Introduction
Mesenchymal stromal cells (MSCs) are multipotent
cells that can be mobilized from the bone marrow and other tissues
and localize to sites of inflammation including tumors and areas of
injury (1–11). They have the capacity to
differentiate into mesenchymal cell types including adipocytes,
chondrocytes and myocytes (5).
Upon incorporation into the stroma, MSCs promote tumor growth and
metastasis (11,12). The molecular mechanisms regulating
the mobilization and homing of MSCs to tumors have not been
completely defined.
p53 is a transcription factor and tumor suppressor
gene that is mutated in >50% of human cancers (13). It regulates a diverse array of
cellular processes including cell cycle control, DNA repair, growth
factor production and cell motility. Recent studies have suggested
a role for stromal p53 in tumor biology. P53-null stromal
fibroblasts promote more efficient tumor growth in a murine
prostate cancer model (14) and
there is a reduced latency of tumor formation in xenografts in
p53-deficient mice compared to animals with wild-type p53 (15). Additionally, administration of MSCs
carrying a p53 mutation decreased the time required to develop
mammary tumors in
ApcMin/+Rag2−/−
mice (16). These observations
were extended to human cancers where loss of heterozygosity in the
TP53 gene in carcinoma-associated fibroblasts was identified in a
number of tumor types (17–21).
In experimental systems, tumors with decreased stromal p53 activity
are more resistant to chemotherapy (22) and have increased tumor growth
(14).
In vitro, tumor cells stimulate MSC motility
and, over time, induce MSCs to adopt a carcinoma-associated
fibroblast phenotype (9,12). Because p53 activity impacts cell
mobility in fibroblasts (23,24)
and may also play a role in the differentiation of mesenchymal
cells (25) it is reasonable to
suggest that p53 status influences the interaction between
neoplastic and stromal cells. One mechanism for p53 regulation of
tumor/stromal interaction is through modulation of CXCL12
production. Induction of MSC migration by tumor cells and
stimulation of tumor growth by fibroblasts are dependent on stromal
CXCL12 production (9,14).
Our data show that p53 regulates MSC motility.
Increased p53 levels inhibit MSC mobility in response to tumor
cells. The influence of p53 on MSC motility may be mediated, in
part, through the transcriptional regulation of CXCL12. Conversely,
MSCs with p53 knock-down show increased migration to tumor cells
in vitro as well as to in vivo tumors. Interestingly,
MSCs with increased p53 do not show increased migration in response
to tumor conditioned media, even in an environment with a high
concentration of exogenous CXCL12. This suggests that p53 is
involved in other aspects of tumor/stromal interaction distinct
from CXCL12 production. Our study demonstrates that stromal p53
status influences important aspects of the response of MSCs to
tumor cells and provides additional insight into the molecular
systems that regulate this interaction.
Materials and methods
Reagents and cell lines
C57BL/6J p53−/− mice were generously
provided by Dr Arnold Levine. C57BL/6J wt mice were purchased from
Charles River Laboratories (Wilmington, MA, USA). Nude mice were
purchased from Taconic Farms (Hudson, NY, USA). All animal
procedures were approved by the Animal Care and Use Committee of
RWJMS. MDA- MB231 cells were obtained from American Type Culture
Collection (Manassas, VA, USA http://www.atcc.org); pooled human MSCs were obtained
from Lonza (Walkersville, MD, USA, http://www.lonza.com) and used in early passage (below
passage 8). MDA-MB231 cells were cultured in DMEM (Invitrogen,
Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS)
and 100 U/ml penicillin G and 100 μg/ml streptomycin at 37°C
in 5% CO2. Human MSCs were expanded in MesenCult media
with hMSC stimulatory supplements (Lonza) and 10% FBS. Antibodies
used in these studies included p53 (SC263) (Santa Cruz
Biotechnology, Santa Cruz, CA, USA); α-tubulin and p21
(Sigma-Aldrich, St. Louis, MO, USA); GAPDH (Trevigen, Gaithersberg,
MD, USA).
Transwell chamber migration assay
A Falcon cell culture insert system along with
companion 24-well tissue culture plate was used for the chemotaxis
assay as described previously (9).
The polyethylene terepthalate membrane, pore size 8 μM, was
selected to allow passage of mammalian cells. The insert was
removed aseptically and placed in the notch of each well using
forceps. MSCs (1–2×104) were plated in 500 μl of
α-MEM (Invitrogen) supplemented with 10% heat-inactivated FBS and
1% penicillin/streptomycin, placed in the insert (top chamber). The
bottom chamber contained either conditioned medium from tumor cells
or control medium (DMEM containing 2% FBS and
penicillin/streptomycin). Migration assays were stopped after 16 h
and cells remaining on the top of the membrane were removed with a
wet cotton swab. MCSs that had migrated through the membrane were
stained with crystal violet. Stained cells were counted under high
power magnification (×40). For some experiments Nutlin-3
(Sigma-Aldrich, St. Louis, MO, USA) or recombinant CXCL12 (R&D
Systems, Minneapolis, MN, USA) were added to both the upper and
lower chambers to the indicated final concentrations.
Knockdown of p53 and CXCL12 in hMSCs
using siRNA and lentiviral short hairpin RNA
Human MSCs (1.5×105 cells) were plated in
α-MEM (Invitrogen) with 10% FBS. After overnight incubation hMSCs
were transfected with small interfering RNA (siRNA) specifically
targeting p53 or CXCL12 or a scrambled sequence serving as a
control (Thermo Scientific) using Lipofectamine 2000 (Invitrogen).
Experiments were performed 2 days after transfection. Expression
constructs containing short hairpin RNA (shRNA) sequences targeting
p53 were obtained from Santa Cruz. hMSCs were infected using the
manufacturer’s protocols and the knockdown of p53 was confirmed by
western blotting. Cells were allowed to recover for 24 h prior to
performing experiments.
Production of conditioned medium from
MDA-MB-231 cells
To obtain tumor conditioned medium, a ratio of
7.5×104 tumor cells/700 μl of DMEM containing 2%
heat-inactivated FBS and 1% penicillin/streptomycin (Invitrogen)
were incubated overnight. Conditioned medium was collected and spun
down at 1,200 rpm to remove cellular debris. Supernatant was
filtered through a 0.45-μm steriflip filter (Millipore,
Billerica, MA, USA) prior to use in experiments.
Western blotting
Human MSCs were cultured to 80% confluence in a
150-mm tissue culture dish. Cells were then treated with MDA-MB231
conditioned medium or control medium with 25 μM Nutlin-3 or
vehicle [dimethyl sulfoxide (Sigma-Aldrich)] for 6 and 24 h.
Following this treatment, cells were scraped from the dish and
re-suspended in 200 μl of radio-immunoprecipitation assay
(RIPA) buffer plus 10 μg/ml aprotinin, 10 μg/ml
leupeptin and 1 mM phenylmethylsulfonyl fluoride (PMSF). After
clearing the cell lysates by centrifugation (16,000 g, 20 min at
4°C), the protein concentration was determined (Pierce, IL, USA).
After boiling for 10 min, lysates (20 μg) were resolved by
SDS-polyacrylamide gel electrophoresis, transferred onto PDVF
membrane (Millipore) and visualized by immunoblotting with
antibodies of interest.
Quantitative reverse transcription-PCR
for SDF-1
Human MSCs were treated with Nutlin-3 as described
above. Cells were then collected and RNA was extracted from MSCs
using the RNeasy Mini kit (Qiagen Inc., Valencia, CA, USA)
following standard procedures and quantified using the NanoDrop
(Thermo Fisher Scientific, Rockford, IL, USA). Messenger RNA (mRNA)
was used to generate complementary DNA (cDNA) which was amplified
with a one-step RT-PCR kit (Applied Biosystems Inc., Foster City,
CA, USA) using the MX4000 Multiplex Quantitative PCR System
(Stratagene, Cedar Creek, TX, USA). CXCL12 cDNA was amplified by a
CXCL12 taqman gene expression assay (Hs00930455, Applied Biosystems
Inc., Foster City, CA, USA) using 100 ng of total RNA as starting
material. 18s rRNA was amplified by the internal control 18s rRNA
taqman assay (Applied Biosystems Inc.) using 1 ng of total RNA.
Each RNA sample was assayed in quadruplicate and relative cDNA
levels were determined after normalization to the internal 18s rRNA
control.
In vivo migration of MSCs to tumors
MSCs were isolated from the bone marrow of C57BL/6J
p53−/− mice as previously described (9). Briefly, mice were euthanized using
bottled CO2 inhalation and the bilateral femurs were
dissected out using sterile technique. The femurs were washed in
phosphate-buffered saline containing 2% fetal bovine serum (FBS).
Bone marrow cells were then obtained by flushing the femurs with
PBS with 2% FBS. Cells were then filtered through a 70-μm
nylon mesh and plated in α-MEM with 10% FBS and
penicillin/streptomycin and cultured for seven days and then used
in experiments. For use as a control, murine MSCs were transfected
with wild-type murine p53. The p53 wild-type (wt) expression
plasmid was the generous gift of Dr Arnold Levine. The p53-wt
plasmid has been previously described and encodes murine p53 and
G418 resistance with an SV40 origin of replication (26). Wild-type MSCs were labeled using
carboxyfluorescein diacetate succinimidyl ester (CFSE) (Invitrogen)
and p53 knockout MSCs were labeled using CellTracker CM-Dil
(Invitrogen) according to the manufacturer’s instructions. The
breast cancer cell line MDA-MB-231 (American Type Culture
Collection) was used in this study. Cells (106) along with Matrigel
(50 μl per injection; BD Biosciences) were injected
subcutaneously into nude mice and tumors were allowed to reach a
size of ∼5 mm in diameter. At this point, CFSE-labeled murine MSCs
expressing human p53 and CM-Dil-labeled murine p53−/−
MSCs were mixed at a ratio of 1:1. Mixed MSCs (5×106)
were then injected subcutaneously 10 mm from each tumor. Seven days
later the mice were sacrificed using CO2 inhalation.
Tumors were dissected from the mice and a single cell suspension of
each tumor was made. Tumors were dissected into small pieces using
a scalpel and dissociated using collagenase (Roche, Mannheim,
Germany) (0.035% wt/vol) in α-MEM at 37°C for 1 h. Tissue was
further dissociated by pipetting several times through a 5cc
pipette and debris was removed using a nylon strainer. The single
cell suspension was them washed with α-MEM with 10% FBS. The
cellular component of each tumor was then analyzed using flow
cytometry.
Statistical analysis
At least three independent experiments were
performed for each in vitro migration assay. Results are
presented as the means ± standard deviation. Statistical
significance was determined using the Student’s t-test and a value
of P<0.05 was considered statistically significant. Microsoft
Excel software was use for statistical analysis.
Results
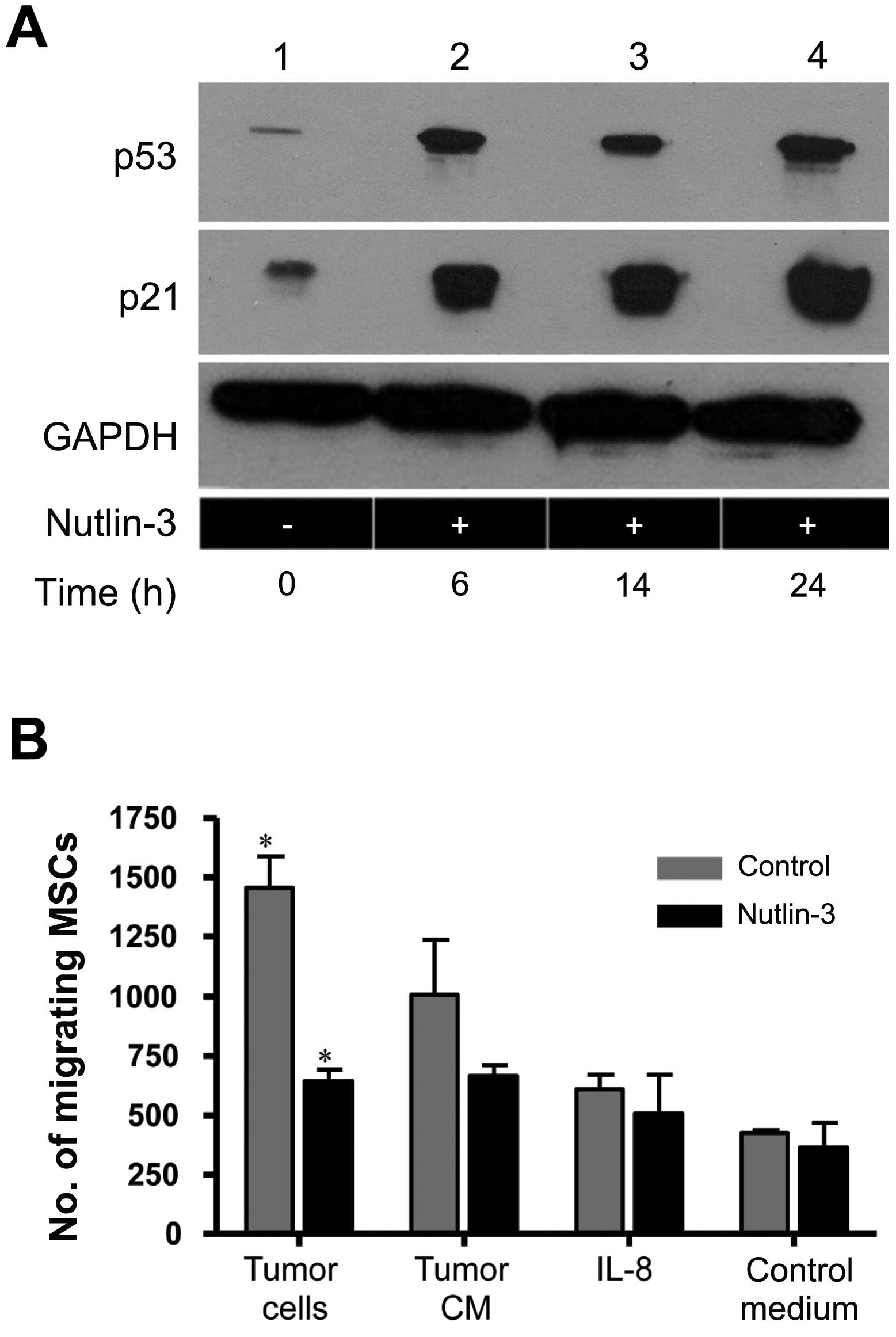
Regulation of MSC migration by p53
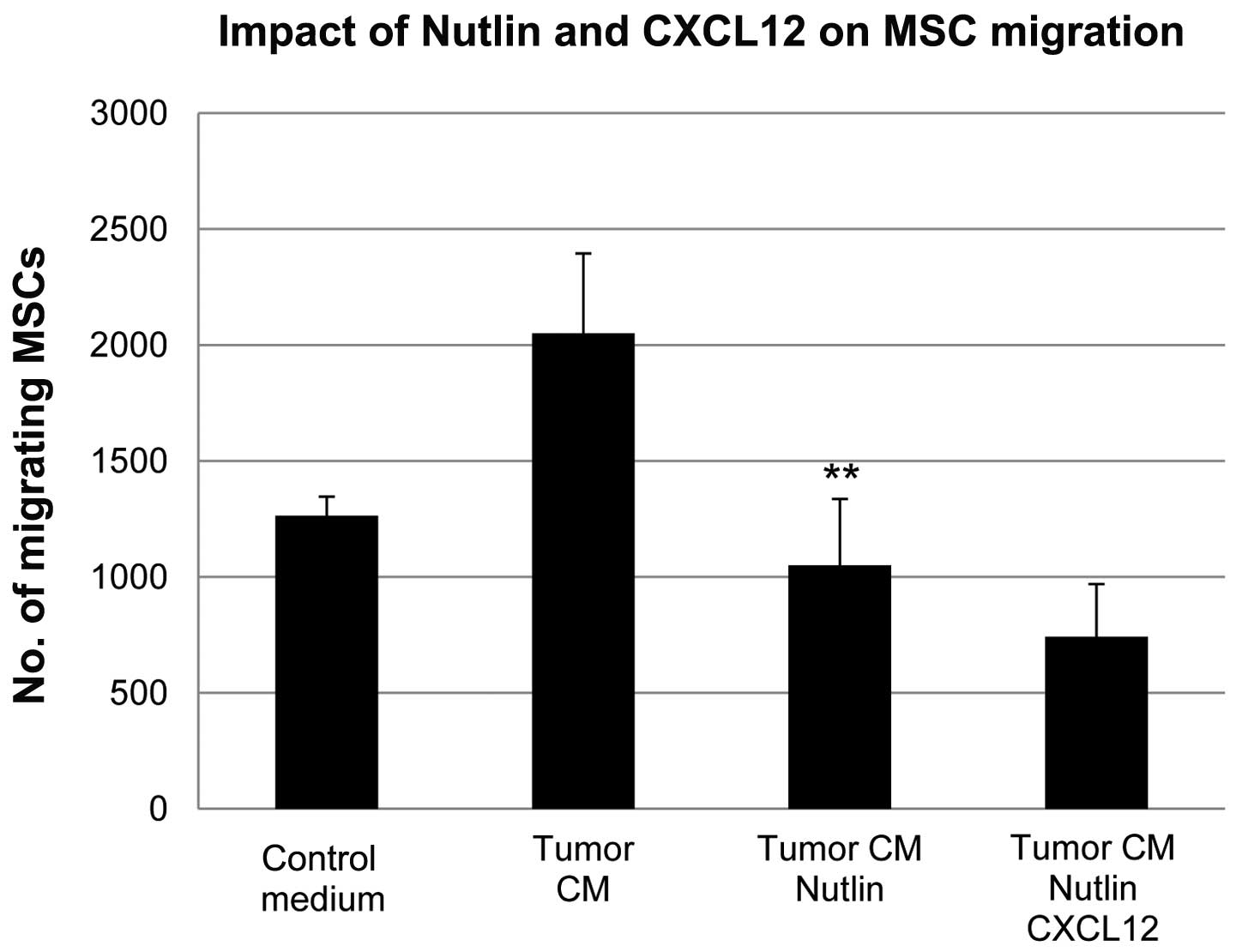
Human MSCs were treated with the murine double
minute 2 (MDM2) antagonist, Nutlin-3, leading to expected increases
in p53 as well as increases in the p53 target p21 (Fig. 1A). In conjunction with increased
p53 levels, the in vitro migration of MSCs in response to
MDA-MB-231 tumor cells was decreased. The migration to tumor
conditioned media showed a non-significant trend toward decreasing
and Nutlin-3 did not inhibit the migration of MSCs in response to
interleukin 8 (IL-8) (Fig. 1B).
There was minimal basal migration of MSCs in response to control
medium and this was not changed by treatment with Nutlin-3. When
levels of p53 were decreased using siRNA (Fig. 1C), MSCs exhibited increased
migration in response to tumor cells (Fig. 1D). These results suggested that p53
plays a role in regulating the response of MSCs to tumor cells.
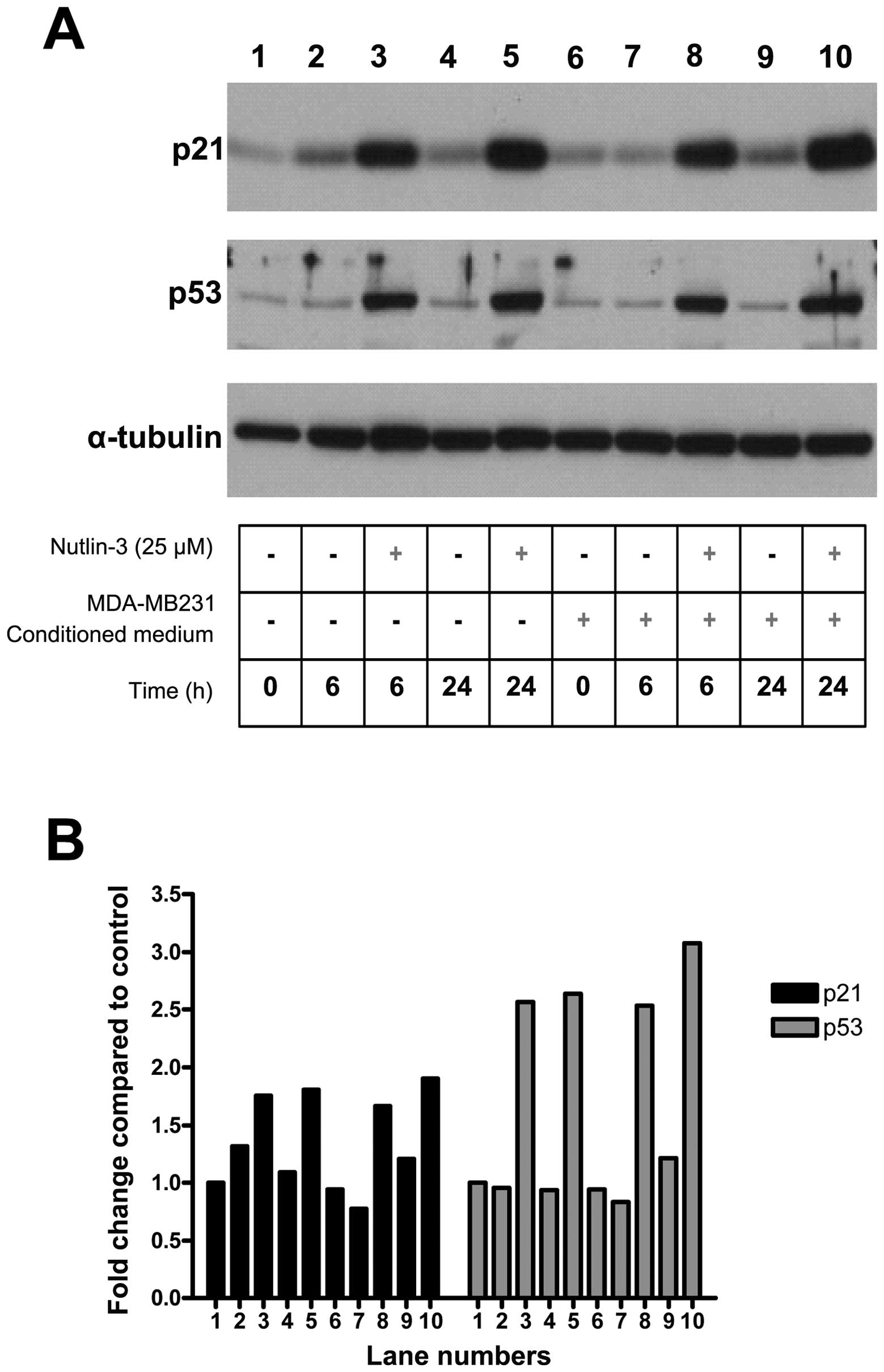
Exposure to tumor-conditioned medium does
not influence MSC p53 activity
hMSCs were exposed to a combination of Nutlin-3 and
MDA-MB-231 conditioned medium. As expected, exposure of MSCs to
nutlin-3 led to increased levels of p53 as well as its target p21.
However, exposure of MSCs to tumor conditioned medium did not
impact p53 levels (Fig. 2). This
result indicates that, while the p53 level influences MSC migration
in response to tumor cells, induction of MSC motility by TCM is not
mediated by changes in p53 activity.
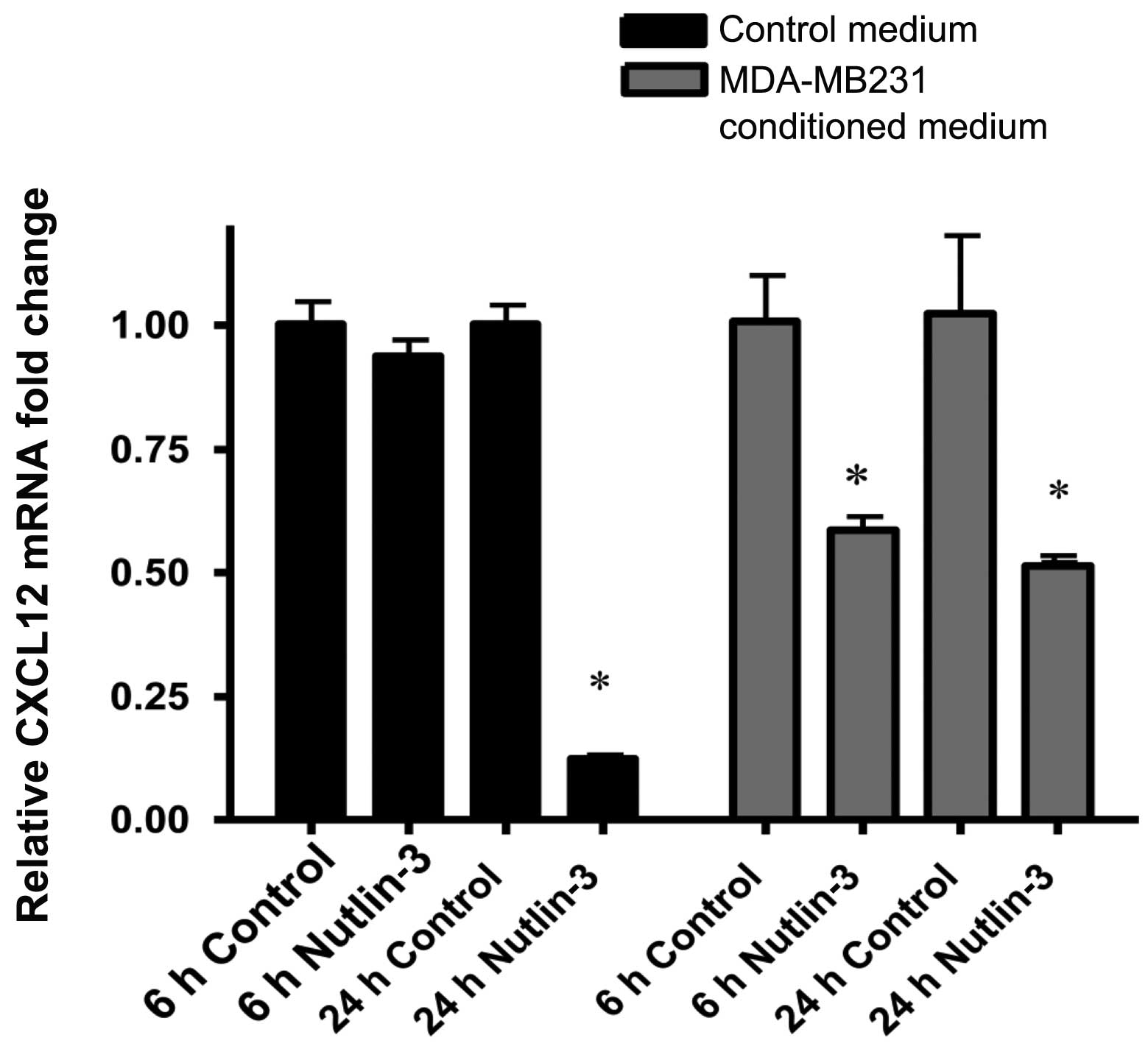
Increased p53 level leads to decreased
CXCL12 production by MSCs
We next explored the mechanism of regulation of MSC
migration by p53. Because production of CXCL12 by MSCs is required
for their migration in response to tumor cells (9), we investigated the effect of
increased p53 on CXCL12 production by MSCs. Nutlin-3 treatment
decreased hMSC CXCL12 mRNA levels after 24 h. CXCL12 mRNA levels in
MSCs exposed to conditioned medium from MDA-MB-231 cells were
decreased by nutlin-3 treatment at both 6- and 24-h time intervals
(Fig. 3). These data suggested
that p53 impacts MSC migration through regulation of CXCL12
transcription.
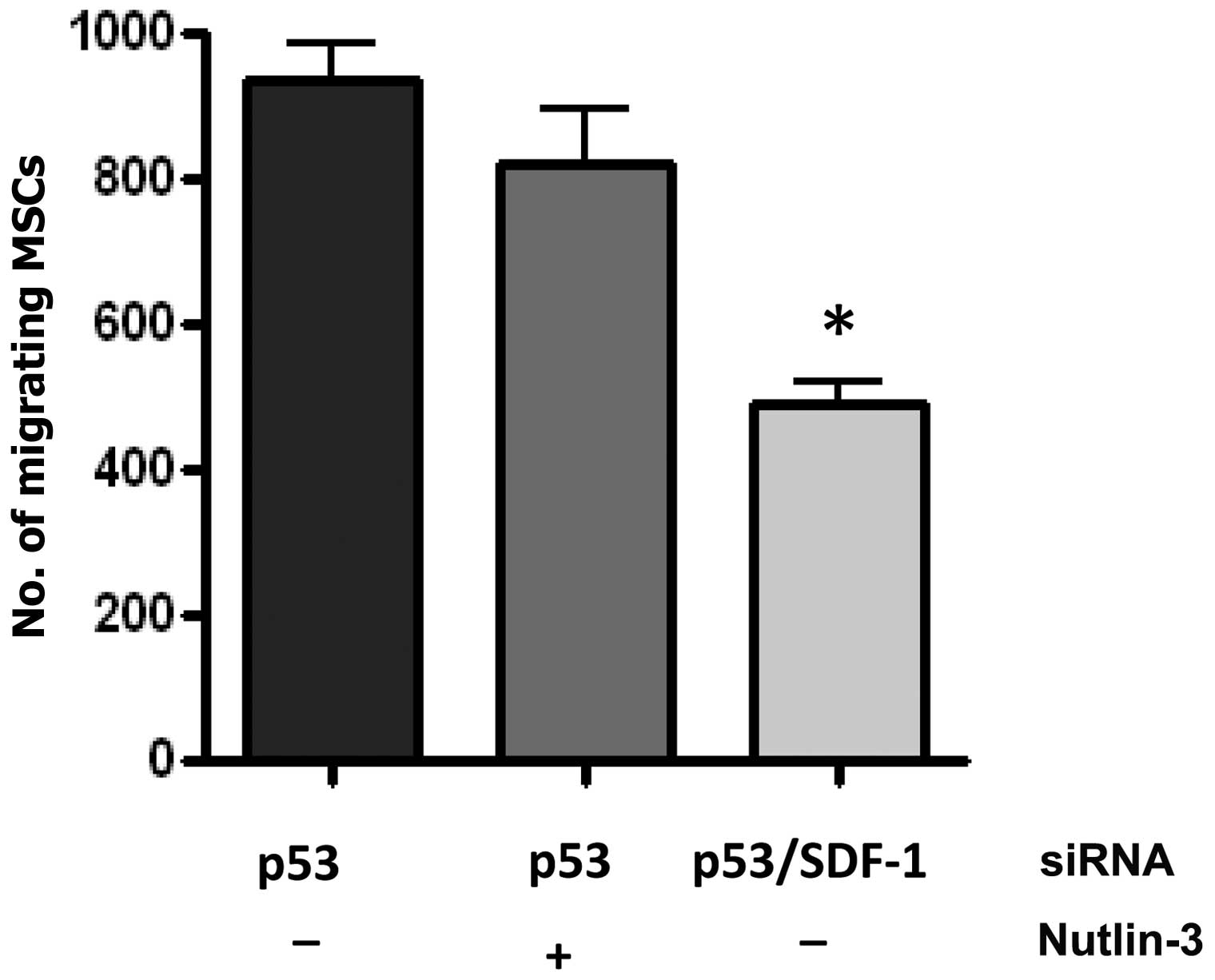
The increased motility of MSCs due to p53
knockdown is dependent on CXCL12
To further demonstrate the mechanism of action for
Nutlin-3 inhibition of MSC chemokinesis, MSCs with p53-knockdown
were treated with Nutlin-3. Nutlin-3 did not diminish the migration
of MSCs with p53 knockdown induced by tumor cells (Fig. 4), indicating that p53 is required
for Nutlin-3-mediated suppression of migration.
In order to determine whether the regulation of
CXCL12 plays a role in the increased motility of MSCs with p53
knockdown, MSCs with both p53 and CXCL12 knockdown were generated
using siRNA. CXCL12 knockdown blocked the increased motility of
MSCs with p53 knockdown (Fig.
4).
P53 regulates MSC migration using
multiple mechanisms
We then sought to determine whether the effect of
p53 levels on MSC migration was exclusively through its role in the
regulation of CXCL12 production. Recombinant CXCL12 protein was
added to human MSCs that had been treated with Nutlin-3. While
exogenous application of CXCL12 does stimulate migration of MSCs in
response to tumor conditioned medium (9), recombinant CXCL12 failed to increase
migration of MSCs treated with Nutlin-3 (Fig. 5). This result suggests that, in
addition to regulating the production of CXCL12, p53 may impact MSC
migration through additional mechanisms.
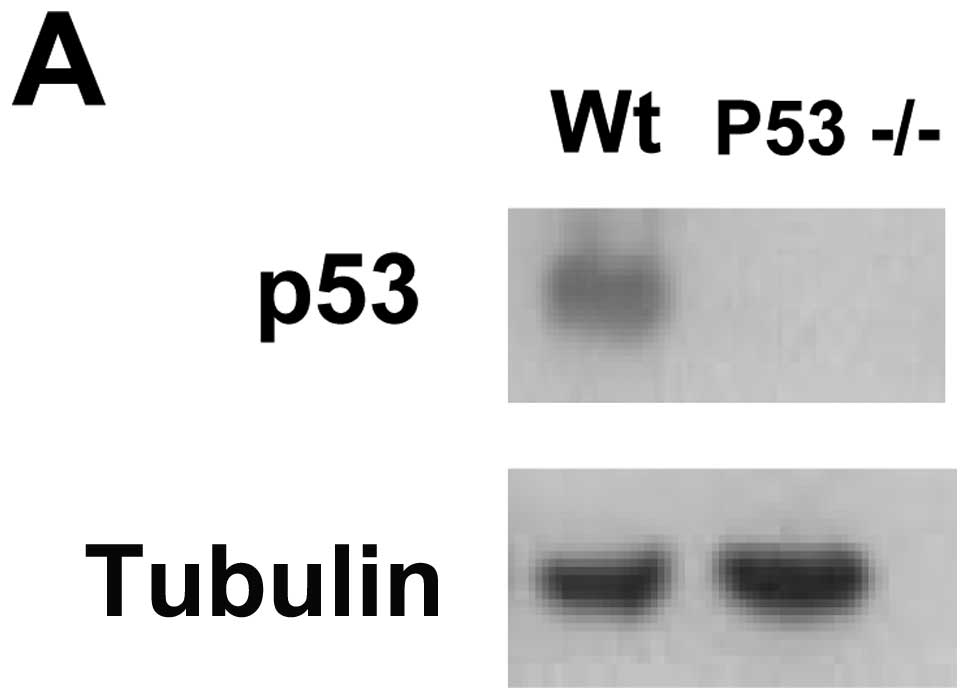
The in vivo homing capability of p53-null
MSCs to tumors is enhanced compared to wild-type
To determine whether p53 regulates homing of MSCs to
tumor sites in vivo, we used bone marrow-derived MSCs
isolated from p53-null mice. The human wild-type p53 gene was
introduced into the MSCs using a lentiviral vector. Western blot
analysis demonstrated that MSCs isolated from p53-null animals were
deficient in p53 protein and that after transfection of wild-type
p53 gene, the expression of the tumor suppressor was detected
(Fig. 6A). Wild-type MSCs were
labeled using CFSE and p53 knockout MSCs were labeled using CM-Dil.
Wild-type and p53−/− MSCs were mixed in a 1:1 ratio and
subcutaneously co-injected into nude mice 5 mm from established
tumors (MDA-MB231). At day 7 after administration of the MSCs,
animals were sacrificed and tumors were collected and used to
generate single cell suspensions. The ratio of wild-type to
p53−/− cells in the tumor was then determined using flow
cytometry. Increased number of p53-null MSCs were found in the
tumors compared to wild-type MSCs, indicating that the in
vivo homing capability of MSCs was enhanced in cells with
decreased p53 activity (Fig.
6B).
Discussion
Carcinoma associated fibroblasts are known as a key
mediator of tumor growth and progression. A better understanding of
signaling pathways underlying communication between neoplastic
cells and MSCs is important to better define their role in tumor
biology. MSCs are mobilized from bone marrow and other tissues and
integrate into the tumor stroma (2,7,9,12).
They impact diverse aspects of tumor progression such as
angiogenesis, tumor growth and metastasis (11,12,27).
While there are likely to be multiple mechanisms for the
intercellular signaling between MSCs and tumor cells, the
chemokine, CXCL12 has been implicated in MSC chemotaxis and homing
to tumors, MSC-mediated stimulation of tumor growth and cellular
tissue invasion (9,14,27,28).
Recently, Addadi and colleagues demonstrated that
CXCL12 production is downregulated in tumor stomal fibroblasts by
p53 and that this change in the stroma has a significant impact on
tumor growth (14). Importantly,
it has also been shown that systemically delivered p53-deficient
MSCs decrease tumor latency (14,16),
providing further evidence that the stromal p53 status is important
in tumor growth.
Clinical studies have reinforced the potential role
of p53-mediated signaling in tumor/stromal interactions. TP53
mutations have been reported within the stroma of sporadic breast
cancers (19) and both TP53
mutation as well as p53 expression in stromal fibroblasts are
associated with lymph node metastasis in breast cancer (19,29).
Others have found that changes in the stromal expression of the p53
target, p21, is associated with increasing malignancy in breast
cancers as well as an increased growth rate of human breast cancer
xenografts when tumors are implanted along with p21 deficient
fibroblasts (30). Interaction
with cancer cells can influence the p53 status of fibroblasts.
Co-culture with the small cell lung cancer cell line H1299 inhibits
the induction of p53 expression by cisplatin (31). However, in alignment with our data,
there was no change in basal p53 levels. These data suggest that
p53-dependent pathways play an important role in tumor stromal
biology.
Our experiments build on this study and demonstrate
that additional functional consequences of the p53 status of MSCs
include changes in migration efficiency both in vitro and
in vivo. Our data also suggest that increased CXCL12
production is not the only important outcome of decreased p53
function in stromal cells. Even in the context of exogenous CXCL12,
increased p53 levels lead to impaired motility of MSCs in response
to tumor cells. It is likely that the increased rates of tumor
formation and progression due to aberrant stromal p53 are a
consequence not only of increased CXCL12, but of multiple changes
in the stroma. The mechanisms of p53-mediated regulation of CXCL12
expression as well as identification of other important targets of
p53 within the tumor stroma are important areas of continued
investigation.
In conclusion, the complex interplay between tumor
cells and the surrounding non-neoplastic cellular components of
solid tumors remains incompletely understood. This study suggests
that stromal p53 is a critical mediator of this interaction through
multiple pathways. Stromal p53 status influences not only CXCL12
signaling with tumor stromal cells, but also impacts the stromal
response to neoplastic cells through other mechanisms.
Acknowledgements
We thank Dr Arnold Levine and Angie
Teresky for providing p53 knockout mice and reagents. The authors
received support from the New Jersey Commission on Cancer Research
NJCCR-10-1964-CCR-EO (DB and JG) and NJCCR-07-1063-CCR-EO (S.D.,
predoctoral fellowship).
References
|
1.
|
Studeny M, Marini FC, Dembinski JL,
Zompetta C, Cabreira-Hansen M, Bekele BN, Champlin RE and Andreeff
M: Mesenchymal stem cells: potential precursors for tumor stroma
and targeted-delivery vehicles for anticancer agents. J Natl Cancer
Inst. 96:1593–1603. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Studeny M, Marini FC, Champlin RE,
Zompetta C, Fidler IJ and Andreeff M: Bone marrow-derived
mesenchymal stem cells as vehicles for interferon-beta delivery
into tumors. Cancer Res. 62:3603–3608. 2002.PubMed/NCBI
|
|
3.
|
Shen FH, Visger JM, Balian G, Hurwitz SR
and Diduch DR: Systemically administered mesenchymal stromal cells
transduced with insulin-like growth factor-I localize to a fracture
site and potentiate healing. J Orthop Trauma. 16:651–659. 2002.
View Article : Google Scholar
|
|
4.
|
Rochefort GY, Delorme B, Lopez A, Herault
O, Bonnet P, Charbord P, Eder V and Domenech J: Multipotential
mesenchymal stem cells are mobilized into peripheral blood by
hypoxia. Stem Cells. 24:2202–2208. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Pittenger MF, Mackay AM, Beck SC, et al:
Multilineage potential of adult human mesenchymal stem cells.
Science. 284:143–147. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Picinich SC, Mishra PJ, Mishra PJ, Glod J
and Banerjee D: The therapeutic potential of mesenchymal stem
cells. Cell- and tissue-based therapy. Expert Opin Biol Ther.
7:965–973. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Nakamizo A, Marini F, Amano T, et al:
Human bone marrow-derived mesenchymal stem cells in the treatment
of gliomas. Cancer Res. 65:3307–3318. 2005.PubMed/NCBI
|
|
8.
|
Mishra PK: Bone marrow-derived mesenchymal
stem cells for treatment of heart failure: is it all paracrine
actions and immunomodulation? J Cardiovasc Med (Hagerstown).
9:122–128. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Menon LG, Picinich S, Koneru R, et al:
Differential gene expression associated with migration of
mesenchymal stem cells to conditioned medium from tumor cells or
bone marrow cells. Stem Cells. 25:520–528. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
McFarlin K, Gao X, Liu YB, et al: Bone
marrow-derived mesenchymal stromal cells accelerate wound healing
in the rat. Wound Repair Regen. 14:471–478. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Karnoub AE, Dash AB, Vo AP, et al:
Mesenchymal stem cells within tumour stroma promote breast cancer
metastasis. Nature. 449:557–563. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Mishra PJ, Mishra PJ, Humeniuk R, Medina
DJ, Alexe G, Mesirov JP, Ganesan S, Glod JW and Banerjee D:
Carcinoma-associated fibroblast-like differentiation of human
mesenchymal stem cells. Cancer Res. 68:4331–4339. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Levine AJ: p53, the cellular gatekeeper
for growth and division. Cell. 88:323–331. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Addadi Y, Moskovits N, Granot D, Lozano G,
Carmi Y, Apte RN, Neeman M and Oren M: p53 status in stromal
fibroblasts modulates tumor growth in an SDF1-dependent manner.
Cancer Res. 70:9650–9658. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Kiaris H, Chatzistamou I, Trimis G,
Frangou-Plemmenou M, Pafiti-Kondi A and Kalofoutis A: Evidence for
nonautonomous effect of p53 tumor suppressor in carcinogenesis.
Cancer Res. 65:1627–1630. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Houghton J, Li H, Fan X, et al: Mutations
in bone marrow-derived stromal stem cells unmask latent malignancy.
Stem Cells Dev. 19:1153–1166. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Fukino K, Shen L, Matsumoto S, Morrison
CD, Mutter GL and Eng C: Combined total genome loss of
heterozygosity scan of breast cancer stroma and epithelium reveals
multiplicity of stromal targets. Cancer Res. 64:7231–7236. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Fukino K, Shen L, Patocs A, Mutter GL and
Eng C: Genomic instability within tumor stroma and
clinicopathological characteristics of sporadic primary invasive
breast carcinoma. JAMA. 297:2103–2111. 2007. View Article : Google Scholar
|
|
19.
|
Patocs A, Zhang L, Xu Y, Weber F, Caldes
T, Mutter GL, Platzer P and Eng C: Breast-cancer stromal cells with
TP53 mutations and nodal metastases. N Engl J Med. 357:2543–2551.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Kurose K, Gilley K, Matsumoto S, Watson
PH, Zhou XP and Eng C: Frequent somatic mutations in PTEN and TP53
are mutually exclusive in the stroma of breast carcinomas. Nat
Genet. 32:355–357. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Paterson RF, Ulbright TM, MacLennan GT, et
al: Molecular genetic alterations in the
laser-capture-microdissected stroma adjacent to bladder carcinoma.
Cancer. 98:1830–1836. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Dudley AC, Shih SC, Cliffe AR, Hida K and
Klagsbrun M: Attenuated p53 activation in tumour-associated stromal
cells accompanies decreased sensitivity to etoposide and
vincristine. Br J Cancer. 99:118–125. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Alexandrova A, Ivanov A, Chumakov P,
Kopnin B and Vasiliev J: Changes in p53 expression in mouse
fibroblasts can modify motility and extracellular matrix
organization. Oncogene. 19:5826–5830. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Guo F, Gao Y, Wang L and Zheng Y:
p19Arf-p53 tumor suppressor pathway regulates cell motility by
suppression of phosphoinositide 3-kinase and Rac1 GTPase
activities. J Biol Chem. 278:14414–14419. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Molchadsky A, Shats I, Goldfinger N, et
al: p53 plays a role in mesenchymal differentiation programs, in a
cell fate dependent manner. PLoS One. 3:e37072008. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Hinds P, Finlay C and Levine AJ: Mutation
is required to activate the p53 gene for cooperation with the ras
oncogene and transformation. J Virol. 63:739–746. 1989.PubMed/NCBI
|
|
27.
|
Orimo A, Gupta PB, Sgroi DC, et al:
Stromal fibroblasts present in invasive human breast carcinomas
promote tumor growth and angiogenesis through elevated SDF-1/CXCL12
secretion. Cell. 121:335–348. 2005. View Article : Google Scholar
|
|
28.
|
Kang H, Watkins G, Parr C, Douglas-Jones
A, Mansel RE and Jiang WG: Stromal cell derived factor-1: its
influence on invasiveness and migration of breast cancer cells in
vitro and its association with prognosis and survival in human
breast cancer. Breast Cancer Res. 7:R402–R410. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Hasebe T, Tamura N, Okada N, et al: p53
expression in tumor-stromal fibroblasts is closely associated with
the nodal metastasis and outcome of patients with invasive ductal
carcinoma who received neoadjuvant therapy. Hum Pathol. 41:262–270.
2010. View Article : Google Scholar
|
|
30.
|
Trimis G, Chatzistamou I, Politi K, Kiaris
H and Papavassiliou AG: Expression of p21waf1/Cip1 in
stromal fibroblasts of primary breast tumors. Hum Mol Genet.
17:3596–3600. 2008.
|
|
31.
|
Bar J, Feniger-Barish R, Lukashchuk N, et
al: Cancer cells suppress p53 in adjacent fibroblasts. Oncogene.
28:933–936. 2009. View Article : Google Scholar : PubMed/NCBI
|