Introduction
Nickel is a ubiquitous environmental metal, widely
used in industrial processes. Nickel compounds are important human
carcinogens. Epidemiologically, chronic and professional exposure
to nickel compounds is associated with increased incidence of
certain human cancer, including lung and nasal cancers (1,2).
Both the soluble and insoluble forms of nickel compounds are
detrimental to health with the insoluble form possesses higher
risk, such as nickel oxide and nickel subsulfide (3,4). In
1990, nickel is classified as established carcinogen to humans
(group 1) by International Agency for Research on Cancer (IARC)
(3,4).
Several mechanisms have been proposed for
nickel-induced carcinogenesis. These include production of reactive
oxygen species (ROS), induction of DNA damage (1,4),
alteration of epigenetic changes such as histone modifications
(5,6), disruption of cellular iron
homeostasis (7), and activation of
oncogenic pathways (8,9). Despite intensive investigation, the
molecular features of nickel-induced oncogenic transformation,
which are essential components in tumor initiation and progression,
remain elusive.
Apoptosis (programmed cell death) is a key mechanism
to maintain normal cell homeostasis, it removes cells that carry
abnormal genetic information and keeps the functional integrity of
organ or cell populations (10).
Aberrant expression of anti-apoptotic protein is a common feature
to many types of malignancy including lung, breast, prostate
cancer, melanoma and leukemia. Their overexpression confers the
cancer cells a growth advantage to escape from physiological
surveillance and promote tumor formation (10,11).
Recent studies also suggest that activation of oncogenic pathways
mediate nickel chloride (NiCl2)-induced epithelial cell
oncogenic transformation accompanied by enhanced Bcl-2, Bcl-xL
protein expression (9).
Bcl-2, and Bcl-xL protein expression has been
implicated in cancer and cell transformation (10,12),
therefore, we hypothesized that their overexpression in transformed
cells may lead to apoptosis resistance contributing to
nickel-induced carcinogenesis. However, it is not clear if a metal
compounds such as nickel might induce apoptosis resistance in
transformed cells and the underlying molecular mechanisms, neither
is it known if these cells are tumorigenic. Therefore, we
investigated the role of Bcl-2, Bcl-xL and ROS scavenging enzyme in
nickel-induced apoptosis resistance and the molecular mechanisms,
which are necessary steps to understand nickel-induced
carcinogenesis.
We demonstrate in this study that increased Bcl-2,
Bcl-xL and ROS scavenging enzyme expression are important in
conferring NiCl2-transformed BEAS-2B cell apoptosis
resistance. The mechanisms are mitochondria-mediated and
caspase-dependent. Transformed cells are also tumorigenic with
accompanied oncogenic protein expression. The results characterize
the oncogenic properties of nickel-transformed human lung
epithelial cells and provide insight into nickel-induced
carcinogenesis.
Materials and methods
Cell culture and reagents
Immortalized normal human bronchial epithelial cell
line, BEAS-2B was purchased from American Type Culture Collection
(ATCC, Manassas, VA). NiCl2-transformed BEAS-2B cells
were isolated from soft agar by low dose NiCl2 exposure
with BEAS-2B cells for 6 months (9). These cells showed
anchorage-independent growth, proliferated faster, but were
morphologically similar to normal BEAS-2B cells (9). BEAS-2B cells that stably express
catalase (OriGene, Rockville, MD), Akt and dominant negative-Akt
(DN-Akt) which is a kinase-dead (K179M) mutant, were generated by
integration of catalase, wild-type Akt (Akt-C), DN-Akt expression
vectors into BEAS-2B cells and selected with G418. Characterization
of these cells has been described before (13,14).
Tissue culture reagents were purchased from Gibco (Invitrogen,
Carlsbad, CA). Cells were maintained in DMEM medium supplemented
with 10% fetal bovine serum and antibiotics at 37°C in a humidified
10% CO2 incubator.
Nickel chloride (NiCl2) was purchased
from Sigma Chemical Company (St. Louis, MO, USA). Polyclonal and
monoclonal antibodies against phospho-Akt at Ser473 (#9275),
phospho-Stat3 (#9145), Bcl-xL (#2764), catalase (#8841), HIF-1α
(#3434), cleaved caspase-3 (#9661), cleaved caspase-7 (#9491),
cleaved PARP (#9542), peroxiredoxin 1 (#8499) were purchased from
Cell Signaling Technology (Danvers, MA). Bcl-2 was from Dako
(Carpinteria, CA). SOD1 (sc-11407), SOD2 (sc-130345), β-actin
(sc-47778), lamin A/C (sc-6215), Akt1 (sc-5298) and NF-κB p50
(sc-7178) were purchased from Santa Cruz Biotechnology (Santa Cruz,
CA).
Assays for apoptosis
Apoptosis was determined by Annexin V/propidium
iodide (PI) staining kit (BD Pharmingen, San Jose, CA). In some
experiments, due to the expression of green fluorescence protein
(GFP) interfering with Annexin V-FITC reading, a double stain
apoptosis detection kit using Hoechst 33342/PI staining (GenScript,
Piscataway, NJ) was chosen to replace Annexin V-FITC. Briefly,
cells (1×106) treated with or without NiCl2
were collected and washed once with cold PBS, they were stained
with 5 μl of Annexin V-FITC or Hoechst 33342 followed by 10 μl of
PI (5 μg/ml) in binding buffer [10 mmol/l HEPES (pH 7.4), 140
mmol/l NaOH, and 2.5 mmol/l CaCl2] for 15 min at room
temperature in dark. The apoptotic cells were determined using a
Becton-Dickinson FACScan cytofluorometer. Both early apoptotic
(Annexin V or Hoechst 33342-positive and PI-negative) and late
apoptotic (Annexin V or Hoechst 33342-positive and PI-positive)
cells were included in cell death determinations.
Western blot analyses
Cell lysates were collected in lysis buffer (50 mM
Tris-HCl, pH 7.4, 1 mM EDTA, 150 mM NaCl, 1% NP-40, 0.25%
Na-deoxycholate, and 1 μg/ml of aprotinin, leupeptin and
pepstatin). Proteins (20 μg) were separated on 10% SDS
polyacrylamide gels. Proteins were subsequently transferred from
gel onto nitrocellulose membrane (Bio-Rad, Hercules, CA). Membranes
were blocked for 1 h at room temperature in Tris-buffered saline
plus 0.01% Tween-20 (TBS-T) in 5% non-fat dry milk (pH 7.4).
Various antibodies were diluted at 1:1,000 in TBS-T with 5% non-fat
dry milk solution, incubated with membrane at 4°C overnight and
washed three times with TBS-T. The secondary antibody, horseradish
peroxidase (HRP)-conjugated antibodies, diluted at 1:2,500 in TBS-T
with 5% non-fat dry milk were incubated with membrane at room
temperature for 2–3 h. Signal was detected with an enhanced
chemiluminescence detection kit (Perkin Elmer Life Sciences,
Boston, MA). In each experiment, either anti-β-actin or anti-lamin
A/C antibody was reprobed to monitor protein loading. Image
quantification was processed by ImageJ software (NIH, Rockville,
MD). In some experiments, mean densitometry values were adjusted
with β-actin and expressed as fold changes over the appropriate
controls.
Quantitative RT-PCR for gene
expression
Total RNA from BEAS-2B cells was extracted using
TRIzol reagents as described previously (15). Reverse transcription of 2 μg of
total cellular RNA was performed in a final volume of 20 μl
containing 5X first strand buffer (Invitrogen), 1 mM of each dNTP,
20 units of placental RNase inhibitor, 5 μM random hexamer and 9
units of Moloney murine leukemia virus reverse transcriptase
(Invitrogen). After incubation at 37°C for 45 min, the samples were
heated for 5 min at 92°C to end the reaction, diluted at 1:4 and
stored at −20°C until PCR. cDNA (2 μl) was subjected to real-time
quantitative PCR using the real-time PCR detection systems
(Bio-Rad, Hercules, CA) with SYBR-Green I (Molecular Probes,
Eugene, OR) as a fluorescent reporter. Threshold cycle number of
duplicate reactions was determined using PCR software and levels of
selected gene mRNA expression were normalized to hypoxanthine
phosphoribosyltransferase (HPRT) levels using the formula
2(Rt-Et); where Rt is the mean threshold
cycle for the reference gene HPRT and Et is the mean
threshold cycle for experimental gene. Data are expressed as
arbitrary units and adjusted to fold changes over the
non-stimulated control cells. Primer sequences are provided in
Table I.
 | Table IPCR primers. |
Table I
PCR primers.
| Gene name | For RT-PCR
(5′→3′) |
|---|
| Bcl-2 | Fwd:
GCCTTCTTTGAGTTCGGTGG |
| Rev:
ATCTCCCGGTTGACGCTCT |
| Bcl-xL | Fwd:
GAGCTGGTGGTTGACTTTCTC |
| Rev:
TCCATCTCCGATTCAGTCCCT |
| Hprt | Fwd:
TTGGAAAGGGTGTTTATTCCTCA |
| Rew:
TCCAGCAGGTCAGCAAAGAA |
Measurement of mitochondrial membrane
potential
Mitochondrial membrane potential (ΔΨm) was monitored
using MitoProbe JC-1 assay kit (Invitrogen).
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine
iodide (JC-1), a lipophilic cationic fluorescence dye, is capable
of selectively entering mitochondria, where it forms monomers, and
emits green fluorescence (FL-1) when ΔΨm is relatively low. At high
ΔΨm, JC-1 aggregates and gives red fluorescence (FL-2) (16). Thus the red and green fluorescence
of JC-1 reflect the change of ΔΨm of the mitochondrial membrane.
Briefly, BEAS-2B or transformed BEAS-2B cells (5×105)
were seeded into 60-mm culture dishes and treated with
NiCl2 for 16 h. Cells were trypsinized, washed in
ice-cold PBS and incubated with 2 μM JC-1 at 37°C for 20 min. Cells
were washed once with PBS and analyzed by FACScan
cytofluorometer.
Caspase activity assay
Caspase activity was assessed using the luminescent
Caspase-Glo® 3/7 assay system (Promega, Madison, WI)
following manufacturer’s instructions. Briefly, BEAS-2B or
transformed BEAS-2B cells were treated with or without
NiCl2 at 1.5 mM for 16 h. Caspase-Glo 3/7 reagent (100
μl) was added into 96-well plates and incubated for 1 h at room
temperature. The luminescence was measured using a Glomax™ 96
microplate luminometer (Promega). Z-VAD-FMK
(carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone),
a cell-permeant pan-caspase inhibitor that irreversibly binds to
the catalytic site of caspase proteases that inhibit induction of
apoptosis was added 30 min before nickel stimulation at 20 μM
solution in DMSO (Promega). Data were collected and expressed as
fold change over the control without NiCl2
treatment.
Small interfering RNA transfection
Transfection procedures followed manufacturer’s
recommended protocol. Small interference RNA (siRNA) for Bcl-2
(sc-29214), Bcl-xL (sc-43630) were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA). Scrambled control and siRNA for
catalase (s-2443) were obtained from Ambion (Austin, TX). Briefly,
siRNA for Bcl-2, Bcl-xL, catalase and controls were incubated with
Lipofectamine™ RNAiMAX in Opti-MEM I medium (Invitrogen) for 20 min
at room temperature. They were then added to cell culture media
without antibiotics. Media were replaced 24 h post-transfection.
Cells were treated with or without NiCl2 for another 24
h. Cell lysates or cells were collected either for western blot or
apoptosis analysis as mentioned above.
Retrovirus infection
GFP labeled p-MIG, p-MIG-Bcl-2, pMIG-Bcl-xL
retroviral vector (17) were kind
gifts from Dr Stanley Korsmeyer, Dana-Farber Cancer Institute, USA
(Addgene, #9044, #3541, #3544). The plasmids were transfected into
293-GPG cell using Lipofectamine 2000 reagent, viruses were
produced in 293-GPG cells with subsequent centrifugation,
filtration and stored at −20°C. BEAS-2B or T-BEAS-2B cells were
then infected with control, Bcl-2, Bcl-xL retroviruses by adding
virus solution into the cells with polybrene for 48 h before they
were used for apoptosis and western blot analysis, virus infection
in cells was monitored by checking GFP under fluorescence
microscope.
Tumor xenograft model
Athymic nude (nu/nu) female mice, 5 weeks old and
weighing about 18 to 20 g were purchased from Jackson Laboratory
(Bar Harbor, MI). Mice were handled following the University of
Kentucky guidelines for care and use of laboratory animals and
approved by the ethical committee. BEAS-2B or transformed BEAS-2B
cells (2×106) were mixed with Matrigel and injected in
both side of flank region in 50 μl volume. Five animals in each
group gave 10 injection sites. Mice were fed normally, the body
weight, and tumor growth was monitored twice weekly until the tumor
size reached 1 × 1 × 1 cm3 in size or 8 weeks post
inoculation.
Statistical analysis
All values are expressed as mean ± standard error
(SEM). Student’s t-test was used to compare the difference between
various controls and NiCl2-treated groups. P-value less
than 0.05 was considered statistically significant.
Results
Transformed BEAS-2B cells have increased
Bcl-2, Bcl-xL protein expression and are resistant to
nickel-induced apoptosis
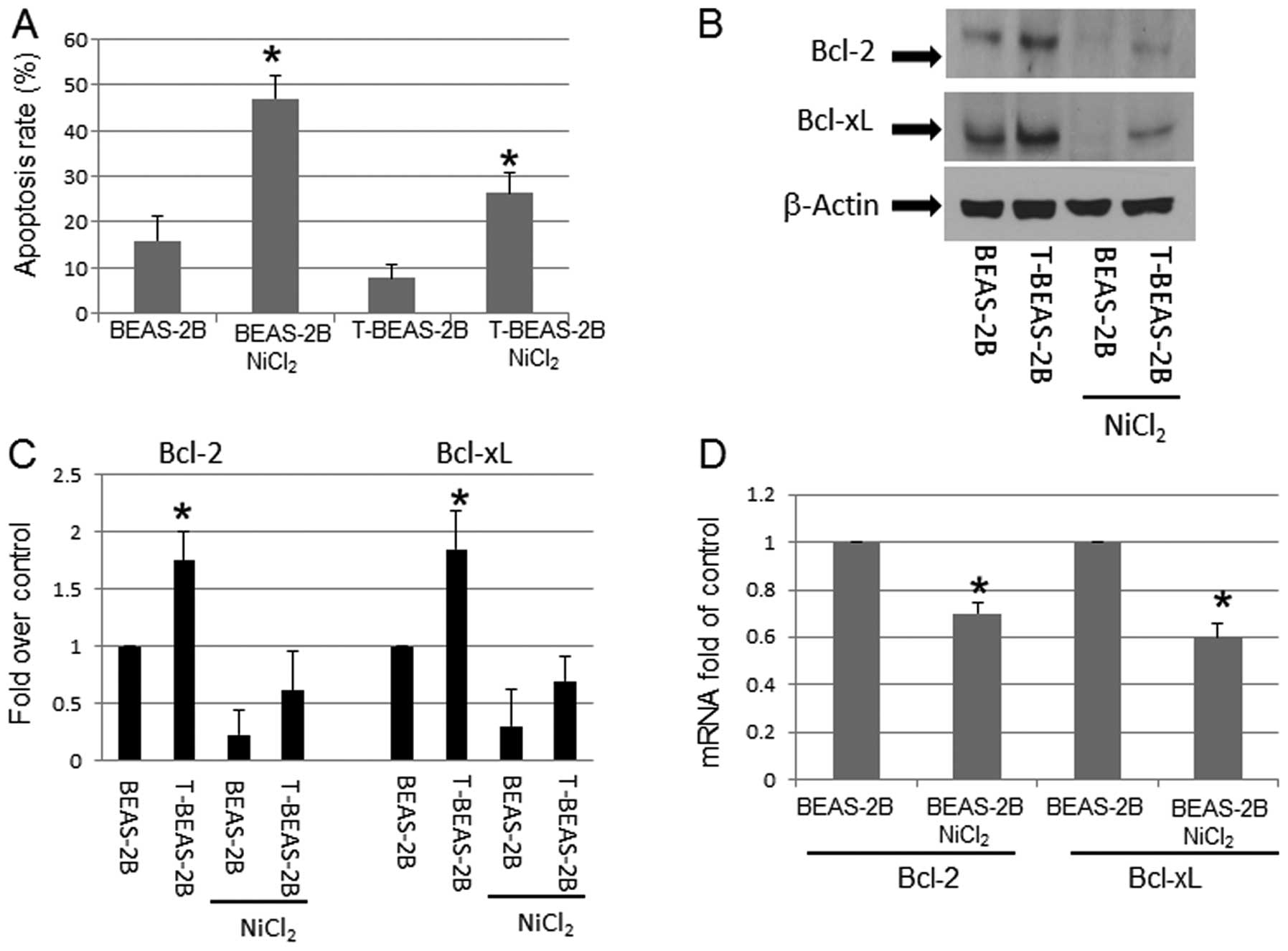
To evaluate Bcl-2, Bcl-xL protein expression and
their role in nickel-induced apoptosis, BEAS-2B and transformed
BEAS-2B (T-BEAS-2B) cells were first treated with or without 0.5, 1
and 1.5 mM of NiCl2 for 24 h for apoptosis induction. At
0.5, and 1 mM doses, only BEAS-2B cells showed apoptosis induction
in 24 h, T-BEAS-2B cell showed little or no apoptosis (data not
shown), therefore, 1.5 mM of NiCl2 was selected for
apoptosis induction in both types of cells.
BEAS-2B and T-BEAS-2B cells were treated with or
without 1.5 mM of NiCl2 for 24 h to analysis apoptosis.
The results indicated that T-BEAS-2B cells had significantly lower
level of apoptosis (Fig. 1A), but
increased Bcl-2, and Bcl-xL protein expression (Fig. 1B and C) when compared with control
BEAS-2B cells. Following NiCl2 exposure, the Bcl-2, and
Bcl-xL protein expression declined in both type of cells, but
control BEAS-2B cells had greater level of reduction than T-BEAS-2B
cells (Fig. 1B and C). To
understand if Bcl-2, and Bcl-xL protein reduction might be due to
NiCl2-induced transcriptional repression, quantitative
RT-PCR assay was performed in both types of cells. Fig. 1D shows that Bcl-2 and Bcl-xL mRNA
expression was repressed in BEAS-2B cells following
NiCl2-treatment. Similar pattern of mRNA expression was
also noted in T-BEAS-2B cells (data not shown). These results
suggested a mechanism of NiCl2-induced gene
transcription repression (Fig.
1D).
We tested if the levels of Bcl-2 and Bcl-xL protein
expression in BEAS-2B and T-BEAS-2B cells might affect
NiCl2-induced apoptosis. Forced overexpression of Bcl-2,
and Bcl-xL protein with plasmids in BEAS-2B cells reduced
NiCl2-induced cell apoptosis in a dose-dependent manner
(Fig. 2A–C). In contrast, siRNA
knockdown of Bcl-2, and Bcl-xL protein expression in T-BEAS-2B
cells enhanced the apoptosis rate (Fig. 2A and D). These results suggested
that the relative levels of Bcl-2, and Bcl-xL protein are important
for apoptosis induction/resistance in both types of cells.
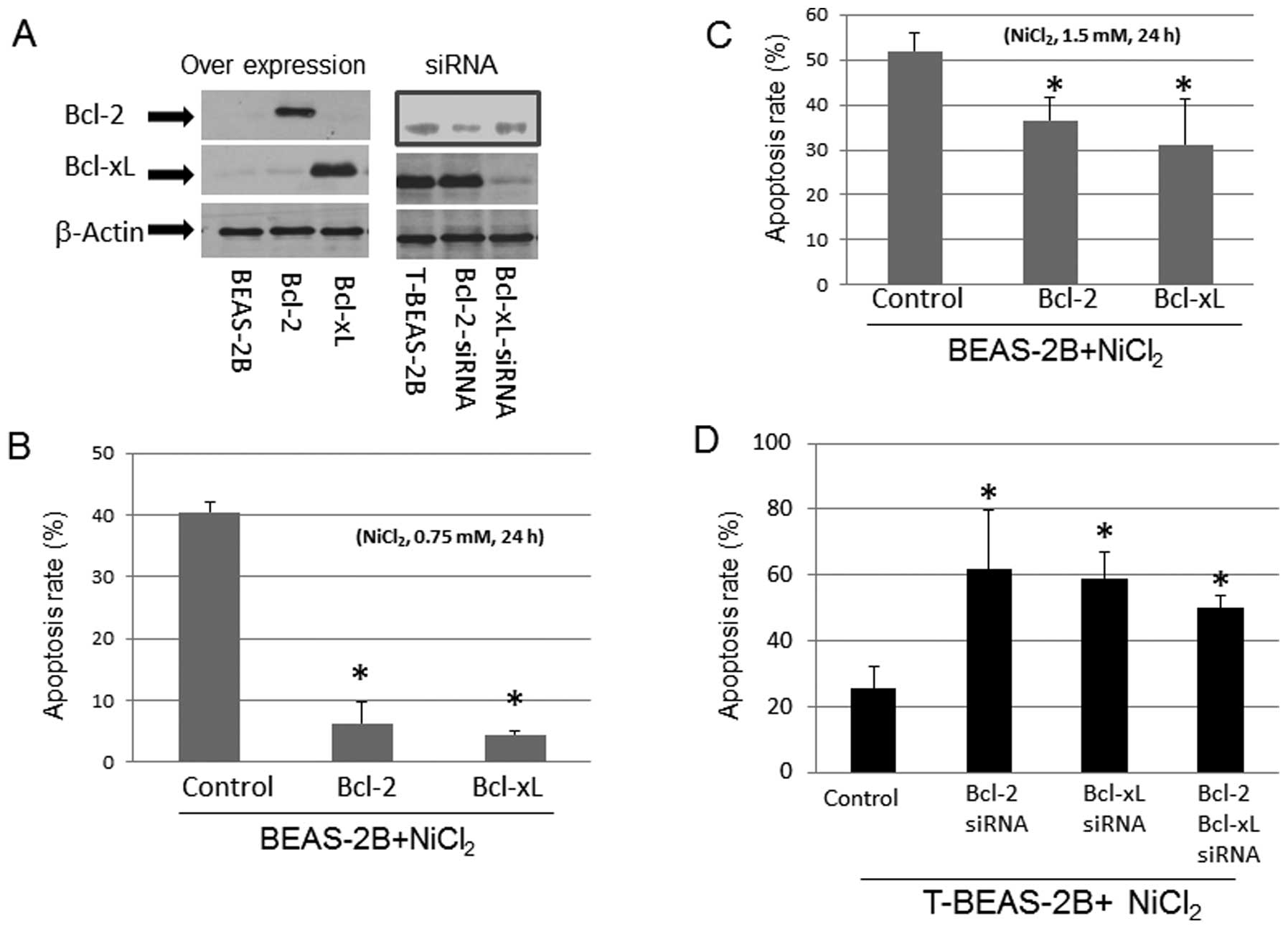 | Figure 2Effects of Bcl-2, and Bcl-xL
expression in NiCl2-induced apoptosis. BEAS-2B cells
were infected with control empty vector, Bcl-2, and Bcl-xL
retrovirus (control, Bcl-2, Bcl-xL), transformed BEAS-2B cells
(T-BEAS-2B) were transfected with control, Bcl-2, and Bcl-xL siRNA
(siRNA). Cells were treated with or without NiCl2 at
0.75 and 1.5 mM for 24 h. To determine Bcl-2, and Bcl-xL specific
protein expression, cell lysates were collected from the retrovirus
infected and siRNA transfected cells (A). Protein (20 μg) was
resolved in 10% SDS-PAGE to detect the respective protein
expression. Blots are representative of 2–3 separate experiments
with similar results, arrows indicate the specific band. Apoptosis
was assayed using either Annexin V/PI (B and C) or Hoechst 33342/PI
staining (D) followed by flow cytometry analysis. Data are mean ±
SEM from three separate experiments, *P<0.05 when
compared with controls. |
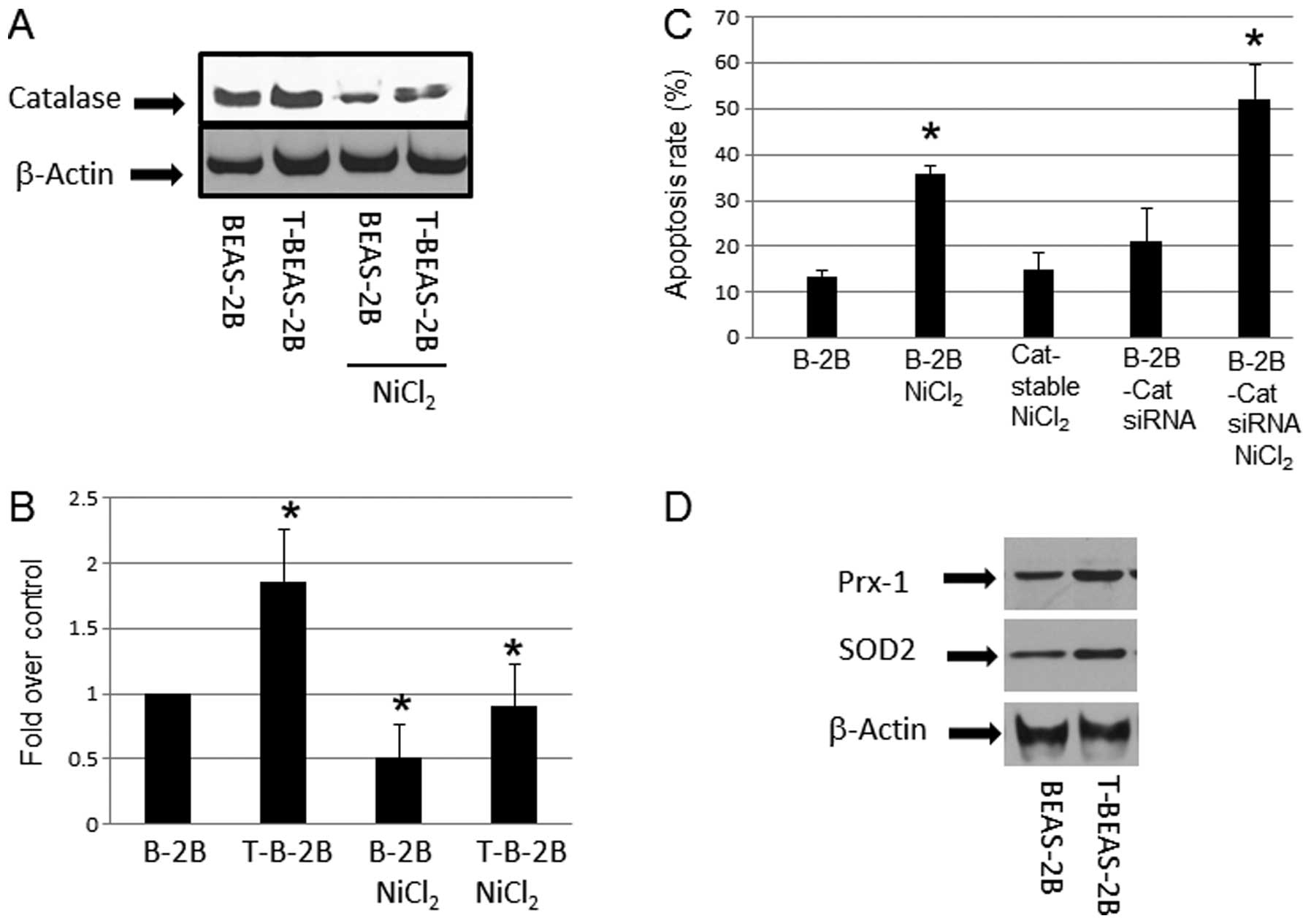
Effects of catalase in
NiCl2-induced apoptosis
After cell transformation, ROS generation is reduced
in transformed cells, a mechanism that attributed to increased ROS
scavenging enzyme expression (18). We examined if the apoptosis
resistance in NiCl2-transformed cells are accompanied by
higher ROS scavenging proteins expression. The results showed
upregulation of catalase in nickel-transformed cells over the
controls (Fig. 3A and B).
Overexpression of catalase by using a stable cell line reduced
NiCl2-induced apoptosis; in contrast, using catalase
siRNA to reduce its expression (data not shown) enhanced apoptosis
(Fig. 3C). These results suggested
that catalase or generation of ROS is important mechanism in
NiCl2-induced apoptosis. Western blot analysis also
indicated that other ROS scavenging enzymes such as SOD2, and Prx-1
protein levels were increased in T-BEAS-2B cells when compared with
control non-transformed BEAS-2B cells (Fig. 3D), while SOD1, and Phox47 protein
expression showed no difference (data not shown). Thus, these
results provide mechanistic explanation of reduced ROS level in
T-BEAS-2B cells as reported previously (9), and these mechanisms may contribute to
the apoptosis resistance in transformed cells.
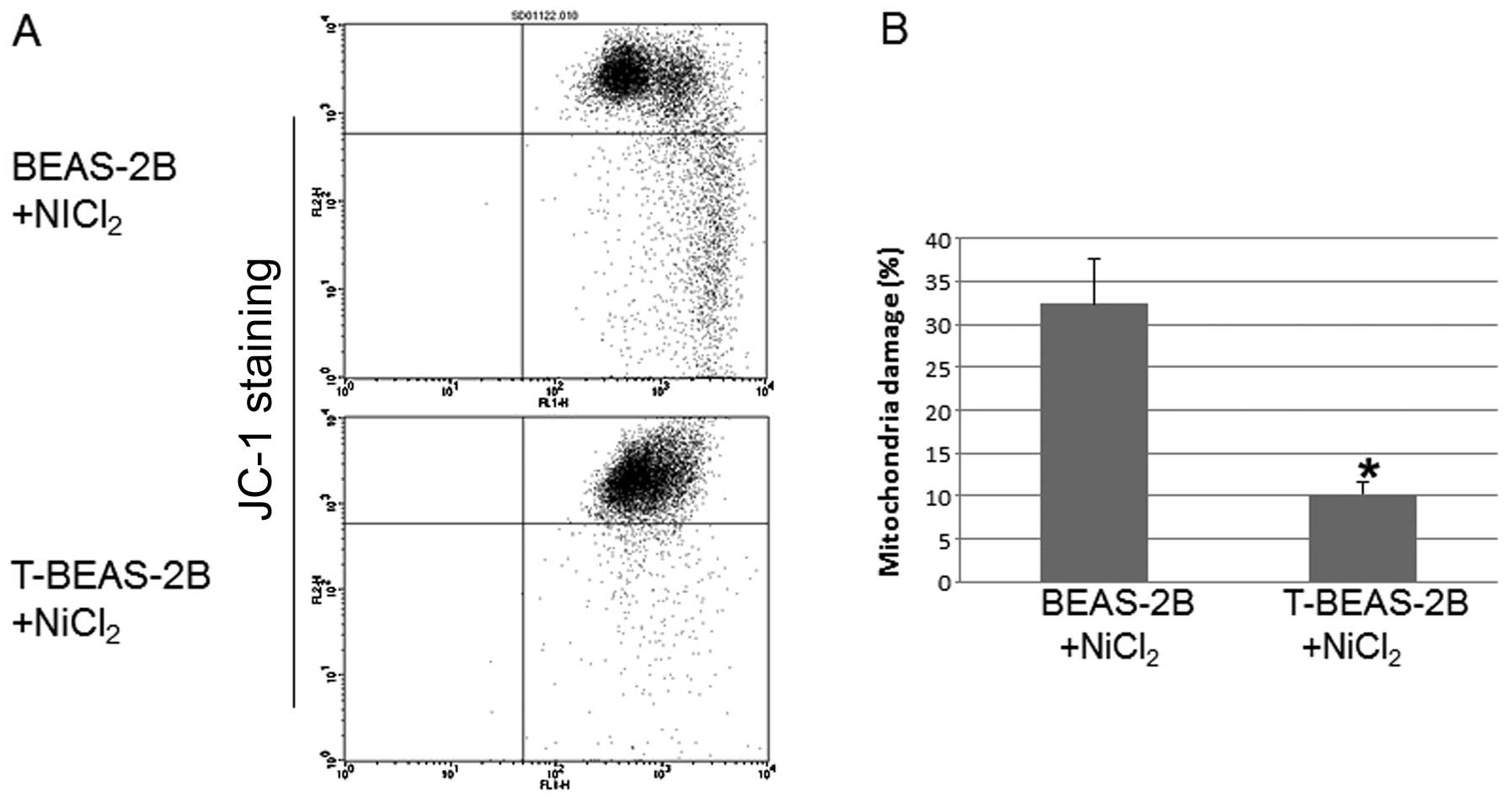
Transformed cells are resistant to
NiCl2-induced mitochondria damage
To investigate whether transformed cells are
resistant to mitochondria damage. We evaluated
NiCl2-induced mitochondria damage in both types of cells
and the role of Bcl-2 and Bcl-xL in these processes. The results
showed that T-BEAS-2B cells are more resistant to
NiCl2-induced mitochondrial damage compared with
non-transformed BEAS-2B cells (Fig. 4A
and B). Forced overexpression of Bcl-2, Bcl-xL and catalase
largely abolished NiCl2-induced mitochondria damage in
BEAS-2B cells (Fig. 4C and D).
Significant difference was noted when the quantitative data were
compared with control cells (*P<0.05). These results
indicated that enhanced Bcl-2, Bcl-xL and catalase protein
expression are important mechanisms for reduced mitochondria damage
in transformed cells.
Caspase activation in nickel-transformed
cells
The above results demonstrated that T-BEAS-2B cells
differ from BEAS-2B cells in NiCl2-induced apoptosis
resistance and mitochondria damage; thus we further studied the
underlying molecular mechanisms. We compared caspase activation in
both types of cells. The result in Fig. 5 clearly indicates that upon
NiCl2 stimulation, BEAS-2B cells had much stronger
caspase activation, presented as increased c-caspase 3, 7 and
c-PARP protein expression when compared with T-BEAS-2B cells
(Fig. 5A). This was accompanied by
increased caspase 3/7 enzymatic activity, while Z-VAD, a
pan-caspase inhibitor, blocked the caspase 3/7 enzymatic activity
(Fig. 5B), reduced c-caspase 3, 7
expression (Fig. 5C), and
attenuated apoptosis (Fig.
5D).
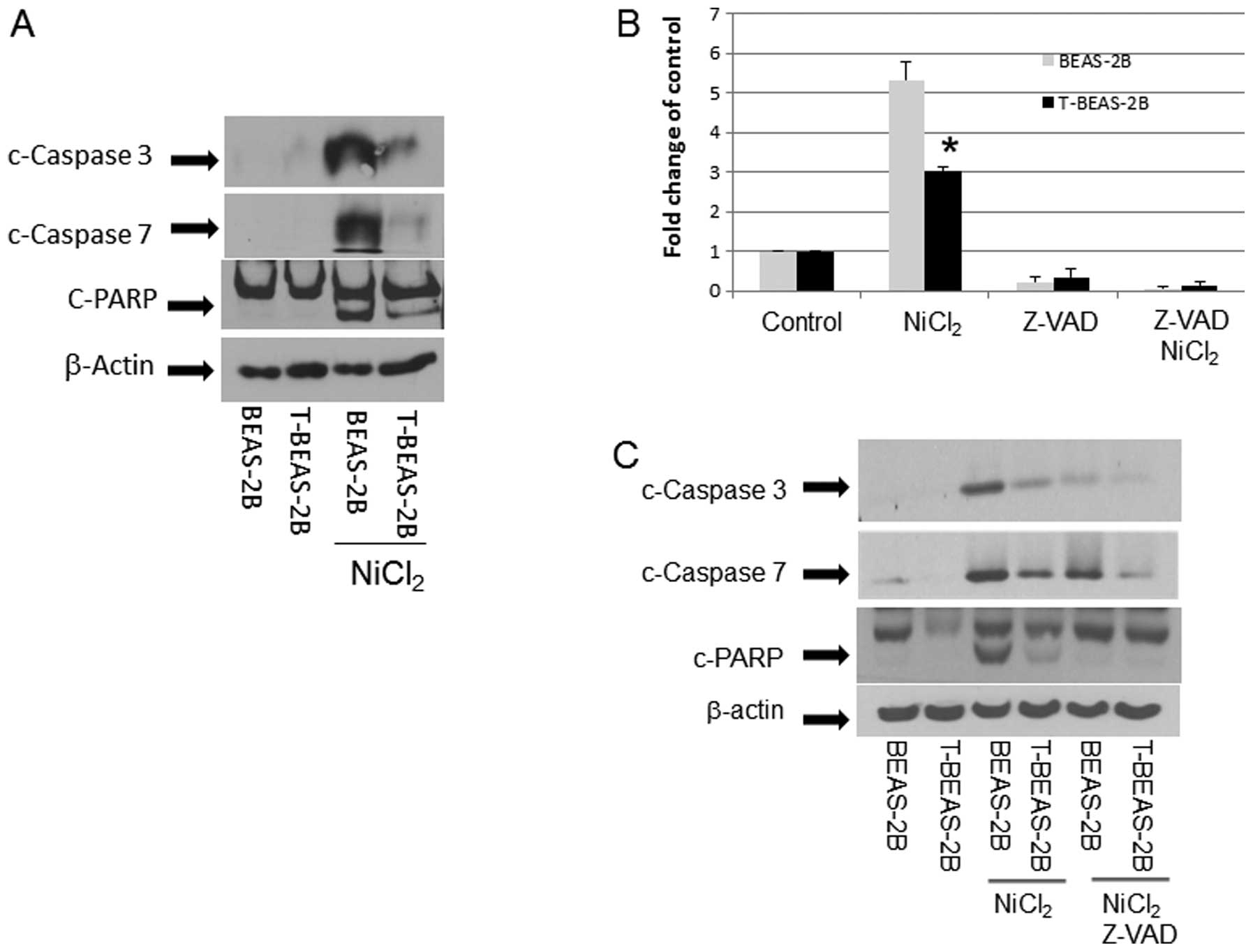 | Figure 5Caspase activation in
nickel-transformed cells. BEAS-2B, transformed BEAS-2B (T-BEAS-2B),
control empty vector, Bcl-2, and Bcl-xL-retrovirus infected BEAS-2B
(Bcl-2, Bcl-xL) and BEAS-2B catalase-stable expressing cells
(Cat-stable) (1×106) were treated with or without
NiCl2 at 1.5 mM for 24 h. Cell lysate was collected and
20 μg of protein resolved in 10% SDS-PAGE to detect respective
protein expression (A, C, E–G). Blots are representative from 3–4
separate experiments with similar results, arrows indicate the
specific band. To determine caspase 3/7 enzymatic activity, BEAS-2B
and T-BEAS-2B cells were treated with or without NiCl2
at 1.5 mM for 16 h and enzymatic activity was assayed as described
in Materials and methods (B). For apoptosis assay, BEAS-2B,
transformed BEAS-2B (T-BEAS-2B) cells were treated with or without
NiCl2 at 1.5 mM and Z-VAD for 24 h, apoptosis was
assayed using the Annexin V/PI staining followed by flow cytometry
(D). Quantitative data are expressed as mean ± SEM from three
separate experiments, *P<0.05 when compared with
respective controls. |
In addition, we evaluated if Bcl-2, Bcl-xL and
catalase proteins that are overexpressed in T-BEAS-2B cell may
antagonize the NiCl2-induced caspase activation. The
results indicated that overexpression of Bcl-2, Bcl-xL and catalase
protein effectively reduced the c-caspase 3, 7 and c-PARP protein
expression (Fig. 5E–G),
respectively, correlating with reduced mitochondria damage and
apoptosis. Therefore, these results demonstrated that upregulation
of Bcl-2, Bcl-xL and catalase in T-BEAS-2B suppresses caspase
activation, which are essential mechanisms underlying the adapted
apoptosis resistance in these cells.
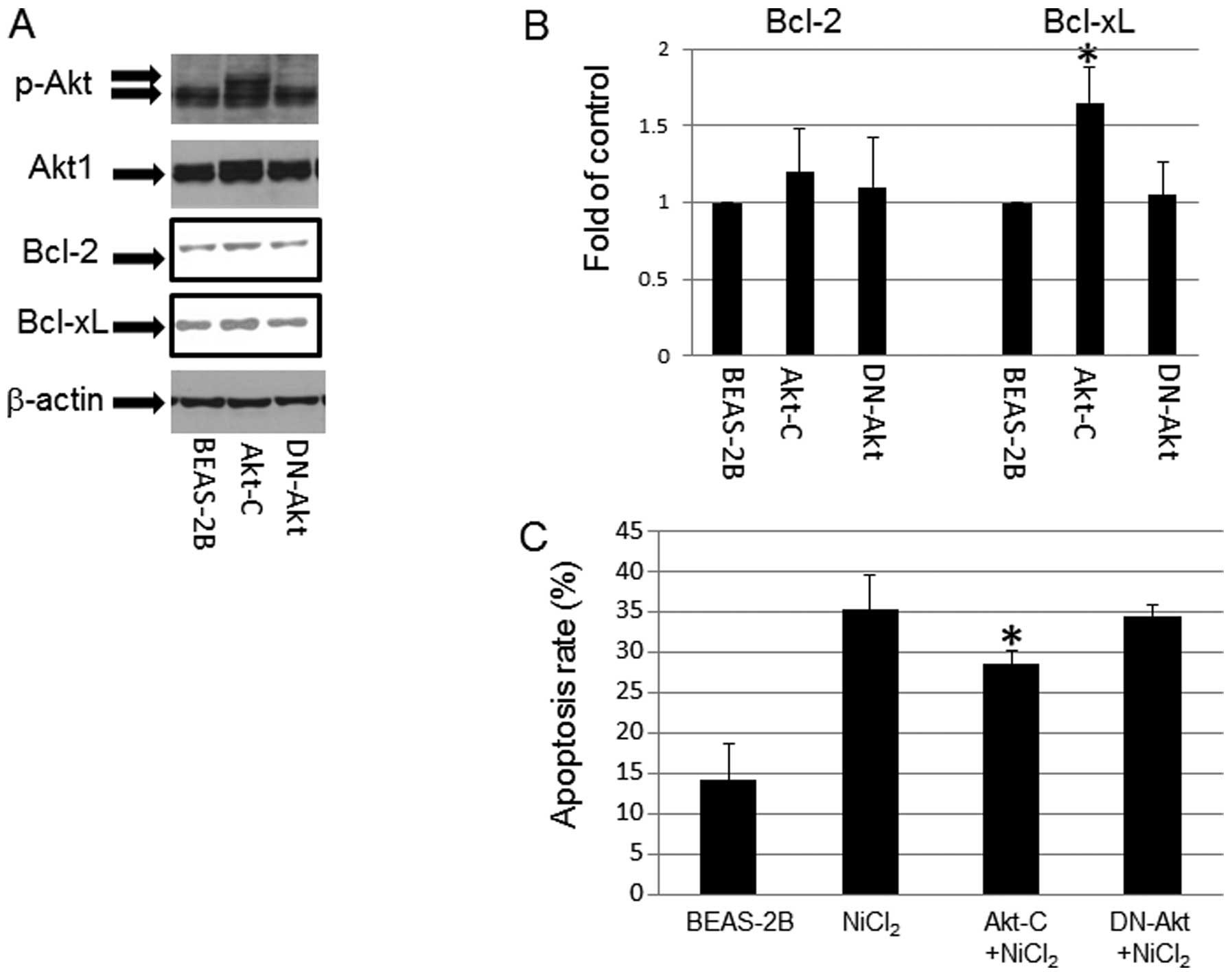
Role of p-Akt in NiCl2
induced-apoptosis
p-Akt expression was upregulated in T-BEAS-2B cells
in a previous study (9). To
understand its role in NiCl2-induced apoptosis, we used
Akt overexpression (Akt-C) and dominant negative Akt (DN-Akt)
stable cell lines to determine the effects of Akt on
NiCl2-induced apoptosis. The results indicated that Akt
stable cells had persistently enhanced p-Akt level (Fig. 6A), and increased Bcl-xL protein
expression, but not Bcl-2 expression; while in DN-Akt cells, Bcl-2,
and Bcl-xL protein expression remain at the basal control level
(Fig. 6A and B). In addition,
NiCl2 challenge induced marginally higher levels of
apoptosis in control and DN-Akt cells over the Akt stable cells
(Fig. 6C), pointing out the
protective roles of Akt in NiCl2-induce BEAS-2B cell
apopotsis.
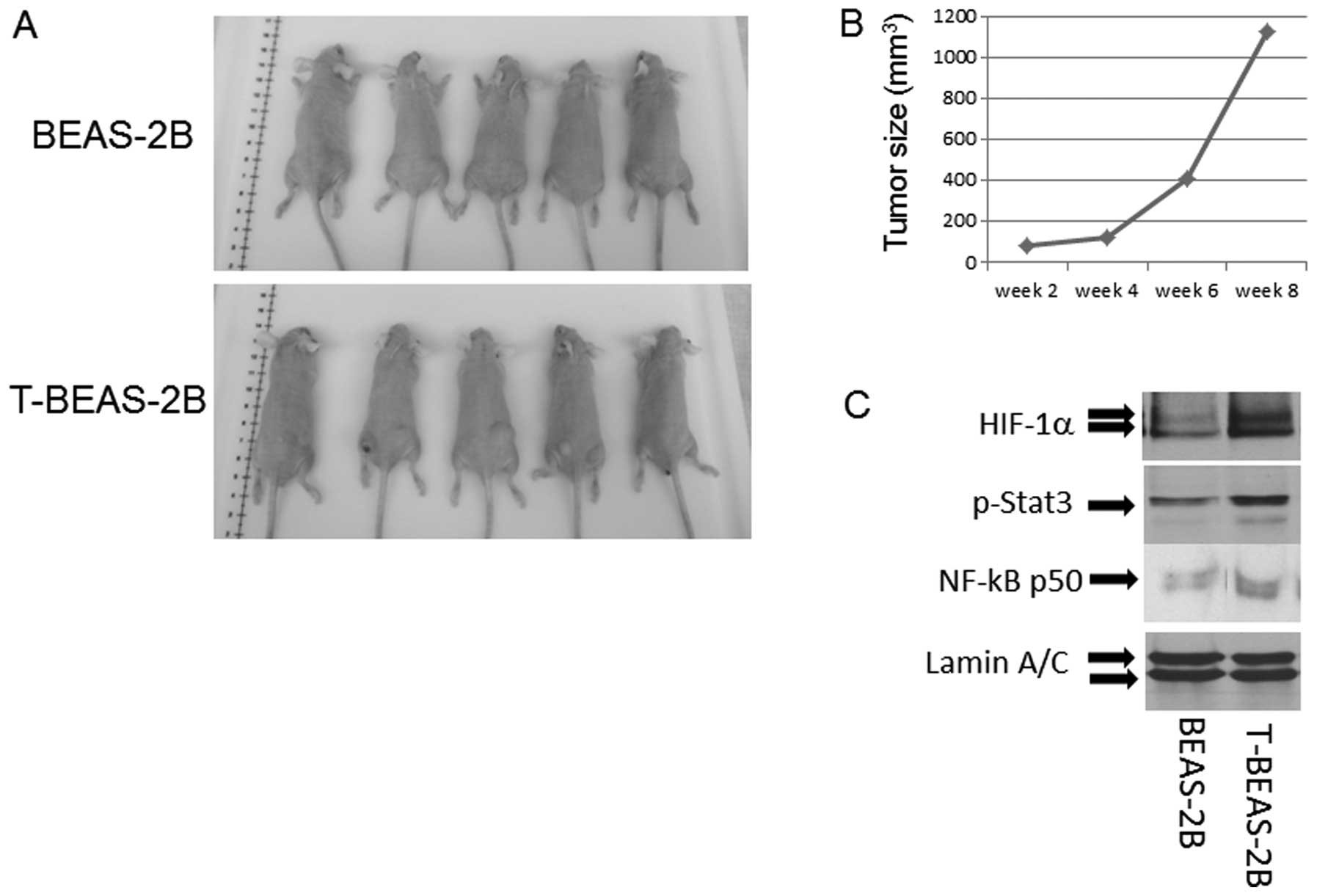
Tumorigenic properties of
NiCl2 transformed cells
To evaluate if the transformed cells are
tumorigenic, we injected both control BEAS-2B and T-BEAS-2B cells
into nude mice and observed the tumor growth. The results indicated
that among five injected mice in each group, only T-BEAS-2B cells
induced tumor growth, and tumors occurred at multiple injection
sites, grew into various sizes within 8 weeks (Fig. 7A and B). Furthermore, western blot
analysis revealed increased HIF-1α NF-κB-p50 subunit and p-Stat3
protein expression in T-BEAS-2B cells when compared with
non-transformed cells, underlying the oncogenic features of the
NiCl2-transformed cells (Fig. 7C).
Discussion
Nickel is a known human carcinogen and induces
genotoxic stress. A number of mechanisms have been proposed for its
carcinogenic effects; such as generation of ROS, induction of DNA
damage, and activation of oncogenic pathways (3). However, little information is
available with regard to the mechanisms of apoptosis resistance in
nickel-transformed cells. In this study, we focused on apoptosis
resistant and tumorigenic property of NiCl2-transformed
human lung epithelial cells, and explored the potential molecular
mechanisms. The results demonstrate that NiCl2
transformed BEAS-2B cells are resistant to apoptosis and
tumorigenic. Higher levels of Bcl-2, Bcl-xL and catalase protein
expression are important mechanisms contributing to the transformed
cell oncogenic properties.
Previously in vitro studies have shown that
water-insoluble nickel compounds such as nickel oxide, nickel
subsulfide are more readily phagocytized than the water-soluble
nickel compounds such as nickel chloride. They can reach the
nucleus of the cell in greater amounts than that of water-soluble
nickel compounds and induce more cell damage (19,20).
In the current study, NiCl2 was selected due to the
BEAS-2B cells being transformed by NiCl2 exposure
(9). One important characteristic
of nickel compounds is the generation of ROS in host cells, and ROS
appear critical in nickel-induced cell damage, such as apoptosis
and oncogenic transformation (1,4).
Increasing evidence has indicated that ROS plays an important role
in inducing apoptosis and carcinogenesis following diverse exposure
to environmental stimuli (21).
However, ROS generation in transformed cells is reduced (9,18),
the mechanism is not clear. In the current study, we confirm that
antioxidant enzymes are increased in transformed cells including
catalase, SOD2 and Prx-1 (22),
suggesting that they are responsible for the reduced ROS generation
in transformed cells (9,18). Higher ROS scavenging enzyme
expression may also contribute to NiCl2-induced
apoptosis resistance in transformed cells by attenuating the
detrimental effects from ROS generated by NiCl2
exposure. However, the biological implications of these changes
during transformation have yet to be explored, as well as the
molecular mechanisms that result in their increased expression in
transformed cells. The data provide evidence that transformation
involves multiple cellular signal alternations, which deserve
further investigation.
Bcl-2 and Bcl-xL proteins are overexpressed in a
variety of human cancers (23),
and are important members of anti-apoptotic proteins family. The
mechanisms of Bcl-2 and Bcl-xL protect cells from apoptosis through
either heterodimerization with pro-apoptotic proteins, or their
direct pore-blocking effects on the outer membrane of mitochondria
(11,23). Mitochondria play a critical role in
apoptosis induction, which involves both caspase-dependent and
-independent pathways. We noted that NiCl2-induced
apoptosis can be affected by both Bcl-2 and Bcl-xL expressing
vector and siRNA transfection, indicating that their relative
levels regulate cell apoptosis resistance in T-BEAS-2B cells. The
question of how transformed cell evolve into higher level of Bcl-2,
and Bcl-xL protein expression remain to be investigated, we
hypothesis that either genetic or epigenetic modifications may be
responsible during the transformation and selection processes.
In our study, NiCl2 treatment reduced the
cellular level of Bcl-2 and Bcl-xL proteins in both transformed and
non-transformed cells (Fig. 1A and
B), but the level of reduction in transformed cells is less
dramatic, which is associated with low apoptotic rate. This effect
also correlates with reduced mitochondrial stress, c-caspase 3, 7
protein level, caspase enzymatic activity and PARP cleavage.
Together, they suggest that transformed cells evolve a mechanism of
increased Bcl-2/Bcl-xL protein expression, reduced mitochondrial
stress and alleviated caspase activation upon NiCl2
exposure, therefore, confer these cells the apoptosis resistant
property.
Several signaling pathways such as PI3K, NF-κB, Akt,
and Sonic hedgehog have been shown to regulate Bcl-2 and Bcl-xL
protein expression and apoptosis in different cell types (12,24,25).
Induction of cyclooxygenesis 2 is also important in nickel induced
apoptosis (26). In the current
study, we focus on the effects of Bcl-2, Bcl-xL, and catalase by
examining their roles in apoptosis resistance in transformed cells,
as they are either the effector proteins downstream of the above
mentioned signal pathways or involved in ROS generation. Akt is a
serine/threonine kinase, also known as protein kinase B (PKB). It
is involved in regulating multiple cellular functions, including
cell growth, proliferation, survival, glucose metabolism and genome
stability (27,28). Aberrant Akt activation has been
noted in breast, prostate, lung, pancreatic, liver, ovarian and
colorectal cancers, and in malignant transformation (27–30).
Akt overexpression enhances Bcl-xL expression, which is correlated
with the fact that Akt-stable expressing cells are more resistant
to NiCl2-induced apoptosis. The results uncovered
anti-apoptotic protein-dependent mechanism in nickel-induced
apoptosis resistance in transformed cells.
It is worth noting that overexpression of Akt could
not completely block the nickel-induced apoptosis (Fig. 6), and even this effect was
marginal. Since there are two major apoptotic pathways, the
death-receptor pathway and the mitochondrial pathway, we consider
other signal pathways might also contribute to the nickel-induced
apoptosis. It has been reported that increased FasL expression,
cell cycle alteration, activation of c-Myc through ERK pathway,
caspase-8/AIF-mediated pathways, and activation of NF-κB/Cox2
pathway all participate in nickel-induced apoptosis (26,31–34).
Therefore, each may initiate and contribute to the pathway or cell
type-specific apoptosis processes in additional to the mechanisms
discussed in the present study. However, increased Bcl-2, Bcl-xL
and catalase protein expression, reduced mitochondria damage and
caspase activation described here appear to be the essential
mechanisms involved in NiCl2-mediated cell death
resistance program in transformed BEAS-2B cells. Future
investigations will be required to clarify if other mechanisms
might also be involved in transformed cell resistance to
apoptosis.
One important feature of transformed cells is their
tumorigenic property. These cells also have enhanced p-Stat3,
NF-κB-p50 subunit, and HIF-1α protein expression. The altered
oncogene expression probably reflects extensive genetic or
epigenetic rearrangements during the transformation processes which
allow transformed cells to adopt and grow in a new environment. It
is also possible that they acquire cancer stem cell trait to allow
cell resistant apoptosis. Future studies focusing on nickel-induced
apoptosis resistance and cell transformation are of great interest
to understand nickel-induced carcinogenic mechanisms and to provide
options for prevention.
Acknowledgements
This research was supported in part by NIH grants
(1R01CA119028) and a Henan provincial government bio-medical key
research grant (132102310444).
References
|
1
|
Costa M: Molecular mechanisms of nickel
carcinogenesis. Annu Rev Pharmacol Toxicol. 31:321–337. 1991.
View Article : Google Scholar
|
|
2
|
Doll R, Morgan LG and Speizer FE: Cancers
of the lung and nasal sinuses in nickel workers. Br J Cancer.
24:623–632. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Costa M, Davidson TL, Chen H, et al:
Nickel carcinogenesis: epigenetics and hypoxia signaling. Mutat
Res. 592:79–88. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lu H, Shi X, Costa M and Huang C:
Carcinogenic effect of nickel compounds. Mol Cell Biochem.
279:45–67. 2005. View Article : Google Scholar
|
|
5
|
Salnikow K and Zhitkovich A: Genetic and
epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis:
nickel, arsenic, and chromium. Chem Res Toxicol. 21:28–44. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Salnikow K and Costa M: Epigenetic
mechanisms of nickel carcinogenesis. J Environ Pathol Toxicol
Oncol. 19:307–318. 2000.
|
|
7
|
Davidson T, Chen H, Garrick MD, D’Angelo G
and Costa M: Soluble nickel interferes with cellular iron
homeostasis. Mol Cell Biochem. 279:157–162. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pan J, Chang Q, Wang X, et al: Reactive
oxygen species-activated Akt/ASK1/p38 signaling pathway in nickel
compound-induced apoptosis in BEAS 2B cells. Chem Res Toxicol.
23:568–577. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pan JJ, Chang QS, Wang X, et al:
Activation of Akt/GSK3β and Akt/Bcl-2 signaling pathways in
nickel-transformed BEAS-2B cells. Int J Oncol. 39:1285–1294.
2011.
|
|
10
|
Cory S and Adams JM: The Bcl2 family:
regulators of the cellular life-or-death switch. Nat Rev Cancer.
2:647–656. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gross A, McDonnell JM and Korsmeyer SJ:
BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pugazhenthi S, Nesterova A, Sable C, et
al: Akt/protein kinase B up-regulates Bcl-2 expression through
cAMP-response element-binding protein. J Biol Chem.
275:10761–10766. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ding SZ, Yang YX, Li XL, et al:
Epithelial-mesenchymal transition during oncogenic transformation
induced by hexavalent chromium involves reactive oxygen
species-dependent mechanism in lung epithelial cells. Toxicol Appl
Pharmacol. 269:61–71. 2013. View Article : Google Scholar
|
|
14
|
Zhou BP, Hu MC, Miller SA, et al:
HER-2/neu blocks tumor necrosis factor-induced apoptosis via the
Akt/NF-kappaB pathway. J Biol Chem. 275:8027–8031. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ding SZ, Fischer W, Kaparakis-Liaskos M,
et al: Helicobacter pylori-induced histone modification,
associated gene expression in gastric epithelial cells, and its
implication in pathogenesis. PLoS One. 5:e98752010. View Article : Google Scholar
|
|
16
|
Cossarizza A, Baccarani-Contri M,
Kalashnikova G and Franceschi C: A new method for the
cytofluorimetric analysis of mitochondrial membrane potential using
the J-aggregate forming lipophilic cation
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine
iodide (JC-1). Biochem Biophys Res Commun. 197:40–45.
1993.PubMed/NCBI
|
|
17
|
Cheng EH, Wei MC, Weiler S, et al: BCL-2,
BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and
BAK-mediated mitochondrial apoptosis. Mol Cell. 8:705–711. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chang Q, Pan J, Wang X, Zhang Z, Chen F
and Shi X: Reduced reactive oxygen species-generating capacity
contributes to the enhanced cell growth of arsenic-transformed
epithelial cells. Cancer Res. 70:5127–5135. 2010. View Article : Google Scholar
|
|
19
|
Dunnick JK, Elwell MR, Radovsky AE, et al:
Comparative carcinogenic effects of nickel subsulfide, nickel
oxide, or nickel sulfate hexahydrate chronic exposures in the lung.
Cancer Res. 55:5251–5256. 1995.PubMed/NCBI
|
|
20
|
Oller AR, Costa M and Oberdorster G:
Carcinogenicity assessment of selected nickel compounds. Toxicol
Appl Pharmacol. 143:152–166. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao J, Shi X, Castranova V and Ding M:
Occupational toxicology of nickel and nickel compounds. J Environ
Pathol Toxicol Oncol. 28:177–208. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim YJ, Ahn JY, Liang P, Ip C, Zhang Y and
Park YM: Human prx1 gene is a target of Nrf2 and is up-regulated by
hypoxia/reoxygenation: implication to tumor biology. Cancer Res.
67:546–554. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yip KW and Reed JC: Bcl-2 family proteins
and cancer. Oncogene. 27:6398–6406. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Catz SD and Johnson JL: Transcriptional
regulation of bcl-2 by nuclear factor kappa B and its significance
in prostate cancer. Oncogene. 20:7342–7351. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bigelow RL, Chari NS, Unden AB, et al:
Transcriptional regulation of bcl-2 mediated by the sonic hedgehog
signaling pathway through gli-1. J Biol Chem. 279:1197–1205. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ding J, Zhang X, Li J, et al: Nickel
compounds render anti-apoptotic effect to human bronchial
epithelial BEAS-2B cells by induction of cyclooxygenase-2 through
an IKKbeta/p65-dependent and IKKalpha- and p50-independent pathway.
J Biol Chem. 281:39022–39032. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lawlor MA and Alessi DR: PKB/Akt: a key
mediator of cell proliferation, survival and insulin responses? J
Cell Sci. 114:2903–2910. 2001.PubMed/NCBI
|
|
28
|
Morley S, Wagner J, Kauppinen K, Sherman M
and Manor D: Requirement for Akt-mediated survival in cell
transformation by the dbl oncogene. Cell Signal. 19:211–218. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bellacosa A, Kumar CC, Di Cristofano A and
Testa JR: Activation of AKT kinases in cancer: implications for
therapeutic targeting. Adv Cancer Res. 94:29–86. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Manning BD and Cantley LC: AKT/PKB
signaling: navigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li Q, Suen TC, Sun H, Arita A and Costa M:
Nickel compounds induce apoptosis in human bronchial epithelial
Beas-2B cells by activation of c-Myc through ERK pathway. Toxicol
Appl Pharmacol. 235:191–198. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ding J, He G, Gong W, et al: Effects of
nickel on cyclin expression, cell cycle progression and cell
proliferation in human pulmonary cells. Cancer Epidemiol Biomarkers
Prev. 18:1720–1729. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim K, Lee SH, Seo YR, Perkins SN and
Kasprzak KS: Nickel(II)-induced apoptosis in murine T cell
hybridoma cells is associated with increased fas ligand expression.
Toxicol Appl Pharmacol. 185:41–47. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhao J, Bowman L, Zhang X, et al: Metallic
nickel nano- and fine particles induce JB6 cell apoptosis through a
caspase-8/AIF mediated cytochrome c-independent pathway. J
Nanobiotechnol. 7:22009. View Article : Google Scholar : PubMed/NCBI
|