Introduction
Colorectal cancer is the third most commonly
diagnosed cancer in men and the second in women, with over 1.2
million new cancer cases and 608,700 deaths estimated to have
occurred worldwide in 2008 (1).
Colorectal cancer is a multistep process, involving progressive
disruption of intestinal epithelium growth (2). The disease arises as a result of the
accumulation of genetic errors, many of which affect the control of
apoptosis (3). Clinical treatment
options for colorectal cancer consist of surgery, radiation and
chemotherapy, but these treatments are often unsatisfactory
(4).
Previous studies have reported that heat shock
protein 90 (HSP90) inhibitors possess significant antitumor
activity (5). HSP90 plays an
essential role as a molecular chaperone, assisting in the correct
folding of nascent and stress-accumulated misfolded proteins and
preventing their aggregation (6).
High HSP expression is a property of, and essential for the
survival of, most cancers. Neutralizing HSPs could, thus, provide
an alternative strategy for anticancer therapy (7).
Induction of cell cycle arrest and apoptosis are
also potential strategies for cancer treatment. Investigators have
widely explored the process of the cell cycle, particularly the
CDK1/cyclin B1 complex, which plays an important role in the
regulation of the G2/M phase. During pro-metaphase,
spindle-assembly checkpoint proteins, such as BubR1, prevent
anaphase and mitotic exit (8,9).
Anti-mitotic drugs that target microtubules have potential clinical
application (10). Several studies
have determined that microtubule-targeting agents act primarily to
suppress spindle-microtubule dynamics, resulting in a blockade of
metaphase/anaphase transition and the triggering of apoptotic cell
death (11). The aurora kinase
family is a collection of closely related serine/threonine kinases
that are key regulators of mitosis (12,13).
Fu et al demonstrated upregulated aurora kinase expression
and activity in cancer cells, indicating that aurora kinase might
serve as a useful target for therapy (14). Three related aurora kinases have
evolved in mammalian cells: aurora A, aurora B, and aurora C
(13,15–18).
Inhibition of aurora A or aurora B activity in tumor cells results
in impaired chromosome alignment, abrogation of the mitotic
checkpoint, polyploidy and subsequent cell death (12,19,20).
Apoptosis is a mechanism by which cells undergo programmed death to
control cell proliferation or in response to DNA damage (21). Biochemical features of apoptosis
include DNA fragmentation, protein cleavage at specific locations,
increased mitochondrial membrane permeability and the appearance of
phosphatidylserine (PS) on the cell membrane surface (22). The induction of apoptosis has
formed the basis of several previous research investigations that
focused on the selective killing of cancer cells (23).
Carbazole alkaloids are well-known natural
compounds, some of which display anticancer activity (24–26).
The β-carboline alkaloids occurring in medicinal plants have gained
attention because of their antitumor effects (27). However, research on the anticancer
activity of α-carboline (isostere of β-carboline) derivatives is
relatively limited. In a prior investigation, we synthesized a
series of α-carboline derivatives and identified several of them as
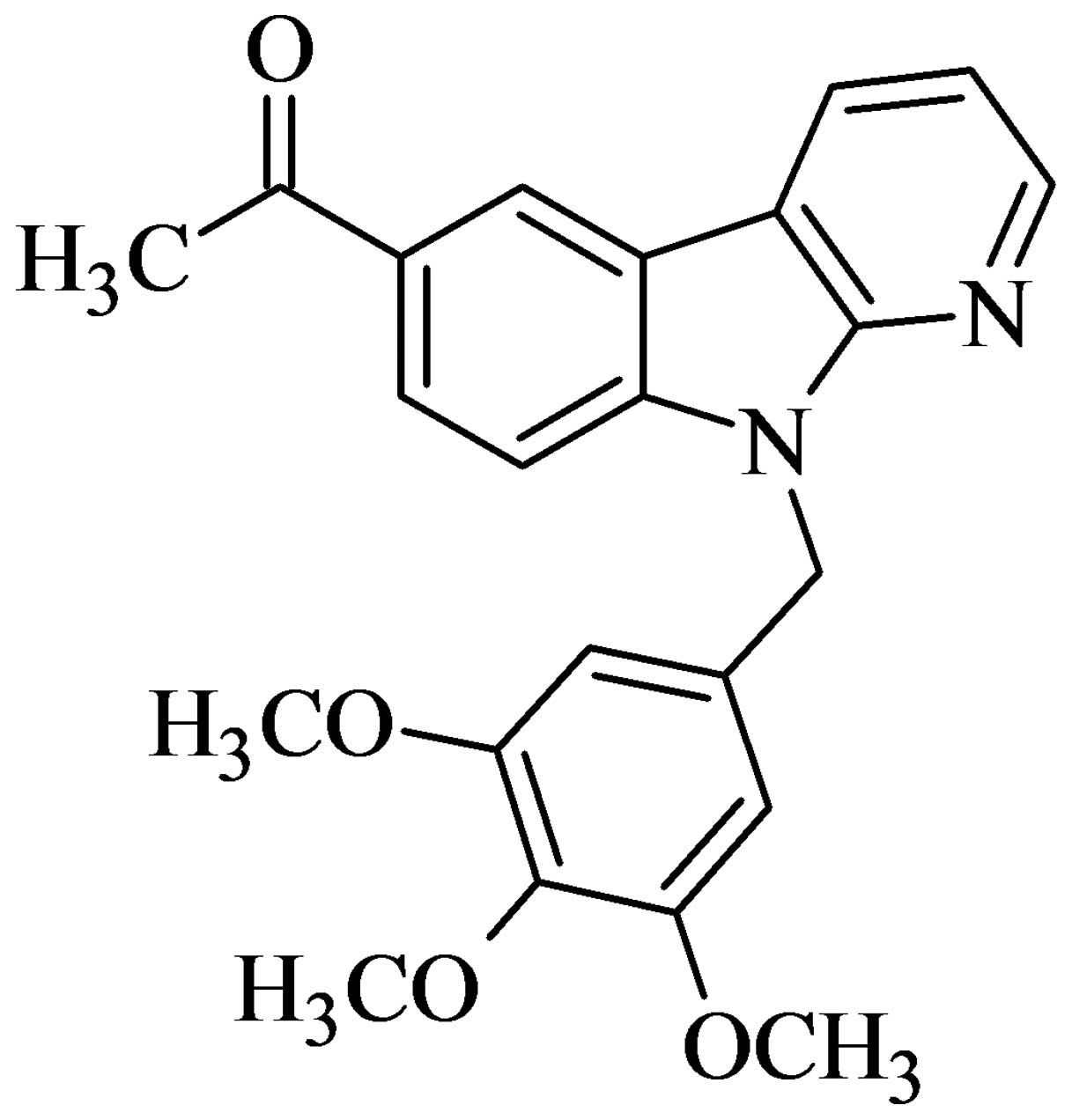
novel anticancer agents. Among these,
6-acetyl-9-(3,4,5-trimethoxybenzyl)-9H-pyrido[2,3-b]
indole (HAC-Y6) (Fig. 1) was the
most potent. In the present study, HAC-Y6 was selected as a target
compound and submitted to the National Cancer Institute (NCI,
Bethesda, MD, USA) for further screening against a panel of 60
human tumor cell lines. Results indicated that HAC-Y6 is a
promising anti-colon cancer agent. Further investigations of the
molecular mechanisms of HAC-Y6’s anti-human colon carcinoma
effects, using COLO 205 cells as a model, were performed, and our
results are presented here.
Materials and methods
Chemicals and reagents
HAC-Y6 was synthesized in the laboratory of the
Graduate Institute of Pharmaceutical Chemistry, China Medical
University (Taichung, Taiwan, R.O.C.). MTT
(3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide),
Hoechst 33258, propidium iodide (PI), Tris-HC1, Triton X-100, and
RNase A were purchased from Sigma Chemical Co. (St. Louis, MO,
USA). RPMI-1640 medium, L-glutamine, fetal bovine serum,
penicillin-streptomycin, and trypsin-EDTA were obtained from
Invitrogen Corp (Carlsbad, CA, USA). Antibodies for AIF, Apaf-1,
aurora A, aurora B, phospho-aurora A, phospho-aurora B, Bcl-xL,
BubR1, caspase-9, caspase-3, Endo G, phospho-H3, α-tubulin,
β-tubulin, and poly(ADP-ribose) polymerase (PARP) were purchased
from Cell Signaling Technology (Beverly, MA, USA). Antibodies for
β-actin, Bax, Bcl-2, cyclin B1, CDK1, cytochrome c,
horseradish peroxidase (HRP)-linked goat anti-mouse IgG, and goat
anti-rabbit IgG were purchased from Santa Cruz Biotechnology (Santa
Cruz, CA, USA). The Annexin V-FITC Apoptosis Detection kit was
obtained from Strong Biotech Corporation (Taipei, Taiwan, R.O.C.).
The Flow Cytometry Mitochondrial Membrane Potential Detection kit
was obtained from BD Biosciences (Los Angeles, CA, USA).
Cell culture
The human colon cancer cell line COLO 205 was
obtained from the Food Industry Research and Development Institute
(Hsinchu, Taiwan, R.O.C.). Cells were seeded into plates or dishes
in RPMI-1640 medium supplemented with 10% fetal bovine serum,
L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin
in a humid atmosphere of 5% CO2 and 95% air at 37°C.
Cells were plated at a density of 2.5×104 cells per well
in 96-well plates for the cell viability assay, 1×105
cells per well in 24-well plates with cover slips for Hoechst
staining, 2×105 cells per well in 12-well plates for
cell cycle assay, 1×106 cells per well in 6-well plates
for mitochondrial membrane potential assay, and 1×107
cells per plate in 10 cm dishes for western blot analysis. Cells
were allowed to adhere for 24 h before use.
Cell morphology
COLO 205 cells were plated at a density of
2.5×105 cells per well in 12-well plates and then
incubated with 0.5 μM of HAC-Y6 for 24 to 72 h. Cells were
directly examined and photographed under a phase contrast
microscope.
Evaluation of cell viability using the
MTT assay
COLO 205 cells were plated at a density of
2.5×104 cells per well in 96-well plates, and cell
survival was evaluated using MTT reduction assays. The reduction
status of the cells was measured by a colorimetric assay,
indicating cell survival (28).
MTT was dissolved in phosphate-buffered saline (PBS, 500 ml
contains 137 mM NaCl, 2.7 mM KCl, 4.3 mM
Na2HPO4, 1.47 mM
KH2PO4, pH 7.4) at a concentration of 1 mg/ml
and filtered. After 48 h exposure to HAC-Y6, 50 μl MTT was
added to each well and incubated for 2 h at 37°C in the dark. When
absorbed by living cells, MTT is converted to a water-insoluble
blue product (formazan). The formazan product was dissolved by
adding 150 μl dimethylsulf-oxide (DMSO) to each well. The
absorption value at 570 nm was determined using an ELISA plate
reader. Data are presented as the percentage of survival relative
to vehicle-treated control culture. All measurements were performed
in triplicate and each experiment was repeated at least three
times.
Hoechst 33258 staining
Nuclei were stained with Hoechst 33258
(bis-benzimide, Sigma) to detect chromatin condensation or nuclear
fragmentation, morphological characteristics of apoptosis. HAC-Y6
treatment cells were stained with 5 μg/ml Hoechst 33258 for
10 min. After washing twice with PBS, cells were fixed with 4%
paraformaldehyde (PFA) in PBS for 10 min at 25°C. Fluorescence of
the soluble DNA (apoptotic) fragments was measured in a Varian
Fluorometer at an excitation wavelength of 365 nm and emission
wavelength of 460 nm.
Flow cytometric analysis for cell cycle
effects
Cells were fixed in 70% ethanol overnight and
re-suspended in PBS containing 20 μg/ml PI, 0.2 mg/ml RNase
A, and 0.1% Triton X-100 in a dark room. After 30 min incubation at
37°C, cell cycle distribution was analyzed using ModFit LT Software
(Verity Software House, Topsham, ME, USA) in a BD FACSCanto flow
cytometer (Becton-Dickinson, San Jose, CA, USA).
Quantification of apoptosis
COLO 205 cells (2×105 cells/well) were
fluorescently labeled for detection of apoptotic and necrotic cells
by adding 100 μl of binding buffer, 2 μl of Annexin
V-FITC, and 2 μl of PI to each sample. Samples were mixed
gently and incubated at room temperature in the dark for 15 min.
Binding buffer (300 μl) was added to each sample immediately
before flow cytometric analysis. A minimum of 10,000 cells within
the gated region were analyzed.
Mitochondrial membrane potential
analysis
Cells were plated on 6-well at 1.0×106
cells/well and treated with 1 μM HAC-Y6 for 6–24 h.
Mitochondrial membranes were stained with 0.5 ml JC-1 working
solution (BD MitoScreen Kit, Becton-Dickinson) to each sample.
Samples were incubated for 10–15 min at 37°C in the dark.
Mitochondrial membrane potential was measured using the BD
FACSCanto flow cytometer (Becton-Dickinson).
Confocal microscopy
After treatment, cells were fixed with 4% PFA,
blocked with 2% bovine serum albumin, stained with anti-tubulin
monoclonal antibody, and then with FITC conjugated anti-mouse IgG
antibody. PI was used to stain the nuclei. Cells were visualized
using a Leica TCS SP2 Spectral Confocal System.
Tubulin assays
Tubulin assembly was measured through turbidimetry,
as described previously (29).
Assay mixtures containing 1.0 mg/ml (10 μM) tubulin and
varying drug concentrations were preincubated for 15 min at 30°C in
the absence of GTP. The samples were then placed on ice, and 0.4 mM
GTP was added. Reaction mixtures were transferred to cuvettes held
at 0°C, and turbidity development was monitored for 20 min at 30°C
after a rapid temperature increase. Drug concentrations that
inhibited the increase in turbidity by 50% relative to a control
sample were determined. Inhibition of the binding of
[3H]colchicine to tubulin was measured as described
previously (29). A total of 1.0
μM tubulin was incubated with 5.0 μM
[3H]colchicine and 5.0 μM inhibitor for 10 min at
37°C. At this time, approximately 40 to 60% of maximum colchicine
binding occurred.
Western blot assay
The treated cells were collected and washed with
PBS. After centrifugation, cells were lysed in a lysis buffer. The
lysates were incubated on ice for 30 min and centrifuged at 12,000
x g for 20 min. Supernatants were collected, and protein
concentrations were then determined using the Bradford assay. After
adding a 5X sample loading buffer containing 625 mM Tris-HCl, pH
6.8, 500 mM dithiothreitol, 10% SDS, 0.06% bromophenol blue, and
50% glycerol, protein samples were electrophoresed on 10%
SDS-polyacrylamide gels and transferred to a nitrocellulose
membrane. Immunoreactivity was detected using the western blot
chemiluminescence reagent system (PerkinElmer, Boston, MA, USA).
β-actin was used as a loading control.
Statistical analysis
Statistical analysis was performed with an analysis
of variance (ANOVA) followed by the Tukey’s test. All data were
expressed as mean ± SD from at least three independent experiments.
*P<0.001 was indicative of a significant
difference.
Results
Growth inhibitory activity of HAC-Y6
against a panel of human cancer cell lines
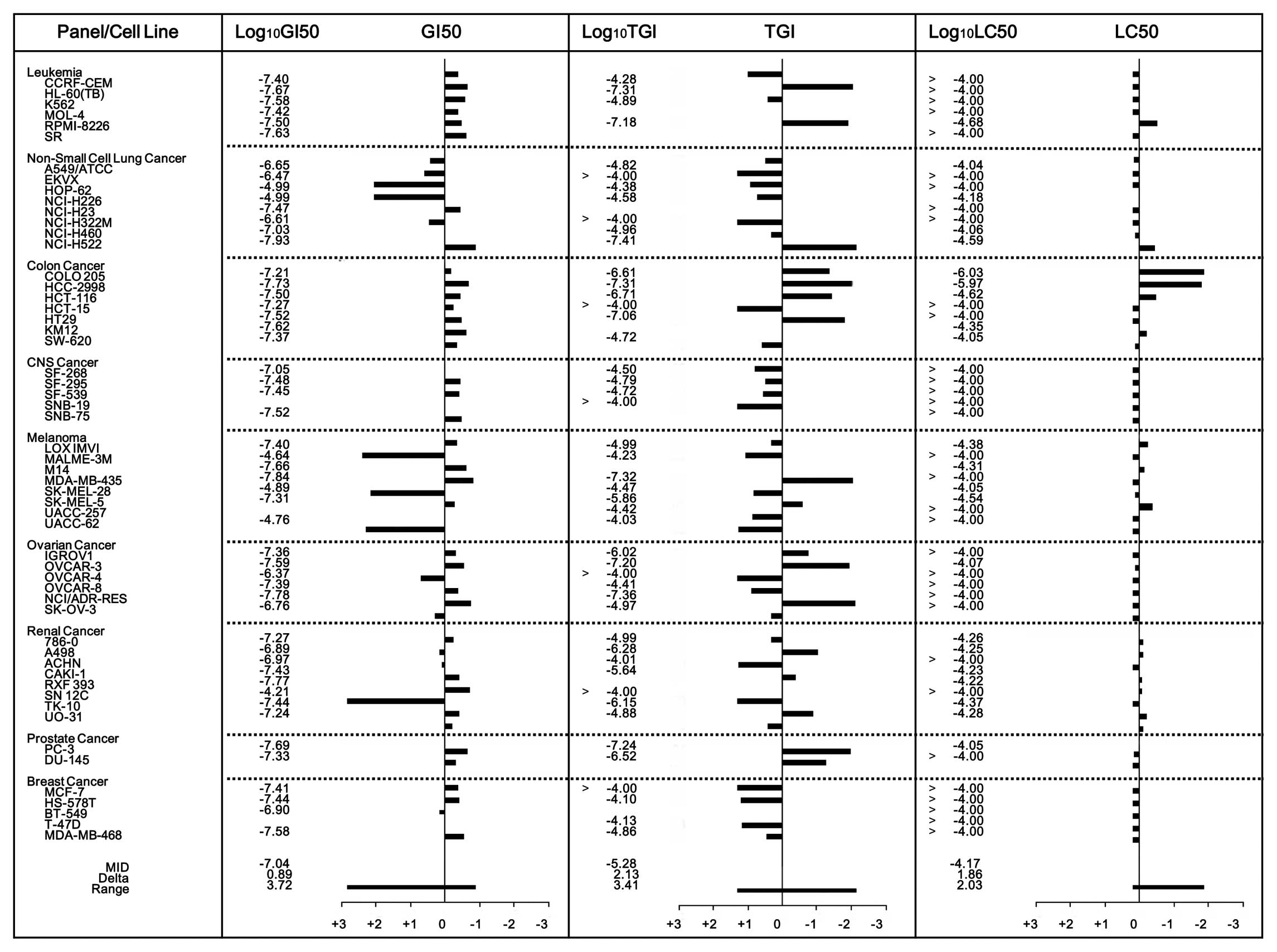
Evaluation of the HAC-Y6 inhibitory activity against
the NCI panel of 60 human cancer cell lines provided the results
shown in Fig. 2. The mean graph
midpoint (MID) values for the log GI50, log TGI, and log
LC50 values were −7.04, −5.26 and −4.17, respectively.
For total growth inhibition (TGI), HAC-Y6 also demonstrated
significant inhibitory activity (log TGI <−6.5) against 12 tumor
cell lines: HL-60, RPMI-8228, NCI-H522, COLO 205, HCC-2998,
HCT-116, HT29, MDA-MB-435, OVCAR-3, NCI/ADR-RES, PC-3 and
DU145.
HAC-Y6 induces toxicity in COLO 205
cells
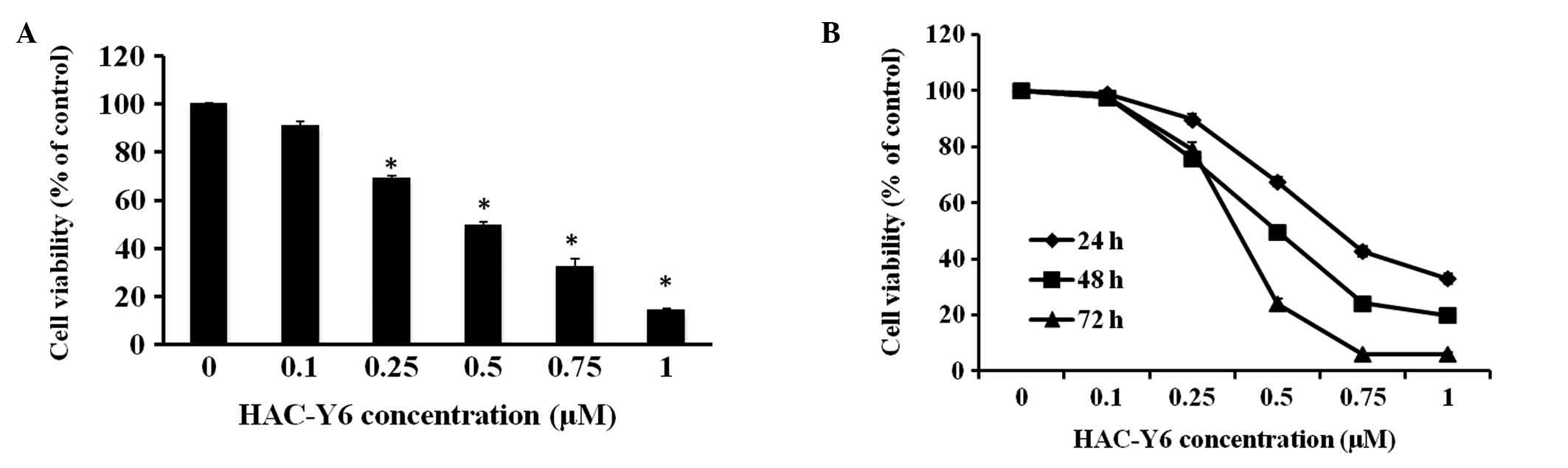
Exposure of COLO 205 cells to HAC-Y6 for 48 h and
performance of MTT metabolism assays confirmed the effects of
HAC-Y6 on cell viability. The IC50 value was 0.52±0.035
μM with HAC-Y6 decreasing COLO 205 cell viability in a
dose-dependent manner. Exposure of COLO 205 cells to 0.1, 0.25,
0.5, 0.75 and 1 μM HAC-Y6 reduced the survival to 90.8±1.9,
69.2±0.9, 49.4±1.7, 32.3±3.4 and 14.1±1.1% of the control (0.1%
DMSO), respectively (Fig. 3A).
HAC-Y6 inhibited COLO 205 cell growth dose- and time-dependently
(Fig. 3B).
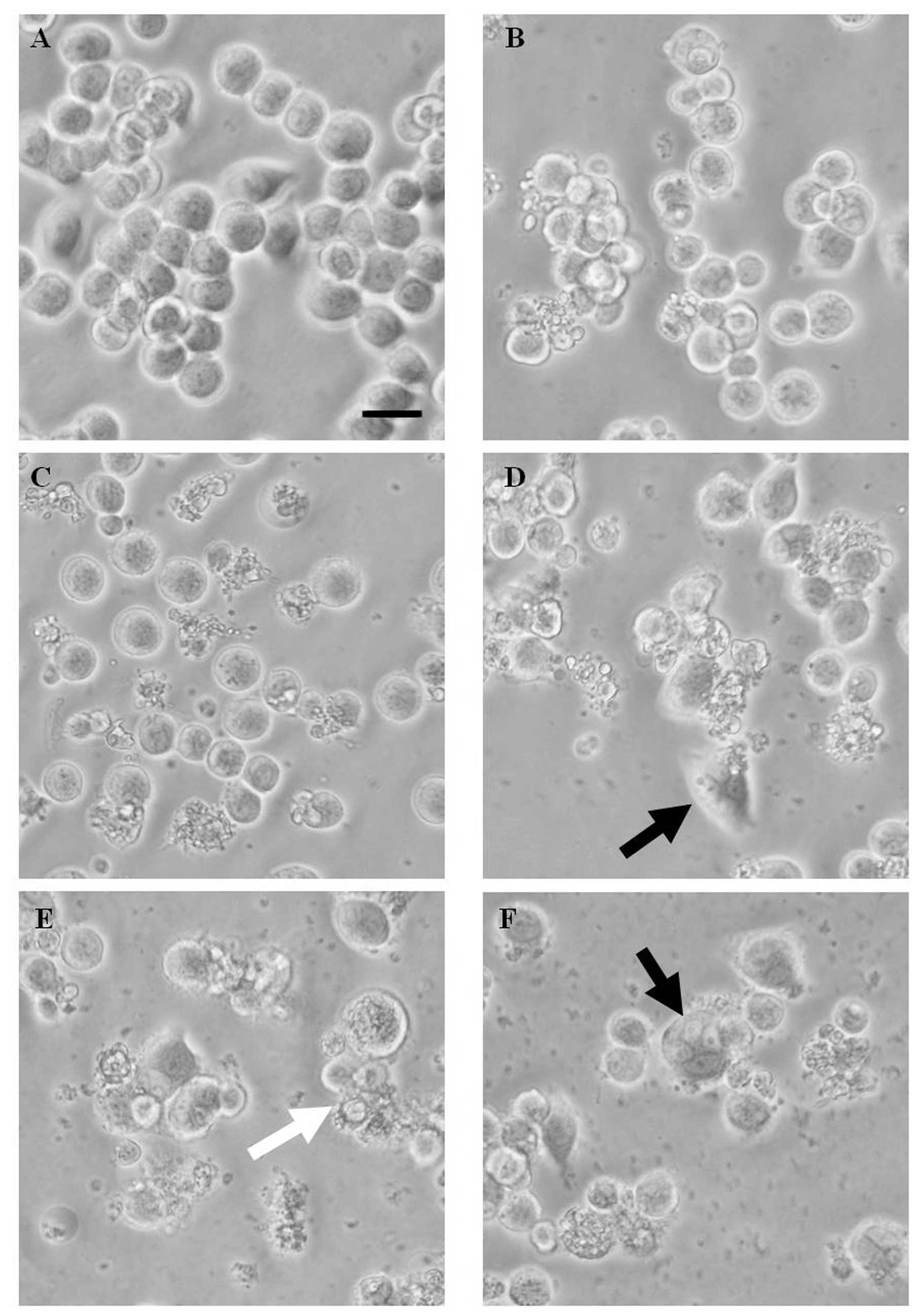
HAC-Y6 induces morphological changes in
COLO 205 cells
Morphological analysis confirmed the HAC-Y6
cytotoxic effects. As shown in Fig.
4, apoptotic morphological changes included cell rounding and
shrinkage after 24 h incubation with 0.5 μM of HAC-Y6 (the
white arrowhead indicates an apoptotic nucleus). Fig. 4D and F show COLO 205 cells with
more than one nucleus per cell (the black arrowheads indicate
multinucleate cells).
HAC-Y6 induces apoptosis in COLO 205
cells
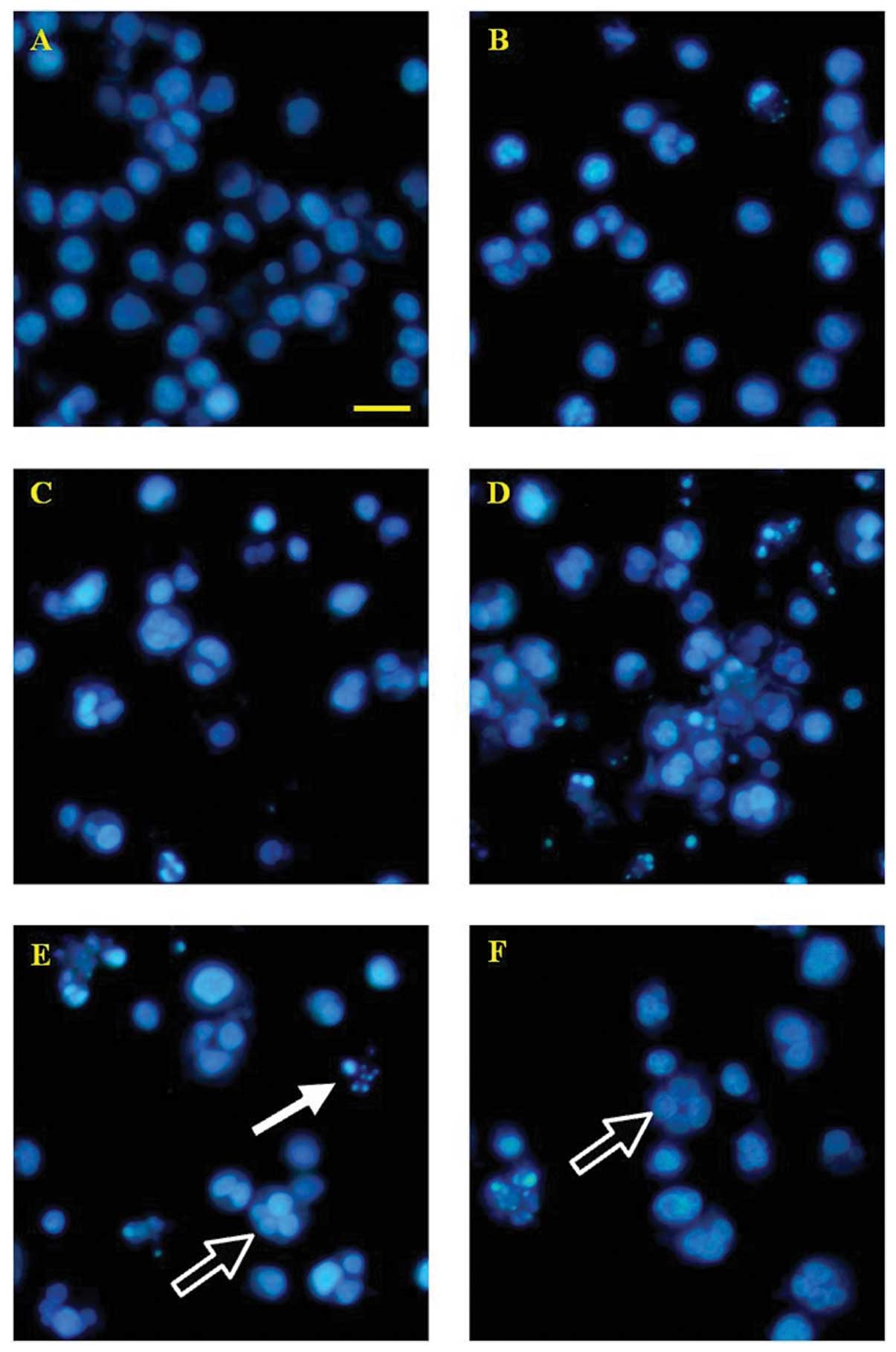
Staining COLO 205 cells with Hoechst 33258 confirmed
apoptosis as the cause of reduced cell viability. As shown in
Fig. 5, control cells without
HAC-Y6 treatment exhibited uniformly dispersed chromatin, normal
organelles, and intact cell membranes. Cells treated with 0.5
μM of HAC-Y6 for 24, 36, 48, 60 and 72 h demonstrated
typical characteristics of apoptosis, including the condensation of
chromatin, the shrinkage of nuclei and the appearance of apoptotic
bodies (the white arrowhead indicates an apoptotic nucleus).
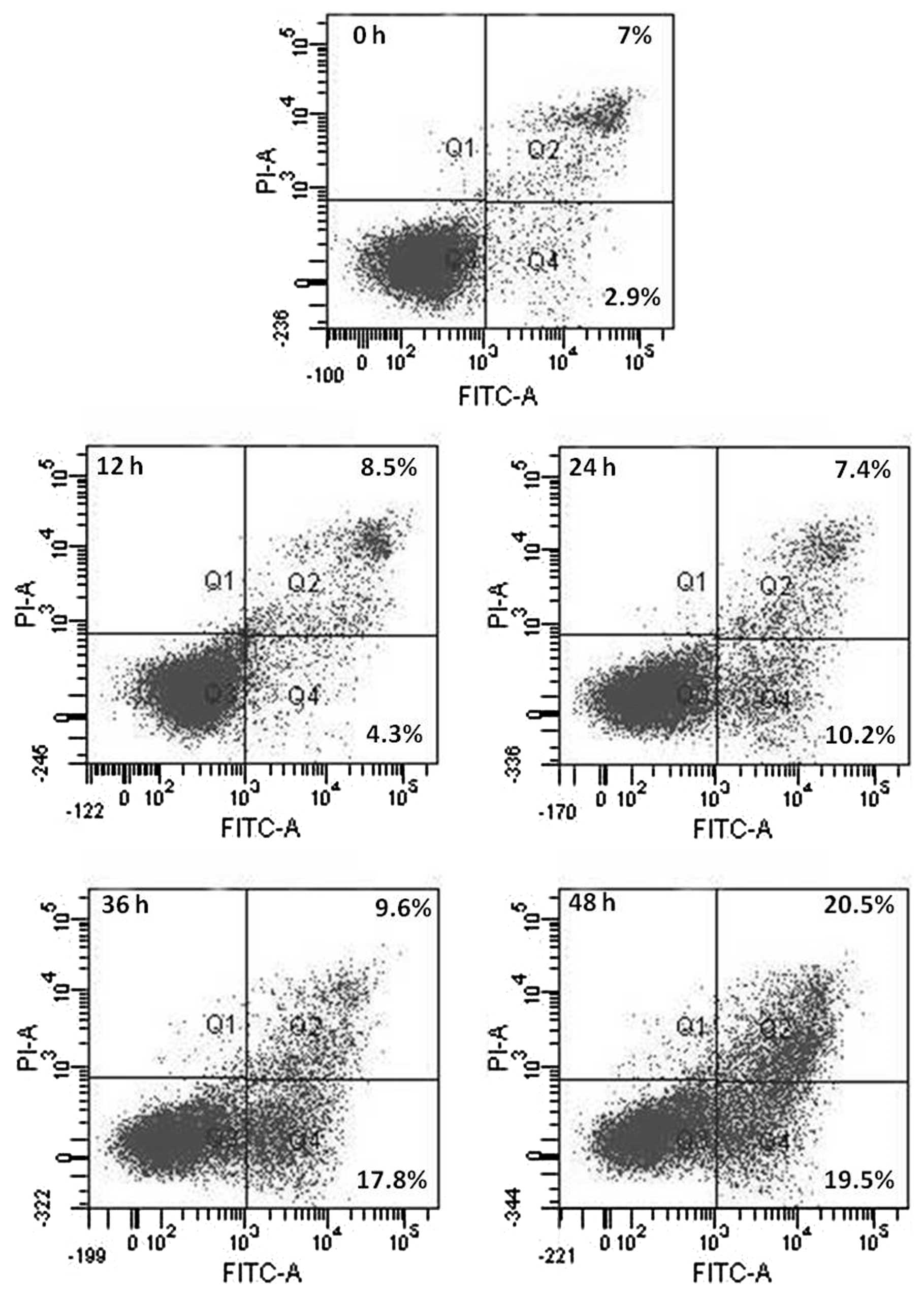
Annexin V-FITC/PI double-labeling detected
phosphatidylserine externalization, a hallmark of the early phase
of apoptosis (Fig. 6). Cells
incubated in the absence of HAC-Y6 for 12, 24, 36 and 48 h were
undamaged and were negative for both Annexin V-FITC and PI staining
(Q3). After incubation with 1 μM HA-Y6 for 24 to 48 h, the
numbers of advanced apoptotic cells stained by positive Annexin
V-FITC and negative PI (Q4) were significantly increased as the
incubation time grew longer. The numbers of advanced apoptotic
cells stained by positive Annexin V-FITC and PI (Q2) also increased
significantly with incubation time.
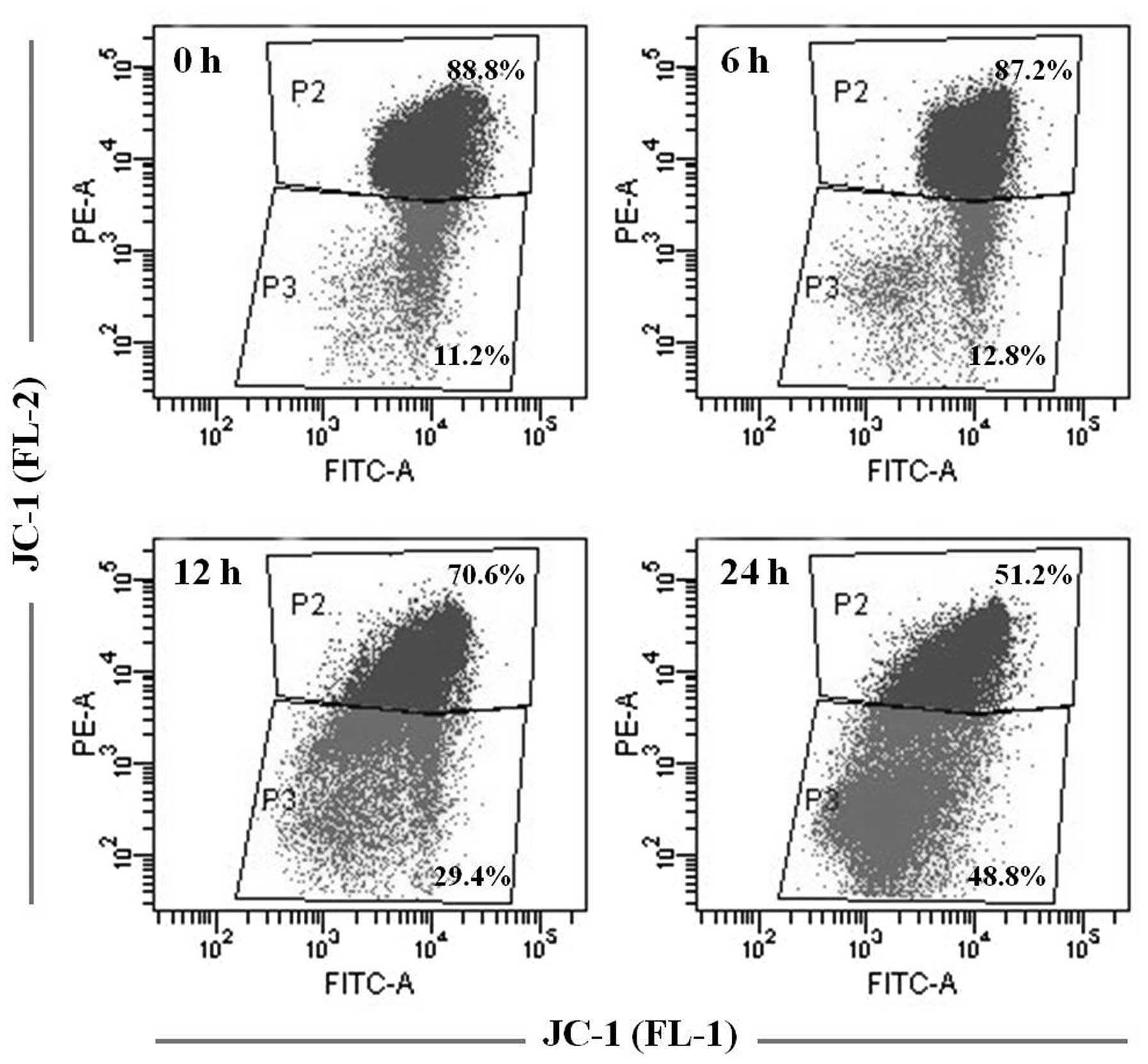
During apoptosis, the mitochondrial membrane
potential (Δψm) decreases. Cells treated with 1 μM of
HAC-Y6 for 6, 12 and 24 h, stained cells with JC-1 confirmed
apoptosis as the cause of decreased Δψm. As shown in
Fig. 7, JC-1 fluorescence is seen
in both the FL2 and FL1 channel (P2) in the control cell population
(0 h). There is a significant increase in the number of cells with
reduced red fluorescence (P3), indicative of a change in the
Δψm, in the population induced to undergo apoptosis (6–24
h). These data demonstrate that HAC-Y6 induced cell apoptosis in
COLO 205 cells.
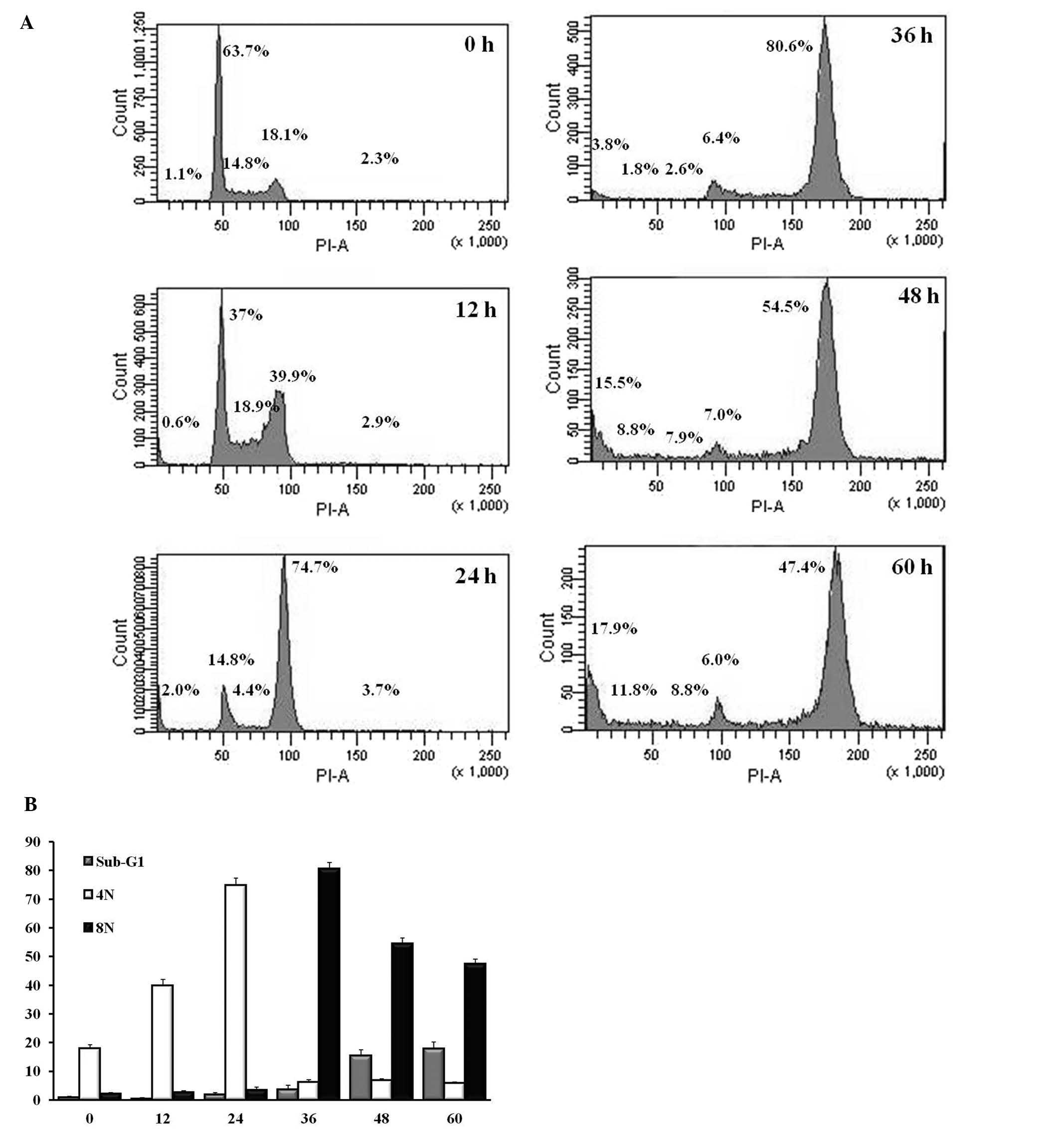
HAC-Y6 induces G2/M arrest,
multinucleation and apoptosis in COLO 205 cells
Treatment of COLO 205 cells with 1 μM of
HAC-Y6 for 0, 12, 24, 36, 48 and 60 h, followed by flow cytometric
analysis to determine cell cycle distribution of the treated cells,
was used to investigate the effects of HAC-Y6 on disruption of the
cell cycle and provide further insights into the apoptotic effects
of the compound. As shown in Fig.
8, HAC-Y6 induced a time-dependent accumulation of
G2/M (4N) cells, as well as causing formation of
octoploid cell (8N) population and apoptotic (sub-G1)
cells. These flow cytometric findings with HAC-Y6-treated COLO 205
cells are in accord with the multinucleated cells presented above
(Figs. 4D, F and 5; the black arrowheads indicate
multinucleate cells).
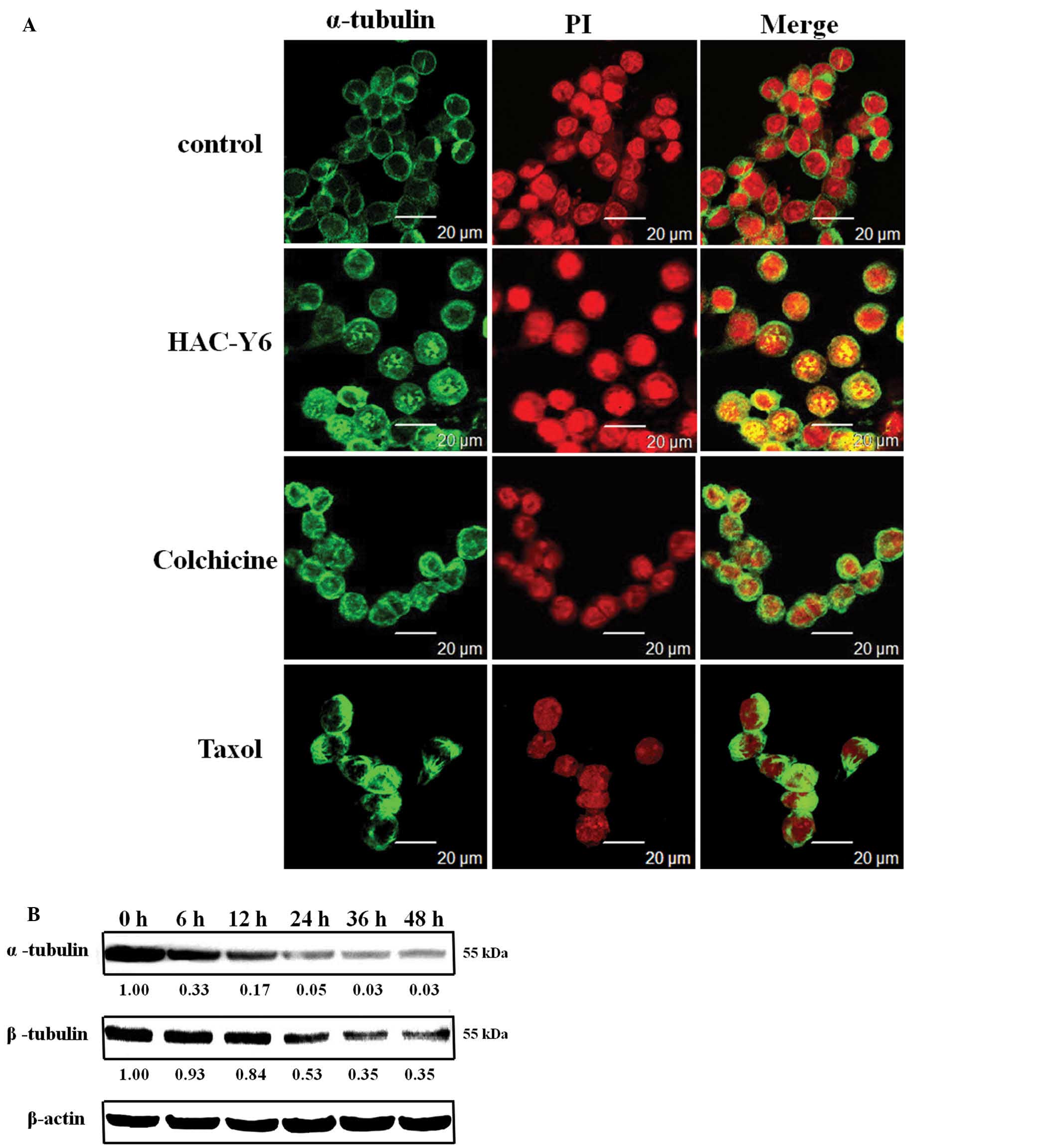
HAC-Y6 inhibites tubulin
polymerization
COLO 205 cells were treated with 1 μM of
HAC-Y6 for 24 h and then stained appropriately and examined in a
confocal microscope to determine the effects of the compound on the
cellular microtubule cytoskeleton. As shown in Fig. 9A, treatment with HAC-Y6 resulted in
microtubule changes similar to those induced by colchicine. Both
compounds caused cellular microtubule depolymerization with short
microtubule fragments scattered throughout the cytoplasm. Taxol, in
contrast, induced significantly increased tubulin polymerization.
As shown in Fig. 9B, after cells
were treated with HAC-Y6 for 6–48 h, HAC-Y6 caused inhibition of
microtubule (α- and β-tubulin) assembly. Therefore, our data
demonstrated that HAC-Y6 induced microtubule depolymerization.
Further examination of HAC-Y6 in tubulin assays and
comparison with combretastatin A-4 (CA-4) provided the results
presented in Table I. HAC-Y6
potently inhibited tubulin assembly, with an IC50 value
of 0.81±0.03 μM. Although HAC-Y6 inhibited tubulin assembly
more potently than CA-4 (IC50 1.3±0.1 μM), it was
slightly less effective than CA-4 in inhibiting colchicine binding
to tubulin.
 | Table I.Anti-tubulin data for HAC-Y6. |
Table I.
Anti-tubulin data for HAC-Y6.
| Compound | Inhibition of
tubulin assemblya
IC50 (μM) ± SD | Inhibition of
colchicine binding with 5 μM inhibitorb % inhibition ± SD |
|---|
| CA-4 | 1.30±0.1 | 98±1 |
| HAC-Y6 | 0.81±0.03 | 90±1 |
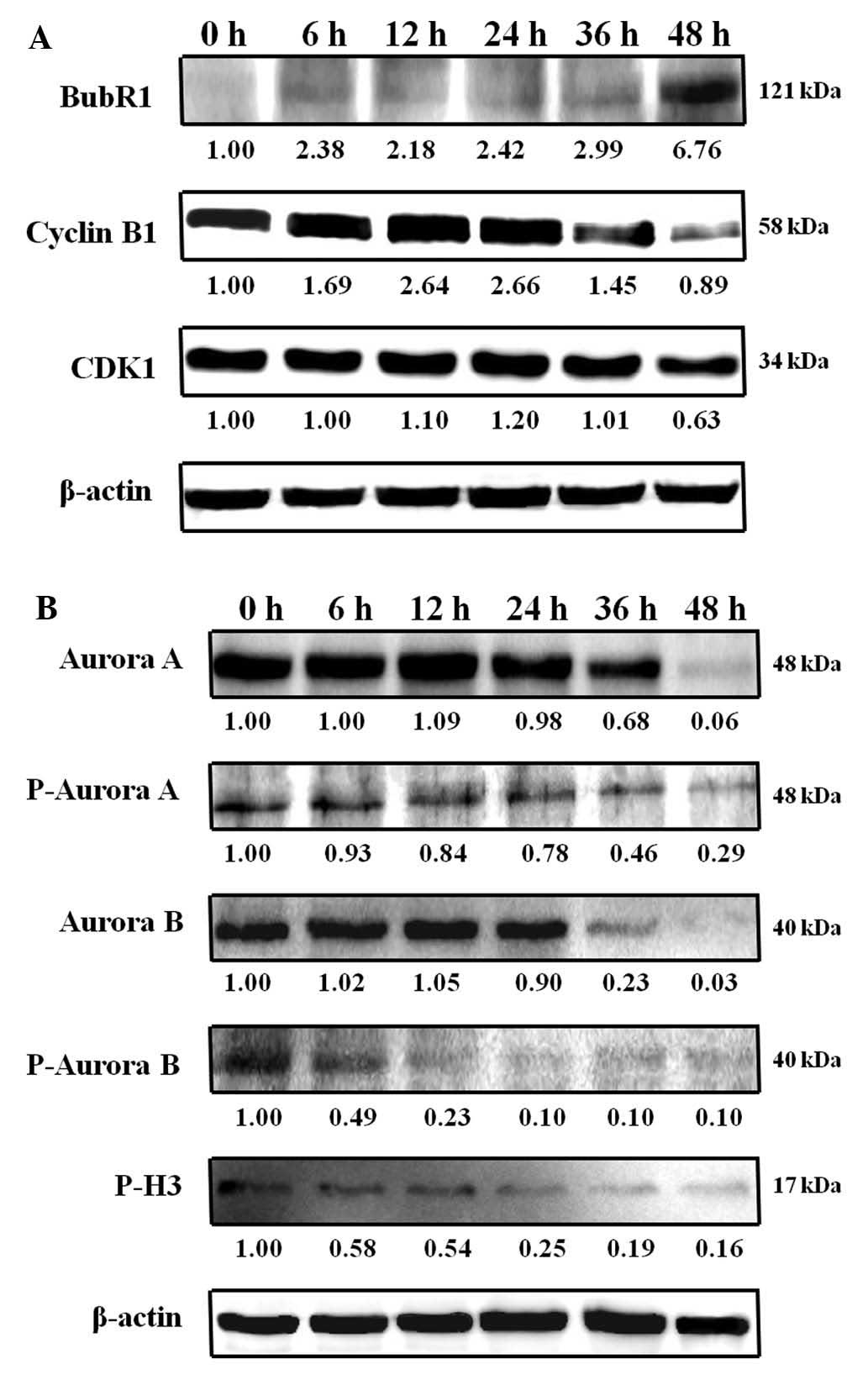
Increased BubR1 and inhibition of aurora
kinase by HAC-Y6 associated with G2/M cell cycle
arrest
Cyclin B1 and CDK1 are markers for induction of
mitotic arrest. Treatment of COLO 205 cells with 1 μM of
HAC-Y6 increased cyclin B1 protein levels (Fig. 10A). BubR1, an essential component
of the mitotic check-point, localizes in kinetochores (8). Treatment of COLO 205 cells with 1
μM HAC-Y6 increased the levels of BubR1 (Fig. 10A). This result suggests that
BubR1 contributed to activation of the mitotic checkpoint induced
by HAC-Y6. The compound directly causes microtubule disassembly,
and the cell cycle protein changes, including increased BubR1
expression, are secondary to the failure to form a spindle, which
results from disruption of tubulin assembly.
Treatment of COLO 205 cells with 1 μM HAC-Y6
was used to investigate the effects of HAC-Y6 on aurora kinase
function. As shown in Fig. 10B,
HAC-Y6 decreased aurora A, phospho-aurora A, aurora B and
phospho-aurora B expression. Histone H3 is one of the substrates of
aurora B kinase. During mitosis, aurora B is required for
phosphorylation of histone H3 on serine 10, and this might be
important for chromosome condensation (15). We therefore examined whether HAC-Y6
inhibited phosphorylation of histone H3 in COLO 205 cells by
western blot analysis. As shown in Fig. 10B, HAC-Y6 decreased phospho-H3
expression after a 6 h treatment. This finding suggests that
inactivation of aurora kinases A and B is involved in
HAC-Y6-induced G2/M arrest and multinucleation.
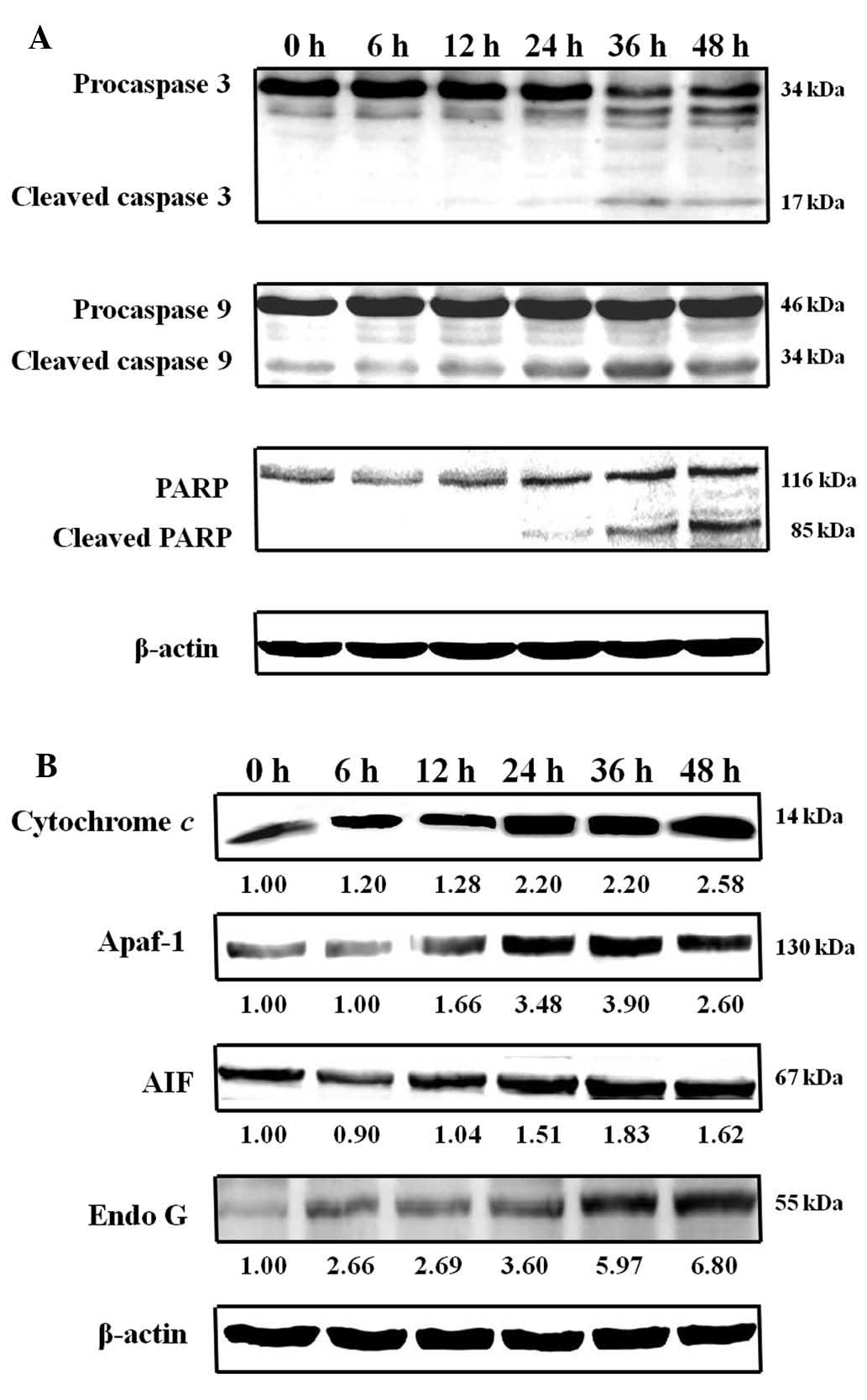
HAC-Y6 induces apoptosis via activation
of mitochondrial signaling pathways and affects Bcl-2 family
proteins in COLO 205 cells
Following our observations that HAC-Y6 caused
apoptosis in COLO 205 cells, we next determined levels of selected
proteins associated with apoptosis. The activation of caspases is a
hallmark of apoptosis. Activated caspases cleave a variety of
target proteins including poly(ADP-ribose) polymerase,
DNA-dependent protein kinase, and sterol regulatory-dependent
binding protein, and thereby disable cellular processes and break
down the cellular structure (23).
To investigate whether HAC-Y6-induced apoptosis was involved in the
activation of caspase cascades, COLO 205 cells were exposed to 1
μM of HAC-Y6 for 6, 12, 24, 36 and 48 h. The activities of
caspase-3 and caspase-9 were then determined using Western blot
analysis, which revealed activation of caspase-3 and caspase-9
within 12 h of HAC-Y6 treatment. Cleavage of PARP, a substrate for
caspase-3, also occurred (Fig.
11A). HAC-Y6 also increased levels of Endo G, AIF, Apaf-1 and
cytochrome c (Fig. 11B).
These results suggest that the mitochondrial signaling pathways of
COLO 205 cells mediate HAC-Y6-induced apoptosis.
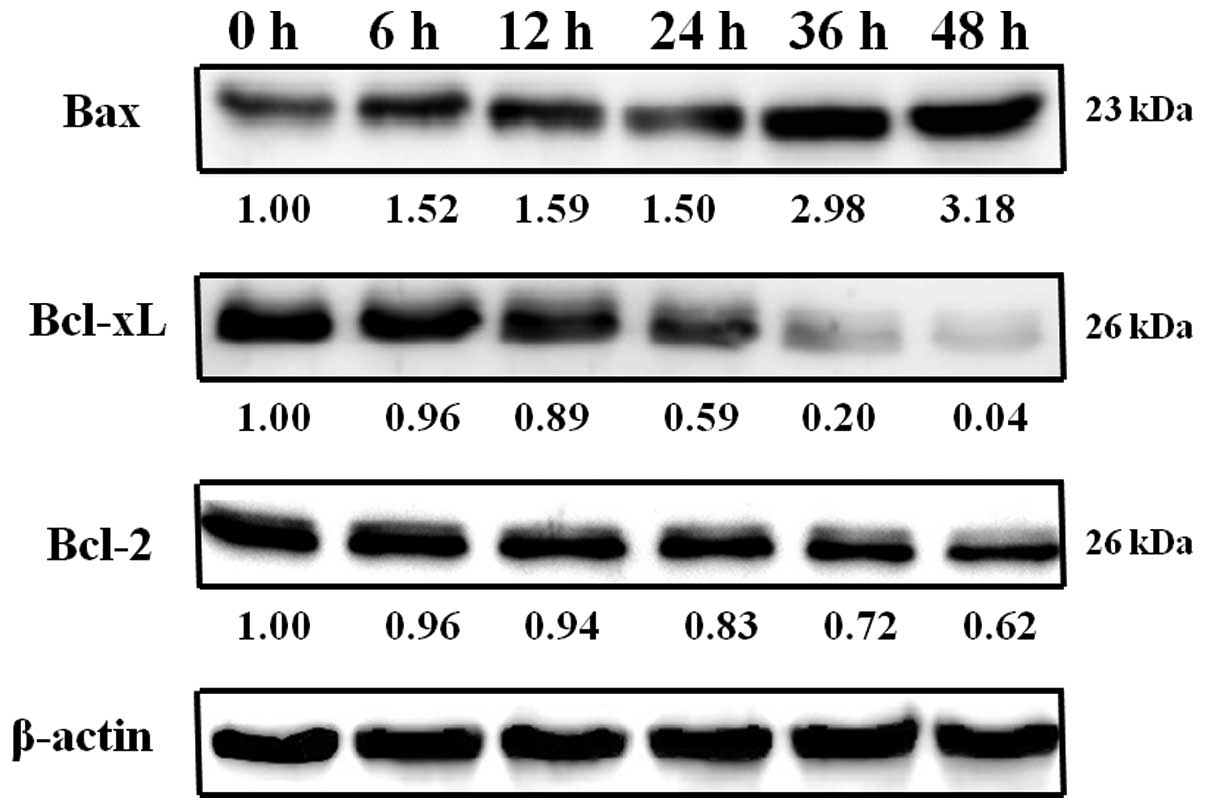
The Bcl-2 family proteins are key regulators of
mitochondrial-related apoptotic pathways (30). Some of these proteins (such as
Bcl-2 and Bcl-xL) are anti-apoptotic, whereas others (such as Bad,
Bax and Bid) are pro-apoptotic. The balance of pro- and
anti-apoptotic bcl-2 proteins influences the sensitivity of cells
to apoptotic stimuli (31).
Exposure of COLO 205 cells to 1 μM of HAC-Y6 for 6, 12, 24,
36 and 48 h verified the involvement of Bcl-2 protein activity in
HAC-Y6-induced apoptosis. As shown in Fig. 12, results indicated that HAC-Y6
reduced anti-apoptotic Bcl-2 and Bcl-xL levels, as well as
increased pro-apoptotic Bax levels and the release of Endo G, AIF,
Apaf-1, cytochrome c and procaspase-9 from the mitochondria
to the cytosol. Release of Apaf-1, and cytochrome c leads to
the activation of caspase-9. Activated caspase-9, in turn, cleaves
and activates caspase-3.
Discussion
Derivation of new drugs from natural products
involves the synthesis of new compounds by modifying the structural
skeletons of the natural products. Several reports have
demonstrated that carbazole alkaloids exhibit anticancer activity
(24–26). α-carboline is a bioisostere of
carbazole, where the benzene ring of carbazole is replaced by a
pyridine ring, and previous studies have described the anticancer
effects of its derivatives (32,33).
In this study, we describe the anticancer mechanisms of the novel
α-carboline derivative HAC-Y6.
The NCI results for HAC-Y6 presented in Fig. 2 indicated potent inhibitory
activity against multiple cancer cell lines. The LC50
values, however, indicated selective inhibition of COLO 205 and HCC
2998 colon cancer cells, with log LC50 values of −6.3
and −5.97, respectively; potencies that are approximately 100 times
the average value (−4.17). These results prompted our further
investigation of the effects of the compound on COLO 205 cells.
Investigation of the anticancer activity of HAC-Y6
in COLO 205 cells provided data indicating that HAC-Y6 induced
cytotoxicity in COLO 205 cells in a dose- and time-dependent manner
(Fig. 3), acting through
G2/M arrest, polyploidy and apoptosis (Fig. 8). Annexin V/PI double staining
demonstrated the presence of apoptotic cells in HAC-Y6-treated COLO
205 cells (Fig. 6). Mitochondrial
membrane potential analysis showed that HAC-Y6 induced
mitochondrial membrane potential in support of apoptosis in COLO
205 cells (Fig. 7).
Microtubules are important cellular targets for
anticancer therapy, because of their key role in mitosis (34). In this study, HAC-Y6 induced the
depolymerization of microtubules in COLO 205 cells. Treatment with
HAC-Y6 for 24 h resulted in microtubule changes similar to those
induced by colchicine (Fig. 9A).
HAC-Y6 caused inhibition of microtubule (α- and β-tubulin) assembly
(Fig. 9B). HAC-Y6 can thus be
classified as a microtubule-depolymerizing agent.
Previous investigations have reported that cyclin
B1/CDK1 complexes are involved in the regulation of G2/M
phase and M-phase transitions (9,35).
HAC-Y6 initiated induction of G2/M phase arrest (4N)
within 12 to 24 h and polyploidy (8N) within 36 to 60 h of
treatment (Fig. 8B). Our data
showed increased levels of cyclin B1 after HAC-Y6 treatment
(Fig. 10A). HAC-Y6 arrested the
growth of COLO 205 cells at the G2/M phase through the
accumulation of cyclin B1. BubR1 is a key component of the mitotic
spindle checkpoint machinery (8,9). In
this study, BubR1 upregulation caused microtubule disruption, as
indicated by a significant increase in the percentage of cell cycle
G2/M arrest. Results demonstrated an increase in BubR1
after HAC-Y6 treatment (Fig.
10A). We suggest that HAC-Y6 induced G2/M arrest and
multinucleation via the accumulation of BubR1 protein.
Aurora kinases play important roles in chromosome
alignment, segregation and cytokinesis during mitosis (14,17,36).
Our data showed decreased aurora A, phospho-aurora A, aurora B,
phospho-aurora B and phospho-H3 expression after HAC-Y6 treatment
(Fig. 10B). HAC-Y6, therefore,
arrested the growth of COLO 205 cells at the G2/M phase
and induced polyploidy through the inactivation of aurora
kinases.
Apoptosis regulators provide the basis for novel
therapeutic strategies aimed at promoting tumor cell death
(21,37). Mitochondria and Bcl-2 largely
control intrinsic pro-apoptotic pathways. When mitochondria receive
an apoptotic signal, their outer membranes become permeabilized,
releasing Endo G, AIF, Apaf-1, cytochrome c and
procaspase-9, and into the cytosol and activating caspase-3 through
caspase-9, leading to apoptosis (38–40).
Our data showed that these effects occurred after HAC-Y6 treatment
(Fig. 11). Proteolytic
degradation of PARP further demonstrated the involvement of caspase
activation. These findings together suggest that HAC-Y6 might
activate intrinsic signaling pathways.
The Bcl-2 family proteins largely mediate the
mitochondrial apoptotic pathway (41,42).
Overexpression of Bcl-2 increases cell survival by suppressing
apoptosis. Bax levels increase in conjunction with Bax inhibition
of Bcl-2, and the cells undergo apoptosis. The present results
showed increased Bax 6 h after HAC-Y6 treatment and decreased
Bcl-xL and Bcl-2 12 h after HAC-Y6 treatment (Fig. 12). HAC-Y6, therefore, induced
apoptosis of COLO 205 cells through Bax activation and Bcl-xL and
Bcl-2 inactivation.
Among our findings, HAC-Y6 significantly decreased
levels of HSP90 in COLO 205 cells after a 2 h treatment (data not
shown). HSP90 plays an essential role as a molecular chaperone for
stress-accumulated misfolded proteins to prevent their aggregation
(7,43,44).
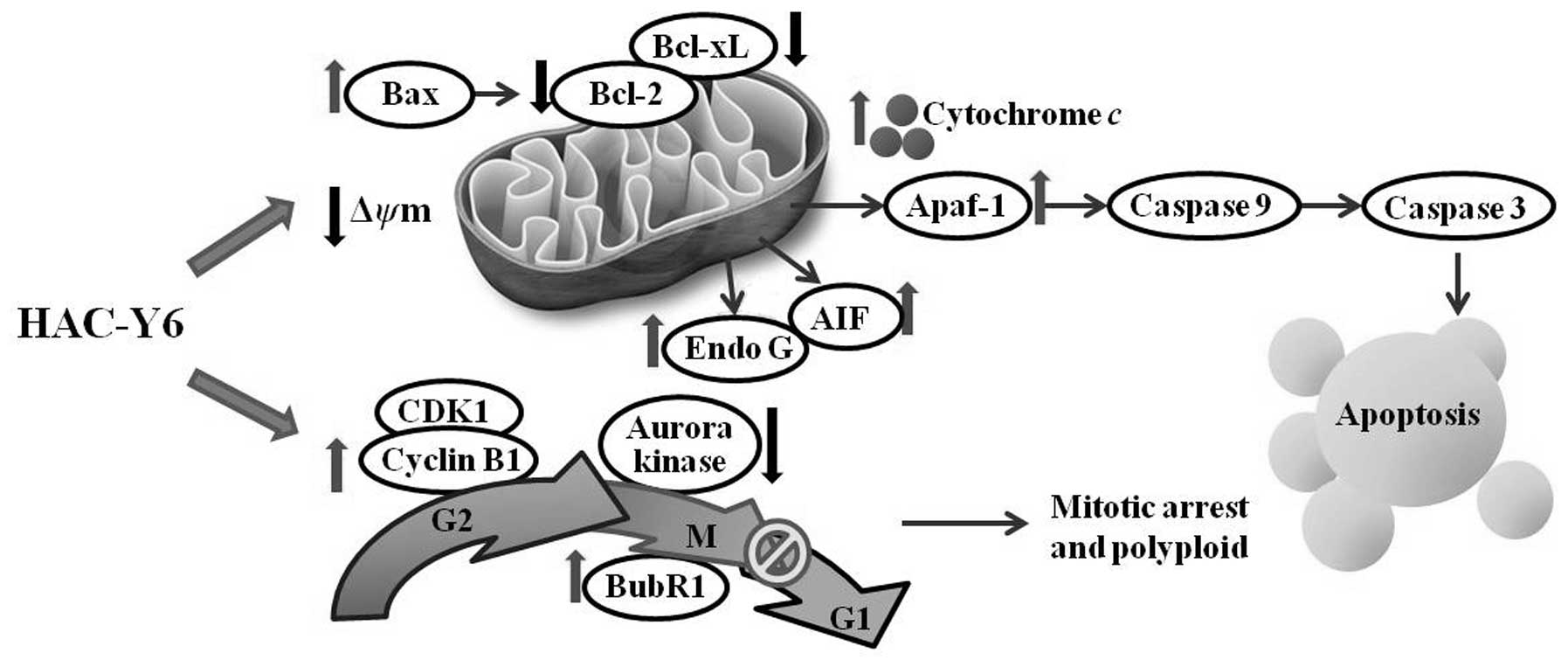
Fig. 13 summarizes
the molecular signaling pathways induced by HAC-Y6. We demonstrated
that HAC-Y6, a novel synthetic HSP90 inhibitor, exerts potent
anticancer activity against COLO 205 cells through microtubule
depolymerization, BubR1 activation, aurora A and aurora B
inactivation, induction of G2/M arrest and polyploidy.
HAC-Y6 also induces apoptosis of COLO 205 cells via intrinsic
signaling pathways. These findings suggest that HAC-Y6 has
potential use as a novel therapeutic agent for the treatment of
human colon carcinoma.
Acknowledgements
This study was supported by a research
grant from the National Science Council of the Republic of China,
awarded to L.-J.H. (NSC 98-2628-B-039-018-MY3). Experiments and
data analysis were performed, in part, through the use of the
Medical Research Core Facilities Center, Office of Research and
Development, China Medical University, Taichung, Taiwan, R.O.C.
References
|
1.
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2.
|
Ceelen W, Van Nieuwenhove Y and Pattyn P:
Surgery and intracavitary chemotherapy for peritoneal
carcinomatosis from colorectal origin. Acta Gastroenterol Belg.
71:373–378. 2008.PubMed/NCBI
|
|
3.
|
Watson AJ: An overview of apoptosis and
the prevention of colorectal cancer. Crit Rev Oncol Hematol.
57:107–121. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Huang WW, Ko SW, Tsai HY, et al:
Cantharidin induces G2/M phase arrest and apoptosis in human
colorectal cancer colo 205 cells through inhibition of CDK1
activity and caspase-dependent signaling pathways. Int J Oncol.
38:1067–1073. 2011.
|
|
5.
|
Sato S: Modulation of Akt kinase activity
by binding to Hsp90. Proc Natl Acad Sci USA. 97:10832–10837. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Sreedhar AS, Sti C and Csermely P:
Inhibition of Hsp90: a new strategy for inhibiting protein kinases.
Biochim Biophys Acta. 1697:233–242. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Schmitt E, Gehrmann M, Brunet M, Multhoff
G and Garrido C: Intracellular and extracellular functions of heat
shock proteins: repercussions in cancer therapy. J Leukoc Biol.
81:15–27. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Morrow CJ: Bub1 and aurora B cooperate to
maintain BubR1-mediated inhibition of APC/CCdc20. J Cell Sci.
118:3639–3652. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Peters JM: The anaphase promoting
complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell
Biol. 7:644–656. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Jordan MA and Wilson L: Microtubules as a
target for anti-cancer drugs. Nat Rev Cancer. 4:253–265. 2004.
View Article : Google Scholar
|
|
11.
|
Bhalla KN: Microtubule-targeted anticancer
agents and apoptosis. Oncogene. 22:9075–9086. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Carvajal RD, Tse A and Schwartz GK: Aurora
kinases: new targets for cancer therapy. Clin Cancer Res.
12:6869–6875. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Pérez Fidalgo J, Roda D, Roselló S,
Rodríguez-Braun E and Cervantes A: Aurora kinase inhibitors: a new
class of drugs targeting the regulatory mitotic system. Clin Transl
Oncol. 11:787–798. 2009.PubMed/NCBI
|
|
14.
|
Fu J, Bian M, Jiang Q and Zhang C: Roles
of aurora kinases in mitosis and tumorigenesis. Mol Cancer Res.
5:1–10. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Keen N and Taylor S: Aurora-kinase
inhibitors as anticancer agents. Nat Rev Cancer. 4:927–936. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Carpinelli P, Ceruti R, Giorgini ML, et
al: PHA-739358, a potent inhibitor of Aurora kinases with a
selective target inhibition profile relevant to cancer. Mol Cancer
Ther. 6:3158–3168. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Andrews PD: Aurora kinases: shining lights
on the therapeutic horizon? Oncogene. 24:5005–5015. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Ruchaud S, Carmena M and Earnshaw WC:
Chromosomal passengers: conducting cell division. Nat Rev Mol Cell
Biol. 8:798–812. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Monaco L: Inhibition of Aurora-B kinase
activity by poly(ADP-ribosyl)ation in response to DNA damage. Proc
Natl Acad Sci USA. 102:14244–14248. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Yasui Y: Autophosphorylation of a newly
identified site of aurora-B is indispensable for cytokinesis. J
Biol Chem. 279:12997–13003. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Ghobrial IM, Witzig TE and Adjei AA:
Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin.
55:178–194. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
O’Brien MA and Kirby R: Apoptosis: a
review of pro-apoptotic and anti-apoptotic pathways and
dysregulation in disease. J Vet Emerg Crit Care. 18:572–585.
2008.
|
|
23.
|
Lawen A: Apoptosis? an introduction.
BioEssays. 25:888–896. 2003. View Article : Google Scholar
|
|
24.
|
Cai Y, Cai B, Cui CB, Zhang DY, Han B,
Wang YG and Wang MW: Apoptosis-inducing effect of carbazole
alkaloid (HY-1) in human erythroleukemia K562 cells. Zhonghua Zhong
Liu Za Zhi. 27:457–460. 2005.(In Chinese).
|
|
25.
|
Roy M: Mechanism of mahanine-induced
apoptosis in human leukemia cells (HL-60). Biochem Pharmacol.
67:41–51. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Roy MK, Thalang VN, Trakoontivakorn G and
Nakahara K: Mahanine, a carbazole alkaloid from Micromelum
minutum, inhibits cell growth and induces apoptosis in U937
cells through a mitochondrial dependent pathway. Br J Pharmacol.
145:145–155. 2005.
|
|
27.
|
Nafisi S, Malekabady ZM and Khalilzadeh
MA: Interaction of β-carboline alkaloids with RNA. DNA Cell Biol.
29:753–761. 2010.
|
|
28.
|
Mosmann T: Rapid colorimetric assay for
cellular growth and survival: application to proliferation and
cytotoxicity assays. J Immunol Methods. 65:55–63. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Chang YH, Hsu MH, Wang SH, et al: Design
and synthesis of
2-(3-benzo[b]thienyl)-6,7-methylenedioxyquinolin-4-one analogues as
potent antitumor agents that inhibit tubulin assembly. J Med Chem.
52:4883–4891. 2009.
|
|
30.
|
Zhai D, Jin C, Huang Z, Satterthwait AC
and Reed JC: Differential regulation of Bax and Bak by
anti-apoptotic Bcl-2 family proteins Bcl-B and Mcl-1. J Biol Chem.
283:9580–9586. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Brunelle JK and Letai A: Control of
mitochondrial apoptosis by the Bcl-2 family. Cell Sci. 122:437–441.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Tsai JY, Lin YC, Hsu MH, Kuo SC and Huang
LJ: Synthesis and cytotoxicity of 1,6,8,9-substituted α-carboline
derivatives. Kaohsiung J of Med Sci. 26:593–602. 2010.
|
|
33.
|
Tasi JY, Hung CM, Bai ST, et al: Induction
of apoptosis by HAC-Y6, a novel microtubule inhibitor, through
activation of the death receptor 4 signaling pathway in human
hepatocellular carcinoma cells. Oncol Rep. 24:1169–1178. 2010.
|
|
34.
|
Perez EA: Microtubule inhibitors:
differentiating tubulin-inhibiting agents based on mechanisms of
action, clinical activity, and resistance. Mol Cancer Ther.
8:2086–2095. 2009. View Article : Google Scholar
|
|
35.
|
Yang J, Chen G, Hsia T, et al: Diallyl
disulfide induces apoptosis in human colon cancer cell line (COLO
205) through the induction of reactive oxygen species, endoplasmic
reticulum stress, caspases casade and mitochondrial-dependent
pathways. Food Chem Toxicol. 47:171–179. 2009. View Article : Google Scholar
|
|
36.
|
Yang J, Ikezoe T, Nishioka C, et al:
AZD1152, a novel and selective aurora B kinase inhibitor, induces
growth arrest, apoptosis and sensitization for tubulin
depolymerizing agent or topoisomerase II inhibitor in human acute
leukemia cells in vitro and in vivo. Blood. 110:2034–2040. 2007.
View Article : Google Scholar
|
|
37.
|
Lowe SW and Lin AW: Apoptosis in cancer.
Carcinogenesis. 21:485–495. 2000. View Article : Google Scholar
|
|
38.
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Eberle J, Fecker LF, Forschner T, Ulrich
C, Rowert-Huber J and Stockfleth E: Apoptosis pathways as promising
targets for skin cancer therapy. Br J Dermatol. 156(Suppl 3):
18–24. 2007. View Article : Google Scholar
|
|
40.
|
Dlamini Z, Mbita Z and Zungu M: Genealogy,
expression, and molecular mechanisms in apoptosis. Pharmacol Ther.
101:1–15. 2004. View Article : Google Scholar
|
|
41.
|
Bagci EZ, Vodovotz Y, Billiar TR,
Ermentrout GB and Bahar I: Bistability in apoptosis: roles of Bax,
Bcl-2, and mitochondrial permeability transition pores. Biophys J.
90:1546–1559. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Antonsson B, Conti F, Ciavatta A, et al:
Inhibition of Bax channel-forming activity by Bcl-2. Science.
277:370–372. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Pratt WB and Toft DO: Regulation of
signaling protein function and trafficking by the hsp90/hsp70-based
chaperone machinery. Exp Biol Med (Maywood). 228:111–133.
2003.PubMed/NCBI
|
|
44.
|
Bai L, Xu S, Chen W, et al: Blocking
NF-kappaB and Akt by Hsp90 inhibition sensitizes Smac mimetic
compound 3-induced extrinsic apoptosis pathway and results in
synergistic cancer cell death. Apoptosis. 16:45–54. 2011.
View Article : Google Scholar : PubMed/NCBI
|