Introduction
Hepatocellular carcinoma (HCC) is one of the most
common malignancies worldwide, with a 5-year survival rate of only
5% (1–5). As such it is a major health problem
with increasing number of new cases diagnosed every year. The major
risk factors for HCC development are largely known (hepatitis B
virus, hepatitis C virus, alcohol, toxic chemicals), but the
specific pathogenesis and progression mechanism is still not well
understood (6–8).
c-Myc oncogene activation is a critical event in the
pathogenesis of a large number of human malignancies, including
HCC, the most common solid tumor (8–12). A
number of c-Myc target genes have been identified, including CAD,
ODC, CDC25A, LDH-A and cyclin E (13–19).
It has previously been shown that c-Myc upregulates CDCA7L mRNA
expression and colocalizes and interacts with CDCA7L. Furthermore,
CDCA7L is able to complement the c-Myc transformation-defective
mutant W135E and potentiate Myc-mediated transformation (20,21).
It has also been confirmed that CDCA7L induces colony formation,
and contributes to Myc-mediated transformation of medulloblastoma
cells, indicating that CDCA7L plays an important role in
medulloblastoma tumor development (21). There is ample evidence indicating
that CDCA7L can suppress monoamine oxidase A (MAO A) by interacting
with the MAO A promoter and inhibiting enzymatic activities, this
effect leads to increased expression of the cell cycle enhancers
E2F1 and cyclin D, demonstrating that both CDCA7L and MAO A are
involved in cell growth and apoptotic signaling pathways (20–23).
Despite many previous studies that have been carried
out in this area, CDCA7L expression in HCC is still not completely
known. Therefore, our objective was to examine the role of CDCA7L
in HCC development. Our results indicate that CDCA7L is markedly
upregulated in HCC and can act to promote HCC cell proliferation
and colony formation both in vitro and in vivo. These
data may provide further insight into HCC development and suggest
that CDCA7L may constitute a potential therapeutic target in
HCC.
Materials and methods
Patients
Sixty pairs of clinical specimens were obtained from
patients who were diagnosed with HCC at the First Affiliated
Hospital of Nanjing Medical University. Adjacent non-HCC tissues
were taken 2 cm away from the primary cancer edge and were
confirmed to be non-HCC tissues by pathological diagnosis. All
patients gave their informed consent and the study was approved by
the institutional ethics committee of Nanjing Medical
University.
Cell lines and cell culture
All HCC and normal human liver cell lines (Huh-7,
SK-hep-1, MHCC-97H, MHCC-97L, LM3, LM6, Hep-3B, Hep-G2, YY-8103,
Focus, WRL-68, L02) used in this study were obtained from the
Chinese National Human Genome Center at Shanghai. All cell lines
were cultured in a 5% CO2, 37°C-humidified incubator in
Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Grand Island, NY,
USA) supplemented with 10% heat-inactivated fetal bovine serum
(FBS) and penicillin (50 U/ml) and streptomycin (50 μg/ml).
RT-PCR and real-time PCR
Total RNA was extracted using TRIzol®
(Invitrogen) according to the manufacturer’s protocol. Reverse
transcription (RT) was performed in a 25 μl reaction mix with 2 μg
RNA using an M-MLV reverse transcriptase kit (Promega). The
sequences of primers used in these experiments were as follows:
CDCA7L-457 bp, 5′-CGACTCGCTACCAG ATCCCT-3′ (forward) and
5′-TTGTTGGCCAGCTTCTT GGT-3′ (reverse); CDCA7L-178 bp,
5′-TTGGCGACTCGCTA CCAGAT-3′ (forward) and 5′-AATGAAAGCGCACATC
CTGC-3′ (reverse); β-actin-230 bp: 5′-AGAGCCTCGCCTTT GCCGATCC-3′
(forward) and 5′-CTGGGCCTCGTCGCCCA CATA-3′ (reverse).
Construction of the CDCA7L expression
vector
The CDCA7L open reading frame (ORF) was amplified
from a human liver cDNA library (GeneBank accession no.:
NM_018719.4) using Prime Star PCR, and then inserted into
pcDNA3.1B-Flag, obtained from the Chinese National Human Genome
Centre at Shanghai. The sequences of the cloning primers were as
follows: CDCA7L-EcoRI forward primer (5′-AGAGAATTC
ATGGAGTTGGCGACTCGCTAC-3′), CDCA7L-BamHI reverse primer
(5′-AGAGGATCCATTGTCTTCTACCAG CTCCTT-3′). The final sequence of the
CDCA7L open reading frame (ORF) was confirmed by DNA
sequencing.
siRNA preparation and shRNA cloning
Small interference RNAs (RNAi) were chemically
synthesized (GenePharma Co.). Two siRNAs against CDCA7L were
designed and their sequences were as follows: siRNA-834,
5′-GCCAGAUUUC UUCCCAGUAdTdT-3′ (sense) and 5′-UACUGGGAAGAA
AUCUGGCdTdT-3′ (antisense); siRNA-1020, 5′-CCGAAGA
AGGAAGACAAUUdTdT-3′ (sense) and 5′-AAUUGUCUUC CUUCUUCGGdTdT-3′
(antisense). A common sequence was used as negative control:
siRNA-NC, 5′-UUCUCCGAACGUG UCACGUdTdT-3′ (sense);
5′-ACGUGACACGUUCGGAGAA dTdT-3′ (antisense). The oligonucleotides
encoding short hairpin RNAs (shRNA) were synthesized and inserted
into pSUPER vector, and designated pSUPER-shNC, pSUPER-sh834, and
pSUPER-sh1020, respectively. Their sequences were: shRNA-NC, sense,
5′-GATCCCCTTCTCCGAACGTGTCAC GTTTCAAGAGAACGTGACACGTTCGGAGAATTTTTG
GAAA-3′; antisense, 5′-AGCTTTTCCAAAAATTCTCGAA
CGTGTCACGTTCTCTTGAAACGTGACACGTTCGGAG AAGGG-3′; shRNA-834, sense,
5′-GATCCCCGCCAGATTT CTTCCCAGTATTCAAGAGATACTGGGAAGAAATCTG
GCTTTTTGGAAA-3′; antisense, 5′-AGCTTTTCCAAAAA
GCCAGATTTCTTCCCAGTATCTCTTGAATACTGGGAA GAAATCTGGCGGG-3′; shRNA-1020,
sense, 5′-GATCCCC CCGAAGAAGGAAGACAATTTTCAAGAGAAATTGTCT
TCCTTCTTCGGTTTTTGGAAA-3′; antisense, 5′-AGCTTT
TCCAAAAACCGAAGAAGGAAGACAATTTCTCTTGAA AATTGTCTTCCTTCTTCGGGGG-3′.
Cell transfection
Cell transfection was performed using Lipofectamine
2000 (Invitrogen) according to the manufacturer’s instructions.
Cells were transfected with RNAi and plasmid at cell density 30–50
and 80–90%, respectively.
Cell proliferation
For the proliferation assay, 2,000–5,000 HCC cells
were plated into 96-well plates and cultured for about a week. Cell
viability was measured using the Cell Counting Kit-8 (CCK-8,
Dojindo Laboratories) according to the manufacturer’s instructions.
Absorbance was measured at 450 nm each day and used to plot a cell
growth curve.
Colony formation assay and soft-agarose
colony formation assay
HCC cells were transfected and then plated onto 10
cm plates, at a seeding density of 10,000–50,000 cells. Transfected
cells were selected using G418 (Life Technologies Inc.) which was
added to the medium at a final concentration of 0.8–1 mg/ml. After
2–3 weeks, all clones were stained with Coomassie Brilliant
Blue.
For the soft agar assay, 2,000–5,000 transfected
cells were plated into culture dishes in complete medium mixed with
0.5% agarose over a bottom layer of complete medium mixed with 1%
agarose. After culture for 2–3 weeks, all clones were photographed
and counted under a microscope.
Western blot analysis
Western blot analysis was performed according to the
manufacturer’s instructions (Santa Cruz Biotechnology). Cell
lysates were prepared in lysis buffer [25 mmol/l Tris (pH 6.8), 1%
SDS, 5 mmol/l EDTA, protease inhibitor cocktail (Sigma)], and total
protein was assayed using a BCA kit. The samples were subjected to
SDS-PAGE on a 10% gel, then transferred to a polyvinylidene
difluoride (PVDF) membrane. Protein blots were blocked with 5% milk
and 0.1% Tween-20 in PBS for 2 h at room temperature, then
incubated with the primary antibody at 4°C overnight. After
washing, blots were incubated with the secondary antibody at room
temperature for 1 h and labeled protein was detected using the
Odyssey Infrared Imaging System (Li-COR). Mouse antibodies to
cyclin D1 and β-actin (Santa Cruz Biotechnology), goat antibody to
CDCA7L (Santa Cruz Biotechnology) and rabbit antibodies to pERK1/2
and total ERK1/2 (Cell Signaling Technology) were used in this
study.
In vivo subcutaneous HCC tumor model
Approximately 5×106 HCC cells were
injected subcutaneously into the flank of male nude mice (4–6 weeks
old). Tumors were measured and weighed 3–4 weeks after injection or
when the tumor mass reached 1,500 mm3. Tumor size was
measured with calipers every 3 days, and tumor volume was
determined using the following formula: volume = 0.5 ×
width2 × length.
Cell cycle analysis and cell
synchronization
Flow cytometry was used to analyze the cell cycle.
First, cells were cultured in serum-free medium for 24 h to induce
cell cycle synchronization. These cells were then transfected with
siRNA or plasmid and then harvested at a different time point. For
DNA content analysis, cells were fixed in 70% ethanol, rehydrated
in PBS, treated with RNase A (10 mg/ml) for 30 min then stained
with propidium iodide (10 μg/ml) for 5 min.
Statistical analysis
All experimental data were analyzed by Student’s
t-test using GraphPad Prism 5 software. In all tests, P<0.05 was
considered significant.
Results
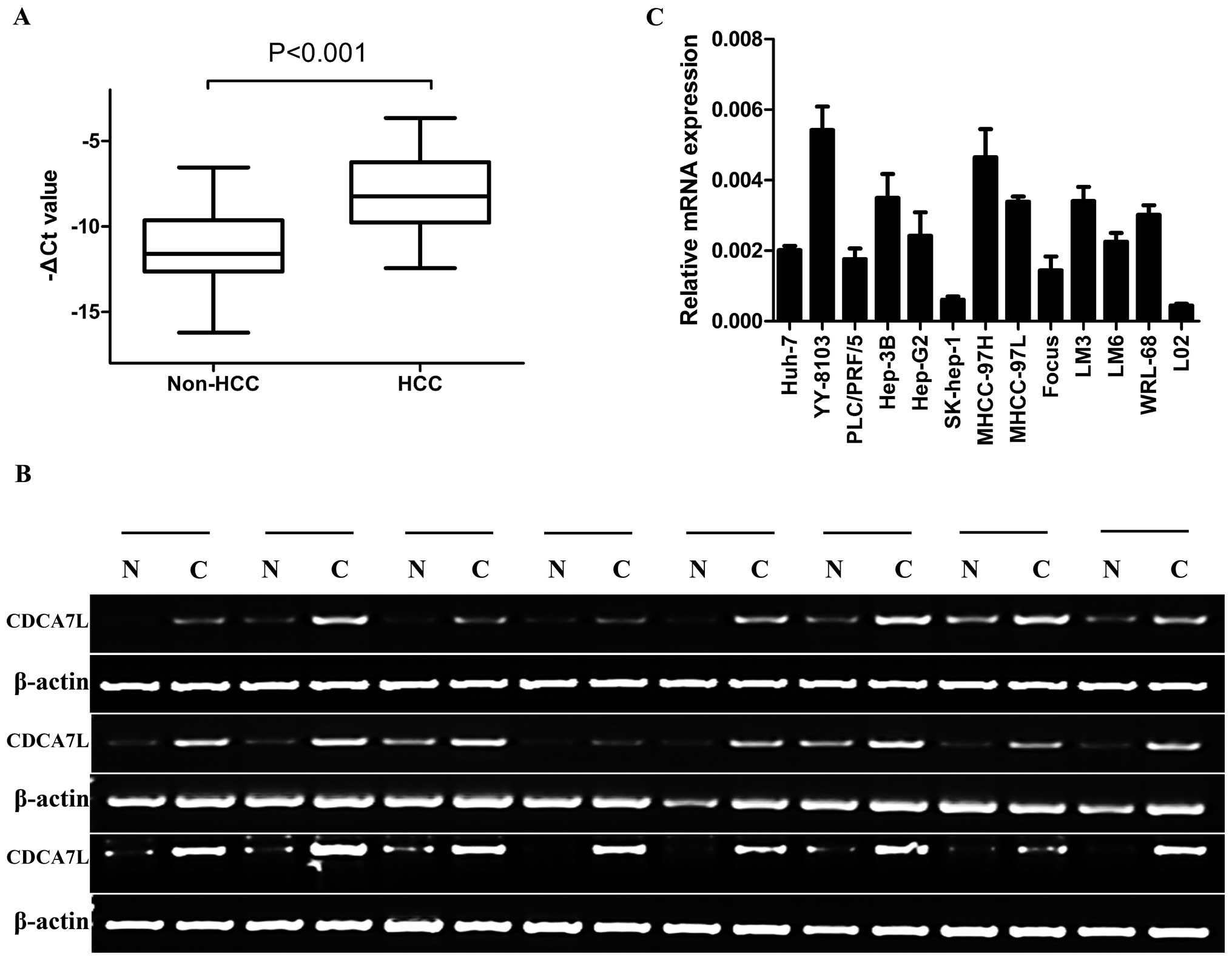
CDCA7L is significantly upregulated in
HCC
The expression level of CDCA7L was evaluated in 60
paired human HCC specimens by quantitative and semi-quantitative
RT-PCR. The results showed that CDCA7L was overexpressed in 42 of
60 HCC tissue specimens compared with the corresponding non-HCC
tissue specimens (Fig. 1A and B).
Furthermore, when the expression level of CDCA7L in HCC cell lines
and a normal human liver cell line was analyzed by real-time PCR,
the results showed that CDCA7L was highly expressed in YY-8103 and
MHCC-97H HCC cell lines but low in SK-hep-1 and Focus HCC cell
lines, while CDCA7L expression in the normal human liver cell line
L02 was lower than in any of the HCC cell lines tested (Fig. 1C).
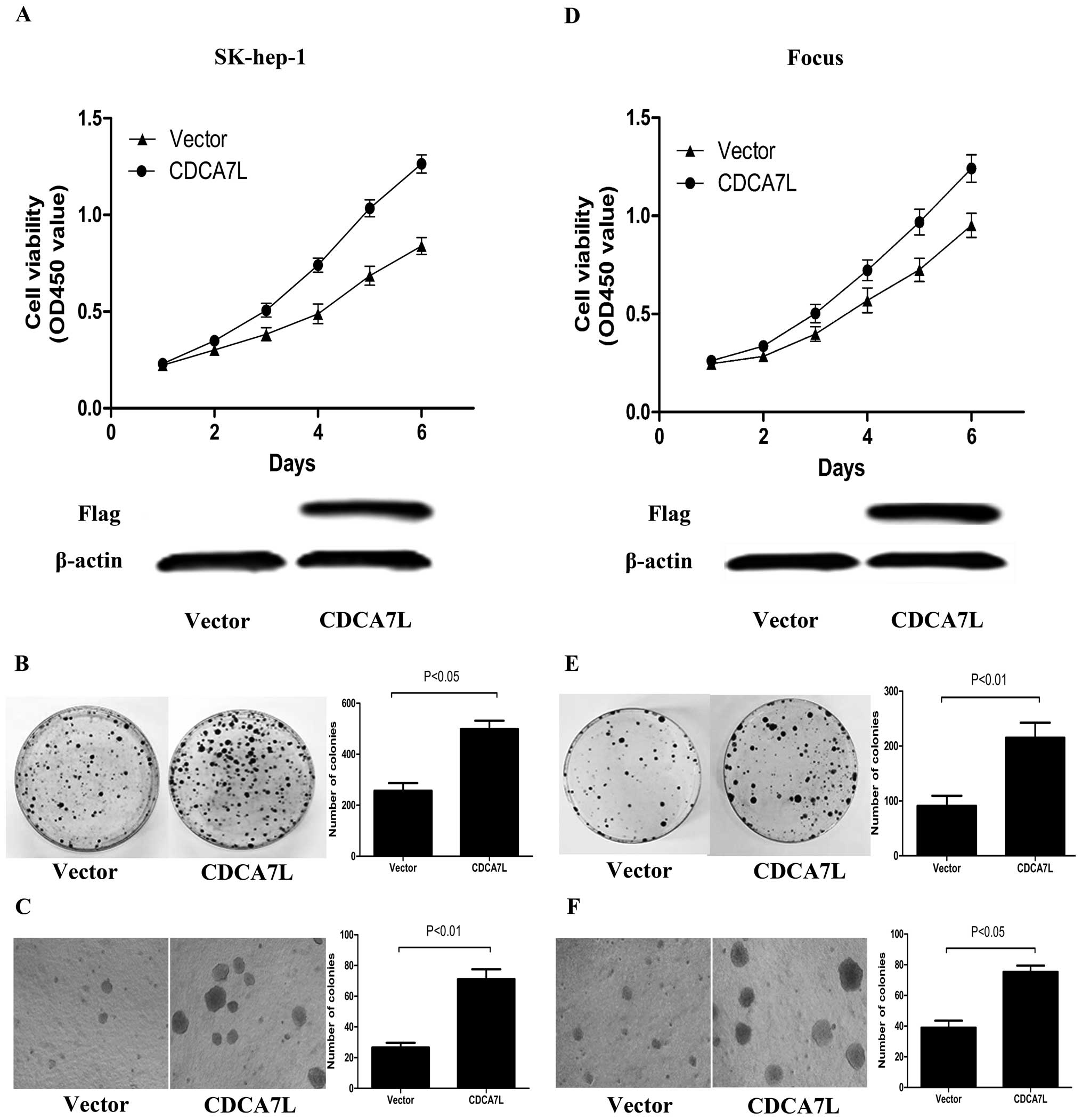
CDCA7L overexpression promotes HCC cell
proliferation and colony formation
To determine the effect of CDCA7L expression in HCC
cells, the recombinant plasmid pcDNA3.1-CDCA7L (CDCA7L) was
transfected into SK-hep-1 and Focus cells, to observe and measure
cellular proliferation, colony formation and soft agar colony
formation. Cells transfected with empty plasmid pcDNA3.1 (vector)
were used as control. The expression of exogenous CDCA7L protein
was identified by western blot analysis. Analysis of cell
proliferation and colony formation revealed that SK-hep-1 and Focus
cells transfected with pcDNA3.1-CDCA7L (CDCA7L) had increased cell
viability and produced more colonies than the corresponding cells
transfected with pcDNA3.1 (vector) (Fig. 2A, B, D and E). In order to
determine the effect of CDCA7L on anchorage-independent growth, a
soft agar colony formation experiment was performed, and again,
compared with control cultures, ectopic CDCA7L resulted in a
significant enhancement of cellular colony formation by SK-hep-1
and Focus cells (Fig. 2C and F).
Taken together, these data indicate that CDCA7L overexpression
enhances HCC cellular proliferation, colony formation and soft agar
colony formation.
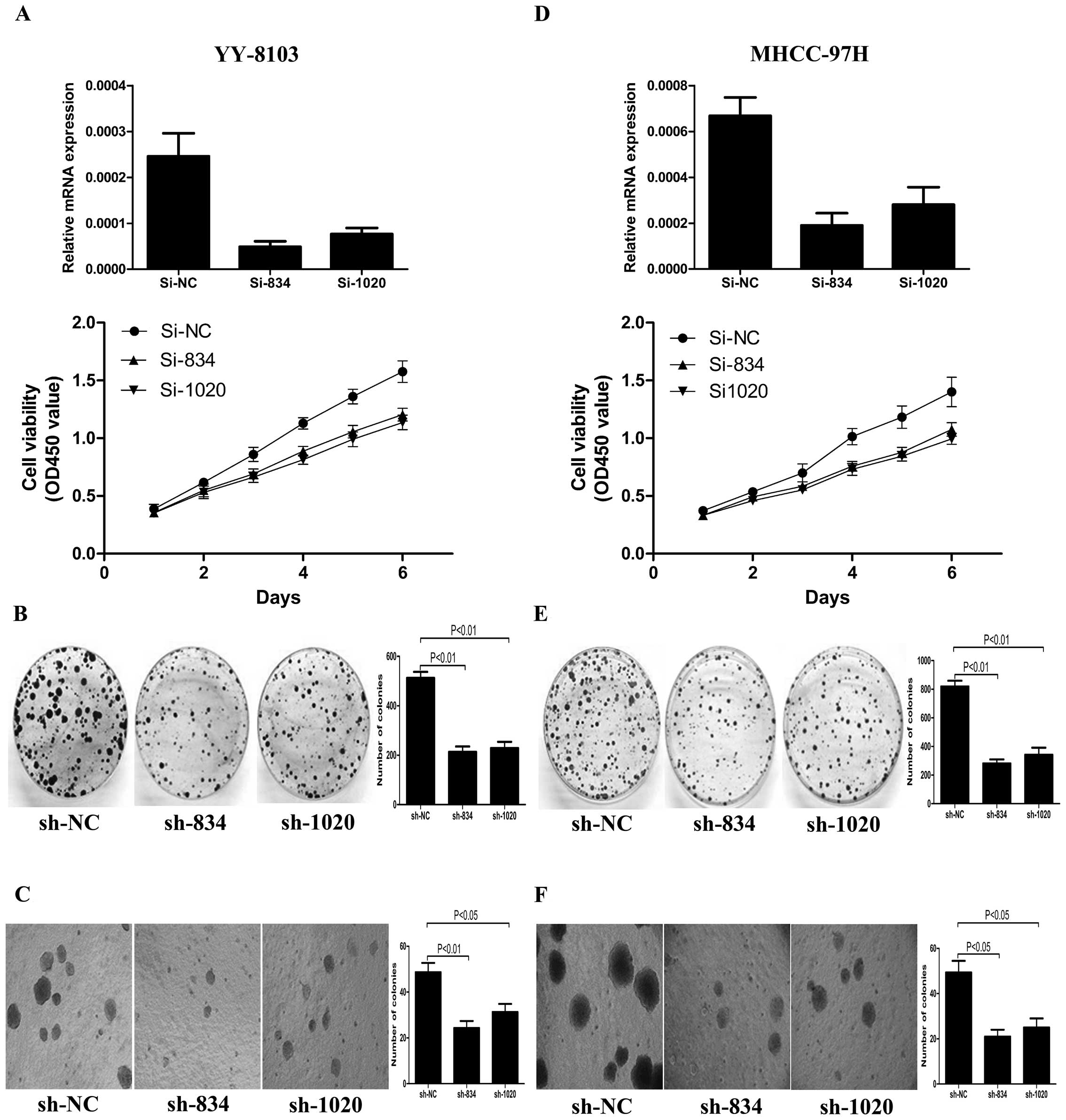
CDCA7L knockdown inhibits HCC cell
proliferation and colony formation
To further investigate the effect of CDCA7L on cell
proliferation, colony formation and soft agar colony formation, we
synthetized three specific siRNAs (si-NC, si834 and si1020) and
constructed the corresponding shRNA plasmids (pSUPER-shNC,
pSUPER-sh834 and pSUPER-sh1020) to knock down endogenous CDCA7L in
selected HCC cell lines. After evaluating the efficiency of siRNA,
si834 and si1020 were considered appropriate for CDCA7L knockdown,
while as negative control, si-NC was not able to knock down
expression of any gene. SiRNA was capable of silencing target gene
expression for a short period, while pSUPER-shRNA was able to
maintain this effect indefinitely. YY-8103 and MHCC-97H cells
transfected with si-NC, si834 or si1020 were used for the cell
proliferation experiment. The same cells transfected with
pSUPER-shNC, pSUPER-sh834 or pSUPER-sh1020 were used for colony
formation and soft agar colony formation experiments. The cell
growth curve revealed that cell viability of YY-8103 and MHCC-97H
cells transfected with si-NC was greater than that of the
corresponding cells transfected with si834 or si1020 (Fig. 3A and D). Likewise, YY-8103 and
MHCC-97H cells transfected with pSUPER-shNC formed more, larger
colonies compared with the corresponding cells transfected with
pSUPER-sh834 and pSUPER-sh1020 (Fig.
3B, C, E and F). These collective data indicate that knockdown
of CDCA7L can inhibit HCC cell proliferation, colony formation and
soft agar colony formation.
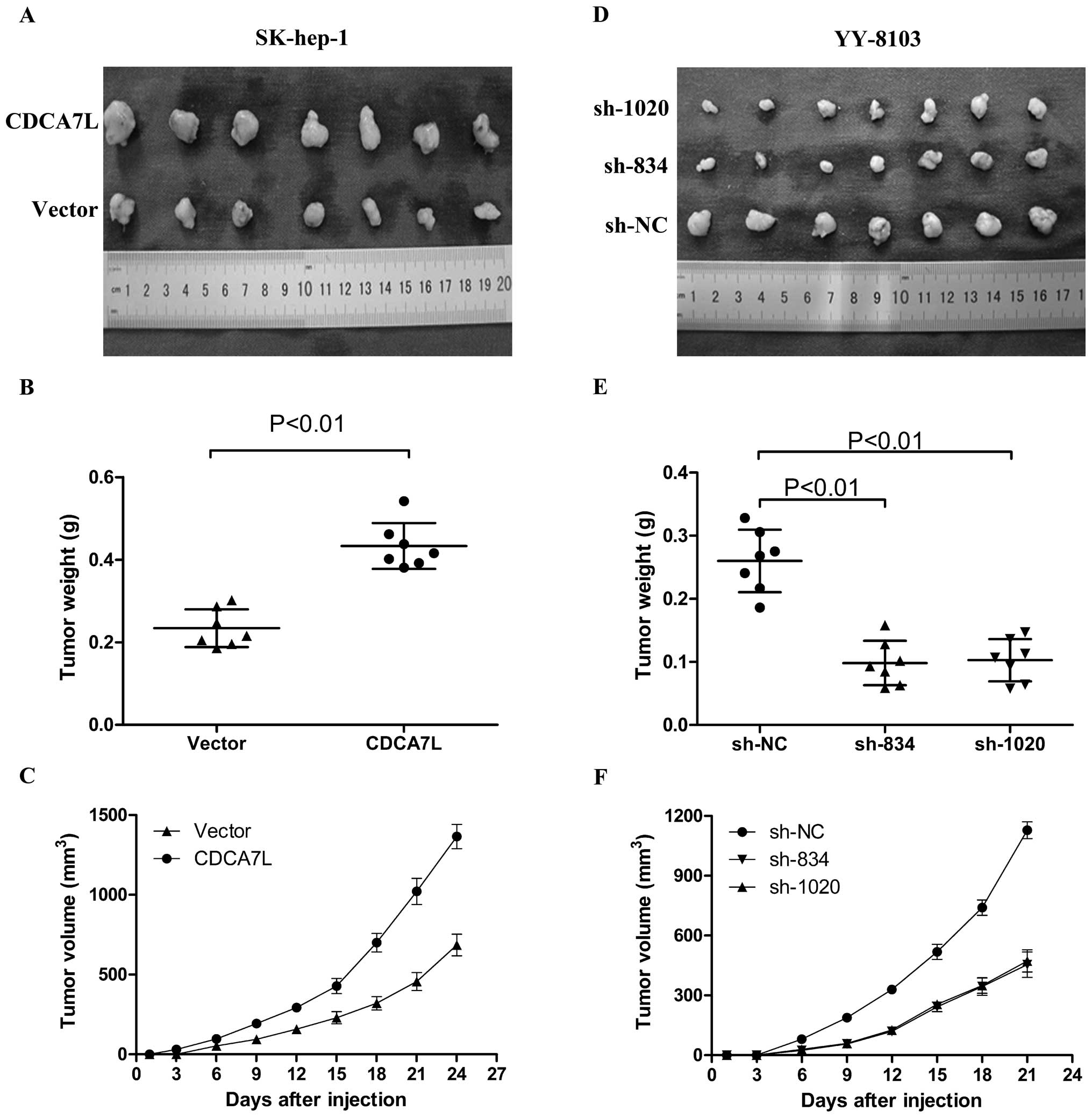
CDCA7L overexpression contributes to
tumorigenicity while CDCA7L knockdown suppresses tumorigenicity and
reduces tumor burden
To further confirm the promotional effect of CDCA7L
on cell proliferation and colony formation, a subcutaneous
xenograft tumor model in male nude mice was used to evaluate the
effect of CDCA7L on tumorigenicity. Compared with SK-hep-1 cells
transfected with empty vector plasmid pcDNA3.1 (vector), injection
of the same cells transfected with pcDNA3.1-CDCA7L (CDCA7L)
resulted in larger tumors in male nude mice under observation about
3 to 4 weeks after injection (Fig.
4A–C). To determine the effect of CDCA7L knockdown on
tumorigenicity in vivo, pSUPER-shRNA plasmids were used to
transfect YY-8103 cells, then these cells were harvested and
injected into the flank of male nude mice. As expected,
pSUPER-sh834 and pSUPER-sh1020 were able to significantly suppress
tumorigenicity, resulting in obvious reductions in tumor weight and
volume compared to the negative controls pSUPER-shNC (Fig. 4D–F). All these data indicate that
CDCA7L plays a role in HCC tumorigenicity.
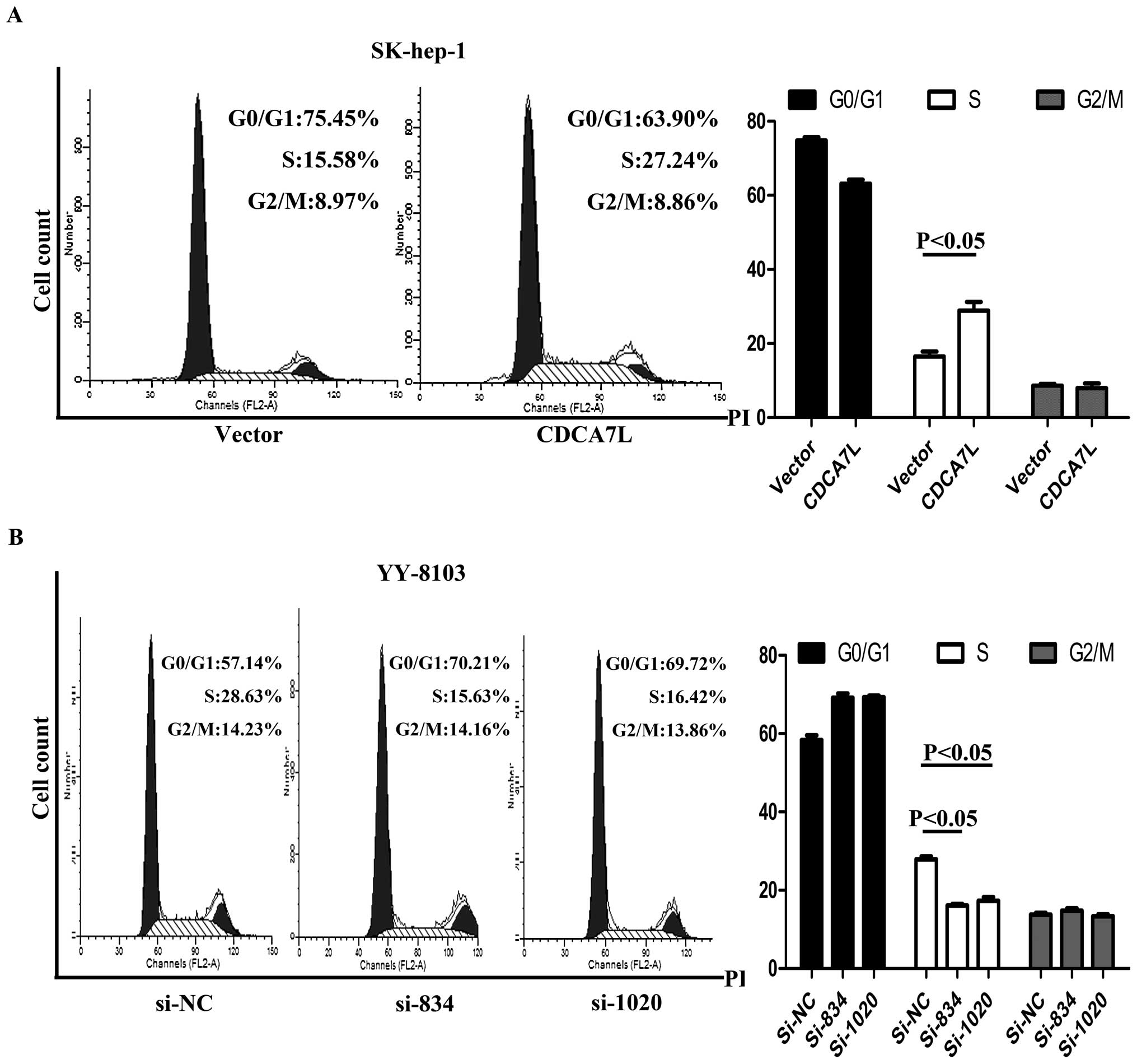
CDCA7L promotes cell cycle
progression
Flow cytometry was used to evaluate the effect of
CDCA7L on the cell cycle. After cell cycle synchronization, these
cells were transfected with plasmids or siRNA and then harvested,
and their cell cycle distribution was analyzed. The results showed
that enforced expression of CDCA7L caused SK-hep-1 cells to
progress from G0/G1 phase and enter into S phase while the same
cells transfected with pcDNA3.1 (vector) did not show the same
progression (Fig. 5A). Conversely,
si834 and si1020 knockdown of endogenous CDCA7L in YY-8103 cells
led to G0/G1 arrest while transfection with negative control si-NC
had no effect (Fig. 5B). These
collective data suggest that CDCA7L could contribute to HCC cell
cycle progression by promoting the entry of cells from G0/G1 phase
into S phase.
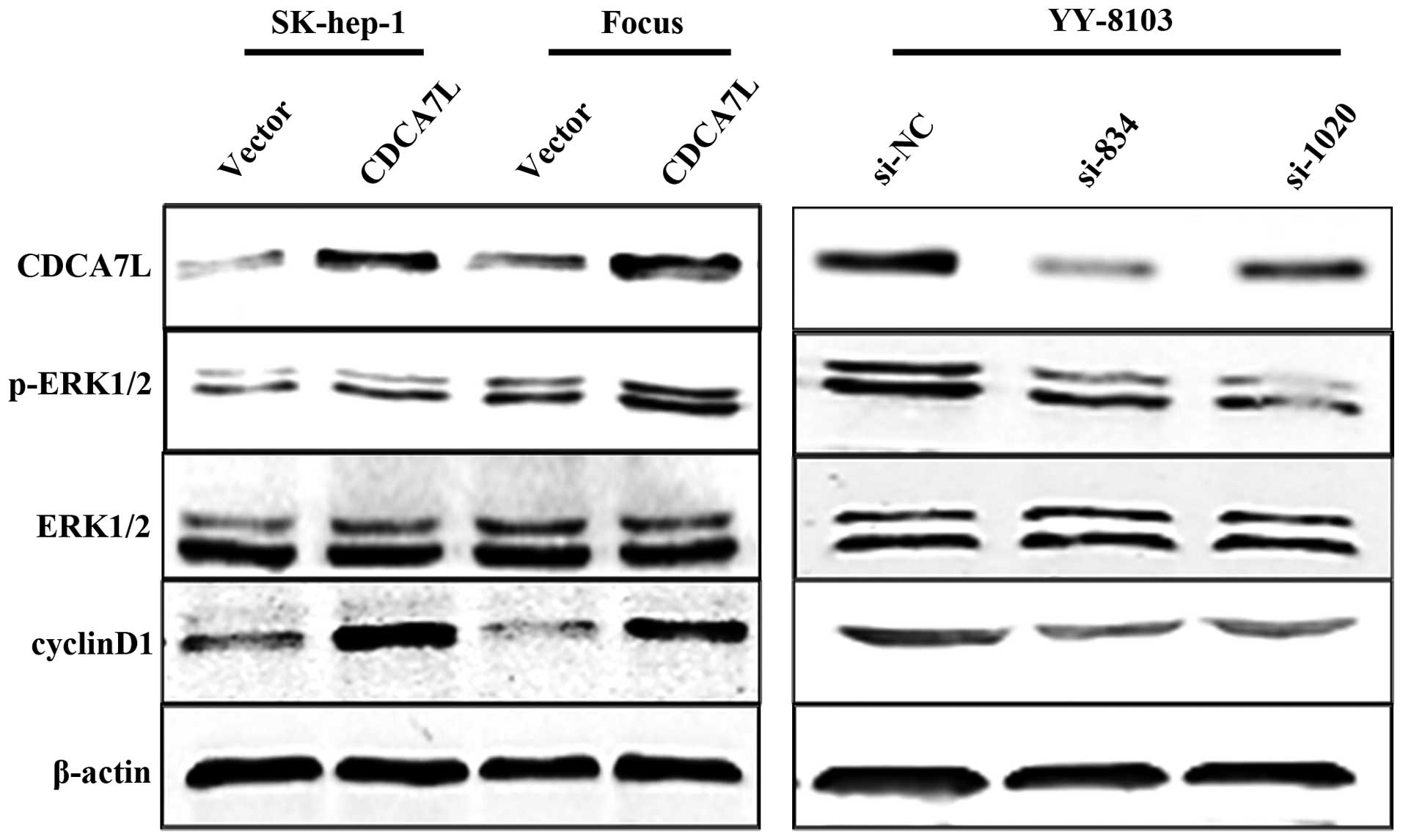
CDCA7L activates the ERK1/2 signaling
pathway and positively regulates cyclinD1 expression
To further investigate the mechanism underlying the
effect of CDCA7L on the cell cycle, we first analyzed protein
expression of cyclin D1 using western blot analysis. Our data
showed that the cyclin D1 protein was significantly upregulated in
SK-hep-1 and Focus HCC cells transfected with pcDNA3.1-CDCA7L
(CDCA7L) compared with the same cells transfected with the pcDNA3.1
negative control (vector). As expected, CDCA7L knockdown caused a
reduction in the level of cyclin D1 protein in YY-8103 cells
compared with the negative control. It is known that activation of
the ERK1/2 signaling pathway can result in upregulation of cyclin
D1 expression, so we next measured the levels of phosphorylated and
total ERK1/2 protein. The results revealed that phosphorylated
ERK1/2 was increased in SK-hep-1 and Focus cells transfected with
pcDNA3.1-CDCA7L (CDCA7L) and reduced in YY-8103 cells treated with
si834 or si1020, while the level of total ERK1/2 was unchanged
(Fig. 6). These data indicate that
CDCA7L is involved in activation of ERK1/2 signaling and regulation
of the cell cycle.
Discussion
HCC is the fifth most common malignancy and the
third highest cause of cancer-related death worldwide, and its
incidence is increasing (24,25).
The c-Myc oncogene is overexpressed in most cases of HCC, and is
known to play an important role in cell proliferation,
differentiation and apoptosis (26). c-Myc acts as a transcriptional
regulator and promotes carcinogenesis by regulating a series of
target genes, but the biological effects of these genes in HCC
development remain unclear. CDCA7 and CDCA7L belong to the JPO
protein family, recently identified as target genes of c-Myc
(27,28). CDCA7 expression is highly elevated
during blast crises in chronic myelogenous leukemia as compared
with the chronic phase, and CDCA7 transgenic mice have been shown
to have a 2-fold increase in the risk of developing lymphoid
malignancies. These findings indicate that CDCA7 is involved in the
development of hematologic malignancies (29). Interestingly, CDCA7L expression is
upregulated, and may promote cellular proliferation, in
medulloblastoma cells. Both CDCA7 and CDCA7L are capable of
interacting with c-Myc and complement and potentiate Myc-mediated
transformation, and could thus further participate in neoplastic
transformation and contribute to tumorigenesis (27–30).
In this study, we first determined the expression of
CDCA7L in HCC, and further investigated its function in HCC cell
proliferation and tumorigenicity both in vitro and in
vivo. CDCA7L and CDCA7 are homologous analogues; they both
belong to the JPO protein family, also called the cell division
cycle associated (CDCA) protein family. We therefore analyzed
CDCA7L expression in HCC cells during the cell cycle and found that
CDCA7L promotes entry of HCC cells into S phase from G0/G1 phase.
Several studies have reported that regulation of the cell cycle
G1/S transition check-point is often abnormal in tumor cells
(31), with the cyclins or CDK
proteins and their upstream regulators changed accordingly
(32–34,37).
It is known that cyclin D1 is required for cell cycle progression
from G0/G1 phase to S phase (35,36),
and that sustained ERK1/2 activity can upregulate cyclin D1
expression (31), so we next
determined the levels of phosphorylated and total ERK1/2 and cyclin
D1 proteins using western blot analysis.
In summary, our findings show that CDCA7L is
significantly upregulated in HCC, while ectopic CDCA7L expression
promotes cell proliferation, colony formation, soft agar colony
formation and tumorigenicity of SK-hep-1 and Focus HCC cells.
Likewise, knockdown of CDCA7L inhibits cell proliferation and
colony formation and reduces tumor burden of YY-8103 and MHCC-97H
HCC cells. Further investigation revealed that CDCA7L stimulates
ERK1/2 signaling and regulates the cell cycle by promoting the
entry of HCC cells into S phase from G0/G1 phase. This study is the
first to discover that CDCA7L expression plays an important role in
HCC progression, but the specific role of CDCA7L in HCC remains
worthy of further investigation.
Acknowledgements
This study was supported by National
Natural Science Foundation, China (grant no. 81170415).
References
|
1.
|
Kan Z, Zheng H, Liu X, et al: Whole genome
sequencing identifies recurrent mutations in hepatocellular
carcinoma. Genome Res. 23:1422–1433. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Wei Y, Doria C and Liu Y: Targeted
therapies in the treatment of advanced hepatocellular carcinoma.
Clin Med Insights Oncol. 7:87–102. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Dai L, Lei N, Liu M and Zhang JY:
Autoantibodies to tumor-associated antigens as biomarkers in human
hepatocellular carcinoma (HCC). Exp Hematol Oncol. 2:15–21. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Shiraha H, Yamamoto K and Namba M: Human
hepatocyte carcinogenesis (Review). Int J Oncol. 42:1133–1138.
2013.PubMed/NCBI
|
|
5.
|
Wang X, Chen J, Li F, et al: MiR-214
inhibits cell growth in hepatocellular carcinoma through
suppression of β-catenin. Biochem Biophys Res Commun. 428:525–531.
2012.PubMed/NCBI
|
|
6.
|
Hakami A, Ali A and Hakami A: Effects of
hepatitis B virus mutations on its replication and liver disease
severity. Open Virol J. 7:12–18. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Jeong SW, Jang JY and Chung RT: Hepatitis
C virus and hepatocarcinogenesis. Clin Mol Hepatol. 18:347–356.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Zhao Y, Jian W, Gao W, et al: RNAi
silencing of c-Myc inhibits cell migration, invasion, and
proliferation in HepG2 human hepatocellular carcinoma cell line:
c-Myc silencing in hepatocellular carcinoma cell. Cancer Cell Int.
13:23–28. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Oster SK, Ho CS, Soucie EL and Penn LZ:
The myc oncogene: MarvelouslY Complex. Adv Cancer Res. 84:81–154.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Pelengaris S and Khan M: The many faces of
c-MYC. Arch Biochem Biophys. 416:129–136. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Facchini LM and Penn LZ: The molecular
role of Myc in growth and transformation: recent discoveries lead
to new insights. FASEB J. 12:633–651. 1998.PubMed/NCBI
|
|
12.
|
Schmidt EV: The role of c-myc in cellular
growth control. Oncogene. 18:2988–2996. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Boyd KE and Farnham PJ: Myc versus USF:
discrimination at the cad gene is determined by core promoter
elements. Mol Cell Biol. 17:2529–2537. 1997.PubMed/NCBI
|
|
14.
|
Bello-Fernandez C, Packham G and Cleveland
JL: The ornithine decarboxylase gene is a transcriptional target of
c-Myc. Proc Natl Acad Sci USA. 90:7804–7808. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Wu S, Pena A, Korcz A, Soprano DR and
Soprano KJ: Over expression of Mxi1 inhibits the induction of the
human ornithine decarboxylase gene by the Myc/Max protein complex.
Oncogene. 12:621–629. 1996.PubMed/NCBI
|
|
16.
|
Shim H, Dolde C, Lewis BC, Wu CS, Dang G,
Jungmann RA, Dalla-Favera R and Dang CV: c-Myc transactivation of
LDH-A: implications for tumor metabolism and growth. Proc Natl Acad
Sci USA. 94:6658–6663. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Hubank M and Schatz DG: Identifying
differences in mRNA expression by representational difference
analysis of cDNA. Nucleic Acids Res. 22:5640–5648. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Perez-Roger I, Solomon DL, Sewing A and
Land H: Myc activation of cyclin E/Cdk2 kinase involves induction
of cyclin E gene transcription and inhibition of p27(Kip1) binding
to newly formed complexes. Oncogene. 14:2373–2381. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Galaktionov K, Chen X and Beach D: Cdc25
cell-cycle phosphatase as a target of c-myc. Nature. 382:511–517.
1996. View
Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Ou XM, Chen K and Shih JC: Monoamine
oxidase A and repressor R1 are involved in apoptotic signaling
pathway. Proc Natl Acad Sci USA. 103:10923–10928. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Huang A, Ho CS, Ponzielli R, et al:
Identification of a novel c-Myc protein interactor, JPO2, with
transforming activity in medulloblastoma cells. Cancer Res.
65:5607–5619. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Chen K, Ou XM, Chen G, et al: R1, a novel
repressor of the human monoamine oxidase A. J Biol Chem.
280:11552–11559. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Johnson S, Stockmeier CA, Meyer JH, et al:
The reduction of R1, a novel repressor protein for monoamine
oxidase A, in major depressive disorder. Neuropsychopharmacology.
36:2139–2148. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Dai CX, Gao Q, Qiu SJ, et al:
Hypoxia-inducible factor-1 alpha, in association with inflammation,
angiogenesis and MYC, is a critical prognostic factor in patients
with HCC after surgery. BMC Cancer. 9:418–428. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Dituri F, Mazzocca A, Peidro FJ, et al:
Differential inhibition of the TGF-b signaling pathway in HCC cells
using the small molecule inhibitor LY2157299 and the D10 monoclonal
antibody against TGF-b receptor type II. PLoS One. 8:67109–67123.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Lin C-P, Liu C-R, Lee C-N, et al:
Targeting c-Myc as a novel approach for hepatocellular carcinoma.
World J Hepatol. 2:16–20. 2010.PubMed/NCBI
|
|
27.
|
Prescott JE, Osthus RC, Lee LA, et al: A
novel c-Myc-responsive gene, JPO1, participates in neoplastic
transformation. J Biol Chem. 276:48276–48284. 2001.PubMed/NCBI
|
|
28.
|
Goto Y, Hayashi R, Muramatsu T, et al:
JPO1/CDCA7, a novel transcription factor E2F1-induced protein,
possesses intrinsic transcriptional regulator activity. Biochim
Biophys Acta. 1759:60–68. 2006. View Article : Google Scholar
|
|
29.
|
Osthus RC, Karim B, Prescott JE, et al:
The Myc target gene JPO1/CDCA7 is frequently overexpressed in human
tumors and has limited transforming activity in vivo. Cancer Res.
65:5620–5627. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Gill RM, Gabor TV, Couzens AL, et al: The
MYC-associated protein CDCA7 is phosphorylated by AKT to regulate
MYC-dependent apoptosis and transformation. Mol Cell Biol.
33:498–513. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Wang H, Wu K, Sun Y, et al: STC2 is
upregulated in hepatocellular carcinoma and promotes cell
proliferation and migration in vitro. BMB Rep. 45:629–634. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Sherr CJ: Cancer cell cycles. Science.
274:1672–1677. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Braun-Dullaeus RC, Mann MJ, Sedding DG,
Sherwood SW, von der Leyen HE and Dzau VJ: Cell cycle-dependent
regulation of smooth muscle cell activation. Arterioscler Thromb
Vasc Biol. 24:845–850. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Braun-Dullaeus RC, Man MJ and Dzau VJ:
Cell cycle progression: new therapeutic target for vascular
proliferative disease. Circulation. 98:82–89. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Tarn WY and Lai MC: Translational control
of cyclins. Cell Div. 6:5–13. 2011. View Article : Google Scholar
|
|
36.
|
Ajchenbaum F, Ando K, DeCaprio JA and
Griffin JD: Independent regulation of human D-type cyclin gene
expression during G1 phase in primary human T lymphocytes. J Biol
Chem. 268:4113–4119. 1993.PubMed/NCBI
|
|
37.
|
Liu Y, Li W, Ye C, et al: Gambogic Acid
induces G0/G1 cell cycle arrest and cell migration inhibition via
suppressing PDGF receptor β tyrosine phosphorylation and Rac1
activity in Rat aortic smooth muscle cells. J Atheroscler Thromb.
17:901–913. 2010.PubMed/NCBI
|