Introduction
Breast cancer is the most frequently diagnosed
cancer in the industrialized world (1). Like other tumors, breast
tumorigenesis involves the accumulation of multiple genetic and
epigenetic changes affecting mechanisms that control cell
proliferation, survival and apoptosis. The balance between
proliferation, differentiation and cell death is critical for
mammary gland development and maintenance (2). The ability of cancer cells to evade
apoptosis is one of the essential alterations in cell physiology
that dictate tumor growth and impact chemo- and radio-resistance
(3,4).
Prostate apoptosis response-4 (Par-4), also known as
PAWR, was first identified as upregulated in rat prostate cells
undergoing apoptosis (5). Par-4
encodes a ubiquitously expressed pro-apoptotic protein that is
localized to the cytoplasm of diverse normal tissues and cell
lines; it is present in both the cytoplasm and the nucleus of many
tumors and cancer cells (6–8).
Endogenous Par-4 itself does not cause cell death, although it is
essential for apoptosis induced by a variety of exogenous insults.
Ectopic Par-4 overexpression alone is sufficient to induce
apoptosis in most cancer cells, although not in normal or
immortalized cells (9).
Experimental evidence indicates that Par-4 plays an
important role in tumor cell survival and may be considered a
candidate for tumor cell selective therapy (10). This selective ability of Par-4 to
directly cause apoptosis in most cancer cells is associated with
its phosphorylation state and nuclear translocation (9,11).
Par-4 is phosphorylated and activated by protein kinase A (PKA),
which has elevated activity in cancer cells relative to normal
cells (11). Consistent with its
tumor suppressor function, Par-4 is downregulated in different
types of cancers, such as neuroblastoma (12), endometrial cancer (13), renal cell carcinoma (14), breast cancer (15). Par-4 knockout mice develop
spontaneous tumors in various tissues (16).
Par-4 acts in both intrinsic and extrinsic apoptotic
pathways. The mechanisms of Par-4 apoptosis induction involve
activation of the Fas-FADD-caspase-8 apoptotic pathway, including
the translocation of Fas/FasL to the plasma membrane and inhibition
of the NF-κB pro-survival pathway (10,17).
Studies indicate that Par-4 also functions as a transcriptional
co-repressor that interacts with WT1, leading to the inhibition of
BCL-2 transcriptional regulation (18). Recently, it was observed that Par-4
protein is also spontaneously secreted by normal and cancer cells
to the extracellular compartment (cell culture-conditioned medium
or circulating in serum) and is able to induce cancer cell-specific
apoptosis by interacting with the cell-surface receptor GRP78 and
activating the FADD/caspase-8/caspase-3 pathway (19). Moreover, transgenic mice that
express systemic Par-4 protein are resistant to the growth of
non-autochthonous tumors and intravenous injection of recombinant
Par-4 inhibits metastasis (20).
In cholangiocarcinoma cells, cell proliferation is associated with
reduced Par-4 expression as cells selectively silenced for Par-4
demonstrated a significant increase in cell proliferation and
reduced apoptosis (21). Recently,
we have demonstrated that the mitogenic factors E2 and insulin-like
growth factor-1 (IGF-1) lead to inhibition of Par-4 expression,
suggesting that this downregulation contributes to breast cancer
cell survival (22). To date,
there is no report in the literature on the role of Par-4 in
modulating breast cancer cell proliferation and apoptosis. The
present study aimed to evaluate the effects of increased or
decreased Par-4 levels on the proliferation and apoptosis of breast
cancer cells. Our results demonstrate for the first time that
increased Par-4 expression decreases cell proliferation rates in
MCF-7 cells compared with control cells and that specific Par-4
silencing by RNAi leads to increased MCF-7 cell proliferation. Our
findings also suggest that Par-4 expression increases the
sensitivity of MCF-7 cells to docetaxel.
Materials and methods
Cell culture
Human MCF-7 breast cancer cells were purchased from
the American Type Culture Collection (ATCC) and maintained in
RPMI-1640 medium (Sigma Chemical Co., St. Louis, MO, USA)
supplemented with 100 U/ml penicillin, 100 μg/ml
streptomycin, 0.25 μg/ml fungizon and 10% fetal bovine serum
(FBS) (Invitrogen Life Technologies, Carlsbad, CA, USA) in a
humidified atmosphere of 5% CO2 at 37°C. For assays
involving drug treatment, cells were grown in medium supplemented
with 5% FBS until they reached 60–70% confluence and the medium was
replaced with RPMI-1640 medium without phenol red and supplemented
with 5% FBS containing 5 or 100 nM docetaxel (Taxotere, Sigma
Chemical Co.) and cells were cultured for an additional 24 h.
Plasmid transfection and clone
selection
MCF-7 cells were stably transfected with plasmid
pcCMV6-XL6-Par-4 expressing a full length Par-4 cDNA, or empty
vector (pcCMV6-XL6-Neo) acquired from OriGene (Rockville, MD, USA).
Transfection assays were performed in 6-well plates using
TurboFectin 8.0 transfection reagent according to a standard
protocol (OriGene). Twenty-four hours after transfection, MCF-7
cells were selected with geneticin 3000 μg/ml (Gibco,
Gaithersburg, MD, USA). The selected clones were maintained in 800
μg/ml geneticin and screened for Par-4 mRNA expression by
real-time PCR, or for Par-4 protein by western blotting.
RNA interference to achieve PAR-4
knockdown
The duplex RNA oligonucleotides for Par-4 and
silencer-negative control (scramble) were purchased from
IDT® (TriFECTa™ kit, IA, USA; siSeq1,
HSC.RNAI.N002583.12.1; siSeq2, HSC.RNAI. N002583.12.2; and siSeq3,
HSC.RNAI.N002583.12.3). In brief, 1×105 cells were
seeded and cultured in 6-well plates until they reached 30–50%
confluence and transiently transfected with the specific Par-4
siRNA or scrambled oligonucleotides using TurboFectin 8.0 according
to the manufacturer’s instructions (OriGene). Cells were harvested
24–120 h after transfection and screened for Par-4 mRNA by
real-time PCR, or for Par-4 protein content by western blotting.
Twenty-four hours after transfection, cells were harvested and
plated in 96-well plates for cell proliferation assays using MTT,
or seeded in 6-well plates for flow cytometric apoptosis
assays.
Quantitative RT-PCR
For quantitative RT-PCR analysis, cells were
harvested and total RNA was extracted using the acid guanidinium
thiocyanate-phenol-chloroform method. cDNA synthesis was performed
using the High Capacity cDNA Archive kit (Applied Biosystems,
Warrington, UK). Real-time PCR (qPCR) was performed using the Power
SYBR Green kit (Applied Biosystems, Foster City, CA, USA) following
the manufacturer’s recommendations. qPCR reactions were processed
in the GeneAmp 7500 Sequence Detection System (Applied Biosystems)
under the following conditions: 50°C for 2 min and 95°C for 10 min,
followed by 40 cycles of 95°C for 15 sec and 55°C for 1 min. The
PCR primers used were: for PAR-4, forward
5′-CCAGAGAAGGGCAAGAGCTCGG-3′; reverse 5′-ATTGCATCTTCTCGTTTCCGC-3′;
GADPH, forward 5′-CCTCCAAAATCAAGTGGGGCG-3′; reverse
5′-GGGGCAGAGATGATGACCCTT-3′; BCL-2, forward
5′-CGACTTCGCCGAGATGTCCAG-3′; reverse 5′-CCGTCC CTGAAGAGCTCCTCC-3′;
BAX, forward 5′-GGCCGG GTTGTCGCCCTTTTC-3′; reverse,
5′-GTCCAGCCCATG ATGGTTCTG-3′; BID, forward
5′-AACTCCTGTGACCAC AACATG-3′; reverse 5′-CAGGAAAGCATCTGGTAA GAA-3′;
BECLIN forward, 5′-ACTTTCCAGAGCTACAAC ATG-3′; reverse
5′-GTCCATGGGGTTAAGAATCAA-3′. Relative expression was calculated by
2−ΔΔCT (CT = fluorescence threshold value; ΔCT = CT of
the target gene − CT of the reference gene (GADPH); ΔΔCT = ΔCT of
the target sample − ΔCT of the reference sample). The average value
of the control cells served as the reference sample. The results
were expressed as n-fold differences in mRNA expression relative to
expression of GAPDH and the reference sample.
Western blot analysis
Cells were harvested and total cell lysates were
prepared in lysis buffer (50 mM Na pyrophosphate, 50 mM NaF, 5 mM
NaCl, 5 mM PMSF, 100 mM Na3VO4), followed by
centrifugation at 13,000 rpm for 15 min at 4°C. Thirty micrograms
of protein lysate were separated on a 10% SDS-PAGE gel and blotted
onto nitrocellulose membranes (Pierce Biotechnology, Rockford, IL,
USA). Blots were incubated with anti-Par-4 monoclonal antibody
(Abcam, Cambridge, MA, USA), anti-p-ERK, anti-ERK (Santa Cruz
Biotechnology, Santa Cruz, CA, USA) and anti-β-actin mouse
monoclonal antibody (Millipore, Temecula, CA, USA) for 2 h at room
temperature. Membranes were washed in TBS-T (25 mM Tris, 125 mM
NaCl and 0.1% Tween-20) and incubated with appropriate
peroxidase-conjugated secondary IgG antibody for 2 h at room
temperature. Incubations were performed in 5% skim milk diluted in
TBS-T. Specific proteins were detected using an enhanced
chemiluminescence system (ECL™ Western Blotting
Detection Reagents, GE Healthcare, Buckinghamshire, UK) and exposed
to Hyperfilm ECL film (GE Healthcare).
Cell proliferation assay
Cell proliferation and viability were measured using
a 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT,
Molecular Probes, Invitrogen) assay following the manufacturer’s
instructions. Cells were seeded in 96-well plates (1×104
cells/well) and maintained in RPMI-1640 medium without phenol red
supplemented with 5 or 0.5% FBS. Cell growth was assessed at 0, 48,
72, 96 and 144 h. At the end of incubation, the absorbance was
measured at 570 nm using the Biotrak II Plate reader (Amersham
Biosciences, Cambridge, UK).
Imunocytochemistry
MCF-7 cells were cultured in 8-well chamber slides
in RPMI-1640 medium without phenol red and supplemented with 5% FBS
or 5% dextran-coated charcoal-treated FBS (stripped serum, ST) for
48 h before treatment. For immunocytochemistry, cells were fixed
using 4% paraformaldehyde and permeabilized with 0.5% Triton X-100.
Next, cells were incubated with primary mouse monoclonal anti-Par-4
antibody 1:50 (Santa Cruz, Biotechnology; catalog sc-1666) and
rabbit polyclonal anti-tubulin antibody 1:300 (OriGene), followed
by the appropriate secondary antibodies conjugated with Alexa Fluor
546 and Alexa Fluor 488 1:300 (Invitrogen, OR, USA). Nuclei were
counterstained with Hoechst 33342 1:3000 (Invitrogen). Cells were
visualized with Carl Zeiss LSM 510 Meta (Oberkochen, Germany) laser
scanning confocal microscopy. All immunofluorescence images were
recorded at magnification ×20 and ×63.
Cell cycle analysis
Floating and adherent cells were collected, washed
once with PBS, fixed with 70% ice-cold ethanol and stored at −20°C
until analysis by flow cytometry. The fixed cells were washed twice
with PBS, resuspended in PBS containing 200 μg/ml RNase A,
0.1% Triton X-100 and 20 μg/ ml propidium iodide (PI) and
immediately analyzed by fluorescence-activated cell sorting using
the Becton-Dickinson FACSort flow cytometer (BD Biosciences, NJ,
USA). The cell cycle analysis program CellQuest (BD Biosciences)
was used to determine the percentage of cells at different stages
of the cycle (G0-G1, S-G2/M) and the percentage of cell death using
sub-G1 cells.
Cell death analysis: Annexin V and
acridine orange staining
Apoptosis was evaluated in MCF-7-pcPar-4 and
MCF-7-pcNeo cells before and after treatment with 5 or 100 nM
docetaxel for 24 h using the Annexin V-FITC Apoptosis Detection Kit
I (BD Pharmigen, CA, USA), following the manufacturer’s
instructions. Cytometric analyses were performed using the
Attune® Acoustic Focusing Cytometer (Life Technologies,
Foster City, CA, USA). Apoptosis was also analyzed using a
double-fluorescence staining technique with Hoechst 33342 and
acridine orange (AO). Briefly, transfected MCF-7 cells treated with
docetaxel (5 or 100 nM) for 24 h were co-stained with AO (8.5
μg/ml) and with nuclear dye Hoechst 33342 (5 μg/ml)
for 10 min in the dark and examined under magnification ×20 and ×63
using a fluorescence microscope. Viewed by fluorescence microscopy,
viable cells appear to have an intact and bright green nucleus,
while apoptotic cells exhibit a bright green nucleus in which
chromatin condensation is observed as dense green areas. Apoptotic
cells were calculated as the ratio of apoptotic cells (with
characteristic apoptotic morphology) to total cells. At least 10
fields were counted for each treatment, using the ImageJ Launcher
software (Image Processing and Analysis in Java, National Institute
of Health, USA).
Statistical analysis
Statistical analyses were performed by Student’s and
Mann-Whitney tests using Statistical Package for the Social
Sciences 20.0 (SPSS Inc., Chicago, IL, USA). P-values were
considered statistically significant at P<0.05. All data are
presented as the mean ± SD of three or more replicate
experiments.
Results
Par-4 expression, cell proliferation
rates and ERK phosphorylation
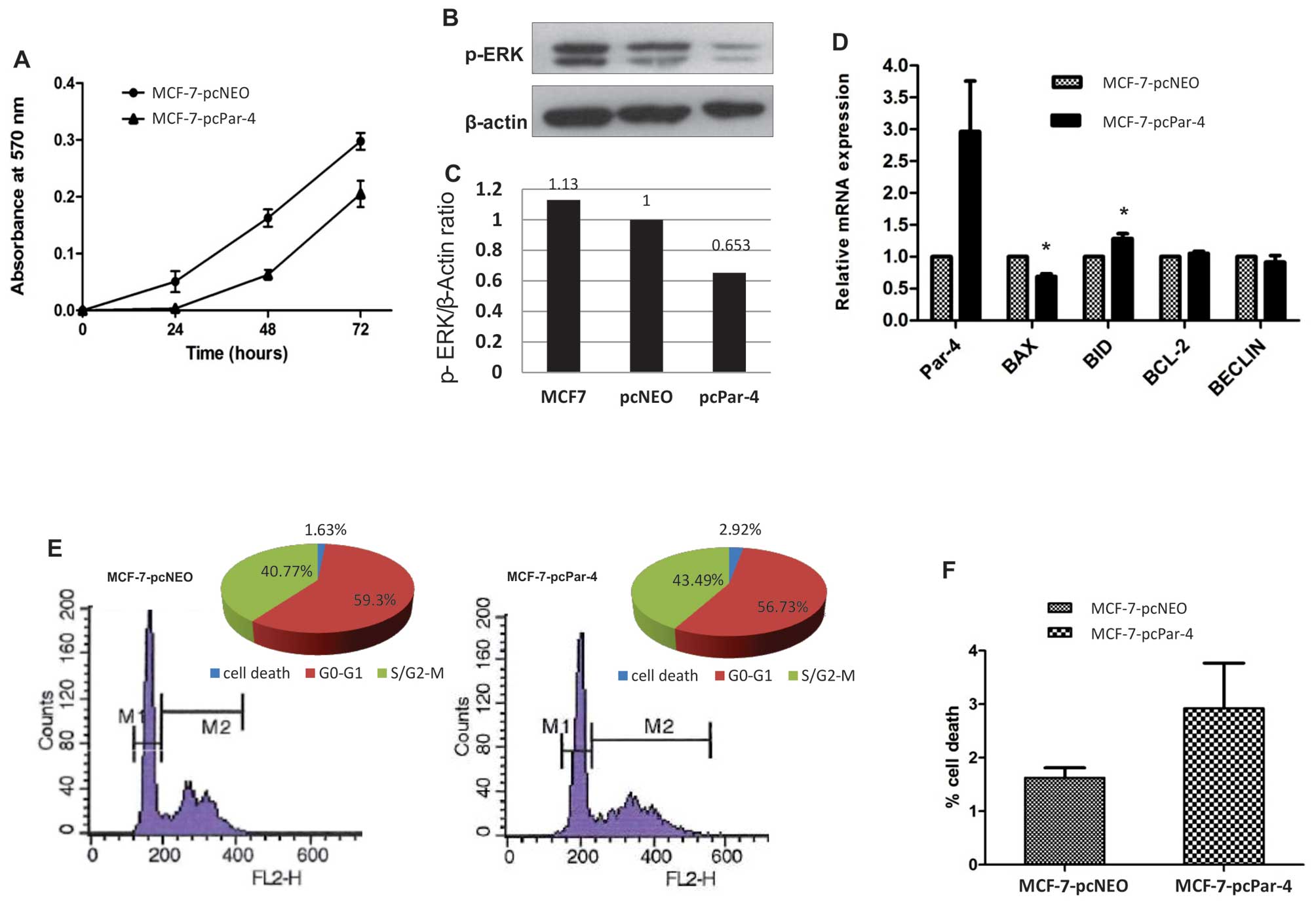
To evaluate the possible effects of Par-4 expression
on cell proliferation, MCF-7 cells were transfected with expression
vector pCMV6-XL6-Par-4 (MCF-7-pcPar-4) or with the empty vector
pCMV6-XL6-Neo (MCF-7-pcNeo). Clones were selected with geneticin
and were characterized by qPCR and western blotting. MCF-7-pcPar-4
tranfectants exhibiting increased Par-4 protein expression compared
with MCF-7-pcNeo transfectant cells showed no significant
morphological differences. Cell proliferation was significantly
inhibited in MCF-7 cells expressing increased Par-4 compared with
control MCF-7-pcNeo transfectants (Fig. 1A). Because the Ras/Raf/MEK/ERK
pathway is a central signaling cascade involved in cell
proliferation (23), we evaluated
whether Par-4 might affects the status of ERK phosphorylation.
Compared with control cells (MCF-7 or MCF-7-pcNeo), MCF-7-pcPar-4
cells with increased Par-4 expression exhibited significantly
reduced ERK phosphorylation (Fig. 1B
and C), suggesting that the proliferation restraint promoted by
Par-4 involves inhibition of the Ras/Raf/MEK/ERK pathway.
We also examined whether increased Par-4 expression
affected the expression profile of genes involved in apoptosis
(BAX, BID, BCL-2) and autophagy (BECLIN). MCF-7-pcPar-4 cells
exhibited slightly reduced BAX expression and marginally increased
BID expression relative to the control cells (Fig. 1D). No detectable alterations were
observed for BCL-2 and BECLIN transcripts expression (Fig. 1D).
Par-4 overexpression did not affect the cell cycle
profile (Fig. 1E). However, MCF-7
cells overexpressing Par-4 display a 1.8-fold greater proportion of
cells in the sub-G1 population (2.92%) compared with control cells
(1.63%) (Fig. 1F).
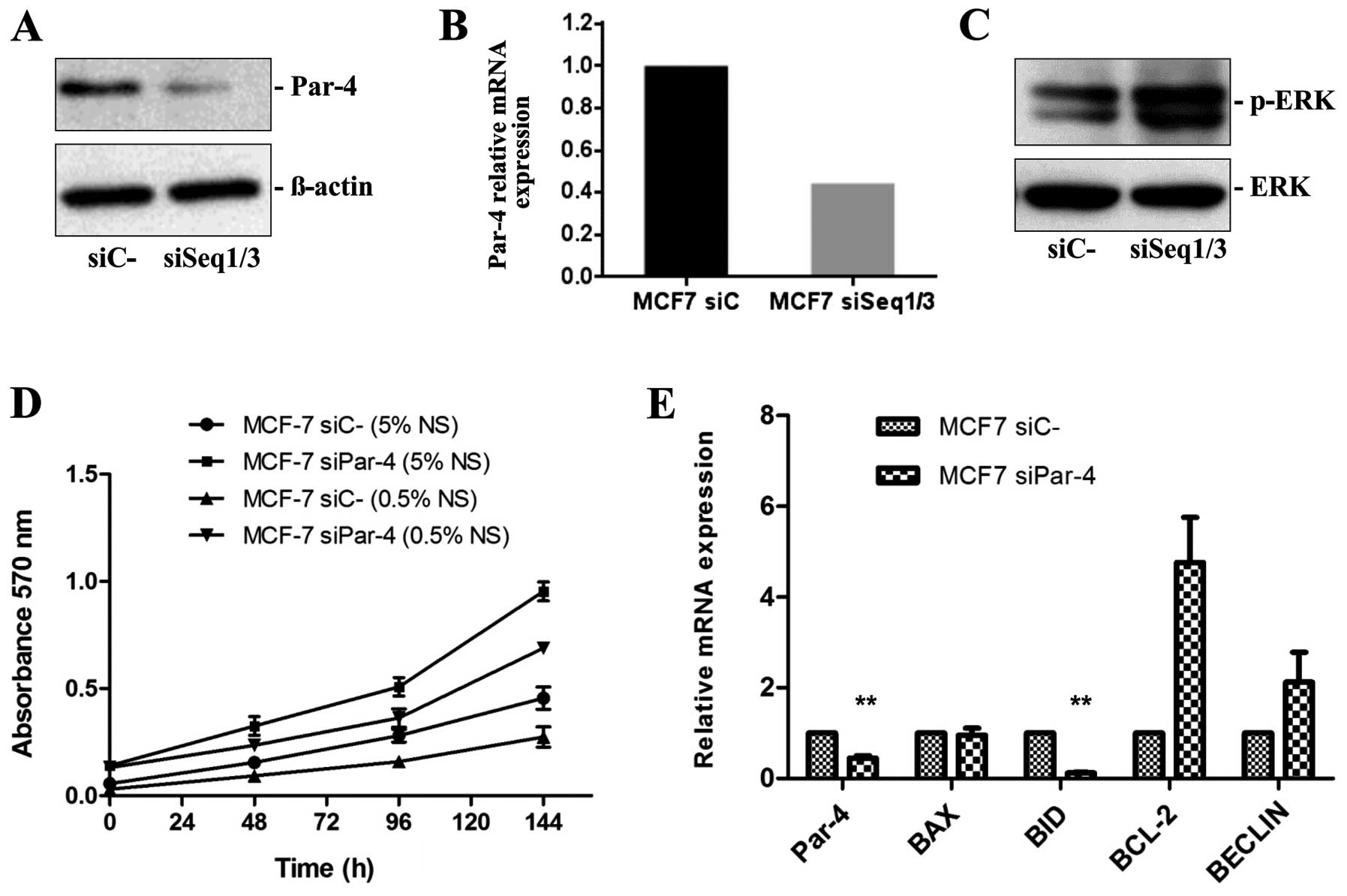
Selective silencing of Par-4 expression,
cell proliferation and ERK phosphorylation
To better characterize the effects of Par-4 on
breast cancer cell proliferation, MCF-7 cells were transiently
transfected with specific siRNA duplexes. Three different siRNA
duplexes synthesized by TriFECTA were tested for Par-4 knockdown
efficiency. Two Par-4 siRNAs (named siSeq1 and siSeq3) efficiently
reduced Par-4 protein and mRNA expression in MCF-7 breast cancer
cells compared with the control scramble siRNAs (Fig. 2A and B).
The following experiments were performed with MCF-7
cells transfected with a mixture of siSeq1 and siSeq3 siRNAs and
control cells transfected with control siRNA. Western blot analysis
revealed that reduced Par-4 expression leads to increased ERK
phosphorylation (Fig. 2C), which
is in agreement with the increased proliferation rate observed in
MTT assays. MCF-7 cells displaying reduced Par-4 expression after
siRNA knockdown exhibited higher proliferation rates, both in
normal serum (5% SN) and when serum-deprived (0.5% NS), compared
with MCF-7 cells transfected with control siRNA (Fig. 2D).
We also evaluated the effect of Par-4 knockdown on
expression of the pro-apoptotic genes BAX and BID, the
anti-apoptotic gene BCL-2 and the pro-autophagic gene BECLIN.
Compared with MCF-7 cells transfected with scrambled siRNA, we
observed significantly decreased BID mRNA expression (8.33-fold,
P≤0.003) and significantly increased BCL-2 protein expression
(4.76-fold) in MCF-7 cells transfected with Par-4-specific siRNAs
(Fig. 2E). MCF-7 cells with
reduced Par-4 expression exhibited increased BECLIN mRNA expression
(2.1-fold) relative to control cells (Fig. 2E).
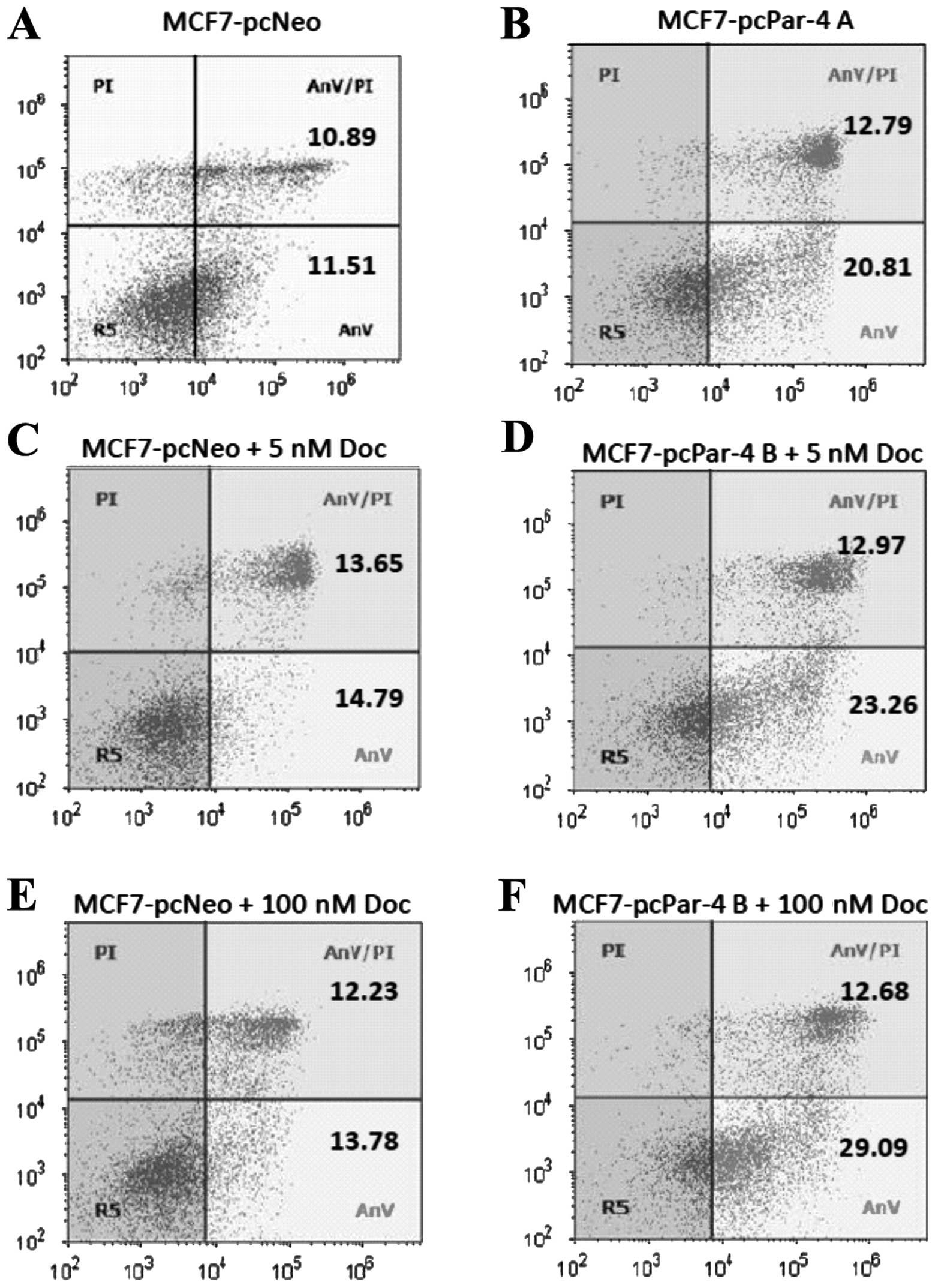
Par-4 expression, apoptosis and
sensitivity to docetaxel
Par-4 overexpression has been associated with
increased sensitivity to apoptotic stimuli in different cell types
(7). To evaluate the effect of
Par-4 expression on apoptosis induction and drug sensitivity,
MCF-7-pcPar-4 and MCF-7-pcNeo cells were evaluated by flow
cytometry using PI/Annexin V-FITC double staining before and after
docetaxel treatment (5 or 100 nM for 24 h). MCF-7-pcPar-4 cells
with increased Par-4 expression exhibited a greater proportion of
early apoptotic cells (stained with Annexin V-FITC) compared with
control cells, in medium containing vehicle (20.81 versus 11.51%)
(Fig. 3A and B), in cells treated
with 5 nM docetaxel (23.26 versus 14.79%) (Fig. 3C and D) and in cells treated with
100 nM docetaxel (29.09 versus 13.78%) (Fig. 3E and F). After treatment with 100
nM docetaxel, 41.77% of cells with increased Par-4 underwent
apoptosis compared with 26.01% of MCF-7 pcNeo cells.
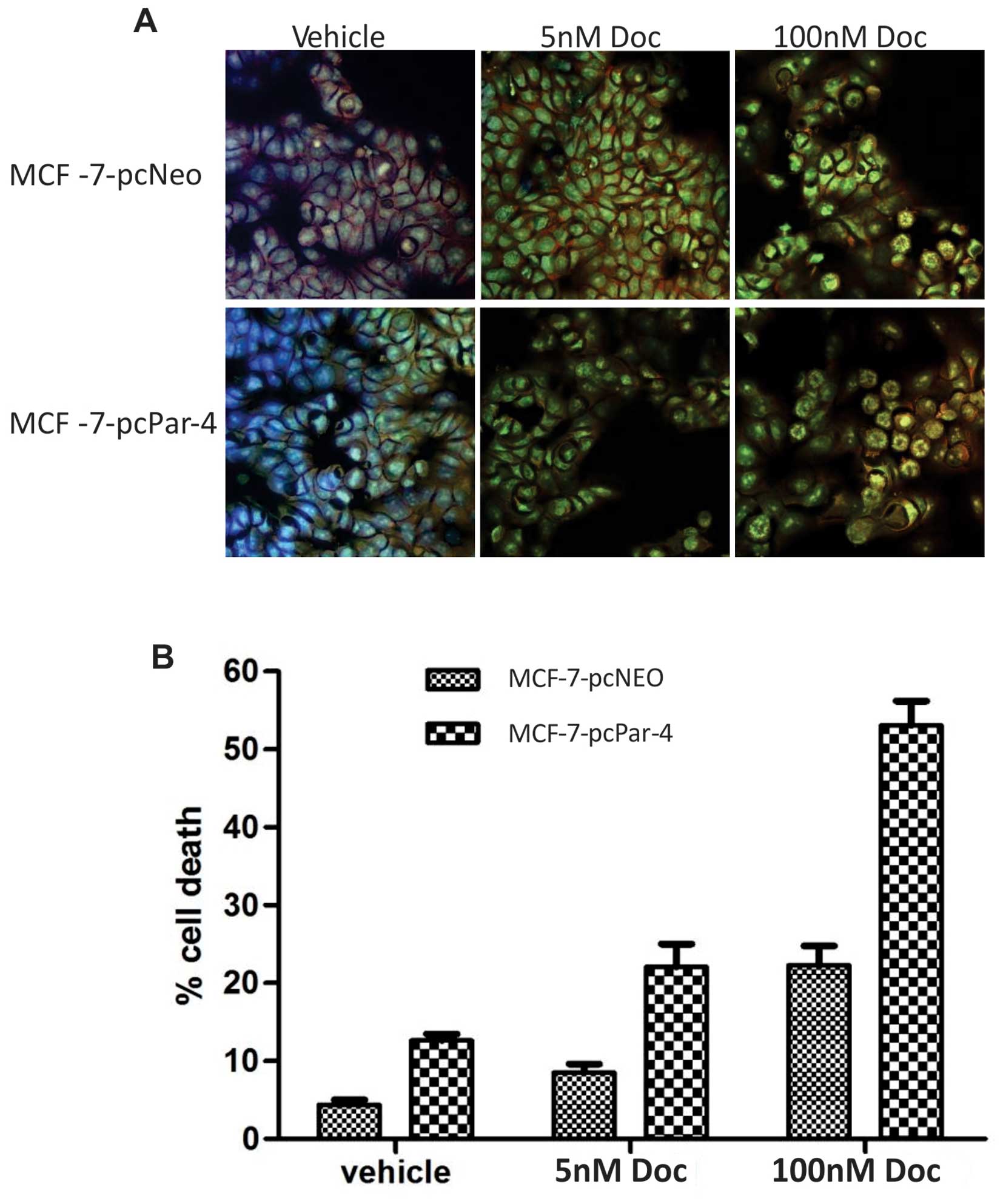
A semi-quantitative analysis of the number of
apoptotic cells was also performed by AO/Hoechst double labeling.
MCF-7-pcPar-4 cells displayed a higher proportion of AO/ Hoechst
double-stained cells exhibiting condensed chromatin, nuclear
fragmentation, nucleolar disappearance, increased nuclear
fluorescence and the appearance of granular apoptotic bodies
compared with control cells (MCF-7-pcNeo) with vehicle (12.61
versus 4.35%, P<0.00001) and in low and high concentrations of
docetaxel (5 nM: 22.09 versus 8.54%, P=0.0004; 100 nM: 53.06 versus
22.28%, P<0.0001) (Fig. 4).
Discussion
The present study provides evidence that Par-4 plays
an important role in cell proliferation and survival in breast
cancer cells. Our findings indicate that elevated levels of
intracellular Par-4 can sensitize MCF-7 cells to apoptosis and
growth inhibition by docetaxel, which is a fundamental and
effective chemotherapeutic agent against primary and advanced
breast cancer, suggesting that intracellular Par-4 are involved in
the pro-apoptotic and growth inhibitory action of docetaxel in
breast cancer cells.
Par-4 acts in both intrinsic and extrinsic apoptotic
pathways. PAR-4 induces apoptosis as a result of its ability to
activate the Fas-FasL-FADD-caspase-8 pathway; by inhibiting the
NF-κB survival pathway, which requires PKA-mediated PAR-4
phosphorylation at residue T155; and by downregulating Bcl-2
expression (11,17,24–26).
Chan et al (27) reported
that Par-4 mRNA and protein levels rapidly and progressively
increase after trophic factor withdrawal in cultured rat
hippocampal neurons and that Par-4 acts early in apoptosis (i.e.,
before mitochondrial dysfunction, caspase activation and nuclear
changes). Previous results from our group also showed that
withdrawal of estrogens and growth factors from the serum led to
significantly increased Par-4 expression compared with the
expression observed in MCF-7 cells maintained in media supplemented
with 5% FBS (22). Furthermore,
our group has demonstrated that Par-4 expression is negatively
regulated by IGF-1 and the hormone 17β-estradiol (22). These findings are in agreement with
results from another study, in which cholangiocarcinoma cells
cultured in the absence of serum exhibited Par-4 protein increased
expression associated with a significant increase in BAX protein
(21).
In the present study, we used RNAi to reduce Par-4
expression and the expression vector pCMV6-XL6-PAR4 to increase
Par-4 expression in MCF-7 cells. MCF-7 cells with reduced Par-4
expression exhibited faster proliferation rates relative to control
cells, as well as an increase in phosphorylated extracellular
signal-regulated kinases-1,2 (p-ERK) protein expression.
Conversely, MCF-7 cells with increased Par-4 expression exhibited
slower proliferation and decreased p-ERK protein expression
compared with control cells. ERK kinase participates in the
Ras/Raf/MEK/ERK signaling pathway, which is activated by mitogens
and growth factors (28). ERK can
phosphorylate, directly or indirectly, many transcription factors
involved in cell cycle control, especially in the activation of
proliferation and cell survival (including Ets-1, c-jun, c-myc and
NF-κB) (23,29). In addition, components of the
ERK-mediated pathway are known to be mutated or aberrantly
expressed in human tumors, including breast cancer (23). These results indicate, for the
first time, a possible role for Par-4 in the proliferation of
breast cancer cells through the ERK1/2 pathway. The involvement of
Par-4 in proliferation has also been demonstrated in
cholangiocarcinoma cells (21).
Specific Par-4 silencing by siRNA-activated cholangiocarcinoma cell
line proliferation demonstrated a significant increase in
proliferating cellular nuclear antigen protein expression,
indicative of induced cell proliferation.
The primary function of Par-4 is related to its
ability to increase cell sensitivity to apoptotic stimuli or direct
induction of apoptosis (30,31).
When we analyzed the expression pattern of apoptosis-related genes,
we found that Par-4 downregulation led to increased BCL-2 and a
significant reduction in BID transcripts. A member of the sub-class
BH3-only, thus possessing only a BCL2 homology-3 (BH3) domain of
the four BCL2 family domains (BH1, BH2, BH3 and BH4), Bid protein
is a key protein in the apoptotic process, because it connects the
extrinsic pathway activated by pro-apoptotic and pro-inflammatory
cytokines to the intrinsic mitochondrial pathway (32,33).
When cleaved and activated by caspase-8, the Bid protein can in
turn cleave downstream caspases, such as caspase-3 and -7, to
execute cell death; alternatively, activated Bid protein interacts
with Bax and Bak, inducing permeabilization of the mitochondrial
membrane to release cytochrome c. Thus, Bid acts by
amplifying the apoptotic signal by also activating the
mitochondrial pathway (34,35).
Some reports in the literature have shown a relationship between
Par-4 and Bid protein. A study with human prostate adenocarcinoma
cells (DU145) demonstrated that cranberry extract increases Par-4
expression, which in turn activates caspase-8, leading to increased
cleavage of Bid protein with consequent induction of apoptosis
(36). Par-4 expression increases
cleavage of caspase-8 and Bid, which translocates to mitochondria
and induces the release of cytochrome c (37).
Interestingly, we observed a significant increase in
the antiapoptotic transcript BCL-2 in MCF-7 breast cancer cells
with reduced Par-4 expression, corroborating the literature
regarding the inverse correlation between Par-4 and BCL-2
expression. This result suggests that silencing Par-4 protects
cells from apoptosis. Qiu et al (25) showed that Par-4 expression leads to
decreased Bcl-2 expression in NIH 3T3 fibroblasts and in PC-3
prostate cancer cells. Qiu et al also demonstrated that
transient Bcl-2 expression prevents Par-4-dependent apoptosis in
fibroblasts cultured in the absence of growth factors, indicating
that the decrease in Bcl-2 is required for Par-4-induced
apoptosis.
Overexpression of Par-4 in neoplastic lymphocytes
decreases Bcl-2 expression, while Bax levels are not modified even
with the addition of chemotherapeutic agents (26). Increased Par-4 expression
sensitizes PC-3 prostate cancer cells to radiation-induced
apoptosis due to inhibition of NF-κB activity, with a consequent
decrease in Bcl-2 expression (38). However, it was observed that
increasing Par-4 did not alter the expression of pro-apoptotic
protein BAX. In this study, we also observed no significant
modulation of BAX by Par-4 expression in MCF-7 cells.
Several reports have provided evidence that Par-4
acts to increase sensitivity to different chemotherapeutic drugs or
chemopreventive agents. In this study, we also sought to
investigate whether Par-4 might be a modulator of antitumor
therapy. We observed for the first time that Par-4 sensitized MCF-7
cells to docetaxel treatment.
Docetaxel (Taxotere) and other taxanes, such as
paclitaxel, are anti-microtubule drugs used as a therapeutic
regimen for patients with breast, lung and ovarian cancer (39). Treatment with docetaxel inhibits
key cellular events (e.g., mitotic division, secretion and
transport) and triggers apoptosis and other forms of cell death
(e.g., mitotic catastrophe, senescence and lytic necrosis)
(40). Docetaxel may act by two
mechanisms depending on its concentration in MCF-7 cells: a low
drug concentration (2–4 nM) induces aberrant mitosis followed by
delayed necrosis, while a high concentration (100 nM) induces
prolonged cell cycle arrest (mitotic arrest) followed by apoptosis
(41). Using AO staining, we
observed a greater percentage of cells with morphological
characteristics of apoptosis (condensed chromatin, nuclear
fragmentation, nucleolar disappearance and the appearance of
granular apoptotic bodies) in both low and high concentrations of
docetaxel in MCF-7 cells with increased Par-4 expression.
In colon cancer cells, Par-4 overexpression led to
increased apoptosis in the presence of 5-fluorouracil chemotherapy
by inhibiting NF-κB activity (42). The increase in Par-4 also increased
the sensitivity of Bcr-Abl-positive myeloid cells to the
chemotherapeutic imatinib (tyrosine kinase inhibitor STI571) and
the histone deacetylase inhibitor LAQ824 (43). Human renal cancer cells (Caki
cells) overexpressing Par-4 were sensitized to apoptosis by
inductor TRAIL and drugs that induce endoplasmic reticulum stress
(i.e., thapsigargin, tunicamycin and etoposide) associated with
decreased levels of XIAP protein and caspase activation (44). Par-4 has been shown to be secreted
by both normal and cancer cells, however, it induces apoptosis only
in cancer cell. Extracellular Par-4 binds to cell surface receptor
GRP78 to induce apoptosis by triggering endoplasmic reticulum
(ER)-stress and the caspase-8/-3 pathway (19). This apoptotic selectivity of Par-4
is due to low levels of GRP78 and lack of a robust ER-stress
response in normal or immortalized cells. Our findings indicate,
for the first time, that apoptosis induction by docetaxel in breast
cancer cells is modulated by Par-4 expression. However, additional
studies are needed to better clarify the mechanism of Par-4 and
ER-stress pathway activation after treatment with docetaxel.
Taken together, these findings suggest that Par-4
expression increases the sensitivity of breast cancer cells to
chemotherapeutic agents and co-parallel elevation of Par-4 may be
an effective strategy to increase the efficacy of docetaxel
treatment in breast cancer.
Our findings demonstrate for the first time that
Par-4 modulates cell proliferation and survival in MCF-7 breast
cancer cells. We show that PAR-4 inhibits MCF-7 cell growth,
possibly through the inhibition of ERK-driven proliferation. Our
results also show that Par-4 expression increases the rate of early
apoptosis and increases the sensitivity of MCF-7 cells to docetaxel
treatment. Our findings have translational relevance as they
suggest that co-parallel elevation of Par-4 may increase the
efficacy of breast cancer therapy.
Acknowledgements
This study was supported by FAPESP -
Fundação de Amparo a Pesquisa do Estado de São Paulo
(2010/16543-5), and partly by CNPq (Conselho Nacional de
Desenvolvimento Científico e Tecnológico; 305408/2009-7).
References
|
1.
|
Macciò A and Madeddu C: Obesity,
inflammation and post-menopausal breast cancer: therapeutic
implications. TSWJ. 11:2020–2036. 2011.
|
|
2.
|
Parton M, Dowsett M and Smith I: Studies
of apoptosis in breast cancer. BMJ. 322:1528–1532. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Makin G and Hickman JA: Apoptosis and
cancer chemotherapy. Cell Tissue Res. 301:143–152. 2000. View Article : Google Scholar
|
|
5.
|
Sells SF, Wood DP Jr, Joshi-Barve SS,
Muthukumar S, Jacob RJ, Crist SA, Humphreys S and Rangnekar VM:
Commonality of the gene programs induced by effectors of apoptosis
in androgen-dependent and -independent prostate cells. Cell Growth
Differ. 5:457–466. 1994.
|
|
6.
|
Sells SF, Han SS, Muthukkumar S, Maddiwar
N, Johnstone R, Boghaert E, Gillis D, Liu G, Nair P, Monnig S,
Collini P, Mattson MP, Sukhatme VP, Zimmer SG, Wood DP Jr,
McRoberts JW, Shi Y and Rangnekar VM: Expression and function of
the leucine zipper protein Par-4 in apoptosis. Mol Cell Biol.
17:3823–3832. 1997.PubMed/NCBI
|
|
7.
|
El-Guendy N and Rangnekar VM: Apoptosis by
Par-4 in cancer and neurodegenerative diseases. Exp Cell Res.
283:51–66. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Goswami A, Ranganathan P and Rangnekar VM:
The phosphoinositide 3-kinase/Akt1/Par-4 axis: a cancer-selective
therapeutic target. Cancer Res. 66:2889–2892. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
El-Guendy N, Zhao Y, Gurumurthy S,
Burikhanov R and Rangnekar VM: Identification of a unique core
domain of par-4 sufficient for selective apoptosis induction in
cancer cells. Mol Cell Biol. 23:5516–5525. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Zhao Y, Burikhanov R, Qiu S, Lele SM,
Jennings CD, Bondada S, Bryson S and Rangnekar VM: Cancer
resistance in transgenic mice expressing the SAC module of Par-4.
Cancer Res. 67:9276–9285. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Gurumurthy S, Goswami A, Vasudevan KM and
Rangnekar VM: Phosphorylation of Par-4 by protein kinase A is
critical for apoptosis. Mol Cell Biol. 25:1146–1161. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Kögel D, Reimertz C, Mech P, Poppe M,
Frühwald MC, Engemann H, Scheidtmann KH and Prehn JH: Dlk/ZIP
kinase-induced apoptosis in human medulloblastoma cells:
requirement of the mitochondrial apoptosis pathway. Br J Cancer.
85:1801–1808. 2001.PubMed/NCBI
|
|
13.
|
Moreno-Bueno G, Fernandez-Marcos PJ,
Collado M, Tendero MJ, Rodriguez-Pinilla SM, Garcia-Cao I,
Hardisson D, Diaz-Meco MT, Moscat J, Serrano M and Palacios J:
Inactivation of the candidate tumor suppressor par-4 in endometrial
cancer. Cancer Res. 67:1927–1934. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Cook J, Krishnan S, Ananth S, Sells SF,
Shi Y, Walther MM, Linehan WM, Sukhatme VP, Weinstein MH and
Rangnekar VM: Decreased expression of the pro-apoptotic protein
Par-4 in renal cell carcinoma. Oncogene. 18:1205–1208. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Nagai MA, Gerhard R, Salaorni S, Fregnani
JH, Nonogaki S, Netto MM and Soares FA: Down-regulation of the
candidate tumor suppressor gene PAR-4 is associated with poor
prognosis in breast cancer. Int J Oncol. 37:41–49. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
García-Cao I, Duran A, Collado M,
Carrascosa MJ, Martín-Caballero J, Flores JM, Diaz-Meco MT, Moscat
J and Serrano M: Tumour-suppression activity of the proapoptotic
regulator Par4. EMBO Rep. 6:577–583. 2005.PubMed/NCBI
|
|
17.
|
Chakraborty M, Qiu SG, Vasudevan KM and
Rangnekar VM: Par-4 drives trafficking and activation of Fas and
Fasl to induce prostate cancer cell apoptosis and tumor regression.
Cancer Res. 61:7255–7263. 2001.PubMed/NCBI
|
|
18.
|
Cheema SK, Mishra SK, Rangnekar VM, Tari
AM, Kumar R and Lopez-Berestein G: Par-4 transcriptionally
regulates Bcl-2 through a WT1-binding site on the bcl-2 promoter. J
Biol Chem. 278:19995–20005. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Burikhanov R, Zhao Y, Goswami A, Qiu S,
Schwarze SR and Rangnekar VM: The tumor suppressor Par-4 activates
an extrinsic pathway for apoptosis. Cell. 138:377–388. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Zhao Y, Burikhanov R, Brandon J, Qiu S,
Shelton BJ, Spear B, Bondada S, Bryson S and Rangnekar VM: Systemic
Par-4 inhibits non-autochthonous tumor growth. Cancer Biol Ther.
12:152–157. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Franchitto A, Torrice A, Semeraro R,
Napoli C, Nuzzo G, Giuliante F, Alpini G, Carpino G, Berloco PB,
Izzo L, Bolognese A, Onori P, Renzi A, Cantafora A, Gaudio E and
Alvaro D: Prostate apoptosis response-4 is expressed in normal
cholangiocytes is down-regulated in human cholangiocarcinoma and
promotes apoptosis of neoplastic cholangiocytes when induced
pharmacologically. Am J Pathol. 177:1779–1790. 2010. View Article : Google Scholar
|
|
22.
|
Casolari DA, Pereira MC, de Bessa Garcia
SA and Nagai MA: Insulin-like growth factor-1 and 17β-estradiol
down-regulate prostate apoptosis response-4 expression in MCF-7
breast cancer cells. Int J Mol Med. 28:337–342. 2011.
|
|
23.
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M,
Tafuri A, Stivala F, Libra M, Basecke J, Evangelisti C, Martelli AM
and Franklin RA: Roles of the Raf/MEK/ERK pathway in cell growth,
malignant transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Johnstone RW, See RH, Sells SF, Wang J,
Muthukkumar S, Englert C, Haber DA, Licht JD, Sugrue SP, Roberts T,
Rangnekar VM and Shi Y: A novel repressor, par-4 modulates
transcription and growth suppression functions of the Wilms’ tumor
suppressor WT1. Mol Cell Biol. 16:6945–6956. 1996.PubMed/NCBI
|
|
25.
|
Qiu G, Ahmed M, Sells SF, Mohiuddin M,
Weinstein MH and Rangnekar VM: Mutually exclusive expression
patterns of Bcl-2 and Par-4 in human prostate tumors consistent
with down-regulation of Bcl-2 by Par-4. Oncogene. 18:623–631. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Boehrer S, Chow KU, Beske F,
Kukoc-Zivojnov N, Puccetti E, Ruthardt M, Baum C, Rangnekar VM,
Hoelzer D, Mitrou PS and Weidmann E: In lymphatic cells par-4
sensitizes to apoptosis by down-regulating bcl-2 and promoting
disruption of mitochondrial membrane potential and caspase
activation. Cancer Res. 62:1768–1775. 2002.PubMed/NCBI
|
|
27.
|
Chan SL, Tammariello SP, Estus S and
Mattson MP: Prostate apoptosis response-4 mediates trophic factor
withdrawal-induced apoptosis of hippocampal neurons: actions prior
to mitochondrial dysfunction and caspase activation. J Neurochem.
73:502–512. 1999. View Article : Google Scholar
|
|
28.
|
Pearson G, Robinson F, Beers Gibson T, Xu
BE, Karandikar M, Berman K and Cobb MH: Mitogen-activated protein
(MAP) kinase pathways: regulation and physiological functions.
Endocr Rev. 22:153–183. 2001.PubMed/NCBI
|
|
29.
|
Nakano H, Shindo M, Sakon S, Nishinaka S,
Mihara M, Yagita H and Okumura K: Differential regulation of
IkappaB kinase alpha and beta by two upstream kinases,
NF-kappaB-inducing kinase and mitogen-activated protein kinase/ERK
kinase kinase-1. Proc Natl Acad Sci USA. 95:3537–3542. 1998.
View Article : Google Scholar
|
|
30.
|
Gurumurthy S and Rangnekar VM: Par-4
inducible apoptosis in prostate cancer cells. J Cell Biochem.
91:504–512. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Shrestha-Bhattarai T and Rangnekar VM:
Cancer-selective apoptotic effects of extracellular and
intracellular Par-4. Oncogene. 29:3873–3880. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Degterev A and Yuan J: Expansion and
evolution of cell death programmes. Nat Rev Mol Cell Biol.
9:378–390. 2008. View
Article : Google Scholar
|
|
33.
|
Youle RJ and Strasser A: The BCL-2 protein
family: opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Simstein R, Burow M, Parker A, Weldon C
and Beckman B: Apoptosis, chemoresistance and breast cancer:
insights from the MCF-7 cell model system. Exp Biol Med.
228:995–1003. 2003.PubMed/NCBI
|
|
35.
|
Galluzzi L, Vitale I, Abrams JM, Alnemri
ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry
WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knight
RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W, Mehlen P, Nuñez
G, Peter ME, Piacentini M, Rubinsztein DC, Shi Y, Simon HU,
Vandenabeele P, White E, Yuan J, Zhivotovsky B, Melino G and
Kroemer G: Molecular definitions of cell death subroutines:
recommendations of the Nomenclature Committee on Cell Death 2012.
Cell Death Differ. 19:107–120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
MacLean MA, Scott BE, Deziel BA, Nunnelley
MC, Liberty AM, Gottschall-Pass KT, Neto CC and Hurta RA: North
American cranberry (Vaccinium macrocarpon) stimulates apoptotic
pathways in DU145 human prostate cancer cells in vitro. Nutr
Cancer. 63:109–120. 2011.
|
|
37.
|
Boehrer S, Kukoc-Zivojnov N, Nowak D,
Bergmann M, Baum C, Puccetti E, Weidmann E, Hoelzer D, Mitrou PS
and Chow KU: Upon drug-induced apoptosis expression of
prostate-apoptosis-response-gene-4 promotes cleavage of caspase-8,
bid and mitochondrial release of cytochrome c. Hematology.
9:425–431. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Chendil D, Das A, Dey S, Mohiuddin M and
Ahmed MM: Par-4, a pro-apoptotic gene inhibits radiation-induced NF
kappa B activity and Bcl-2 expression leading to induction of
radiosensitivity in human prostate cancer cells PC-3. Cancer Biol
Ther. 1:152–160. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Crown J and O’Leary M: The taxanes: an
update. Lancet. 355:1176–1178. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Morse DL, Gray H, Payne CM and Gillies RJ:
Docetaxel induces cell death through mitotic catastrophe in human
breast cancer cells. Mol Cancer Ther. 4:1495–1504. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Hernández-Vargas H, Palacios J and
Moreno-Bueno G: Molecular profiling of docetaxel cytotoxicity in
breast cancer cells: uncoupling of aberrant mitosis and apoptosis.
Oncogene. 26:2902–2913. 2007.PubMed/NCBI
|
|
42.
|
Kline CL, Shanmugavelandy SS, Kester M and
Irby RB: Delivery of PAR-4 plasmid in vivo via nanoliposomes
sensitizes colon tumor cells subcutaneously implanted into nude
mice to 5-FU. Cancer Biol Ther. 8:1831–1837. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Brieger A, Boehrer S, Schaaf S, Nowak D,
Ruthardt M, Kim SZ, Atadja P, Hoelzer D, Mitrou PS, Weidmann E and
Chow KU: In bcr-abl-positive myeloid cells resistant to
conventional chemo-therapeutic agents, expression of Par-4
increases sensitivity to imatinib (STI571) and histone
deacetylase-inhibitors. Biochem Pharm. 68:85–93. 2004. View Article : Google Scholar
|
|
44.
|
Lee TJ, Lee JT, Kim SH, Choi YH, Song KS,
Park JW and Kwon TK: Overexpression of Par-4 enhances
thapsigargin-induced apoptosis via down-regulation of XIAP and
inactivation of Akt in human renal cancer cells. J Cell Biochem.
103:358–368. 2008. View Article : Google Scholar : PubMed/NCBI
|