Introduction
Despite its discovery in 1996, the mechanistic role
of estrogen receptor (ER) β in breast cancer remains incompletely
understood. Five ERβ isoforms exist, formed by alternative splicing
of exon 8 (1). Of these, ERβ1 is
regarded as the wild-type isoform regulating gene transcription in
response to estradiol (2). ERβ1 is
constitutively expressed in the normal mammary gland but frequently
down-regulated in breast cancer (3,4)
where it may function as a tumour-suppressor (5,6).
Loss of ERβ1 might be the result of genetic modifications, such as
the homozygous deletion of both copies of the ERβ gene
(ESR2) or from loss of heterozygosity (LOH) together with
mutation (7). Alternatively,
down-regulation of ERβ1 expression in the absence of genetic
mutations might be the result of epigenetic modifications.
Epigenetic regulation of total ERβ expression has
previously been reported in various cancers (8–11).
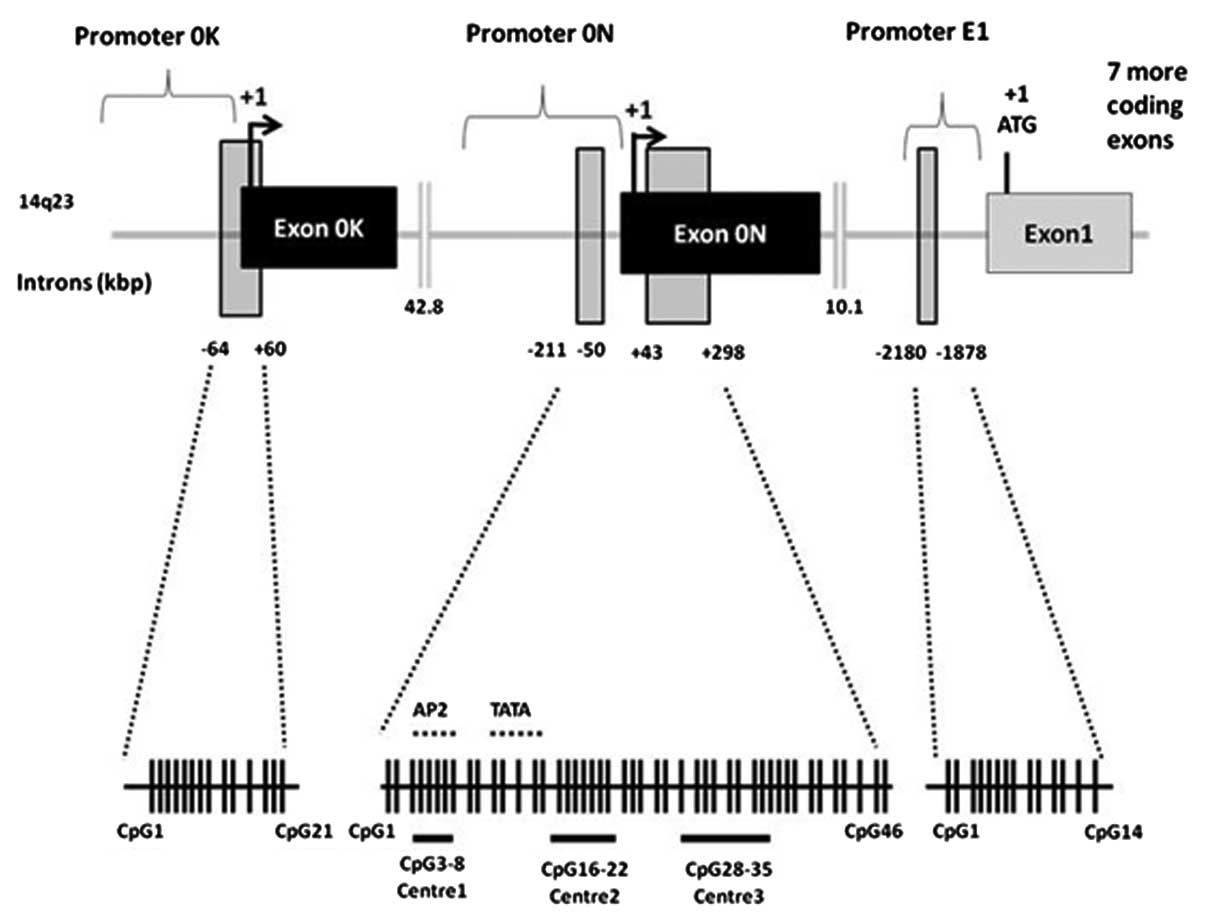
The ERβ promoter region contains several CpG islands, depicted
schematically in Fig. 1.
Transcription from two different ERβ promoters, termed promoters 0N
and 0K, generates ERβ mRNA isoforms that diverge in their
5′-untranslated regions (UTRs) by including the alternative
untranslated exons 0N or 0K. Evidence suggests that total ERβ
expression and, more specifically, the expression of ERβ1, may be
regulated by hypermethylation of CpG islands located within
promoter 0N or exon 0N in various primary tumours and tumour cell
lines (4,10–12).
Interestingly, we have reported that ERβ untranslated exons are
differentially associated with mRNAs for each ERβ isoform (13), adding weight to the current
evidence suggesting epigenetic events at specific sites might
influence the expression of individual ERβ isoforms.
Unlike genetic alterations, changes in DNA
methylation are potentially reversible and the transcriptional
reactivation of tumour suppressor genes through promoter
de-methylation represents an attractive strategy for anticancer
treatment currently being evaluated in clinical trials (14). ERβ1, -β2 and -β transcripts derived
from promoter 0N can be re-expressed in breast cancer cell lines
following treatment with DNA methyltransferase (DNMT) inhibitors
(10). DNA de-methylation and
histone deacetylase (HDAC) inhibition have been associated with
re-expression of total ERβ in prostate and ovarian cancer cell
lines (9,15). However, the effects of combination
therapy on the re-expression of ERβ1 in breast cancer cells have
yet to be explored.
Here, we aimed to determine the underlying
mechanisms of ERβ1 de-regulation, performing LOH analysis to
examine the influence of genetic modifications in the silencing of
ERβ1 expression in breast cancer and examining whether aberrant
methylation of the CpG islands, located in ERβ promoters (0K and
0N) and a 5′-untranslated region (exon 0N) was involved in the
regulation of ERβ1 expression in breast cancer cell lines and in
primary breast cancer. We also examined the effects of a
combination of DNA methylation and HDAC inhibition on the
re-expression of ERβ1 mRNA and ERβ mRNAs containing untranslated
exons (0N or 0K) in ERβ1-negative breast cancer cell lines.
Materials and methods
Case selection
Following ethical approval from the Leeds (East)
Research Ethics Committee (06/Q1206/180), 51 snap frozen tumour and
adjacent matched normal tissues were selected from the Leeds Breast
Tissue Bank. The cohort comprised 10 grade 1, 17 grade 2 and 24
grade 3 tumours; 25 were lymph node positive and 26 were node
negative. Samples were harvested prior to freezing by specialised
breast histopathologists (AMH/SL) who ensured that tumour samples
contained at least 80% of tumour cells. Immunohistochemical
analysis of ERβ1 in matched formalin-fixed paraffin-embedded cases
and gene expression of ERβ1 in frozen tissue was conducted as
previously described (16,17).
Tissue culture
BT-20, MDA-MB-453 and T47D breast cancer cell lines
were maintained in RPMI-1640 medium, supplemented with 5%
heat-inactivated fetal bovine serum (FBS; both Invitrogen), in a 5%
CO2 humidified incubator at 37°C. Bimonthly
Mycoplasma checks (MycoAlert Mycoplasma detection assay,
Lonza) were consistently negative. Short tandem repeat profiles
confirmed cell identity.
DNA extraction and LOH analysis
DNA was extracted using standard phenol/chloroform
methods. Multiplex PCR was performed in a reaction volume of 10 μl
containing 10 ng sample DNA, 1 pmol/μl of each primer pair
(fluorescently labelled forward primer), 1.5 mM MgCl2,
0.2 mM dNTPs, PCR buffer, 1.25 U Taq polymerase (Promega) and
molecular grade water. LOH was determined using four microsatellite
markers D14S1026, AL359235, D14S63 and AL122035, which span the
chromosome 14q22–24 region. Primer sequences were obtained from the
Genome Database (http://gdbwww.gdb.org/) or Ensembl (http://www.ensembl.org). Cycle conditions were: 95°C
for 5 min, 95°C for 30 sec, 58°C for 30 sec, 72°C for 30 sec for 35
cycles and a final extension of 72°C for 10 min. Resulting products
were sequenced (ABI 377 Perkin-Elmer). Allele ratios of tumour and
normal samples were calculated from the peak heights obtained from
the electrophoretograms and a tumour/normal ratio calculated. A
value of <0.5 indicated LOH (18). LOH was correlated to
immunohistochemical expression of ERβ1.
DNA extraction, bisulphite modification
and methylation analysis
All extraction kits were from Qiagen and the
manufacturer’s instructions were followed. Primers for bisulphite
PCR (BSP) and methylation-specific PCR (MSP) are shown in Table I. DNA (1 µg) was extracted from
frozen breast cancer tissues or breast cell lines (DNeasy Blood and
Tissue Kit). This was bisulphite modified (EpiTect Bisulphite Kit).
BSP was performed (Multiplex PCR Kit). Following initial melting at
95°C for 10 min, cycle conditions were: i) 45 cycles of 95°C, 30
sec, 56°C for 45 sec, 72°C for 45 sec (promoter 0N and exon 0N),
ii) 40 cycles of 95°C, 1 min, 63°C for 1 min, 72°C for 1 min
(promoter 0K), both followed by a final extension at 72°C for 10
min. PCR products were analysed on a 1.5% agarose gel containing
0.5 μg/ml ethidium bromide and visualized under UV illumination.
Bands were excised from the gel and purified (QIAquick Gel
Extraction Kit). Purified DNA was directly sequenced. MSP was
performed using the EpiTect MSP Kit. Cycle conditions were: 95°C
for 10 min, 45 cycles of 95°C for 30 sec, 56–62°C for 45 sec, 72°C
for 45 sec and a final extension at 72°C for 10 min. PCR products
were run and analysed on a 1.5% ethidium bromide-stained agarose
gels as described above. MSP primers could equally bind both
unmethylated and methylated DNA (19). To limit PCR bias that could occur
during amplification, MSP primers were optimized using touchdown
PCR with gradient annealing temperature. As positive controls, a
human control DNA set containing bisulphite methylated-converted,
unmethylated DNA and unmethylated-unconverted DNA were used
(EpiTect PCR Control DNA Set). The optimum annealing temperature
for methylated specific primer was chosen at an annealing
temperature which favoured the methylated specific primer to bind
specifically to the methylated template control but not to
unmethylated template control and unconverted unmethylated DNA
control and vice versa.
 | Table I.Primer sequences for BSP, MSP and
QRT-PCR. |
Table I.
Primer sequences for BSP, MSP and
QRT-PCR.
| Primer sets | Forward (5′→3′) | Reverse (5′→3′) | Size (bp) |
|---|
| BSP | | | |
| Promoter 0K |
GTTGGGGTTATTTCGGGGTTGTT |
CCTCCAACAAAACAAACACATTCA | 295 |
| Promoter 0N and
exon 0N |
GTTATTATTTTTGTGGGTGGATTGG |
ACCTTACCTTCTCTAAAATAC | 500 |
| MSP | | | |
| Promoter 0N-W |
CCCAGACTGGCTGTATCAGTGTCGG |
TGACCTCTAAGTGGGAGCACCCTCG | 178 |
| Promoter 0N-M |
TTTAGATTGGTTGTATTAGTGTCGG |
TAACCTCTAAATAAAAACACCCTCG | 178 |
| Promoter 0N-U |
TTAGATTGGTTGTATTAGTGTTGG |
CCTCTAAATAAAAACACCCTCAAA | 174 |
| Exon 0N-W |
GGAGGGACCACCCGAGCTGC |
CCACCTGTTGAGGAAAGCGAGCG | 102 |
| Exon 0N-M |
GGAGGGATTATTCGAGTTGC |
CCACCTATTAAAAAAAACGAACG | 102 |
| Exon 0N-U |
GGGAGGGATTATTTGAGTTGTG |
CCACCTATTAAAAAAAACAAACAC | 101 |
| QRT-PCR | | | |
| ERβ1 |
TGGGCACCTTTCTCCTTTAGTGG |
GCTTCACACCAGGGACTCTTTTGAG | 87 |
| ERβ (exon 0N) |
CGGGAGACCCCCCCTAATGC |
CTCAAAGATTCGTGGGCAAGTATAATG | 105 |
| ERβ (exon 0K) |
AGTTACTGAGTCCGATGAATGTGCTTG |
CTCAAAGATTCGTGGGCAAGTATAATG | 108 |
Pharmacological restoration of ERβ1 mRNA
and ERβ 5′-UTR expression using DNA methyl transferase (DNMT) and
histone deacetylase (HDAC) inhibitors
BT-20 and MDA-MB-453 cells were seeded in 6-well
plates at 3×104 cells/cm2. After overnight
attachment, the DNMT inhibitor, 5-aza-2′-deoxycytidine (5-aza-dC)
was added at final concentration of 5 μM (BT-20) and 1 μM
(MDA-MB-453) for 7 days. Fresh medium containing 5-aza-dC was added
every two days. Trichostatin A (TSA) was added when required at 300
nM for the last 24 h. Cells were harvested on day 8 for RNA and DNA
extraction. Controls cells received DMSO vehicle. QRT-PCR was
performed for ERβ1 and ERβ mRNAs containing untranslated exons (0K
or 0N). Primer sequences are in Table
I.
Results
LOH analysis
To determine whether genetic modifications influence
ERβ1 expression, we performed LOH on 27 breast tumours from our
cohort of 51 (Fig. 2). LOH was
identified in 2/12 (17%) cases at the AL359235 locus. No LOH was
observed in 23 cases that were informative for the marker D14S63,
directly adjacent to the ERβ gene. Similarly only 1/20 (5%) cases
showed LOH at the D14S1026 marker located within the ERβ gene.
However, LOH was more frequently observed at the AL122035 locus, in
5 of 24 informative cases (21%) screened. Comparison of LOH data
with ERβ1 immunohistochemistry showed no correlation between LOH
and ERβ1-negative tumours. Since LOH did not appear to be
associated with loss of ERβ1 expression, we performed MSP to
determine the methylation status of promoter 0N and untranslated
exon 0N in a subset of cases. Methylation was seen in 5/12 samples,
all from ERβ1-negative tumours suggesting epigenetic rather than
genetic events are important in the regulation of ERβ1
expression.
Correlation of ERβ1 mRNA expression with
methylation status of its promoters using BSP and MSP
The location of ERβ promoters, untranslated exons,
exon 1 and CpG islands is depicted schematically in Fig. 1. BSP (Fig. 3A) was performed on promoter 0N,
exon 0N (3 CpG islands), and promoter 0K. ERβ1-positive T47D cells
were predominantly unmethylated at both promoter 0N and exon 0N,
whereas BT-20 (ERβ1-negative) and MDA-MB-453 (low ERβ1 expression)
were mainly methylated (Fig. 3B).
Promoter 0K was unmethylated in all cell lines. Parallel MSP
analysis showed BT-20 and MDA-MB-453 cells were methylated at
promoter 0N and exon 0N, whereas T47D was unmethylated at promoter
0N with partial methylation at exon 0N (Fig. 3C). Methylation at ERβ promoter 0N
and exon 0N was negatively associated with ERβ1 expression in
breast cancer cell lines (Fig.
3D). As promoter 0K was unmethylated in our cell line panel we
did not undertake MSP for promoter 0K. These data suggest that
epigenetic modification at promoter 0N and exon 0N is a key
regulator of ERβ1 expression in breast cancer cells.
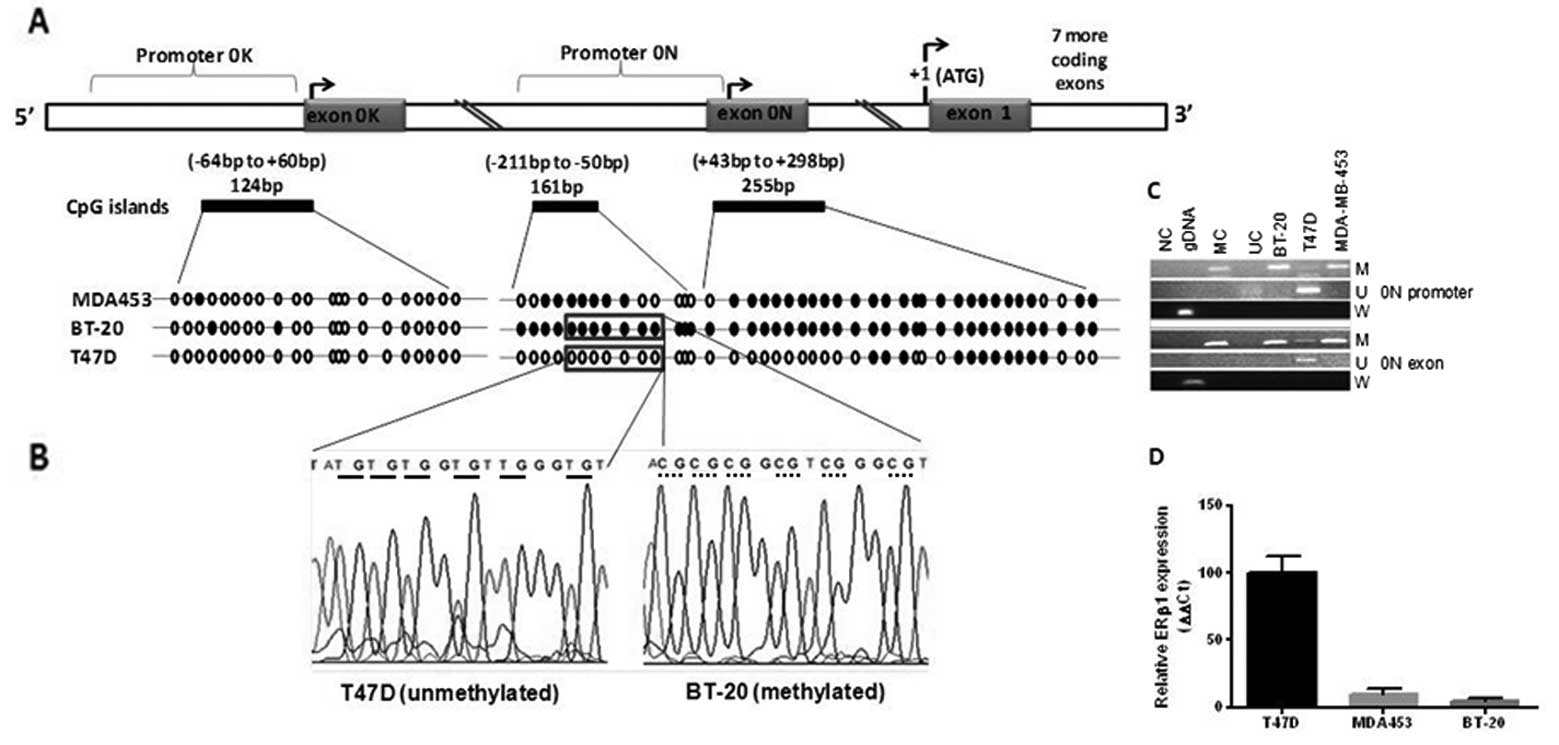 | Figure 3.ERβ1 mRNA expression and methylation
status of its promoters in breast cell lines. Example of BSP (A),
in 3 breast cancer cell lines. Circles represent CpG sites (solid,
methylated; open, unmethylated). Complete methylation of promoter
0N was seen in BT-20, partial methylation in MDA-MB-453 and mostly
unmethylated in T47D cells. Promoter 0K was mainly unmethylated in
all cell lines. (B), Representative DNA sequencing of promoter 0N
from methylated (BT20, dotted underline) and unmethylated (T47D,
solid underline) cells are shown. (C), MSP shows methylation of
promoter 0N in BT20 and MDA-MB-453 cells but not in T47D. Exon 0N
was methylated in all 3 cell lines. U, PCR products amplified with
unmethylated primers. M, amplified products with methylated
primers. Wild-type (W) primer set served as a control for annealing
to methylated or unmethylated DNA without bisulphite modification.
NG, negative control; GDNA, genomic DNA; MC, methylated converted
DNA control; UC, unmethylated converted DNA control. (D), ERβ1 mRNA
expression in ERβ1+ T47D and ERβ1- BT-20 and
ERβ1-low MDA-MB-453 breast cancer cells is shown. |
Relationship between ERβ1 mRNA expression
and promoter methylation patterns in primary human breast tumours
and matched normal material
Having detected aberrant methylation of ERβ promoter
0N and exon 0N in human breast cancer cell lines, MSP analysis was
subsequently conducted on primary breast tumours and tissue from 3
matched normal tumour pairs to determine the methylation pattern.
MSP was used as whole tissue extracts were analyzed containing a
mixture of both cancerous and non-cancerous cells, making the
detection of changes specific to cancerous cells challenging. The
sensitivity of MSP allows for detection of aberrantly methylated
alleles even if they contribute relatively little to the overall
DNA in a sample (21). As shown by
a representative MSP analysis (Fig.
4), promoter 0N (A) and exon 0N (B) were differentially
methylated. As promoter 0K was unmethylated in our cell line panel
we did not study this in clinical samples. Hypermethylation of ERβ
promoter 0N was observed in 7/24 (29%) of breast tumour samples,
whereas hypermethylation of ERβ exon 0N was found in 16/24 cases
(66%). No evidence of promoter 0N and exon 0N methylation was
observed in 17/24 (71%) and 8/24 (33%) breast cancer specimens,
respectively. Promoter 0N or exon 0N methylation was either
undetectable (n=4) or weakly detectable (n=2) in normal breast
tissue adjacent to tumours from the same patients (data not shown).
These results suggested that promoter 0N and exon 0N methylation in
the ERβ gene is a common feature of breast carcinoma and may
account for the frequent down-regulation of ERβ1. The relationship
between ERβ promoter 0N and exon 0N methylation, and ERβ1 mRNA
expression in breast tumours is shown in Fig. 5. ERβ1 mRNA expression was
significantly reduced in tumour versus normal samples, with no
significant difference in ERβ1 expression between the promoter 0N
methylated and unmethylated groups. In contrast, breast cancers
with methylated exon 0N sequences exhibited a significant
down-regulation of ERβ1 mRNA expression, compared to the
unmethylated exon 0N samples (P=0.03; Mann-Whitney). This suggests
methylation at exon 0N, rather than promoter 0N, may be an
important regulatory event leading to ERβ1 silencing in primary
breast cancers.
Pharmacological restoration of ERβ1
expression after in vitro DNA demethylation and histone
deacetylation
To determine if ERβ promoter methylation was
functionally correlated with ERβ1 silencing, ERβ1-negative or -low
breast cancer cell lines, BT-20 and MDA-MB-453, were treated with
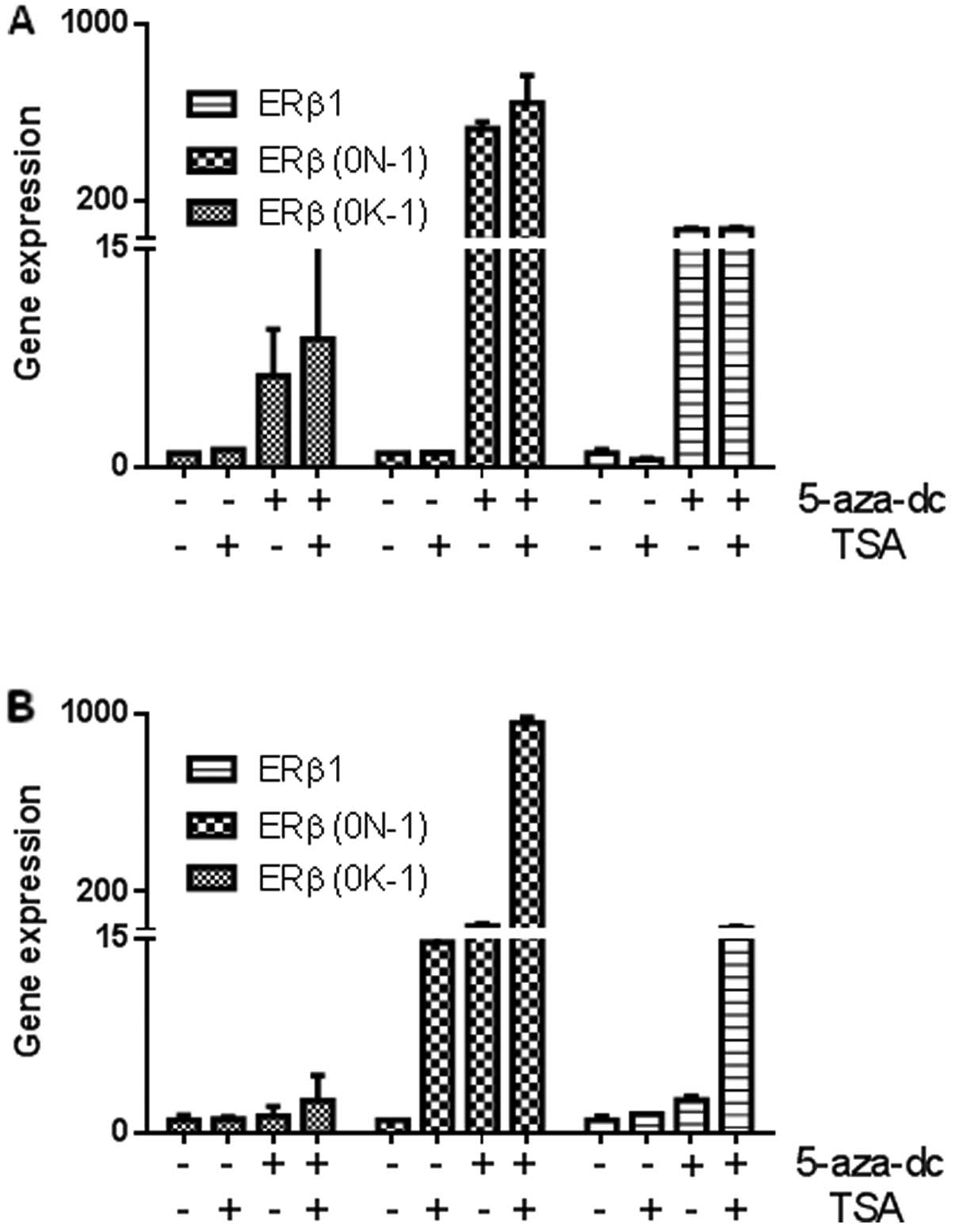
either 5-aza-dC, TSA or both. As shown in Fig. 6A, both cell lines expressed low
levels of ERβ1 mRNA, and ERβ mRNAs containing untranslated exons
(0N or 0K). MDA-MB-453 cells had partial methylation of promoter 0N
and exon 0N and treatment with 5-aza-dC was sufficient to induce
ERβ1 and mRNAs containing untranslated exons (0N or 0K) with no
additional re-expression seen following combination treatment with
TSA (Fig. 6B). When BT-20 cells
(complete methylation of both ERβ promoter 0N and exon 0N), were
exposed to 5-aza-dC and TSA, ERβ1 mRNA expression was restored
(Fig. 6B). Interestingly, our
results are the first to show that the combination of 5-aza-dC and
TSA had a synergistic effect in these breast cells and greatly
enhanced expression of ERβ1 and transcripts containing the
untranslated exon 0N with negligible effects on mRNAs containing
exon 0K.
Discussion
It is well recognized that ERβ1 is frequently
down-regulated in breast cancer compared with normal tissue
(3,4), however little is known about the
mechanisms responsible for this down-regulation. Here, we present
evidence that ERβ1 expression is down-regulated in breast cancer
cells epigenetically but not by LOH. We have shown that aberrant
methylation of CpG islands, located in promoter 0N and exon 0N, is
involved in the regulation of ERβ1 expression in epithelial breast
cancer cell lines and primary breast cancers. We have also
demonstrated that a combination of de-methylating agents and HDAC
inhibitors can have a synergistic effect, greatly enhancing
expression of ERβ1 mRNAs derived from promoter 0N. Importantly, our
data suggest that this combination might offer a therapeutic
approach for the treatment and/or chemoprevention of some
ERβ1-negative breast tumours.
BSP and MSP were used to examine the methylation
status of ERβ promoters (0N and 0K) and the downstream adjacent
untranslated exon 0N in breast cancer cell lines. Our results
showed that CpG islands located in promoter 0N and exon 0N were
differentially methylated in breast cells with differing ERβ1
statuses. Promoter 0K was unmethylated in our cell line panel. ERβ1
mRNA expression was inversely associated with the methylation
status of both promoter 0N and exon 0N, but not promoter 0K in
these breast cell lines, suggesting that CpG islands located in
promoter 0N and exon 0N are important regulatory sites for the
regulation of ERβ1 expression. Zhao et al showed a similar
negative association between the expression of ERβ1 and -β2 mRNA
and the methylation status of exon 0N in breast cells (10). Likewise, others have shown a
negative correlation between total ERβ expression and methylation
of promoter 0N and exon 0N in various human cancers (8,11,22).
Interestingly, we have shown that ERβ1 mRNA transcripts in some
breast cells predominantly contain the untranslated exon 0N rather
than exon 0K (13), indicating
that promoter 0N and exon 0N may be critical regulatory regions for
the control of this specific ERβ isoform.
Next, we examined whether a correlation exists
between the methylation status of either ERβ promoter 0N and/or
untranslated exon 0N and ERβ1 expression in primary breast cancers.
ERβ1 expression was significantly down-regulated in breast tumours
compared with normal breast tissue, therefore we examined whether
this might have been caused by the hypermethylation of ERβ promoter
0N and/or the untranslated exon 0N. MSP analysis of promoter 0N and
exon 0N revealed that CpG islands within these two regions were
differentially methylated. Our data showed a total of 7/24 (29%)
cases had detectable promoter 0N methylation, whereas 16/24 (67%)
cases had detectable exon 0N methylation. No methylation of
promoter 0N or exon 0N was found in four cases of normal breast
tissue, whereas two normal samples showed a weak methylation signal
(data not shown). Data for the methylation status in normal breast
tissue remains inconclusive due to the small cohort size used in
our study. We also found that ERβ1 expression was significantly
down-regulated in methylatedexon 0N breast tumours compared with
unmethylated breast tumours, confirming that DNA hypermethylation
might have caused epigenetic silencing of ERβ1 expression in these
primary breast cancers. This compliments a recent study that used
MSP to measure ERβ methylation in DNA extracted from primary
invasive ductal breast tumours and circulating DNA in a cohort of
Indian patients (23). In late
stage cancers they showed a significant correlation between
methylation status and loss of expression of total ERβ protein. Our
data are also consistent with previous reports, which have
estimated the methylation of exon 0N by direct BSP in breast
clinical samples (10) and various
other cancers (8,11,12,22,24,
25). Hierarchical clustering
previously identified three methyl ation ‘hotspots’ within two ERβ
CpG islands located within promoter 0N and exon 0N in prostate
cancer cells (11). This study
suggested two mechanisms responsible for methylation at promoter 0N
and exon 0N. The first involves methylation seeding to first
establish methylation at promoter 0N and exon 0N as a stochastic
phenomenon at low levels in normal and tumour cells (11). This may provide an explanation for
the weak methylation signal detected in two cases of normal breast
tissue in our study. The second mechanism involves methylation
spreading, which first occurs at the CpG island located within exon
0N and then extends to promoter 0N (11). Thus, it has been suggested that DNA
methyltransferases (DNMTs) have a lower opportunity to access the
CpG islands at promoter 0N due to steric hindrance from
transcription-initiation complexes and upstream enhancer sequences,
which are occupied with the binding of transcription factors. In
contrast, the CpG sites within exon 0N are more accessible to DNMTs
and therefore have a greater opportunity to be methylated first
(11). These suggestions are in
agreement with our findings in clinical samples, where exon 0N had
a higher frequency of methylation compared with promoter 0N and
suggest that aberrant methylation at exon 0N is the key regulatory
site for ERβ1 expression.
Next, we examined whether expression of ERβ1 mRNA
could be pharmacologically restored following in vitro DNA
de-methylation and histone acetylation. We used cell lines with
zero or minimal ERβ1 expression (BT-20, MDA-MB-453, respectively),
which showed high frequencies of methylation at promoter 0N and
exon 0N, to examine whether 5-aza-dC and TSA have a synergistic
effect on ERβ1 re-expression. In MDA-MB-453 cells, 5-aza-dC alone
was sufficient to reactivate expression of ERβ1 mRNA derived from
promoter 0N, but not from promoter 0K, with no additional
significant restoration observed following TSA treatment.
Importantly, in BT-20 cells TSA alone had little effect on
re-expression of ERβ1 mRNAs derived from promoter 0N. In contrast,
5-aza-dC greatly enhanced re-expression of ERβ1 mRNA derived from
promoter 0N only. Strikingly, a combination of de-methylating
agents and HDAC inhibitors had a synergistic effect on the
restoration of ERβ1 mRNA derived from promoter 0N, re-activating
ERβ1 more than with either agent alone. To our knowledge, we are
the first to show that this combination can enhance expression of
the ERβ1 isoform in breast cancer. It is worth noting that it
remains unknown if re-expressed ERβ1 mRNA is translated into
functional protein. In particular, we have shown that untranslated
exons have potent and differential influences on expression acting
at the level of translation in a cell-specific manner (13). We were unable to address this issue
in the present study due to technical difficulties with
ERβ1-specific antibodies preventing us from assessing ERβ1 protein
expression in parallel by western blot analysis.
In conclusion, our results add to the growing body
of evidence showing that ERβ1 is regulated at multiple levels in
breast cancer (13,20,26,27).
Our data indicate that epigenetic mechanisms involving DNA
hypermethylation and/or histone acetylation, at sites adjacent to
promoter 0N, play key roles in the regulation of ERβ1 expression.
Importantly, our data indicate that a combination of de-methylating
agents and HDAC inhibitors might provide an epigenetic approach for
the treatment and/or chemoprevention of some ERβ1-negative breast
cancers. While the prospect of introducing epigenetic therapy to
the clinic presents several clinical and translational challenges,
our results warrant further preclinical investigation; in
particular to define the precise mechanism of action of these
agents and to consider their potential development for future
clinical trials. This is already ongoing for some types of
haematological (28) and solid
(14,29) malignancies.
Acknowledgements
This study was supported by grants
from Breast Cancer Campaign (to V.S., T.A.H. and A.M.H.) and the
government of Saudi Arabia (H.A.N.).
References
|
1.
|
Moore J, McKee D, Slentz-Kesler K, Moore
LB, Jones S, Horne E, Su J, Kliewer S, Lehmann J and Willson T:
Cloning and characterization of human estrogen receptor β isoforms.
Biochem Biophys Res Commun. 247:75–78. 1998.
|
|
2.
|
Leung Y, Mak P, Hassan S and Ho S:
Estrogen receptor (ER)-β isoforms: a key to understanding ER-β
signaling. Proc Natl Acad Sci USA. 103:13162–13167. 1996.
|
|
3.
|
Speirs V, Skliris GP, Burdall SE and
Carder PJ: Distinct expression patterns of ERα and ERβ in normal
human mammary gland. J Clin Pathol. 55:371–374. 2002.
|
|
4.
|
Skliris GP, Munot K, Bell SM, Carder PJ,
Lane S, Horgan K, Lansdown MR, Parkes AT, Hanby AM, Markham AF and
Speirs V: Reduced expression of oestrogen receptor β in invasive
breast cancer and its re-expression using DNA methyl transferase
inhibitors in a cell line model. J Pathol. 201:213–220. 2003.
|
|
5.
|
Matthews J and Gustafsson J: Estrogen
signaling: a subtle balance between ERα and ERβ. Mol Interv.
3:281–292. 2003.PubMed/NCBI
|
|
6.
|
Bardin A, Boulle N, Lazennec G, Vignon F
and Pujol P: Loss of ERβ expression as a common step in
estrogen-dependent tumor progression. Endocr Relat Cancer.
11:537–551. 2004.
|
|
7.
|
Enmark E, Pelto-Huikko M, Grandien K,
Lagercrantz S, Lagercrantz J, Fried G, Nordenskjold M and
Gustafsson JA: Human estrogen receptor beta: gene structure,
chromosomal localization, and expression pattern. J Clin Endocrinol
Metab. 82:4258–4265. 1997.PubMed/NCBI
|
|
8.
|
Suzuki F, Akahira J, Miura I, Suzuki T,
Ito K, Hayashi H, Sasano H and Yaegashi N: Loss of estrogen
receptor β isoform expression and its correlation with aberrant DNA
methylation of the 5′-untranslated region in human epithelial
ovarian carcinoma. Cancer Sci. 99:2365–2372. 2008.
|
|
9.
|
Yap OW, Bhat G, Liu L and Tollefsbol TO:
Epigenetic modifications of the estrogen receptor β gene in
epithelial ovarian cancer cells. Anticancer Res. 29:139–144.
2009.
|
|
10.
|
Zhao C, Lam EW, Sunters A, Enmark E, De
Bella MT, Coombes RC, Gustafsson JA and Dahlman-Wright K:
Expression of estrogen receptor β isoforms in normal breast
epithelial cells and breast cancer: regulation by methylation.
Oncogene. 22:7600–7606. 2003.
|
|
11.
|
Zhu X, Leav I, Leung YK, Wu M, Liu Q, Gao
Y, McNeal JE and Ho SM: Dynamic regulation of estrogen receptor-β
expression by DNA methylation during prostate cancer development
and metastasis. Am J Pathol. 164:2003–2012. 2004.
|
|
12.
|
Rody A, Holtrich U, Solbach C, Kourtis K,
von Minckwitz G, Engels K, Kissler S, Gätje R, Karn T and Kaufmann
M: Methylation of estrogen receptor β promoter correlates with loss
of ER-β expression in mammary carcinoma and is an early indication
marker in premalignant lesions. Endocr Relat Cancer. 12:903–916.
2005.
|
|
13.
|
Smith L, Brannan RA, Hanby AM, Shaaban AM,
Verghese ET, Peter MB, Pollock S, Satheesha S, Szynkiewicz M,
Speirs V and Hughes TA: Differential regulation of oestrogen
receptor β isoforms by 5′ untranslated regions in cancer. J Cell
Mol Med. 14:2172–2184. 2010.
|
|
14.
|
Zambrano P, Segura-Pacheco B,
Perez-Cardenas E, Cetina L, Revilla-Vazquez A, Taja-Chayeb L,
Chavez-Blanco A, Angeles E, Cabrera G, Sandoval K, Trejo-Becerril
C, Chanona-Vilchis J and Duenas-González A: A phase I study of
hydralazine to demethylate and reactivate the expression of tumor
suppressor genes. BMC Cancer. 5:442005. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Walton TJ, Li G, Seth R, McArdle SE,
Bishop MC and Rees RC: DNA demethylation and histone deacetylation
inhibition co-operate to re-express estrogen receptor β and induce
apoptosis in prostate cancer cell-lines. Prostate. 68:210–222.
2008.PubMed/NCBI
|
|
16.
|
Shaaban AM, Green AR, Karthik S, Alizadeh
Y, Hughes TA, Harkins L, Ellis IO, Robertson JF, Paish EC, Saunders
PT, Groome NP and Speirs V: Nuclear and cytoplasmic expression of
ERβ1, ERβ2, and ERβ5 identifies distinct prognostic outcome for
breast cancer patients. Clin Cancer Res. 14:5228–5235. 2008.
|
|
17.
|
Cummings M, Iremonger J, Green CA, Shaaban
AM and Speirs V: Gene expression of ERβ isoforms in laser
microdissected human breast cancers: implications for gene
expression analyses. Cell Oncol. 31:467–473. 2009.
|
|
18.
|
Cawkwell L, Bell SM, Lewis FA, Dixon MF,
Taylor GR and Quirke P: Rapid detection of allele loss in
colorectal tumours using microsatellites and fluorescent DNA
technology. Br J Cancer. 67:1262–1267. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Cottrell SE, Distler J, Goodman NS, Mooney
SH, Kluth A, Olek A, Schwope I, Tetzner R, Ziebarth H and Berlin K:
A real-time PCR assay for DNA-methylation using
methylation-specific blockers. Nucleic Acids Res. 32:e102004.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Smith L, Coleman LJ, Cummings M, Satheesha
S, Shaw SO, Speirs V and Hughes TA: Expression of estrogen receptor
β isoforms is regulated by transcriptional and post-transcriptional
mechanisms. Biochem J. 429:283–290. 2010.
|
|
21.
|
Herman JG, Graff JR, Myohanen S, Nelkin BD
and Baylin SB: Methylation-specific PCR: a novel PCR assay for
methylation status of CpG islands. Proc Natl Acad Sci USA.
93:9821–9826. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Xue Q, Lin Z, Cheng YH, Huang CC, Marsh E,
Yin P, Milad MP, Confino E, Reierstad S, Innes J and Bulun SE:
Promoter methylation regulates estrogen receptor 2 in human
endometrium and endometriosis. Biol Reprod. 77:681–687. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Mirza S, Sharma G, Parshad R, Srivastava
A, Gupta SD and Ralhan R: Clinical significance of promoter
hypermethylation of ERβ and RARβ2 in tumor and serum DNA in Indian
breast cancer patients. Ann Surg Oncol. 19:3107–3115. 2012.
|
|
24.
|
Nojima D, Li LC, Dharia A, Perinchery G,
Ribeiro-Filho L, Yen TS and Dahiya R: CpG hypermethylation of the
promoter region inactivates the estrogen receptor-β gene in
patients with prostate carcinoma. Cancer. 92:2076–2083.
2001.PubMed/NCBI
|
|
25.
|
Zhai RL, Wang GB, Cai KL, Tao KX, Xu F,
Zhang WL and Zang ZY: Transcriptional inactivation of ERβ gene is
mediated by the induction of promoter hypermethylation in a rat
colonic epithelial cell model. J Surg Res. 155:306–310. 2009.
|
|
26.
|
Hamilton-Burke W, Coleman LJ, Cummings M,
Green CA, Holliday DL, Horgan K, Maraqa L, Peter MB, Pollock S,
Shaaban AM, Smith L and Speirs V: Phosphorylation of estrogen
receptor β at serine 105 is associated with good prognosis in
breast cancer. Am J Pathol. 177:1079–1086. 2010.
|
|
27.
|
Al-Nakhle H, Burns PA, Cummings M, Hanby
AM, Hughes TA, Satheesha S, Shaaban AM, Smith L and Speirs V:
Estrogen receptor β1 expression is regulated by miR-92 in breast
cancer. Cancer Res. 70:4778–4784. 2010.
|
|
28.
|
Bernstein I, Byun HM, Mohrbacher A, Douer
D, Gorospe G III, Hergesheimer J, Groshen S, O’Connell C and Yang
AS: A phase I biological study of azacitidine (Vidaza™) to
determine the optimal dose to inhibit DNA methylation. Epigenetics.
5:750–757. 2010.
|
|
29.
|
Yeo W, Chung HC, Chan SL, Wang LZ, Lim R,
Picus J, Boyer M, Mo FK, Koh J, Rha SY, Hui EP and Jeung HC:
Epigenetic therapy using belinostat for patients with unresectable
hepatocellular carcinoma: a multicenter phase I/II study with
biomarker and pharmacokinetic analysis of tumors from patients in
the Mayo Phase II Consortium and the Cancer Therapeutics Research
Group. J Clin Oncol. 30:3361–3367. 2012.
|