Introduction
Lung cancer is the leading cause of cancer-related
death worldwide and accounts for one quarter of all cancer
mortalities in the US (1).
Non-small cell lung cancer (NSCLC) accounts for approximately 80%
of all lung cancer cases and can be classified by histotypes as
adenocarcinoma (AC), squamous cell carcinoma (SCC), and large-cell
lung cancer (LCLC). The high mortality rate of lung cancer is
mainly attributed to the disease not being diagnosed until it is in
advanced stages. Chemotherapy with platinum-based drugs in
combination with taxanes, camptothecins, or vinca alkaloids, the
first-line treatment for patients with NSCLC, has made little
progress in improving prognoses in recent decades (1).
Similar to other malignancies, tumorigenesis in
NSCLC depends on the clustering of gene dysfunction as a result of
genetic susceptibility and/or the accumulation of noxious
environmental factors. The discoveries of recurrent mutations in
the epidermal growth-factor receptor (EGFR) kinase and fusions,
such as EML4-ALK, involving anaplastic lymphoma kinase (ALK)
led to a dramatic change in the treatment of lung AC (2,3).
Recent data suggest that substance CI1040 can bind to MEK and
mutated BRAF, resulting in the shrinkage of lung ACs that harbor
mutated KRAS and BRAF, respectively (4). Other recent data show that targeting
mutations in AKT1, ERBB2 and PIK3CA and
fusions involving ROS1 and RET may also be successful
(5). Unfortunately, activating
mutations in EGFR, EML4-ALK fusions, and mutations in
KRAS are only detected in lung AC, and are not present in
the second most-common type of lung cancer, SCC (6). Thus, targeted agents developed for
lung AC are largely ineffective against lung SCC (7).
Lung SCC accounts for 45% of NSCLC, and is therefore
a main cause of lung cancer mortality. Lung SCC is different from
AC in terms of its clinical features, response to therapies, and,
most importantly, its genetic-variation profiles. Research on the
molecular mechanisms of lung SCC is limited with few encouraging
outcomes. Previous candidate-gene studies of lung SCC reported
recurring mutations in several genes including TP53,
NFE2L2, KEAP1, BAI3, FBXW7,
GRM8, MUC16, RUNX1T1, STK11 and
ERBB4(8,9). Other recent data showed that lung SCC
with FGFR1 amplification and DDR1 mutations would be
responsive to targeted agents (10–12).
We performed whole-exome sequencing of lung SCC
tissue and adjacent normal lung tissue from one patient to identify
new mutations involved in lung SCC tumorigenesis. We annotated our
results by comparing them with those of previous matched
tumor/normal sequencing studies in the Catalogue of Somatic
Mutation in Cancer (COSMIC) database.
Materials and methods
Sample collection and DNA extraction
We obtained 98 paired tumor-tissue and adjacent
normal-tissue samples including 44 lung SCCs, 49 lung ACs, and 5
LCLCs from patients diagnosed with NSCLC who underwent definitive
surgical resection prior to receiving chemotherapy or radiation at
the First Hospital Affiliated to Bengbu Medical College or at
Ruijin Hospital Affiliated to Shanghai Jiaotong University School
of Medicine. The Ethics Committee of Ruijin Hospital approved the
study and we also provided written informed consent. We performed
all our experiments according to the Helsinki Declaration. We
conducted a pathology review of each sample to establish a
histologic diagnosis. The median age of the patients was 53 years
(range 27–83). We extracted genomic DNA from the tissue samples
using the Automatic Nucleic Acid Isolation System (QuickGene-610L,
Fujifilm Life Science, Tokyo, Japan). We selected tumor and
adjacent normal-tissue samples from one 55-year-old male patient
with lung SCC for whole-exome sequencing.
Targeted sequence capture
We captured the genomic DNA on a NimbleGen 2.1M
human-exome array according to the manufacturer’s protocols
(Roche/NimbleGen). We aimed to capture most of the human exome from
the DNA sample with the NimbleGen chip, which contains 24 Mb CCDS
(~85% of the US National Center for Biotechnology Information CCDS
Database) region across approximately 17,000 genes in 34 Mb
targeted nucleotides. The DNA was sheared by sonication and the
adaptors ligated to the fragments. The adaptor-ligated templates
were fractioned by agarose-gel electrophoresis and the fragments
were excised to the desired size. We hybridized the extracted DNA
to the capture array at 42°C using the manufacturer’s buffer. The
array was washed twice at 47.5°C and three more times at room
temperature (20–25°C) with the manufacturer’s buffers. The bound
genomic DNA was eluted in 125 mM NaOH for 10 min at room
temperature. The selected DNA fragments were amplified by
ligation-mediated PCR, purified and sequenced on the Illumina
platform.
The single-nucleotide variants (SNVs) and INDELs
discovered by the whole-exome sequencing was confirmed by
sequencing the PCR amplification with specific primers on ABI3703
(data no shown).
Alignment, SNV/INDEL calling and quality
control
We aligned the paired-end reads to the reference
human genome (hg19, http://genome.ucsc.edu/) using third-party software,
BWA, with the default parameters. The average sequencing depth of
the case and control samples was more than 50X, and the coverage of
the target area was approximately 80% (data no shown).
Approximately 70% of the nucleotides within the coding region were
covered by at least 10 different reads.
We re-aligned the INDEL regions of the bam file
using GATK software (version 1.1–30). The SNVs and INDELs were
extracted using the unified genotyper function in accordance with
the default parameters. To call an SNV or an INDEL, the mapping
quality had to be no less than 40, the mutation had to be measured
at least five times, and the allelic heterozygosity had to be
>12.5%.
Mutation annotation based on COSMIC
We confirmed the mutations by ABI 3730 sequencing
and annotated them using the COSMIC database. The latest version of
the COSMIC database contains 14,819 articles on tumor
somatic-mutation research, including 2,556 whole-genome sequencing
studies of tumor tissues which scanned 22,170 genes for mutations,
and a total of 773,098 tissue samples. The database contains
405,271 mutation sites with 224,649 single-site mutations (there is
no reproducible variation in the tumor samples), 8,931 fusion-gene
variants, and 7,503 genomic rearrangements.
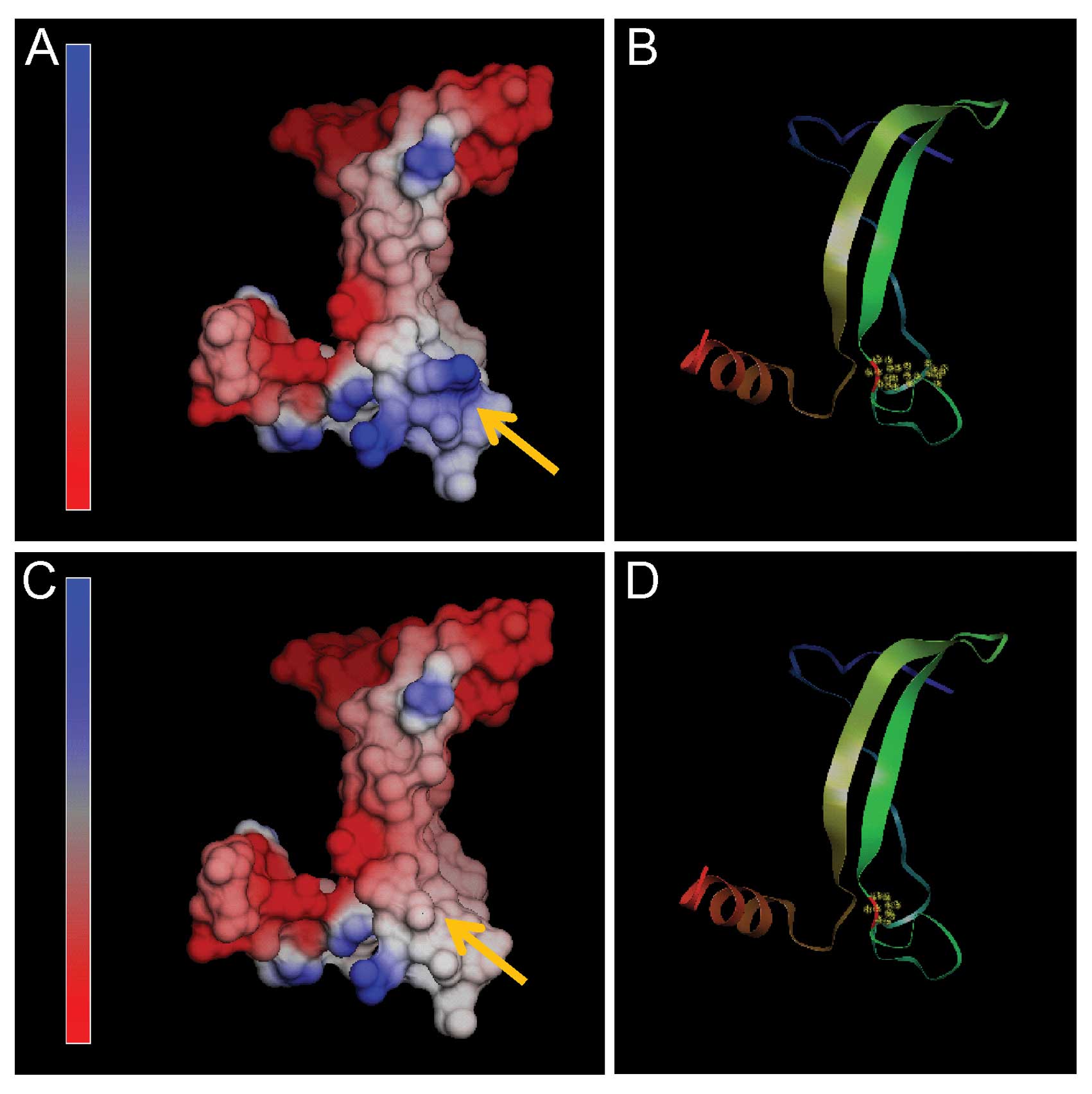
Molecular modeling of TP53
To further investigate the influence of the R249S
mutation on the TP53 structure, a three-dimensional computer model
was constructed with the NOC program. TP53 (residues 219–292) was
modeled with the SWISS-MODEL software (http://swissmodel.expasy.org/) using the crystal
structure of human TP53 (PDB accession code 2qxa, chain B) as a
template (13).
Validation of sequencing results
Four genes (EP300, CADM2, CEP63
and MAP3K3) that harbored amino acid replacements in the
whole-exome sequencing sample were selected based on their
functions and sequenced their exons and exon/intron junctions in
the 98 paired lung cancer/normal tissue samples. EP300 and
MAP3K3 are, respectively, involved in the TP53 and RAS
singnal pathway, which plays an important role in the pathogenesis
of lung cancer. CEP63 binds to and recruits Cdk1 to
centrosomes, and thus regulates mitotic entry (14). Although the function of
CADM2 remained elusive, it has been reported that CADM2 was
recurrently disrupted in prostate cancer (15).
Results
Identification of somatic mutations from
lung SCC
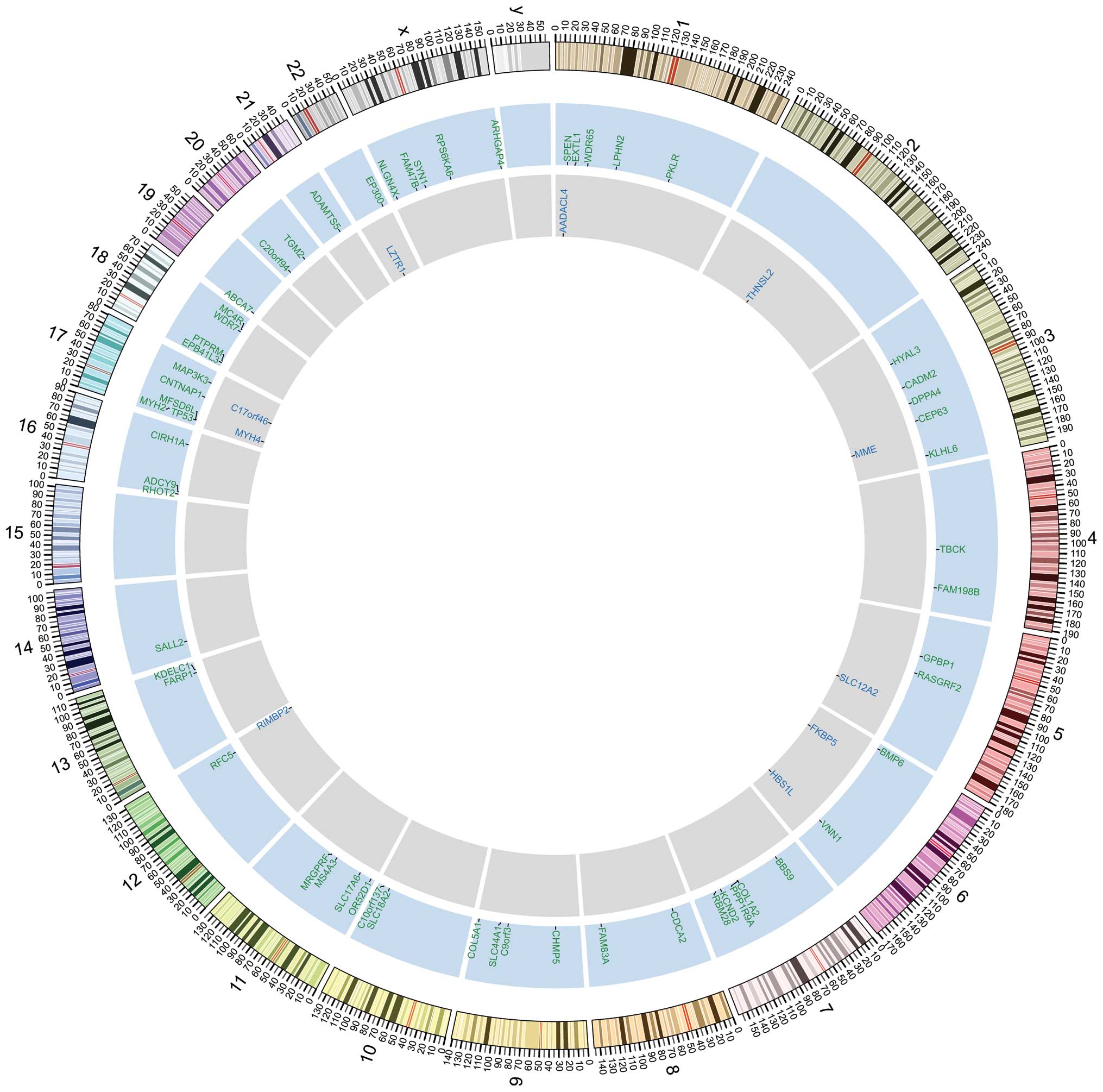
By comparing the whole-exome sequencing data between
the tumor and normal lung tissues from a single patient with lung
SCC, we identified 293 somatic SNVs and 62 INDELs (29 deletions and
33 insertions) (Fig. 1). The
majority of the SNVs were located in inter-genic regions or
introns. We identified 101 SNVs, including 77 non-synonymous SNVs
(67 missense mutations and 10 nonsense mutations) and 11 INDELs, in
the coding regions of genes (data no shown). We also found four
SNVs in splicing sites (within three nucleotides of a splicing
adaptor or receptor) (data no shown).
Confirmation of the somatic
non-synonymous variants in lung SCC
We designed specific primers to verify the 77
non-synonymous SNVs, 11 INDELS, and 4 splicing-site mutations by
sequencing on ABI3730. We confirmed 51 missense mutations, 10
nonsense mutations and 1 splicing-site mutation to be somatic
mutations in the lung SCC tissue by ABI 3730 sequencing (Table I). We also verified 10 of the 11
INDELs as somatic mutations in lung SCC tissue (Table I and Fig. 1).
 | Table IThe 62 confirmed somatic SNVs and 10
INDELs in lung cancer tissues and the effect of missense mutation
on protein function. |
Table I
The 62 confirmed somatic SNVs and 10
INDELs in lung cancer tissues and the effect of missense mutation
on protein function.
| Chromosome | Positiona | Exon | Wild-type
sequence | Mutant
sequence | Amino acid
variation | Mutation type | Certification | SIFT | Gene |
|---|
| 1 | 16258094 | 11 | GCA | CCA | A1787P | Missense | Y | Tolerated | SPEN |
| 1 | 26357058 | 4 | GTG | GCG | V358A | Missense | Y | Tolerated | EXTL1 |
| 1 | 43651012 | 5 | GAG | GAC | E318D | Missense | Y | Tolerated | WDR65 |
| 1 | 82431854 | 10 | CAG | CAC | Q693H | Missense | Y | Damagingb | LPHN2 |
| 1 | 119467295 | 4 | AAC | GAC | N117D | Missense | N | Not scored | TBX15 |
| 1 | 155263101 | 9 | CGG | TGG | R435W | Missense | Y | Not scored | PKLR |
| 2 | 48809658 | 2 | CAG | CGG | Q629R | Missense | Y | Tolerated |
STON1-GTF2A1L |
| 2 | 173429766 | 5 | ATA | ACA | I219T | Missense | N | Damaging | PDK1 |
| 3 | 50332472 | 2 | GCC | TCC | A188S | Missense | Y | Not scored | HYAL3 |
| 3 | 85851331 | 2 | TTT | ATT | F68I | Missense | Y | Tolerated | CADM2 |
| 3 | 109049529 | 5 | GGG | GTG | G174V | Missense | Y | Not scored | DPPA4 |
| 3 | 134269082 | 12 | GAC | TAC | D454Y | Missense | Y | Damaging | CEP63 |
| 3 | 164758812 | 18 | CGC | CAC | R692H | Missense | N | Not scored | SI |
| 3 | 183211938 | 5 | GGA | TGA | G427_ | Nonsense | Y | Not scored | KLHL6 |
| 3 | 197597029 | 17–18 | - | - | - | Splice | N | - | LRCH3 |
| 4 | 23815373 | 8 | TGT | TAT | C578Y | Missense | N | Not scored |
PPARGC1A |
| 4 | 107154130 | 17 | TGG | TTG | W535L | Missense | Y | Not scored | TBCK |
| 4 | 159052121 | 5 | CGC | CAC | R398H | Missense | Y | Not scored | FAM198B |
| 4 | 177084348 | 23 | GCA | GAA | A989E | Missense | N | Damaging | WDR17 |
| 5 | 56526793 | 3 | GGA | TGA | G69_ | Nonsense | Y | N/A | GPBP1 |
| 5 | 80511767 | 24 | CTT | TTT | L1143F | Missense | Y | Damaging | RASGRF2 |
| 6 | 7862569 | 4 | GGC | TGC | G348C | Missense | Y | Damaging | BMP6 |
| 6 | 71508430 | 6 | ACA | AGA | T189R | Missense | N | Tolerated | SMAP1 |
| 6 | 133014234 | 4 | TCA | TAA | S252_ | Nonsense | Y | Not scored | VNN1 |
| 7 | 33312787 | 8 | TGT | TCT | C289S | Missense | Y | Tolerated | BBS9 |
| 7 | 94057137 | 49 | AGA | GGA | R1156G | Missense | Y | Damaging | COL1A2 |
| 7 | 94897906 | 13 | GCA | CCA | A904P | Missense | Y | Damaging | PPP1R9A |
| 7 | 119915111 | 1 | CGA | CTA | R142L | Missense | Y | Not scored | KCND2 |
| 7 | 127954893 | 17 | GAG | TAG | E657_ | Nonsense | Y | Not scored | RBM28 |
| 7 | 143018515 | 4 | TGG | TAG | W164_ | Nonsense | N | N/A | CLCN1 |
| 8 | 25325856 | 6 | GTA | GCA | V221A | Missense | Y | Tolerated | CDCA2 |
| 8 | 87645122 | 10–11 | - | - | - | Splice | N | - | CNGB3 |
| 8 | 124195471 | 1 | GAG | GAT | E125D | Missense | Y | Tolerated | FAM83A |
| 9 | 21206945 | 1 | TCC | TTC | S51F | Missense | N | Not scored | IFNA10 |
| 9 | 33280855 | 8 | TAG | CAG | _220Q | Missense | Y | Not scored | CHMP5 |
| 9 | 97563158 | 4 | CAA | CGA | Q413R | Missense | Y | Tolerated | C9orf3 |
| 9 | 104187214 | 8 | CGG | TGG | R304W | Missense | N | Not scored | ALDOB |
| 9 | 108061571 | 2 | ATC | ACC | I36T | Missense | Y | Tolerated | SLC44A1 |
| 9 | 137710511 | 55 | GGC | CGC | G1414R | Missense | Y | Damaging | COL5A1 |
| 10 | 119013997 | 6 | ATG | ACG | M230T | Missense | Y | Damaging | SLC18A2 |
| 10 | 127409947 | 2 | CTT | TTT | L95F | Missense | Y | Tolerated |
C10orf137 |
| 11 | 674771 | 10 | CTG | CAG | L423Q | Missense | N | Not scored | DEAF1 |
| 11 | 5510732 | 1 | CGC | TGC | R266C | Missense | Y | Damaging | OR52D1 |
| 11 | 6023708 | 1 | TGT | TAT | C224Y | Missense | N | Not scored | OR56A4 |
| 11 | 14991481 | 3 | AGC | ATC | S76I | Missense | N | - | CALCA |
| 11 | 22396340 | 9 | ATT | TTT | I361F | Missense | Y | Tolerated | SLC17A6 |
| 11 | 55432790 | 1 | AGT | GGT | S50G | Missense | N | Damaging | OR4C6 |
| 11 | 59830067 | 3 | GGT | TGT | G95C | Missense | Y | Damaging | MS4A3 |
| 11 | 68773662 | 3 | GCG | GTG | A39V | Missense | Y | Not scored | MRGPRF |
| 11 | 70256067 | 5–6 | - | - | - | Splice | Y | - | CTTN |
| 12 | 118462797 | 6 | CAT | CGT | H188R | Missense | Y | Tolerated | RFC5 |
| 13 | 99098929 | 26 | CCC | ACC | P972T | Missense | Y | Damaging | FARP1 |
| 13 | 103438665 | 9 | GAG | TAG | E470_ | Nonsense | Y | Not scored | KDELC1 |
| 14 | 21992519 | 2 | AGT | ATT | S448I | Missense | Y | Not scored | SALL2 |
| 16 | 718670 | 4 | GAC | TAC | D65Y | Missense | Y | Damaging | RHOT2 |
| 16 | 4016671 | 11 | AGC | ATC | S1056I | Missense | Y | Not scored | ADCY9 |
| 16 | 69170741 | 3 | GGA | GTA | G101V | Missense | Y | Damaging | CIRH1A |
| 17 | 7577534 | 7 | AGG | AGT | R249S | Missense | Y | Not scored | TP53 |
| 17 | 7950699 | 11–12 | - | - | - | Splice | Y | - | ALOX15B |
| 17 | 8701111 | 1 | GGG | GAG | G443E | Missense | Y | Not scored | MFSD6L |
| 17 | 10426681 | 38 | GAG | TAG | E1841_ | Nonsense | Y | Not scored | MYH2 |
| 17 | 40821598 | 12 | TTG | TTC | L685F | Missense | N | Not scored | PLEKHH3 |
| 17 | 40844603 | 17 | GAC | AAC | D873N | Missense | Y | Tolerated | CNTNAP1 |
| 17 | 56811546 | 9 | ACC | AAC | T365N | Missense | N | Tolerated | RAD51C |
| 17 | 61771046 | 17 | CGG | CTG | R628L | Missense | Y | Damaging | MAP3K3 |
| 18 | 5395661 | 20 | CAG | GAG | Q1007E | Missense | Y | Not scored | EPB41L3 |
| 18 | 7949302 | 6 | CGC | TGC | R263C | Missense | Y | Tolerated | PTPRM |
| 18 | 54424249 | 15 | GCC | TCC | A809S | Missense | Y | Tolerated | WDR7 |
| 18 | 58038807 | 1 | GCC | GAC | A259D | Missense | Y | Not scored | MC4R |
| 19 | 1059027 | 40 | TAC | TAG | Y1802_ | Nonsense | Y | N/A | ABCA7 |
| 20 | 10601998 | 7 | GCT | TCT | A148S | Missense | Y | Tolerated |
C20orf94 |
| 20 | 18296358 | 4 | CAC | CGC | H287R | Missense | N | Damaging | ZNF133 |
| 20 | 36770575 | 7 | CGC | AGC | R296S | Missense | Y | Not scored | TGM2 |
| 21 | 28327109 | 2 | GTG | CTG | V396L | Missense | Y | Not scored | ADAMTS5 |
| 22 | 41568590 | 28 | GAA | AAA | E1514K | Missense | Y | Damaging | EP300 |
| X | 5821872 | 5 | GCC | ACC | A283T | Missense | Y | Not scored | NLGN4X |
| X | 34962529 | 1 | TAC | TAA | Y527_ | nonsense | Y | N/A | FAM47B |
| X | 47432323 | 13 | GAG | GAC | E686D | Missense | Y | Not scored | SYN1 |
| X | 83362646 | 13 | GCA | ACA | A366T | Missense | Y | Not scored | RPS6KA6 |
| X | 96136620 | 5 | CAA | GAA | Q164E | Missense | N | Tolerated | DIAPH2 |
| X | 153175487 | 19 | GAG | TAG | E777_ | Nonsense | Y | Not scored | ARHGAP4 |
|
| 1 | 12711337 | 2 | GC | G | NA | Frameshift | Y | Not scored | AADACL4 |
| 2 | 88472701 | 2 | CACGGGT
CAACTTT | C | NA | Frameshift | Y | Not scored | THNSL2 |
| 3 | 154802107 | 2 | AC | A | NA | Frameshift | Y | Not scored | MME |
| 5 | 127474317 | 8 | CG | C | NA | Frameshift | Y | Not scored | SLC12A2 |
| 6 | 35610514 | 2 | C | CT | NA | Frameshift | Y | Not scored | FKBP5 |
| 6 | 135314894 | 8–9 | AC | A | NA | Splice-5 | Y | - | HBS1L |
| 12 | 130898840 | 14 | GC | G | NA | Frameshift | Y | Not scored | RIMBP2 |
| 17 | 10348353 | 37 | AT | A | NA | Frameshift | Y | Not scored | MYH4 |
| 17 | 43332710 | 4 | AGG | A | NA | Frameshift | Y | Not scored |
C17orf46 |
| 18 | 47091805 | 2 | GC | G | NA | Frameshift | N | Not scored | LIPG |
| 22 | 21346634 | 10 | GC | G | NA | Frameshift | Y | Not scored | LZTR1 |
We further analyzed the 51 confirmed missense
mutations by searching the SIFT database (16) to predict their effects on protein
structure. We found that 15 of the missense mutations could
dramatically affect protein functions (Table I).
Comparison with COSMIC database
In the samples of lung cancer tissues and other
types of solid tumors in the COSMIC database, we found previously
identified mutations in all of the genes, except for MRGPRF,
containing the 62 SNVs and 10 INDELs confirmed in our study by ABI
3730 sequencing. Fifteen of the genes with non-synonymous SNVs and
one with an INDEL (LZTR1) had previously identified
mutations in at least one sample of different pathological types of
lung cancer; four of those genes (LPHN2, TP53,
MYH2 and TGM2) were mutated in close to or more than
10% of the tumor tissues available in the COSMIC database.
TP53 and LPHN2 were sequenced in more than 500 tumor
samples, and their mutation frequencies were 18.3% (12,142/66,304)
and 8.32% (49/589), respectively (Table II). TP53, a well-known
oncogene that plays an important role in lung cancer pathogenesis,
is mutated in 62.3% (1,404/2,252) of the lung cancer tissue samples
in the COSMIC database.
| Table IIGenes with reoccurring mutations in
lung cancer tissues in the COSMIC database. |
Table II
Genes with reoccurring mutations in
lung cancer tissues in the COSMIC database.
| Chromosome | Gene | ACC | SCC | SCLC | Solid tumors | Hematol.
cancer |
|---|
| 1 | SPEN | - | 1/11 | - | - | - |
| 1 | LPHN2 | 1/57 | 2/63 | - | 44/465 | 2/4 |
| 2 | PDK1 | 1/253 | 2/70 | - | 4/586 | - |
| 3 | CADM2 | - | 1/10 | - | 10/304 | 2/2 |
| 4 |
PPARGC1A | - | 2/10 | - | 14/308 | - |
| 10 |
C10orf137 | 1/57 | 1/63 | - | 22/557 | - |
| 11 | MS4A3 | 1 | - | - | 6/96 | - |
| 17 | TP53 | 952/1,386 | 452/866 | - | 8,756/58,462 | 1,982/5,590 |
| 17 | MYH2 | - | 1/1 | 36/218 | - | |
| 17 | CNTNAP1 | - | 1/63 | - | 19/404 | - |
| 18 | MC4R | - | 1/63 | - | 4/392 | - |
| 20 | TGM2 | - | - | 1/1 | 13/166 | - |
| 22 | EP300 | - | 1/63 | - | 57/1,495 | 28/525 |
| X | RPS6KA6 | 1/16 | in | LCC | 9/292 | - |
| X | DIAPH2 | 1/1 | - | - | 18/132 | - |
| 22 | LZTR1 | 2/188 | - | - | 14/104 | - |
We also identified two missense mutations in
C10orf137 and MS4A3, respectively, that also appear
in different lung cancer tissues in the COSMIC database. It is
worth noting that the mutation in C10orf137 was investigated
in more than 500 solid tumor tissues and its frequency is
approximately 3.55% (24/677) in COSMIC database (Table II). We identified a somatic
mutation in EP300 and found that 4.2% (85/2,020) of the
tumor tissues in the COSMIC database also had mutations in
EP300 (Table II).
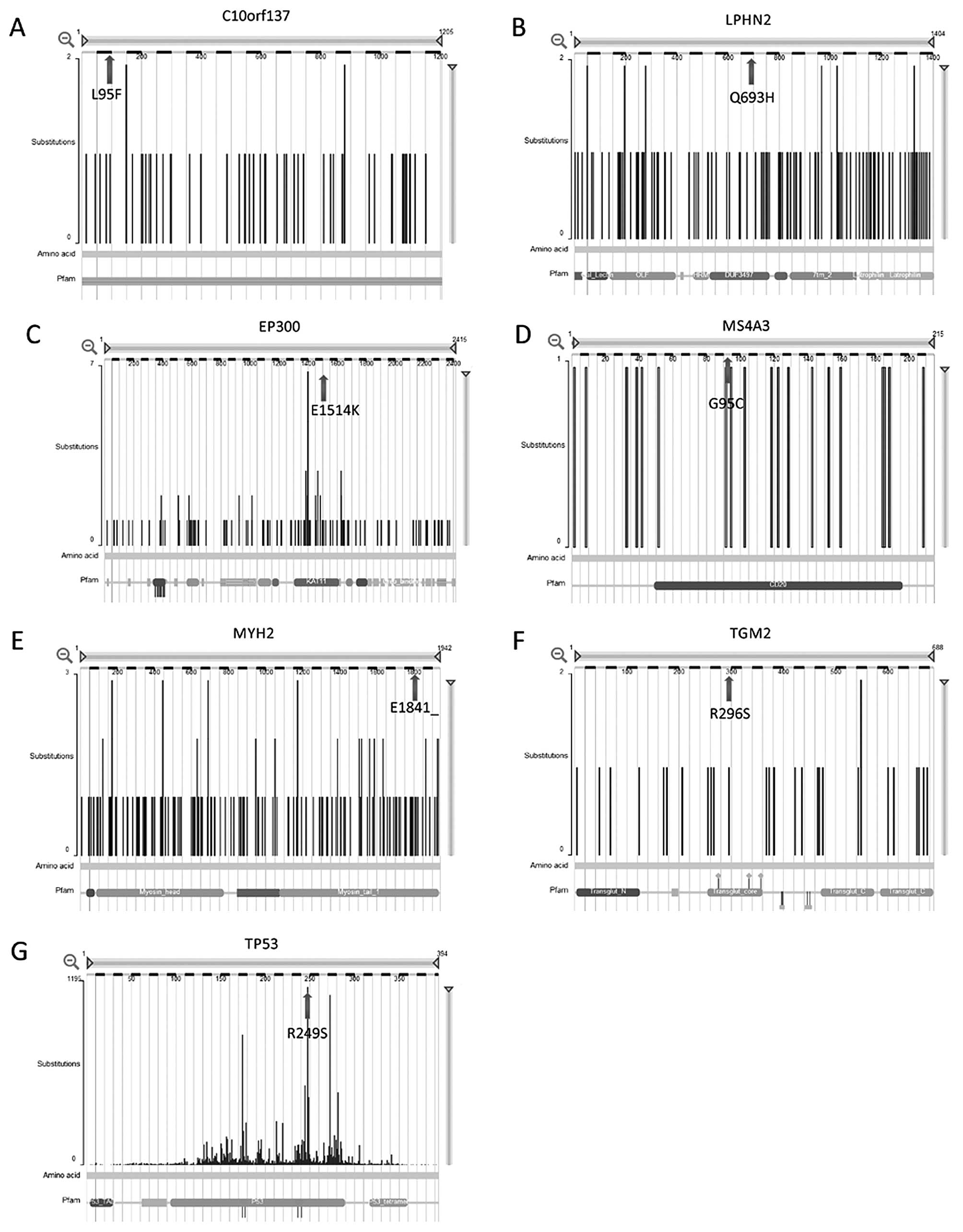
Based on the comparisons of our whole-exome
sequencing results with the previously identified mutations in the
COSMIC database, we identified seven genes (LPHN2,
TP53, MYH2, TGM2, C10orf137,
EP300 and MS4A3) as possible drivers of lung cancer
pathogenesis (Table II and
Fig. 2).
Computer modeling and analysis of
TP53
Our study is the first, however, to identify a
C>A substitution in squamous cell lung cancer tissue changing
Arg to Ser at amino acid position 249 (R249S) in TP53. By molecular
modeling, a charged basic amino acid (Arg) was replaced by an
neutral amino acid (Ser) at codon 249, which caused an abnormal
electrostatic-charge distribution in the DNA-binding domain of TP53
(Fig. 3).
Validation of sequencing results
We sequenced all of the exons of four genes
(MAP3K3, CEP63, CADM2 and EP300) in 98
additional lung cancer samples; including 44 lung SCCs, 49 ACs, and
5 LCLCs; and found no mutations in the coding regions. We found a
deletion of 2–4 cytosine residues in the 5′UTR of CEP63 in
three of the samples, but the mutations did not change the protein
sequences. We also identified a C>G variant located at
nucleotide position 3207 of MAP3K3 (NM_2033351) in one
patient; the variant was located in the 3′UTR, but did not change
the protein sequence.
Discussion
We used whole-exome sequencing to identify 72
somatic mutations, including 62 SNVs (51 missense mutations, 10
nonsense mutations, and 1 splicing-site mutation) and 10 INDELs, in
the coding regions of different genes from a single case of lung
SCC. We found somatic mutations in 71 of the genes in at least one
additional tumor sample in the COSMIC database. We found mutations
in 16 of the genes in at least one additional lung cancer patient.
Four genes (LPHN2, TP53, MYH2 and TGM2)
were mutated in approximately 10% of the tumor samples in the
COSMIC database.
We found the most mutations in TP53: 68.7%
(952/1,386) of lung AD cases and 52.2% (452/866) of lung SCC cases.
Although TP53 is frequently mutated in tumor tissues from
patients with lung cancer, our study is the first to describe the
R249S somatic missense mutation in lung cancer tissues. SIFT
analysis showed that the R249S mutation in TP53 could
dramatically influence the structure of the TP53 protein (16). It worth noting that the R249S
mutation in TP53 is frequently found in HBV-induced
hepatic-cell carcinoma, accounting for 90% of the TP53 mutations
identified in liver cancer (17).
In hepatocellular-carcinoma cell lines, the R249S mutation
abolishes the capacity for TP53 to bind p53 response elements and
trans-activate p53 target genes. Moreover, in a p53-null Hep3B cell
line that constitutively expresses both the R249S variant of TP53
and the hepatitis-B virus antigen HBx (PLC/PRF/5), the silencing of
either R249S TP53 or HBx by RNA interference inhibited cellular
proliferation, but without additive effects when both genes were
silenced (17). Taken together
with the previous results, our results suggest that the R249S
mutation in TP53 may be play a key role in lung cancer
pathogenesis.
LPHN2 was previously sequenced in more than
500 tumor samples and found to be mutated in 9.46% (44/465) of
solid-tumor samples. Moreover, somatic mutations in LPHN2
occurred in 3.17% of lung SCC (2/63) samples in the COSMIC
database. LPHN2 encodes a member of the latrophilin
subfamily of G-protein coupled receptors (GPCR), and genome-wide
association analysis found a significant association between SNVs
of LPHN2 and paclitaxel sensitivity in NCI60 cancer cell
lines (18).
MYH2 and TGM2 were mutated in 16.5%
(36/218) and 7.8% (13/166) of solid tumors; neither mutation,
however, was previously investigated in SCC samples.
We identified two missense mutations in
C10orf137 and MS4A3, respectively; both were
previously identified in different lung cancer tissues. Mutations
in C10orf137 were previously investigated in more than 500
solid-tumor samples and found in 3.55% (24/677) of the samples.
Recently, Gylfe et al identified one missense germ-line
mutation in C10orf137 in 45 familial patients with
colorectal cancers, while none of the 890 population-matched
healthy controls had the same mutation (19). The function of C10orf137,
however, is still unknown. MS4A is a member of the
four-transmembrane protein family. MS4A proteins execute diverse
functions, acting as cell-surface signaling molecules and
intracellular adapter proteins. Tissue microarray analysis showed
MS4A3 expression in a wide variety of ACs including breast,
prostate, and ovarian cancers (20). Moreover, previous studies showed
that MS4A3 forms a functionally relevant complex with
cyclin-dependent kinase-associated phosphatase and CDK2 (21), suggesting that MS4S3 may be a novel
modulator of the cell cycle. Further study is needed to explain the
role of MS4A3 in lung cancer pathogenesis.
We identified a somatic mutation in EP300
that was previously identified in 4.2% (85/2,020) of the tumor
samples in the COSMIC database. Recurrent mutations clustered
around the histone acetyltransferase domain in EP300 were
recently described in small-cell lung cancers (22). EP300 plays an important role in
cell proliferation and differentiation by regulating gene
transcription via chromatin remodeling (23–25).
EP300 is also an important modulator of the TP53 signaling pathway;
it helps to maintain TP53 stability by regulating the
ubiquitination and degradation of TP53 through both MDM2-dependent
and MDM2-independent mechanisms (26,27).
Moreover, EP300 is required for the TP53-mediated transactivation
of target genes because of its co-activator function and its
acetylation of histones (28–30).
Together with the previous results, our data suggest that EP300 may
be a driver gene in lung cancer tumorigenesis.
Several recent whole-genome or exome-sequencing
studies aimed at characterizing the genomic and epigenomic
landscapes of different histopathological types of lung cancer
(ACC, SCC and small-cell cancer) (31–34).
The results included a large number and variety of DNA alterations
with a mean of more than 150 exonic non-synonymous mutations per
lung cancer type (31–34). Analyses by different algorithms
identified some genes with significantly elevated mutational
frequencies in different histological types of lung cancer
(P<0.05; false-discovery rate ≤0.1). Among these genes,
TP53 was confirmed as a tumorigenesis gene and had the
highest mutational frequency (29–81%) in all of the independent
studies. Four other genes; EGFR, KRAS, KEAP1
and RB1; were also implicated as important tumorigenesis
genes in two independent studies (31–34).
A somatic mutation in KEAP1 was repeatedly identified in
independent studies performed on cohorts of lung SCCs and lung ACs
(31,32). Somatic mutations in RB1 were
confirmed in patients with SCLC and lung SCCs (31,33).
Most of the significantly mutated genes, however, were identified
in only one cohort (31–34). These results suggest that genomic
variants in lung cancer tissues are complex; the somatic mutations
in distinct genes underscore the differences between subgroups of
lung cancer, even within a single histological type. More
whole-exome sequencing studies with large sample sizes are needed
to increase the available somatic-mutation data.
In summary, our results show that whole-exome
sequencing is an effective way to detect novel mutations related to
lung cancer. Our study indicates seven genes, TP53,
EP300, LPHN2, C10orf137, MYH2,
TGM2 and MS4A3, that may be drivers of lung cancer
tumorigenesis.
Acknowledgements
We thank all the patients who participated in this
study. This study was supported in part by the National Natural
Science Foundation of China (81071925 and 30900503) and the
Shanghai Science and Technology Committee (10ZR1418300).
References
|
1
|
Borczuk AC, Toonkel RL and Powell CA:
Genomics of lung cancer. Proc Am Thorac Soc. 6:152–158. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Satouchi M, Negoro S, Funada Y, et al:
Predictive factors associated with prolonged survival in patients
with advanced non-small-cell lung cancer (NSCLC) treated with
gefitinib. Br J Cancer. 96:1191–1196. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shaw AT, Yeap BY, Mino-Kenudson M, et al:
Clinical features and outcome of patients with non-small-cell lung
cancer who harbor EML4-ALK. J Clin Oncol. 27:4247–4253. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ji H, Wang Z, Perera SA, et al: Mutations
in BRAF and KRAS converge on activation of the mitogen-activated
protein kinase pathway in lung cancer mouse models. Cancer Res.
67:4933–4939. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Felip E, Gridelli C, Baas P, Rosell R and
Stahel R; Panel Members. Metastatic non-small-cell lung cancer:
consensus on pathology and molecular tests, first-line,
second-line, and third-line therapy. Ann Oncol. 22:1507–1519. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rekhtman N, Paik PK, Arcila ME, et al:
Clarifying the spectrum of driver oncogene mutations in
biomarker-verified squamous carcinoma of lung: lack of EGFR/KRAS
and presence of PIK3CA/AKT1 mutations. Clin Cancer Res.
18:1167–1176. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
James J, Ruggeri B, Armstrong RC, et al:
CEP-32496: a novel orally active BRAF(V600E) inhibitor with
selective cellular and in vivo antitumor activity. Mol Cancer Ther.
11:930–941. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shibata T, Ohta T, Tong KI, Kokubu A,
Odogawa R, Tsuta K, Asamura H, Yamamoto M and Hirohashi S: Cancer
relatedmutations in NRF2 impair its recognition by Keap1-Cul3 E3
ligase and promote malignancy. Proc Natl Acad Sci USA.
105:13568–13573. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kan Z, Jaiswal BS, Stinson J, et al:
Diverse somatic mutation patterns and pathway alterations in human
cancers. Nature. 466:869–873. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dutt A, Ramos AH, Hammerman PS, et al:
Inhibitor-sensitive FGFR1 amplification in human non-small cell
lung cancer. PLoS One. 6:e203512011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hammerman PS, Sos ML, Ramos AH, et al:
Mutations in the DDR2 kinase gene identify a novel therapeutic
target in squamous cell lung cancer. Cancer Discov. 1:78–89. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Weiss J, Sos ML, Seidel D, et al: Frequent
and focal FGFR1 amplification associates with therapeutically
tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl
Med. 2:62ra932010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Arnold K, Bordoli L, Kopp J and Schwede T:
The SWISS-MODEL Workspace: A web-based environment for protein
structure homology modeling. Bioinformatics. 22:195–201. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Löffler H, Fechter A, Matuszewska M, et
al: Cep63 recruits Cdk1 to the centrosome: implications for
regulation of mitotic entry, centrosome amplification, and genome
maintenance. Cancer Res. 71:2129–2139. 2011.PubMed/NCBI
|
|
15
|
Berger MF, Lawrence MS, Demichelis F, et
al: The genomic complexity of primary human prostate cancer.
Nature. 470:214–220. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ng PC and Henikoff S: SIFT: predicting
amino acid changes that affect protein function. Nucleic Acids Res.
31:3812–3814. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gouas DA, Shi H, Hautefeuille AH, et al:
Effects of the TP53 p. R249S mutant on proliferation and clonogenic
properties in human hepatocellular carcinoma cell lines:
interaction with hepatitis B virus X protein. Carcinogenesis.
31:1475–1482. 2010. View Article : Google Scholar
|
|
18
|
Eng L, Ibrahim-Zada I, Jarjanazi H, Savas
S, Meschian M, Pritchard KI and Ozcelik H: Bioinformatic analyses
identifies novel protein-coding pharmacogenomic markers associated
with paclitaxel sensitivity in NCI60 cancer cell lines. BMC Med
Genomics. 4:182011. View Article : Google Scholar
|
|
19
|
Gylfe AE, Sirkiä J, Ahlsten M, Järvinen H,
Mecklin JP, Karhu A and Aaltonen LA: Somatic mutations and germline
sequence variants in patients with familial colorectal cancer. Int
J Cancer. 127:2974–2980. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kutok JL, Yang X, Folkerth R and Adra CN:
Characterization of the expression of HTm4 (MS4A3), a cell cycle
regulator, in human peripheral blood cells and normal and malignant
tissues. J Cell Mol Med. 15:86–93. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Donato JL, Ko J, Kutok JL, et al: Human
HTm4 is a hematopoietic cell cycle regulator. J Clin Invest.
109:51–58. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Peifer M, Fernández-Cuesta L, Sos ML, et
al: Integrative genome analyses identify key somatic driver
mutations of small-cell lung cancer. Nat Genet. 44:1104–1110. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ogryzko VV, Schiltz RL, Russanova V,
Howard BH and Nakatani Y: The transcriptional coactivators p300 and
CBP are histone acetyltransferases. Cell. 87:953–959. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kawasaki H, Eckner R, Yao TP, Taira K,
Chiu R, Livingston DM and Yokoyama KK: Distinct roles of the
co-activators p300 and CBP in retinoic-acid-induced F9-cell
differentiation. Nature. 393:284–289. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yao TP, Oh SP, Fuchs M, et al: Gene
dosagedependent embryonic development and proliferation defects in
mice lacking the transcriptional integrator p300. Cell. 93:361–372.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Grossman SR, Deato ME, Brignone C, Chan
HM, Kung AL, Tagami H, Nakatani Y and Livingston DM:
Polyubiquitination of p53 by a ubiquitin ligase activity of p300.
Science. 300:342–344. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Grossman SR, Perez M, Kung AL, Joseph M,
Mansur C, Xiao ZX, Kumar S, Howley PM and Livingston DM: p300/MDM2
complexes participate in MDM2-mediated p53 degradation. Mol Cell.
2:405–415. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lill NL, Grossman SR, Ginsberg D, DeCaprio
J and Livingston DM: Binding andmodulation of p53 by p300/CBP
coactivators. Nature. 387:823–827. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Espinosa JM and Emerson BM:
Transcriptional regulation by p53 through intrinsic DNA/chromatin
binding and site-directed cofactor recruitment. Mol Cell. 8:57–69.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Avantaggiati ML, Ogryzko V, Gardner K,
Giordano A, Levine AS and Kelly K: Recruitment of p300/CBP in
p53-dependent signal pathways. Cell. 89:1175–1184. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cancer Genome Atlas Research Network.
Comprehensive genomic characterization of squamous cell lung
cancers. Nature. 489:519–525. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Imielinski M, Berger AH, Hammerman PS, et
al: Mapping the hallmarks of lung adenocarcinoma with massively
parallel sequencing. Cell. 150:1107–1120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rudin CM, Durinck S, Stawiski EW, et al:
Comprehensive genomic analysis identifies SOX2 as a frequently
amplified gene in small-cell lung cancer. Nat Genet. 44:1111–1116.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Govindan R, Ding L, Griffith M, et al:
Genomic landscape of non-small cell lung cancer in smokers and
never-smokers. Cell. 150:1121–1134. 2012. View Article : Google Scholar : PubMed/NCBI
|