Introduction
The tumor suppressor genes, tuberous sclerosis
complex 1 (TSC1) and TSC2, encode hamartin and
tuberin, respectively, which are key upstream negative regulators
of the rapamycin-sensitive mTOR complex 1 (mTORC1). Hamartin and
tuberin form a complex and function as a GTPase-activating protein
(GAP) for the small G protein Rheb, a direct activator of mTORC1
(1,2). Mutations of TSC1 or
TSC2 induce abnormal activation of mTORC1 that contributes
to the development of tuberous sclerosis complex (TSC) including
seizure, mental retardation, and formation of benign tumors in
multiple organs such as brain, kidney, heart, lung and skin. In
animal models, Tsc1- or Tsc2-deficiency causes renal
tumor development in heterozygous mutants. In TSC patients,
neurological manifestations vary from patient to patient including
subependymal giant-cell astrocytoma (SEGA), severe and refractory
epilepsy, psychiatric disease and mental retardation. Standard
therapy for SEGA, which develops in up to 20% of young patients
(3,4), has been surgical resection.
Currently, administration of rapamycin has therapeutic potential
for TSC including SEGA (5,6), however, there is limited efficacy
since it cannot completely eliminate all of the symptoms.
Therefore, better understanding of the mTOR signaling pathway and
downstream molecules of hamartin/tuberin will lead to development
of novel, more efficient treatments for TSC.
mTOR is a member of the phosphoinositide-3 kinase
(PI3K) family and is a highly conserved serine/threonine kinase
that integrates signals from growth factors, nutrients and
stresses. It regulates multiple processes such as mRNA translation,
cell cycle progression, autophagy, cell proliferation, growth and
survival (7,8). mTOR forms two structurally and
functionally distinct complexes referred as mTORC1 and mTORC2
(7). mTORC1 consists of mTOR,
GβL/mLST8 and raptor, and regulates cell growth and proliferation
by phosphorylating its downstream targets, S6 kinase (S6K) and
4E-binding protein 1 (4E-BP1), key regulators of protein synthesis.
By contrast, mTORC2 is composed of mTOR, GβL/mLST8 and rictor, and
directly activates Akt by phosphorylating it at Ser473 (7). mTORC1, but not mTORC2, is directly
inhibited by rapamycin (9). As
compared to mTORC1, the function of mTORC2 is less understood,
although some contribution to the regulation of cytoskeleton has
been reported (10).
mTORC1 negatively regulates autophagy by
phosphorylating ATG1 and ATG13 (11). Autophagy is thought to be a
fundamental process by which cytoplasmic proteins and organelles
are degraded. Autophagy is induced by starvation or stress, whereby
double membrane vesicles (autophagosomes) engulf damaged proteins
or organelles before fusion with lysosomes to form autolysosomes
which degrade their contents to regenerate nutrients (12). Impairment of autophagy is tightly
associated with the development of various pathologic conditions
including tumorigenesis and neurodegenerative disease (13,14).
By contrast, there are reports demonstrating that autophagy is
required for the development and progression of tumors, possibly to
support their aggressive growth (15). Similarly, in TSC-associated lesions
treated with rapamycin, re-activation of autophagy may play some
cyto-protective role (16). As
recently demonstrated using model animals, the inhibition of
autophagy could be a novel treatment option for TSC-related
pathology, although side-effects are a concern (17).
Several recent studies have reported the involvement
of hamartin and/or tuberin in cell motility control through the
regulation of the Rho family of small GTPases, including RhoA, Rac
and Cdc42, which are key regulators of actin cytoskeleton
remodeling, cell adhesion and migration. RhoA regulates the
formation of stress fibers that promotes focal adhesions, while Rac
and Cdc42 promote actin polymerization, which induce formation of
lamellipodia and filopodia formation, respectively (18,19).
Overexpression of tuberin in Tsc2-null rat leiomyoma cells
activates Rac1 and inhibits Rho in an mTOR-independent manner
(20). An antagonistic role of
tuberin on Rac1-inhibitory function of hamartin was suggested
(20). A later study showed that
knockdown of TSC2 in colon cancer cells resulted in
decreased cell motility accompanied by reduced activity of Rac1
(21). A contribution of
mTORC1-induced negative feedback regulation of PI3K pathway in Rac1
inhibition was suggested (21). On
the other hand, it has been shown that mTORC2 controls the actin
cytoskeleton through regulation of Rac1 and RhoA in fibroblast
cells (10). Another study
indicated that mTORC2 bound to PIP3- dependent Rac exchange factor
(P-Rex1) activates Rac1 and cell migration (22). P-Rex1 and Rac1 are known to be
involved in neural differentiation and migration, which may be
relevant to TSC-associated neural disorders (23,24).
However, the precise signal transduction system regulated by
hamartin and/or tuberin in Rac1 activation is controversial.
Here, we report that tuberin regulates Rac1 activity
and distribution in an mTORC1-dependent manner in
Tsc2-deficient renal tumor cells. We also report that the
punctate cytoplasmic Rac1 distribution might be related to a
degradation system involving p62/sequestosme1 (SQSTM1) and
ubiquitin.
Materials and methods
Plasmid construction
To establish stable cell lines, plasmid vectors in
the pRevTet-Off system (Clontech, Mountain View, CA) were used.
pRevTRE-rTsc2 for expression of wild-type tuberin was previously
described (25). pRevTRE-LacZ was
generated by inserting a LacZ coding DNA fragment into the
pRevTRE vector. The expression vector for the flag-tagged wild-type
tuberin was previously described (26). For construction of the Rac1
expression vector, rat Rac1 cDNA was amplified by reverse
transcription-polymerase chain reaction (RT-PCR) using primers,
RACF1 (5′-CCGAATTCCAGGCCATCAAG TGTGTGGT-3′) and RACMYCR
(5′-CCCCTCGAGTTACA ACAGCAGGCATTTTCTCT-3′). After digestion with
EcoRI and XhoI, the Rac1 cDNA fragment was subcloned
into the pCAG-GS vector (26) that
had been modified to introduce an N-terminal Myc tag.
Generation of cell lines and culture
conditions
Transfection of prTA-IRES-Puro, a
tetracycline-controlled transactivator (rTA) expression plasmid,
into Tsc2-deficient mouse renal tumor (MKOC1-277) cells and
selection of rTA-expressing cells were as previously described
(25). Subsequently, retro-viruses
derived from pRevTRE-rTsc2 or pRevTRE-LacZ and generated by Ecopack
293 producer cells (Clontech) were used to infect the
rTA-expressing cells, and tuberin- (WT) or
β-galactosidase-expressing (LacZ) cell lines were selected using
200 μg/ml hygromycin B (Clontech) and 100 ng/ml doxycycline
(DOX) (Sigma-Aldrich, St. Louis, MO). MKOC1-277 cells were
maintained in RPMI-1640 medium (Sigma-Aldrich) containing 10% fetal
calf serum (FCS), 100 U/ml penicillin, and 100 μg/ml
streptomycin. LacZ and WT cells, as well as previously established
E8 and T2-5 cell lines (25), were
maintained in RPMI-1640 medium containing 10% FCS, 100 U/ml
penicillin, 100 μg/ml streptomycin, 200 μg/ml
hygromycin B (Clontech), 0.6 μg/ml puromycin (Sigma-Aldrich)
and 100 ng/ml DOX. Analyses were performed with maximal induction
of tuberin in the absence of doxycycline as described below. For
rapamycin treatment, the cells were additionally incubated for 4 h
in the medium containing 20 nM rapamycin.
Western blot analysis
Protein concentrations were determined by DC-protein
assay (Bio-Rad, Hercules, CA). Proteins were separated by
SDS-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to
Immobilon membranes (Millipore, Billerica, MA), blocked in 1% skim
milk in Tris-buffered saline containing 0.05% Tween-20, and probed
with the appropriate antibodies. The following primary antibodies
were used: mouse monoclonal antibodies (mAb) against Rac1 (Abcam,
Cambridge, MA) and β-galactosidase (Promega, Madison, WI); rabbit
polyclonal antibodies from Santa Cruz Biotechnology (Santa Cruz,
CA) against S6K, tuberin, and Rac1; and rabbit mAbs from Cell
Signaling Technology (Beverly, MA) against phospho-S6K (Thr389),
phospho-Akt (Ser473), β-actin, Akt, raptor and rictor. To probe
with both rabbit and mouse antibodies, EnVision System (Dako,
Carpinteria, CA) was used as previously described (27). ECL reagents (GE Healthcare) were
used for detection and visualization of the bands.
Rac1 activation assay
To assess the amount of activated Rac1, GST pulldown
assay was performed using the GST-tagged p21 binding domain of PAK1
(PAK-PBD) according to the manufacturer’s instructions
(Cytoskeleton, Denver, CO). Briefly, cells were seeded at a density
of 1.0x106 cells in 10-cm dishes, cultured overnight in
the presence of DOX, and then incubated in the absence of DOX for
48 h. Subsequently, cells were lysed in cell lysis buffer
containing protease inhibitors (provided by the kit). Protein
concentrations were determined and adjusted with ice-cold cell
lysis buffer for equal protein loading. To equal volume of each
sample, 20 μl of PAK-PBD beads were added and incubated for
60 min at 4°C with rotation. Beads were washed once with 500
μl of wash buffer (provided by the kit), resuspended in 20
μl of 2X SDS-PAGE sample buffer, boiled, and loaded onto a
12% gel for SDS-PAGE. The amount of activated and total Rac1 in
each sample was analyzed using the mouse monoclonal anti-Rac1
antibody by western blot analysis.
RNA interference
For gene silencing of raptor and rictor, the cells
were incubated with the appropriate siRNA for 48 h in the absence
of DOX. Transfection of 25 nM siRNA was performed with
Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA) and OPTI-MEM
(Gibco/Invitrogen, Carlsbad, CA) according to the manufacturer’s
protocols, and 48 h after transfection, cells were lysed for
further analysis. The following siRNA sequences, which reflect
sense strands without 3′-overhang, were used: raptor,
5′-GCGUUCCUUCUGUGGUCAA-3′; rictor, 5′-GGUU GGAAAUGAUGGGCUU-3′; and
control, 5′-GCUGCAAUCG AUUGAUAGC-3′.
Immunofluorescence and confocal
microscopy
Cells were seeded at a density of 2×104
cells in glassbottom dishes (Greiner Bio-One International AG,
Frickenhausen, Germany) and cultured as described for Rac1
activation assay. Cells were fixed and permeabilized with 4%
paraformaldehyde and 0.25% Triton X-100 for 30 min at 4°C. This was
followed by incubation with the appropriate primary antibody for 1
h at room temperature and secondary antibody (Alexa-Fluor
488-labeled IgG; Alexa-Fluor 568-labeled IgG, Invitrogen) together
with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI,
Invitrogen) for 1 h at room temperature. Actin was stained using
phallocidin (Invitrogen). The following primary antibodies were
used: mouse mAbs against ubiquitin (Millipore), early endosome
antigen 1 (BD Transduction Laboratories, Franklin Lakes, NJ),
transferrin receptor (Invitrogen), and Flag (Sigma); rabbit
polyclonal antibody against Myc (Sigma); rat mAb against
lysosomal-associated membrane protein 1 (Santa Cruz Biotechnology);
and guinea pig polyclonal antibody against p62 (ProGen, Toowong,
Australia). The secondary antibody used for p62 was Alexa-Fluor
594-labeled goat anti-guinea pig IgG (Invitrogen). Stainings were
examined using TCS-SP5 laser confocal microscopy (Leica
Microsystems, Wetzlar, Germany).
Transient transfection
MKOC1-277 cells were seeded at a density of
2×104 cells in glass bottom dishes (Greiner Bio-One
International AG) and cultured overnight. Plasmids were transfected
using Lipofectamine 2000 (Invitrogen) according to the
manufacturer’s protocol, and 48 h after transfection, cells were
fixed and permeabilized for confocal microscopy as described
above.
Electron microscopy and immunoelectron
microscopy using ultrathin cryosections
Cells were fixed in 2% glutaraldehyde-2%
paraformaldehyde (PA) buffered with 0.1 M phosphate buffer (PB) (pH
7.2) for ordinary electron microscopy, and in 4% PA buffered with
PB (pH 7.2) for immunoelectron microscopy (28). In the former procedure, the cells
were postfixed with 1% OsO4 and embedded in Epon 812
after dehydration. Ultrathin sections, 60 nm, were cut with an
ultramicrotome (UC6, Leica) and stained with uranium acetate and
lead citrate. In the latter procedure, fixed cells were rotated in
2.3 M sucrose in PB overnight and plunged into liquid nitrogen.
Sections approximately 60 nm thick were cut with a
cryo-ultramicrotome (UC7/FC7, Leica) and reacted overnight at 4°C
with rabbit anti-Rac1 (1:10) or p62 (Wako; 1:10) and mouse
anti-ubiquitin (clone: FK2) (Enzo Life Sciences; 1:10) and then for
1 h with goat anti-rabbit and mouse IgG conjugated with 10- and
5-nm colloidal gold particles (British Biocell International). The
specimens were examined with a Hitachi H-7100 or HT7700 electron
microscope.
Results
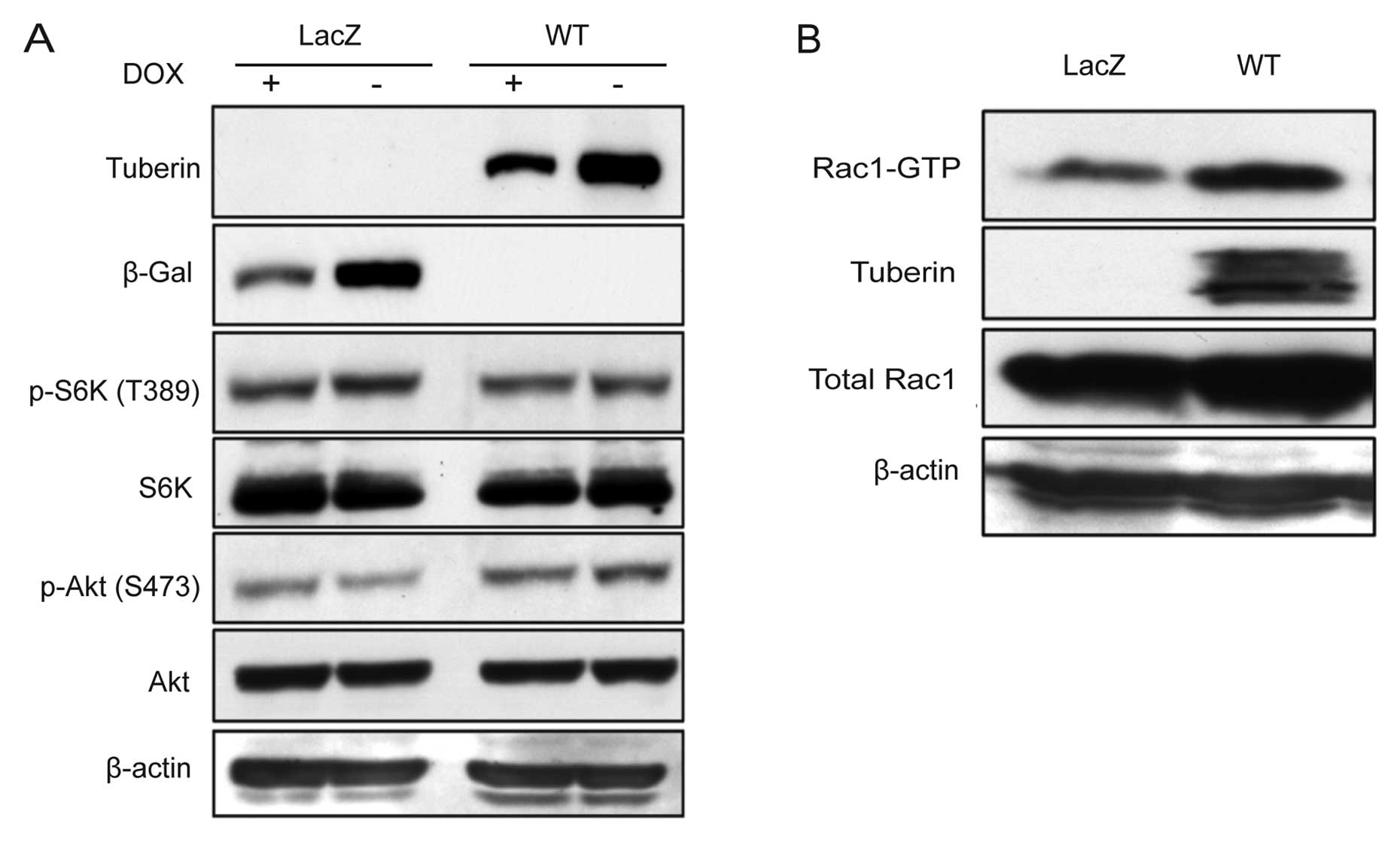
Restoration of tuberin activates Rac1 in
Tsc2-deficient cells
To explore novel functions of tuberin, we
established Tsc2-deficient mouse renal tumor cells in which
expression of wild-type tuberin was restored (WT cells, Fig. 1A) (25). Although we utilized the
conventional Tet-off system, suppression of expression by DOX in
established cells was limited and tuberin was still expressed even
in the presence of 1 μg/ml DOX (data not shown). Thus, we
established and used β-galactosidase-expressing cells as
tuberin-negative control cells (Fig.
1A). The levels of phosphorylation on p70 S6K and Akt were
decreased and increased in WT cells, respectively (Fig. 1A). To test whether tuberin
regulates Rac1 activity, LacZ and WT cells were analyzed using
GST-pulldown assay. Consistent with previous findings (20,21),
the amount of activated Rac1 was significantly increased in WT
cells compared with LacZ cells (Fig.
1B). Similar results were obtained using previously established
tuberin-restored (T2-5) and control (E8) cells (data not shown)
(25). These results suggest that
tuberin regulates Rac1 activity as well as the mTORC1 pathway when
restored in Tsc2-deficient cells.
Tuberin affects Rac1 distribution
We also investigated Rac1 distribution in LacZ and
WT cells by confocal microscopy. Intriguingly, Rac1 appeared to
form large dots within the cytoplasm in LacZ cells, whereas WT
cells showed diffusely distributed Rac1 (Fig. 2A). We then performed transient
co-transfection with expression plasmids for FLAG-tuberin and
Myc-Rac1 in parental Tsc2-null cells (MKOC1-277). As
expected, Rac1 appeared to form dots in the cytoplasm in the
absence of co-expressed tuberin. By contrast, Rac1 was diffusely
distributed in the presence of tuberin (Fig. 2B). Taken together, these results
suggest that the regulatory mechanism of Rac1 distribution is
controlled by tuberin.
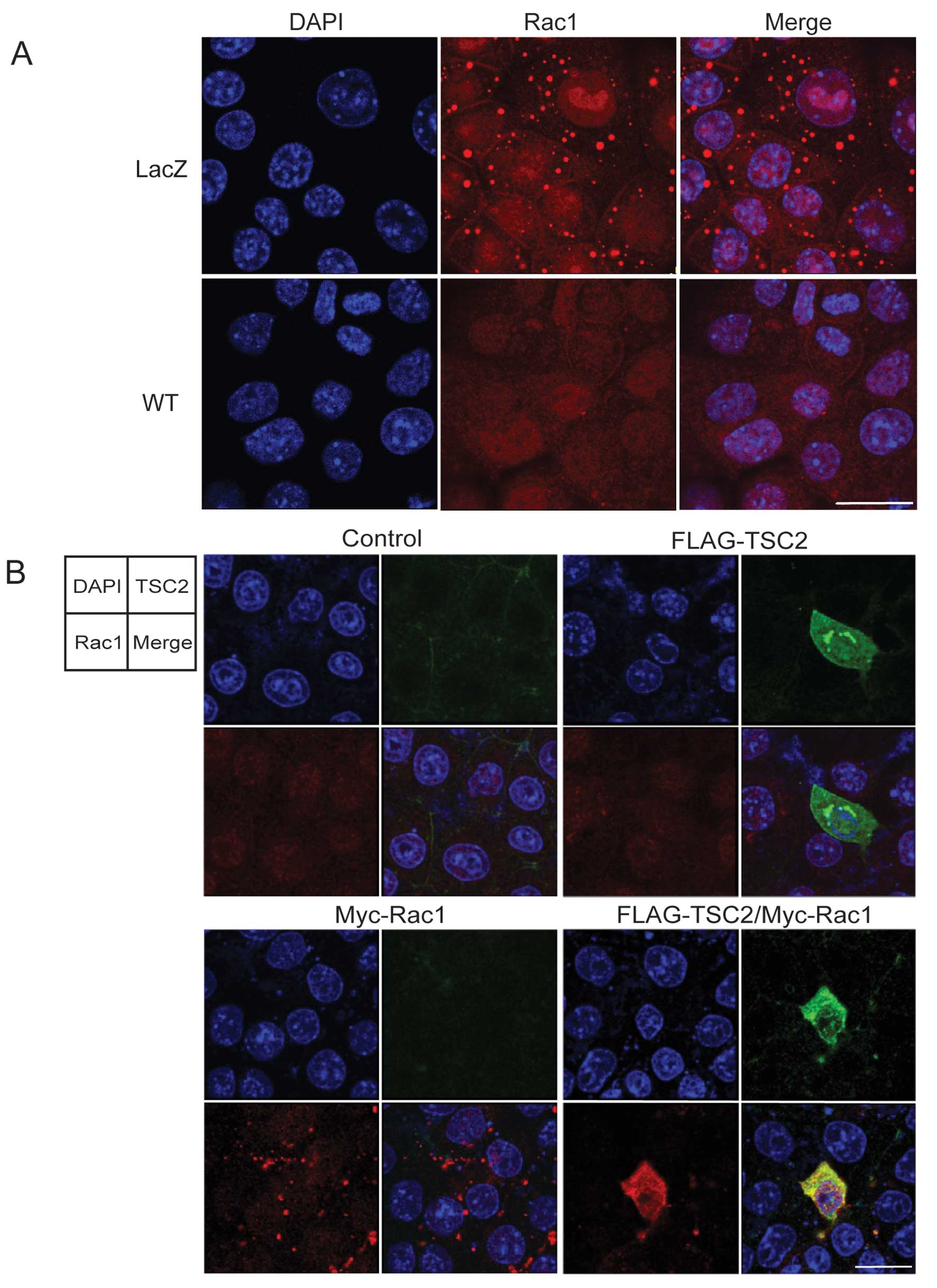 | Figure 2.Wild-type tuberin expression
modulates the distribution of Rac1 in Tsc2-deficient cells. (A)
Immunocytological analysis of Rac1. LacZ and WT cells were grown in
standard growth medium without DOX for 48 h, then fixed and
subjected to immunofluorescence with anti-Rac1 antibody. Results of
DAPI (blue, left images), Rac1 staining (middle panels, red), and
merged images (right mages) are shown. Bar, 20 μm. (B)
Transient expression of tuberin in Tsc2-deficient cells. MKOC1-277
cells were transfected with expression plasmids for FLAG-tagged
tuberin and/or Myc-tagged Rac1, and 48 h after transfection, cells
were fixed and examined for localization of FLAG-tuberin and
Myc-Rac1 by double immunofluorescence. The panels show the results
of FLAG-tuberin (upper right), Myc-Rac1 (lower left), both
FLAG-tuberin and Myc-Rac1 expression (lower right), and control
empty vectors (upper left). The arrangement of images in each panel
is indicated: DAPI (blue); FLAG-tuberin (green); Myc-Rac1 (red).
Bar, 20 μm. |
Rac1 activity as well as distribution are
regulated by mTORC1
We examined whether the regulation of Rac1 by
tuberin is functionally related to the mTORC1 signaling pathway.
First, MKOC1-277 cells were treated with vehicle (DMSO) or
rapamycin (20 nM) for 4 h and analyzed for Rac1 activity and
distribution. By this treatment, phosphorylation of S6K was
completely suppressed while Rac1 activation was increased (Fig. 3A) and its distribution became
diffuse (Fig. 3B). To further test
the contribution of mTORC1 as well as mTORC2 in Rac1 regulation, we
employed gene silencing technology to knockdown raptor or rictor in
both LacZ and WT cells. Since S6K and Akt are downstream targets of
mTORC1 and mTORC2, respectively, phosphorylation of these proteins
was decreased following knockdown of raptor and rictor,
respectively. Consistent with the results of rapamycin treatment,
increased activation of Rac1 was observed in raptor siRNA-treated
LacZ and WT cells compared with control siRNA-treated cells
(Fig. 4A and data not shown).
While the amount of activated Rac1 following rictor siRNA treatment
was similar to control siRNA-treated WT cells, on the other hand,
rictor knockdown in LacZ cells showed a slight decrease in Rac1
activation as compared to control siRNA treatment (Fig. 4A), which may be partly due to the
elevated raptor expression in these cells. These results suggest
that Rac1 activity is negatively regulated by mTORC1. We also
obtained similar results from E8 and T2-5 cells (data not shown).
Distribution of Rac1 became diffuse after raptor, but not rictor or
control, siRNA treatment in LacZ cells (Fig. 4B–D). In WT cells, Rac1 was
diffusely distributed under all conditions regardless of siRNA
treatment (data not shown). Taken together, these results indicate
that Rac1 activity correlates with its distribution and is
regulated by mTORC1.
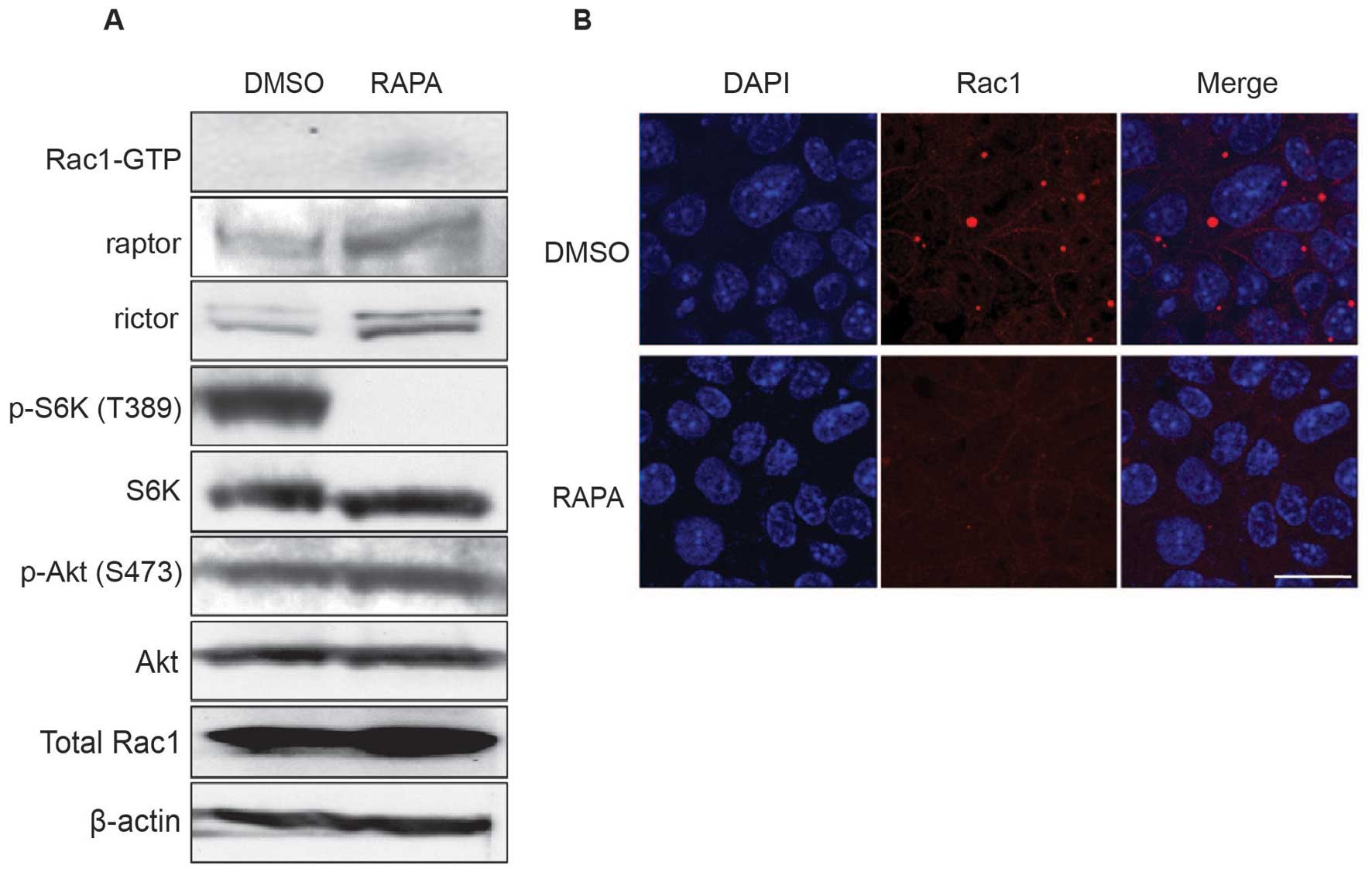 | Figure 3.Rapamycin restores the activation and
distribution of Rac1 in Tsc2-deficient cells. (A) Rac1 activity
following rapamycin treatment. MKOC1-277 cells were grown in the
absence of DOX for 48 h, then treated with vehicle (DMSO) or
rapamycin (RAPA, 20 nM) for 4 h. The amount of activated Rac1 was
analyzed using the pulldown assay. Intriguingly, when
Tsc2-deficient cells were treated with rapamycin, the amount of
activated Rac1 increased (upper panel). (B) Distribution of Rac1
following rapamycin treatment. Cells were treated with drugs as in
(A), then fixed and subjected to immunofluorescence as described in
Fig. 2A. Results of DAPI (blue,
left images), Rac1 staining (red, middle images), and merged images
(right images) are shown. DMSO, vehicle only; RAPA, rapamycin
treatment. Bar, 20 μm. |
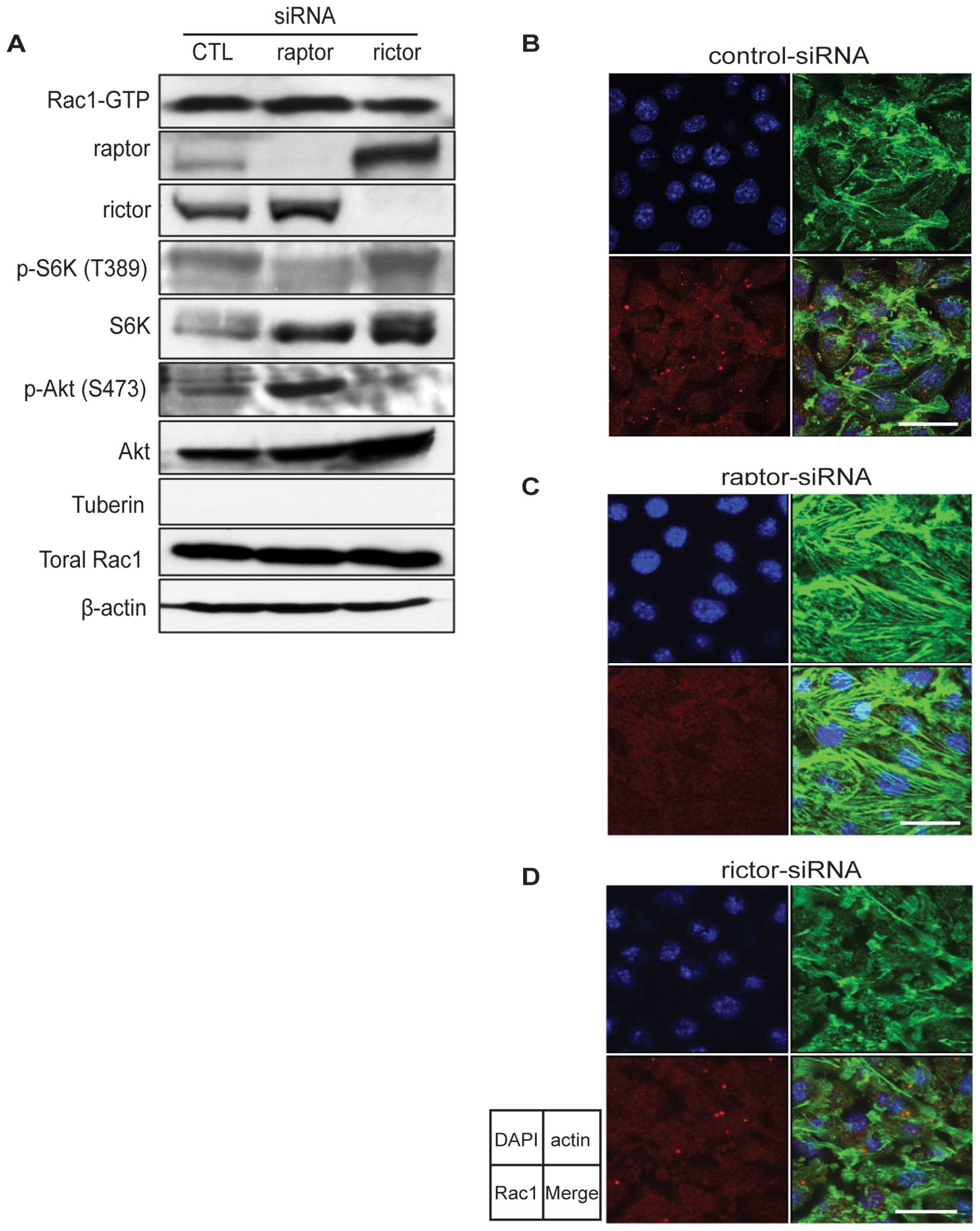 | Figure 4.Knockdown of raptor enhances Rac1
activity and disperses dots in LacZ cells. (A) Rac activation
assay. LacZ cells were incubated with raptor, rictor or control
(CTL) siRNA in the absence of DOX for 48 h. The amount of activated
Rac1 was analyzed using the GST-pulldown assay as described in
Fig. 1B. In addition, total
lysates were analyzed by immunoblot analysis for raptor and rictor
to monitor knockdown. (B–D) Double immunofluorescence of LacZ cells
treated with (B) control, (C) raptor, or (D) rictor siRNA. Cells
were cultured and treated with siRNA as in (A), then subjected to
immunofluorescence for Rac1 and phallocidin to detect F-actin. The
separated panels show the following images: left upper, DAPI
(blue); right upper, phallocidin (actin, green); left lower, Rac1
(red); right lower, merged image. Bars, 20 μm. |
Actin stress fiber organization is
correlated with Rac1 activity and distribution
Furthermore, actin stress fiber formation and
organization, which are downstream effects of Rac1 (29,30),
were assessed following raptor or rictor knockdown in LacZ and WT
cells (Fig. 4B–D and data not
shown). Aberrantly distributed actin fibers became well-ordered
when LacZ cells were treated with raptor siRNA (Fig. 4B and C). In the case of rictor
siRNA treatment, the intensity of actin staining seemed to become
weaker without apparent changes in fiber organization (Fig. 4B and D). In WT cells, distribution
of actin fibers was still well-ordered in all siRNA treatments
(data not shown). Similar results were obtained using E8 and T2-5
cells (data not shown). These results suggest that mTORC1 inhibits
stress fiber formation, at least in part, by inhibiting Rac1 in
Tsc2-deficient renal tumor cells.
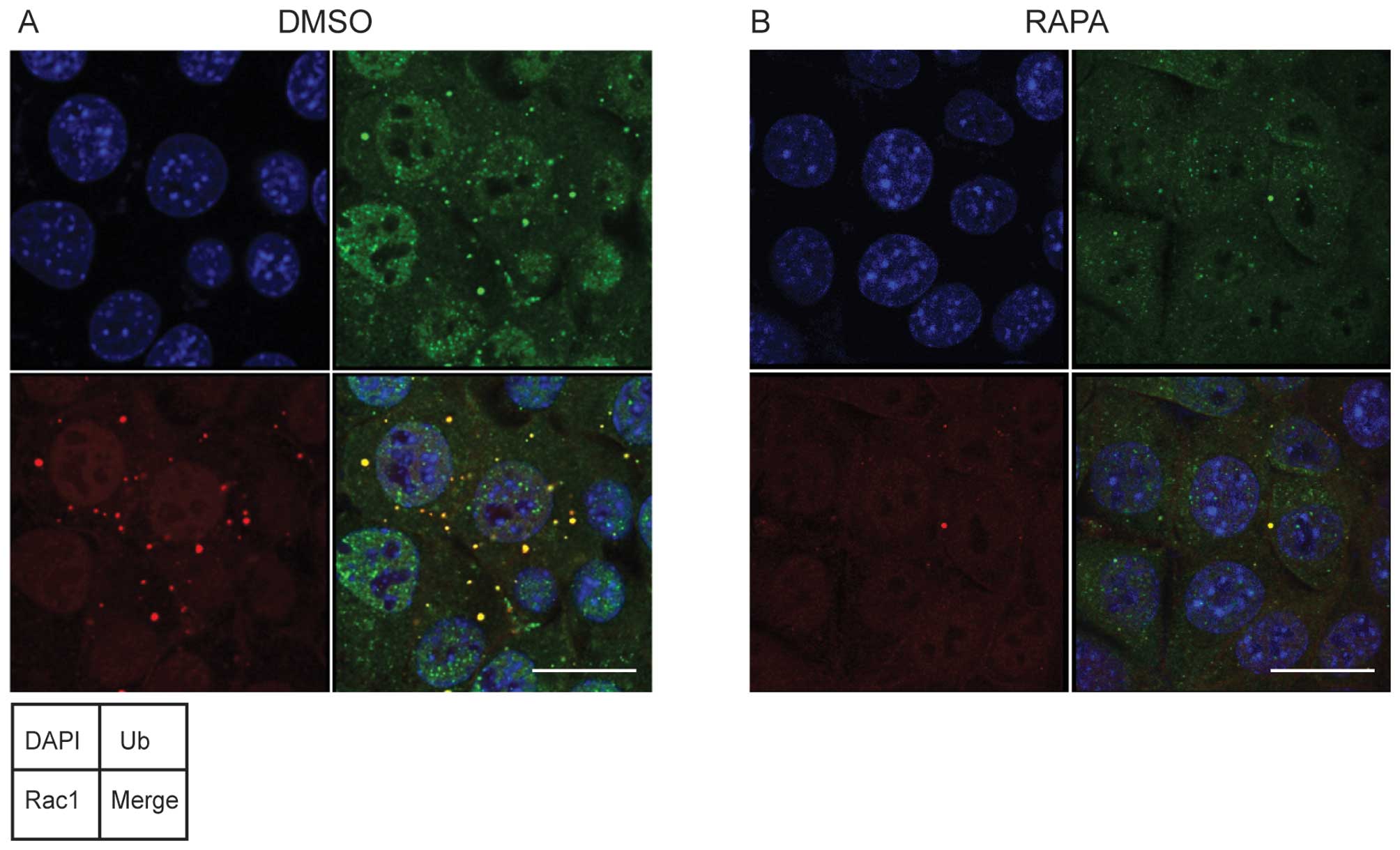
Rac1 dots were colocalized with p62 and
ubiquitin, but not LAMP1, EEA1 and TR
From the observations described above, dot formation
seemed to be connected with inactivation of Rac1. Thus, we focused
our investigation on protein degradation and intracellular membrane
compartment systems (31). First,
to elucidate which system is involved in Rac1 dot formation,
MKOC1-277 cells were co-stained for Rac1 and membrane compartment
markers such as lysosomal-associated membrane protein 1 (LAMP1,
lysosome), early endosomal antigen 1 (EEA1, early endosome), and
transferrin receptor (late endosome) (Fig. 5A–C). Colocalization of ubiquitin
and Rac1 was also examined. As shown in Fig. 6A, ubiquitin colocalized with Rac1
dots, whereas EEA1, transferrin receptor and LAMP1 did not. We then
performed staining of MKOC1-277 cells for p62, which is generally
degraded by autophagy (32,33),
a process that has been shown to be inhibited by mTORC1 and
downregulated in Tsc2-null cells (16,34).
Interestingly, p62 colocalized with Rac1 in MKOC1-277 cells
(Fig. 6B). Ubiquitin also
colocalized with p62, consistent with a previous report that found
that these two proteins interact through the ubiquitin associated
domain of p62 (Fig. 6C) (32). By conventional electron microscopy,
we found aggregate structures in the cytoplasm of
Tsc2-deficient cells (Fig.
6D). By immunoelectron microscopy we also confirmed aggregates
doubly positive for ubiquitin and Rac1 or p62 in
Tsc2-deficient cells (Fig. 6E
and F). We observed that the number and size of ubiquitin dots
in MKOC1-277 cells were significantly reduced by rapamycin
treatment, similarly to those of Rac1 dots (Fig. 7). These findings suggest that in
Tsc2-null cells Rac1 is incorporated into ubiquitin- and
p62-positive aggregate, resulting in the inactivation and
degradation of Rac1.
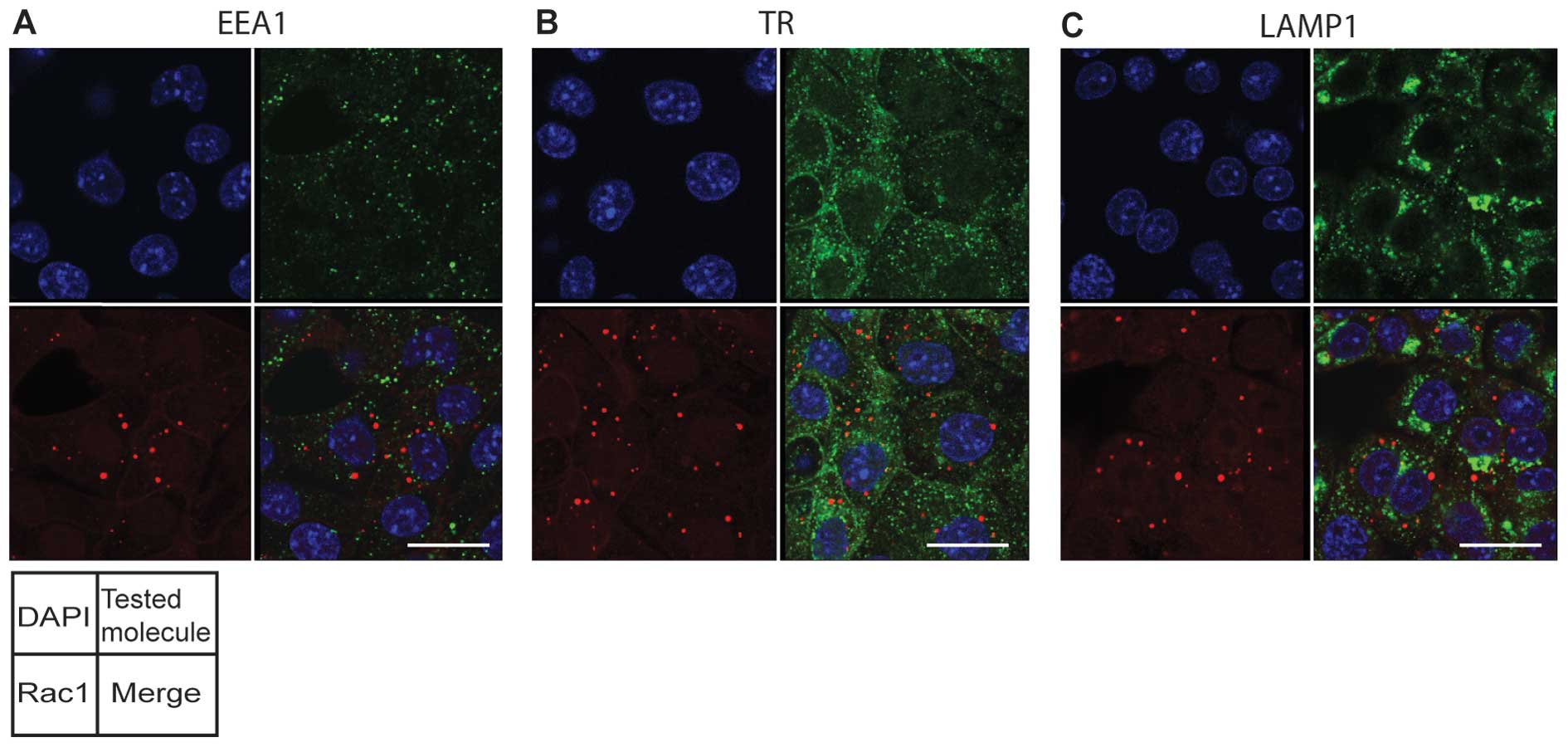 | Figure 5.Rac1 dots are not co-localized with
EEA1, transferrin receptor, and LAMP1 in Tsc2-deficient cells.
(A–C) MKOC1-277 cells were grown in DOX-free medium for 48 h, then
fixed and double immunofluorescence of Rac1 with (A) EEA1, (B)
transferrin receptor (TR), and (C) LAMP1 was performed. The
separated panels show the following images: left upper, DAPI
(blue); left lower, Rac1 (red); right upper, tested molecule
(green); right lower, merged image. Bars, 20 μm. |
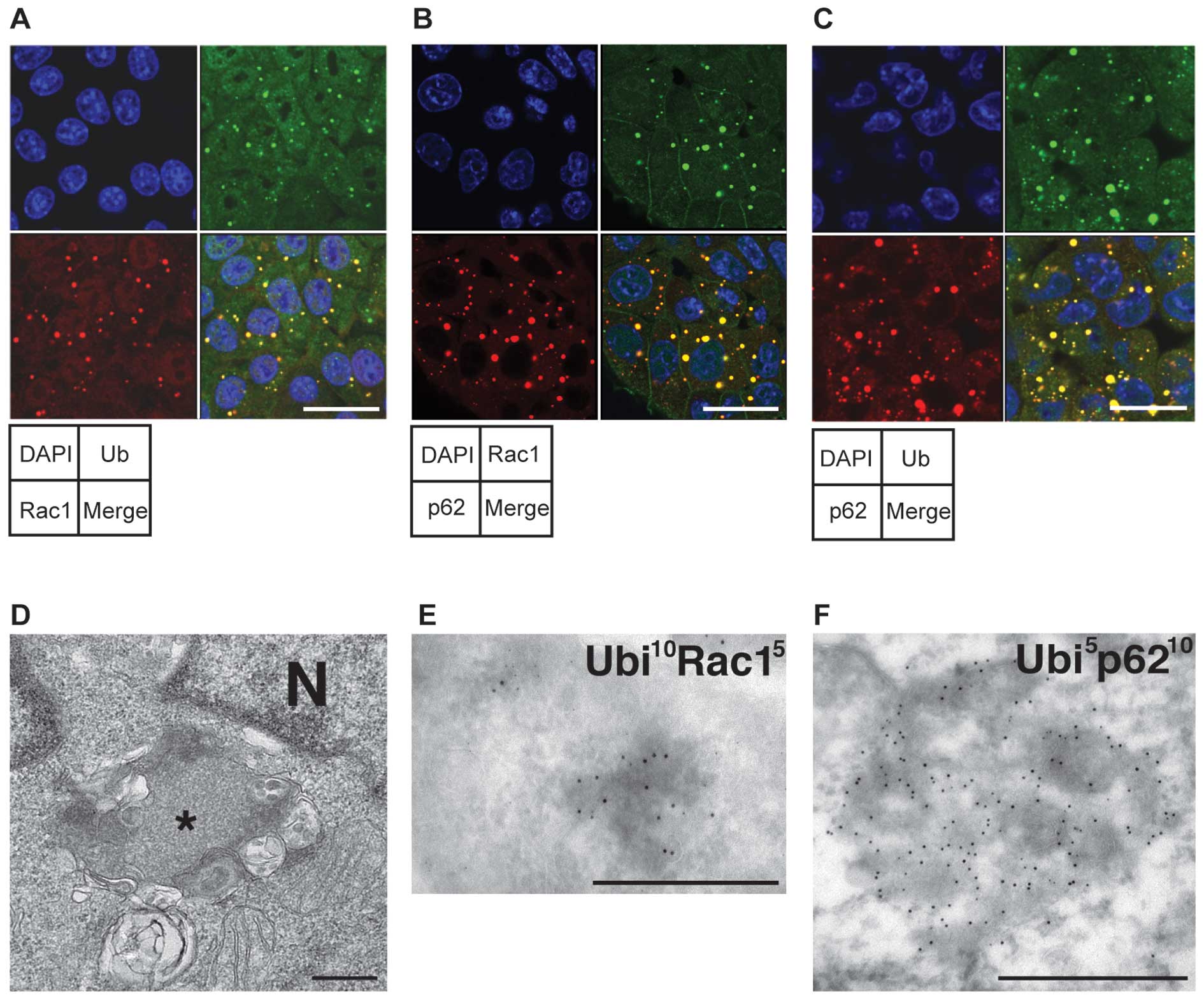 | Figure 6.Ubiquitin and p62 are colocalized
with Rac1 dots in Tsc2-deficient cells. (A and B) Double
immunofluorescence of Rac1 with (A) ubiquitin or (B) p62. Double
immunofluorescence was performed as described in Fig. 5. The separated panels show the
following images: left upper, DAPI (blue); left lower, Rac1 (red);
right upper (green); (A) ubiquitin or (B) p62; right lower, merged
image. Bars, 20 μm. (C) Co-localization of ubiquitin with
p62 in Tsc2-deficient cells. MKOC1-277 cells were analyzed for
co-localization of ubiquitin (Ub, green, upper right) with p62
(red, lower left). The upper left and lower right images show DAPI
staining (blue) and merged images, respectively. Bar, 20 μm.
(D–F) Ultrastructural characterization of the aggregate structure
in Tsc2-deficient cells. (D) Ordinary electron microscopy.
Asterisk, aggregate. N, nucleus. (E and F) Immunoelectron
microscopy using ultrathin cryosections. In Tsc2-deficient cells
gold particles indicating (E) ubiquitin (10 nm) and Rac1 (5 nm) or
(F) ubiquitin (5 nm) and p62 (10 nm) specifically co-localize in
the aggregates. Bars, 0.5 μm. |
Discussion
In this study, we aimed to elucidate the downstream
signals of tuberin that regulate Rac1 activity. Rac1 plays an
important role in cytoskeletal dynamics by regulating actin
remodeling, thereby promoting cell migration, invasion and
metastasis (35,36). Regulation of Rac1 activity is also
known to be important for the neuronal cell differentiation
process, such as neurite development (19,37).
Thus, we believe that the signal transduction pathway involving
tuberin and Rac1 has relevance to not only clinical manifestation
of TS, but also cancer development and progression including
nervous system tumors.
Previous reports have illuminated various aspects of
tuberin’s role on Rac1 regulation. Goncharova et al reported
that hamartin inhibits Rac1 activity, whereas tuberin counteracts
the effects of hamartin to re-activate Rac1 and subsequently
inhibit Rho (20). Rac1 and Rho
activities may be regulated by hamartin and tuberin in a
coordinated fashion in an mTOR-independent manner (20). However, so far, the precise
mechanisms of Rac1 regulation involving the downstream pathways of
tuberin have not been elucidated. Consistent with previous reports,
we showed here that tuberin positively regulated Rac1 activity in
our experimental cell system, which was further demonstrated to be
dependent on mTORC1. Our findings agree with a recent study which
found that mTORC1-dependent regulation of cell migration, polarity
and activation of Rac1 and/or CDC42 was through tuberin signaling
(21). This link between mTORC1
and Rac1 may provide important clues into the evaluation of the
efficacy of rapamycin and side-effects on TS and cancers.
We show for the first time that Rac1 forms
cytoplasmic dots in Tsc2-null cells. In addition, Rac1 dots
co-localized with ubiquitin and p62. Furthermore, the number and
size of the ubiquitin dot structures were significantly decreased
and reduced in Tsc2-null cells treated with rapamycin. The
amount of activated Rac1 was increased and stress fiber formation
was restored by knockdown of raptor, but not rictor, in
Tsc2-null cells. These observations suggest that Rac1
activity is correlated with its distribution and is regulated by
protein degradation in an mTORC1-dependent manner.
Impairment of the protein degradation systems,
including the autophagy-lysosome and ubiquitin-proteasome pathways,
is involved in various pathogenesis (12–14,38).
Autophagy targets cellular proteins, protein aggregates and
organelles for degradation in lysosomes (12). Protein complexes or organelles are
engulfed by autophagosomes and bind to lysosomes that digest target
proteins (12). Autophagy is
thought to be important for cellular response to starvation as well
as normal turnover of cytoplasmic constituents; starvation induces
and mTORC1 suppresses autophagy (34). By contrast, the
ubiquitin-proteasome pathway is another process that degrades
misfolded proteins in the nucleus and cytoplasm via the E1-E2-E3
ubiquitin conjugation system (39). Recent studies suggested that, like
many other proteins, the Rho family of GTPases are degraded by
ubiquitinylation (40). However,
there is no clear consensus as to the relationship between mTOR and
regulation of ubiquitinylation.
The adaptor protein p62 (also known as sequestosome
1) is thought to be a scaffold protein initially identified as the
atypical protein kinase C (aPKC)-binding protein (41). p62 binds to LC3 on the
autophagosome membrane to carry ubiquitylated target proteins for
degradation through its ubiquitin associated domain (32). We demonstrated that Rac1
co-localized with both p62 and ubiquitin in Tsc2-null cells,
which suggest that Rac1 may be degraded through either the
autophagy-lysosome or ubiquitin-proteasome pathway. Autophagosomes
generally require fusion with lysosomes to form autolysosomes for
degradation of proteins not required. In our study, the Rac1 dots
did not co-localize with EEA1, transferrin receptor or LAMP1. This
result may be explained by the increased mTOR activity in
Tsc2-null cells, which may have blocked fusion of
autophagosomes and lysosomes. The proposed ubiquitin- and
p62-mediated Rac1 degradation system may be arrested by the above
mechanism and not due to downregulation of Rac1 in
Tsc2-deficient cells. Further studies are needed to clarify
the presence of such degradation system and the links between
distribution, degradation and activity of Rac1.
In summary, our study revealed that tuberin
positively regulates Rac1 activity and controls its intracellular
distribution through the mTORC1 signaling pathway. In addition,
Rac1 dots in Tsc2-null cells might reflect the presence of
as yet unidentified ubiquitin- and p62-dependent Rac1 degradation
system.
Abbreviations:
|
mTOR
|
mammalian target of rapamycin;
|
|
mTORC1
|
mTOR complex 1;
|
|
mTORC2
|
mTOR complex 2;
|
|
TSC
|
tuberous sclerosis complex
|
Acknowledgements
We would like to thank Mr. M. Yoshida
at Laboratory of Ultrastructure Research of the BioMedical Research
Center at Juntendo University Graduate School of Medicine for
excellent assistance with our electron microscopy studies. This
study was supported in part by the following grants: the High
Technology Research Center Grant and Grants-in-Aid for Scientific
Research from the Ministry of Education, Culture, Sports, Science
and Technology (MEXT), Japan; MEXT-Supported Program for the
Strategic Research Foundation at Private Universities;
Grants-in-Aid for Scientific Research from the Japan Society for
the Promotion of Science, Japan; and Grants-in-Aid for Scientific
Research from the Ministry of Health, Labour and Welfare, Japan.
This study was also supported by the Research Institute for Disease
of Old Age, Institute for Environmental and Gender-Specific
Medicine and Sportology Center.
References
|
1.
|
Inoki K, Li Y, Xu T and Guan KL: Rheb
GTPase is a direct target of TSC2 GAP activity and regulates mTOR
signaling. Genes Dev. 17:1829–1834. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Nellist M, van Slegtenhorst MA, Goedbloed
M, van den Ouweland AM, Halley DJ and van der Sluijs P:
Characterization of the cytosolic tuberin-hamartin complex. Tuberin
is a cytosolic chaperone for hamartin. J Biol Chem.
274:35647–35652. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Adriaensen ME, Schaefer-Prokop CM, Stijnen
T, Duyndam DA, Zonnenberg BA and Prokop M: Prevalence of
subependymal giant cell tumors in patients with tuberous sclerosis
and a review of the literature. Eur J Neurol. 16:691–696. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Nabbout R, Santos M, Rolland Y, Delalande
O, Dulac O and Chiron C: Early diagnosis of subependymal giant cell
astrocytoma in children with tuberous sclerosis. J Neurol Neurosurg
Psychiatry. 66:370–375. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Guertin DA and Sabatini DM: An expanding
role for mTOR in cancer. Trends Mol Med. 11:353–361. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Krueger DA, Care MM, Holland K, et al:
Everolimus for subependymal giant-cell astrocytomas in tuberous
sclerosis. N Engl J Med. 363:1801–1811. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Guertin DA and Sabatini DM: Defining the
role of mTOR in cancer. Cancer Cell. 12:9–22. 2007. View Article : Google Scholar
|
|
8.
|
Sarbassov DD, Ali SM and Sabatini DM:
Growing roles for the mTOR pathway. Curr Opin Cell Biol.
17:596–603. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Loewith R, Jacinto E, Wullschleger S, et
al: Two TOR complexes, only one of which is rapamycin sensitive,
have distinct roles in cell growth control. Mol Cell. 10:457–468.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Jacinto E, Loewith R, Schmidt A, et al:
Mammalian TOR complex 2 controls the actin cytoskeleton and is
rapamycin insensitive. Nat Cell Biol. 6:1122–1128. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Hosokawa N, Hara T, Kaizuka T, et al:
Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200
complex required for autophagy. Mol Biol Cell. 20:1981–1991. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Mathew R, Karp CM, Beaudoin B, et al:
Autophagy suppresses tumorigenesis through elimination of p62.
Cell. 137:1062–1075. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Ross CA and Poirier MA: Protein
aggregation and neurodegenerative disease. Nat Med. 10(Suppl):
S10–S17. 2004. View
Article : Google Scholar
|
|
15.
|
Maiuri MC, Tasdemir E, Criollo A, et al:
Control of autophagy by oncogenes and tumor suppressor genes. Cell
Death Differ. 16:87–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Ng S, Wu YT, Chen B, Zhou J and Shen HM:
Impaired autophagy due to constitutive mTOR activation sensitizes
TSC2-null cells to cell death under stress. Autophagy. 7:1173–1186.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Yu J, Parkhitko A and Henske EP:
Autophagy: An ‘Achilles’ heel of tumorigenesis in TSC and LAM.
Autophagy. 7:1400–1401. 2011.
|
|
18.
|
Burridge K and Wennerberg K: Rho and Rac
take center stage. Cell. 116:167–179. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Etienne-Manneville S and Hall A: Rho
GTPases in cell biology. Nature. 420:629–635. 2002. View Article : Google Scholar
|
|
20.
|
Goncharova E, Goncharov D, Noonan D and
Krymskaya VP: TSC2 modulates actin cytoskeleton and focal adhesion
through TSC1-binding domain and the Rac1 GTPase. J Cell Biol.
167:1171–1182. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Larson Y, Liu J, Stevens PD, et al:
Tuberous sclerosis complex 2 (TSC2) regulates cell migration and
polarity through activation of CDC42 and RAC1. J Biol Chem.
285:24987–24998. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Hernandez-Negrete I, Carretero-Ortega J,
Rosenfeldt H, et al: P-Rex1 links mammalian target of rapamycin
signaling to Rac activation and cell migration. J Biol Chem.
282:23708–23715. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Yoshizawa M, Hoshino M, Sone M and
Nabeshima Y: Expression of stef, an activator of Rac1, correlates
with the stages of neuronal morphological development in the mouse
brain. Mech Dev. 113:65–68. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Yoshizawa M, Kawauchi T, Sone M, et al:
Involvement of a Rac activator, P-Rex1, in neurotrophin-derived
signaling and neuronal migration. J Neurosci. 25:4406–4419. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Piao X, Kobayashi T, Wang L, et al:
Regulation of folliculin (the BHD gene product) phosphorylation by
Tsc2-mTOR pathway. Biochem Biophys Res Commun. 389:16–21. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Cao Y, Kamioka Y, Yokoi N, et al:
Interaction of FoxO1 and TSC2 induces insulin resistance through
activation of the mammalian target of rapamycin/p70 S6K pathway. J
Biol Chem. 281:40242–40251. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Fukuda T, Tani Y, Kobayashi T, Hirayama Y
and Hino O: A new Western blotting method using polymer
immunocomplexes: detection of Tsc1 and Tsc2 expression in various
cultured cell lines. Anal Biochem. 285:274–276. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Mori Y, Koike M, Moriishi E, et al: Human
herpesvirus-6 induces MVB formation, and virus egress occurs by an
exosomal release pathway. Traffic. 9:1728–1742. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Guo F, Debidda M, Yang L, Williams DA and
Zheng Y: Genetic deletion of Rac1 GTPase reveals its critical role
in actin stress fiber formation and focal adhesion complex
assembly. J Biol Chem. 281:18652–18659. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Hall A: Rho GTPases and the actin
cytoskeleton. Science. 279:509–514. 1998. View Article : Google Scholar
|
|
31.
|
Rubinsztein DC: The roles of intracellular
protein-degradation pathways in neurodegeneration. Nature.
443:780–786. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Moscat J and Diaz-Meco MT: p62 at the
crossroads of autophagy, apoptosis, and cancer. Cell.
137:1001–1004. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Bjorkoy G, Lamark T, Brech A, et al:
p62/SQSTM1 forms protein aggregates degraded by autophagy and has a
protective effect on huntingtin-induced cell death. J Cell Biol.
171:603–614. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Yu L, McPhee CK, Zheng L, et al:
Termination of autophagy and reformation of lysosomes regulated by
mTOR. Nature. 465:942–946. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Vega FM and Ridley AJ: Rho GTPases in
cancer cell biology. FEBS Lett. 582:2093–2101. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Lozano E, Betson M and Braga VM: Tumor
progression: small GTPases and loss of cell-cell adhesion.
Bioessays. 25:452–463. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Ng J, Nardine T, Harms M, et al: Rac
GTPases control axon growth, guidance and branching. Nature.
416:442–447. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Hoeller D and Dikic I: Targeting the
ubiquitin system in cancer therapy. Nature. 458:438–444. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Hershko A and Ciechanover A: The ubiquitin
system. Annu Rev Biochem. 67:425–479. 1998. View Article : Google Scholar
|
|
40.
|
Nethe M and Hordijk PL: The role of
ubiquitylation and degradation in RhoGTPase signalling. J Cell Sci.
123:4011–4018. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Sanchez P, De Carcer G, Sandoval IV,
Moscat J and Diaz-Meco MT: Localization of atypical protein kinase
C isoforms into lysosome-targeted endosomes through interaction
with p62. Mol Cell Biol. 18:3069–3080. 1998.PubMed/NCBI
|