Dey BR, Terry C, Albert P,
Nissley P and Furlanetto RW: Interaction of 14-3-3 proteins with
the insulin-like growth factor I receptor (IGFIR): evidence for a
role of 14-3-3 proteins in IGFIR signaling. Biochem Biophys Res
Commun. 312:1060–1066. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Fu H, Subramanian RR and Masters SC:
14-3-3 proteins: structure, function, and regulation. Annu Rev
Pharmacol Toxicol. 40:617–647. 2000. View Article : Google Scholar
|
|
27.
|
Craparo A, Freund R and Gustafson TA:
14-3-3 (epsilon) interacts with the insulin-like growth factor I
receptor and insulin receptor substrate I in a
phosphoserine-dependent manner. J Biol Chem. 272:11663–11669. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Takashima T, Fujiwara Y, Higuchi K, et al:
PPAR-γ ligands inhibit growth of human esophageal adenocarcinoma
cells through induction of apoptosis, cell cycle arrest and
reduction of ornithine decarboxylase activity. Int J Oncol.
19:465–471. 2001.
|
|
29.
|
Yang WL and Frucht H: Activation of the
PPAR pathway induces apoptosis and COX-2 inhibition in HT-29 human
colon cancer cells. Carcinogenesis. 22:1379–1383. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Hall JM and McDonnell DP: The molecular
mechanisms underlying the proinflammatory actions of
thiazolidinediones in human macrophages. Mol Endocrinol.
21:1756–1768. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Chawla A, Barak Y, Nagy L, Liao D,
Tontonoz P and Evans RM: PPAR-gamma dependent and independent
effects on macrophage-gene expression in lipid metabolism and
inflammation. Nat Med. 7:48–52. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Marx N, Kehrle B, Kohlhammer K, et al:
PPAR activators as anti-inflammatory mediators in human T
lymphocytes: implications for atherosclerosis and
transplantation-associated arteriosclerosis. Circ Res. 90:703–710.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Shankaranarayanan P and Nigam S: IL-4
induces apoptosis in A549 lung adenocarcinoma cells: evidence for
the pivotal role of 15-hydroxyeicosatetraenoic acid binding to
activated peroxisome proliferator-activated receptor gamma
transcription factor. J Immunol. 170:887–894. 2003. View Article : Google Scholar
|
|
34.
|
Eichele K, Ramer R and Hinz B:
R(+)-methanandamide-induced apoptosis of human cervical carcinoma
cells involves a cyclo oxygenase-2-dependent pathway. Pharm Res.
26:346–355. 2009.
|