Introduction
Cervical cancer is the second most common cause of
cancer death in women worldwide and ∼500,000 new cases of cervical
cancer are diagnosed each year, with 280,000 deaths (1). Cervical squamous cell carcinoma (SCC)
is the most frequent type of cervical cancer and the most important
risk factor for cervical-SCC is persistent human papilloma virus
(HPV) infection (2–4). High-risk HPVs contain oncoproteins,
i.e., E6 and E7, which contribute to the oncogenesis of cervical
SCC by silencing tumor-suppressive p53 and Rb proteins and several
cancer-related genes (5).
Therefore, recent research on cervical SCC has focused on E6 and E7
oncoproteins. However, the molecular mechanisms of cervical SCC
initiation, development, and metastasis have not yet been fully
elucidated.
The discovery of non-coding RNAs in the human genome
was an important conceptual breakthrough in the post-genome
sequencing era (6). A growing body
of evidence indicates that miRNAs are key regulators that
contribute to the initiation and development of various types of
cancer (7). In cancer pathways,
normal regulatory mechanisms are disrupted by altered expression of
tumor-suppressive or oncogenic miRNAs. Therefore, identification of
differentially expressed miRNAs is an important step to
understanding human oncogenesis.
Based on this, our research group has elucidated the
miRNA expression signatures of various types of human cancers
(8–12). Recent studies of miRNA expression
signatures of hypopharyngeal SCC and maxillary SCC have indicated
that expression of miRNA-29 family miRNAs
(miR-29a/b/c) is significantly reduced in cancer tissues,
suggesting that these miRNAs may contribute to the oncogenesis and
metastasis of cervical SCC (13,14).
Expression analysis of miR-29 family miRNAs
in cervical SCC clinical specimens showed that miR-29a was
the most highly downregulated miRNA in the clinical specimens,
thus, we focused on miR-29a in this study. The aim of the
present study was to investigate the functional significance of
miR-29a and to identify the molecular target genes regulated
by miR-29a in cervical SCC cells. Genome-wide gene
expression data and in silico database analysis showed that
the heat-shock protein 47 (HSP47) gene, also known as serpin
peptidase inhibitor clade H, member 1 (SERPINH1), was a
promising candidate target of miR-29a.
Materials and methods
Clinical specimens
A total of 18 primary cervical SCC specimens and 11
non-cancer specimens were collected from patients who had undergone
surgical treatment at Chiba University Hospital. The samples were
processed and stored in liquid nitrogen until RNA extraction.
Patient information is summarized in Table I. Our study was approved by the
Bioethics Committee of Chiba University; prior written informed
consent and approval was given by each patient. HPV status was
examined by L1 consensus primers and type-specific real-time PCR
primers, as described previously (15).
 | Table I.Characteristics of cervical SCC
specimens and non-cancer specimens. |
Table I.
Characteristics of cervical SCC
specimens and non-cancer specimens.
| Cervical SCC
specimens |
|---|
|
|---|
| Patient no. | Age | FIGO stage | Tumor size
(cm2) | Lymph node
metastasis | HPV status |
|---|
| 1 | 58 | IIB | 1.7×1.9 | − | 16 |
| 2 | 64 | IIB | No data | − | 16 |
| 3 | 37 | IIB | 3.5×3.0 | + | 16 |
| 4 | 41 | IB2 | 8.3×3.3 | − | 16 |
| 5 | 39 | IB1 | 3.5×3.4 | − | 16 |
| 6 | 34 | IB1 | 3.2×2.2 | − | 16 |
| 7 | 43 | IB2 | 4.0×8.0 | − | 18 |
| 8 | 56 | IIIB | 3.0×3.1 | + | 16, 18 |
| 9 | 77 | IIB | 3.0×2.7 | − | 16 |
| 10 | 62 | IB1 | 3.0×2.0 | − | 16 |
| 11 | 56 | IIIA | 4.5×2.2 | + | 16 |
| 12 | 56 | IIA | 4.0×4.0 | − | 16 |
| 13 | 60 | IB1 | 4.0×4.0 | − | 16 |
| 14 | 32 | IIB | 6.0×3.0 | + | 16 |
| 15 | 38 | IB2 | 6.8×4.6 | + | 16 |
| 16 | 44 | IB1 | 3.5×2.2 | − | 16 |
| 17 | 40 | IB1 | 3.0×2.0 | − | 16 |
| 18 | 63 | IB1 | 2.7×2.4 | − | 16 |
| Non-cancer
specimens |
|---|
|
|---|
| Patient no. | Age | HPV status |
|---|
| 1 | 44 | - |
| 2 | 77 | - |
| 3 | 75 | - |
| 4 | 45 | - |
| 5 | 47 | - |
| 6 | 69 | - |
| 7 | 40 | - |
| 8 | 48 | - |
| 9 | 41 | - |
| 10 | 41 | - |
| 11 | 34 | - |
RNA isolation
Total RNA was isolated using TRIzol reagent
(Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s
protocol. RNA concentrations were determined
spectrophotometrically. RNA quality was confirmed using a NanoDrop
1000 Spectrophotometer (Thermo Fisher Scientific, USA).
Quantitative real-time RT-PCR
Stem-loop RT-PCR (TaqMan MicroRNA assays; P/N,
002112 for miR-29a; P/N, 000413 for miR-29b; P/N,
000587 for miR-29c; Applied Biosystems, Foster City, CA,
USA) was used to quantify miRNAs according to earlier published
conditions (14). To normalize the
data for quantification of miR-29-family sequences, we used
RNU48 (Assay ID, 001006; Applied Biosystems) as a control.
The ΔΔCt method was used to calculate the fold-change.
Mature miRNA and siRNA transfections
Cervical cancer cell lines were transfected with
Lipofectamine RNAiMAX transfection reagent (Invitrogen) and
Opti-MEM (Invitrogen) with 10 nM mature miRNA or siRNA molecules.
The following RNA species were used in this study: mature miRNA,
mirVana miRNA mimic for hsa-miR-29a-3p (Product ID, MC12499;
Applied Biosystems), negative control miRNA (P/N, AM17111; Applied
Biosystems), small-interfering RNA (Stealth siRNAs, si-SERPINHl;
P/N, HSS101423 and HSS189522; Invitrogen) and negative control
siRNA (Stealth RNAi Negative Control Medium GC, P/N, 12935-300;
Invitrogen).
Cell proliferation, migration and
invasion assays
Cell proliferation was determined using XTT assays
(Roche Applied Science, Tokyo, Japan) according to the
manufacturer’s instructions. Cell migration assays were performed
using modified Boyden Chambers (Transwells, Corning/Costar no.
3422, USA). Cells were transfected with 10 nM miRNA by reverse
transfection and plated in 10-cm dishes at 8×l05
cells/dish. After 48 h, 1×105 cells were added to the
upper chamber of each migration well and were allowed to migrate
for 48 h. After gentle removal of the non-migratory cells from the
filter surface of the upper chamber, the cells that migrated to the
lower side were fixed and stained with Diff-Quick (Sysmex Corp.,
Japan). The number of cells migrating to the lower surface was
determined microscopically by counting four areas of constant size
per well. Cell invasion assays were carried out using modified
Boyden chambers in 24-well tissue culture plates at
1×105 cells per well (BD Biosciences, USA). All
experiments were performed in duplicate.
Target gene search for miR-29a
A genome-wide screen was performed to identify
miR-29a-target genes using miR-29a-transfected CaSKi
cells. A SurePrint G3 Human GE 8×60K Microarray (Agilent
Technologies, Santa Clara, CA, USA) was used for expression
profiling of miR-29a transfectants in comparison with
miRNA-negative control transfectants. TargetScan release 6.2
(http://www.targetscan.org/) was used to
identify predicted target genes and their miRNA binding site seed
regions. Gene expression data for clinical cervical SCC specimens
were obtained from the GEO database (accession no. GSE6791).
Western blot analysis
Cells were harvested and lysed 72 h after
transfection. Each cell lysate (50 μg of protein) was
separated using Mini-Protean TGX gels (Bio-Rad, Hercules, CA, USA),
followed by subsequent transfer to PVDF membranes. Immunoblotting
was performed with polyclonal anti-HSP47 antibodies (sc-5293; Santa
Cruz Biotechnology, Santa Cruz, CA, USA). Anti-GAPDH antibodies
(ab8245; Abeam, UK) were used as an internal control.
Plasmid construction and dual-luciferase
reporter assays
Partial sequences (191 bp) of the HSP47 3′
untranslated region (3′UTR) that contain the miR-29a target
site (GGTGCTA) were inserted between the XhoI and
PmeI restriction sites in the 3′UTR of the hRluc gene in the
psiCHECK-2 vector (Promega, Madison, WI, USA). The deletion of the
miR-29a target site was cloned and constructed as
deletion-vector in this study. HeLa cells were then transfected
with 5 ng vector and 10 nM mature miRNA.
Immunohistochemistry
We performed immunostaining using a tissue
microarray containing 60 specimens: 10 normal cervical tissues, 10
inflammation tissues, 10 cervical intraepithelial neoplasia (CIN)
tissues and 30 SCC tissues (CR 602; US Biomax, Rockville, MD, USA).
Detailed information on all tumor specimens can be found at
http://www.biomax.us/tissue-arrays/Uterus/CR602. The
tissue microarray was incubated overnight with primary mouse
monoclonal antibodies against HSP47 (1:50, sc-5293, Santa Cruz
Biotechnology). Next, the sample was treated with anti-mouse biotin
antibodies (1:2,000, 115-065-003, Jackson ImmunoResearch
Laboratories, Inc., West Grove, PA, USA) for 1 h and then treated
with an ABC kit (K0377, Dako, Carpinteria, CA, USA) for 30 min.
Counterstaining was performed using a DAB kit (425011, Nichirei
Bioscience Inc., Tokyo, Japan). Immunostaining was evaluated
according to previously described scoring methods (12).
Statistical analysis
The relationships between two variables and
numerical values were analyzed using the Mann-Whitney U test and
the relationships between three variables and numerical values were
analyzed using the Bonferroni-adjusted Mann-Whitney U test. Expert
StatView analysis software (ver. 4; SAS Institute Inc., Cary, NC,
USA) was used in both analyses. In the comparison of three
variables, an unadjusted statistical level of significance of
P<0.05 corresponded to the Bonferroni-adjusted level of
P<0.0167.
Results
Expression of miR-29-family miRNAs in
clinical cervical SCC specimens
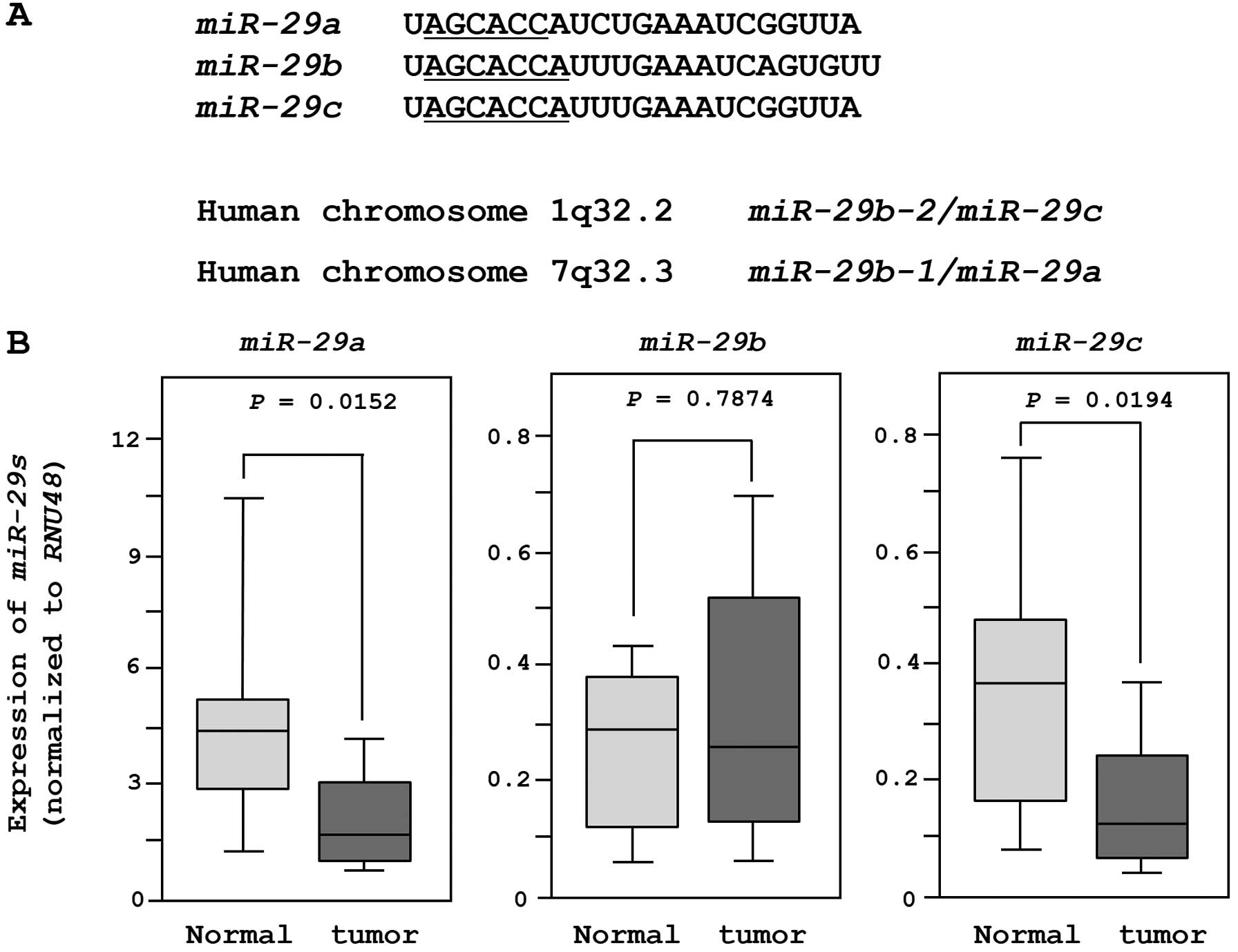
The sequences and chromosomal locations of
miR-29-family miRNAs (miR-29a/b/c) in the human
genome are shown in Fig. 1A. These
miRNAs were clustered at two different human genomic loci,
miR-29b-1 and miR-29a at 7q32.3 and miR-29b-2
and miR-29c at lq32.2.
We evaluated the expression of miR-29-family
miRNAs in 18 clinical specimens and 11 non-cancer tissues. The
expression levels of miR-29a and miR-29c were
significantly lower in tumor tissues than in non-cancer tissues.
However, there was no significant difference in the expression of
miR-29b (Fig. 1B). When we
compared two miRNAs (miR-29a and miR-29c) after
normalization to the expression of RNU48, miR-29a was more
abundantly expressed in both normal and cancer tissues.
Effects of restoring miR-29a on cell
proliferation, migration, and invasion in cervical SCC cell
lines
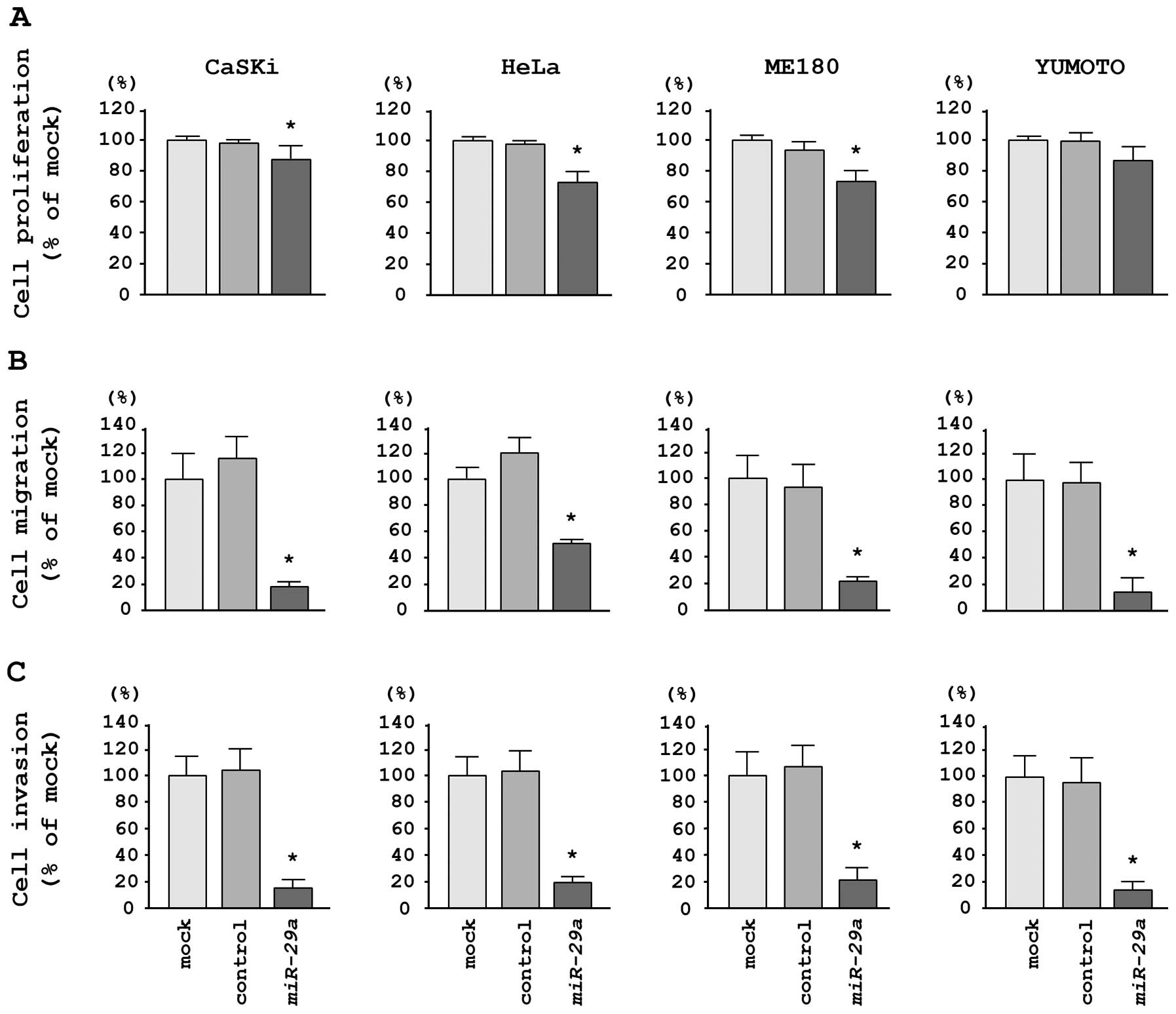
To investigate the functional effects of
miR-29a, we performed gain-of-function studies using miRNA
transfection in four cervical cancer cell lines. XTT assays
demonstrated that cell proliferation was significantly inhibited in
miR-29a transfectants in comparison with mock- or
miR-control-transfected CaSKi, HeLa and ME180 cells; no inhibition
was observed in Yumoto cells in this assay (Fig. 2A). We observed the following
changes in proliferation, expressed as a percentage of
mock-transfected cells: i) CaSKi, mock, 100.0±8.2%; miR-control,
97.9±6.6%; miR-29a, 87.6±5.9%; P=0.0032; ii) HeLa, mock,
100.0±7.5%; miR-control, 97.6±4.1%; miR-29a, 73.8±4.8%;
P<0.0001; iii) ME180, mock, 100.0±4.5%; miR-control, 93.4±5.9%;
miR-29a, 75.1±4.7%; P<0.0001, and iv) Yumoto, mock,
100.0±8.0%; miR-control, 99.9±10.6%; miR-29a, 85.0±9.9%; P=
0.0287 (Fig. 2A).
Migration assays demonstrated that miR-29a
transfection significantly inhibited cell migration compared with
mock- or miR-control-transfected cells. We observed the following
changes in migration activity, expressed as a percentage of
mock-transfected cells: i) CaSKi, mock, 100.0±14.0%; miR-control,
116.1+19.3%; miR-29a, 31.7±5.7%; P<0.0001; ii) HeLa,
mock, 100.0±11.3%; miR-control, 124.0±14.8%; miR-29a,
55.3±10.6%; P<0.0001; iii) ME180, mock, 100.0±12.4%;
miR-control, 89.8±16.8%; miR-29a, 20.3±6.4%; P<0.0001;
iv) Yumoto, mock, 100.0±8.1%; miR-control, 95.4±15.8%;
miR-29a, 13.0±6.3%; P<0.0001 (Fig. 2B).
Matrigel invasion assays demonstrated that cell
invasion was significantly inhibited in miR-29a
transfectants in comparison with mock- or miR-control-transfected
cells for all cell lines tested. We observed the following changes
in invasion activity, expressed as a percentage of mock-transfected
cells: i) CaSKi, mock, 100.0±12.9%; miR-control, 103.4±6.3%;
miR-29a, 16.6±7.5%; P<0.0001; ii) HeLa, mock,
100.0±16.5%; miR-control, 102.7±14.6%; miR-29a, 31.8+13.2%;
P<0.0001; iii) ME180, mock, 100.0±22.7%; miR-control,
109.5±37.6%; miR-29a, 23.8±6.6%; P<0.0001; iv) Yumoto,
mock, 100.0±11.9%; miR-control, 90.9±10.1%; miR-29a,
5.9±3.3%; P<0.0001 (Fig.
2C).
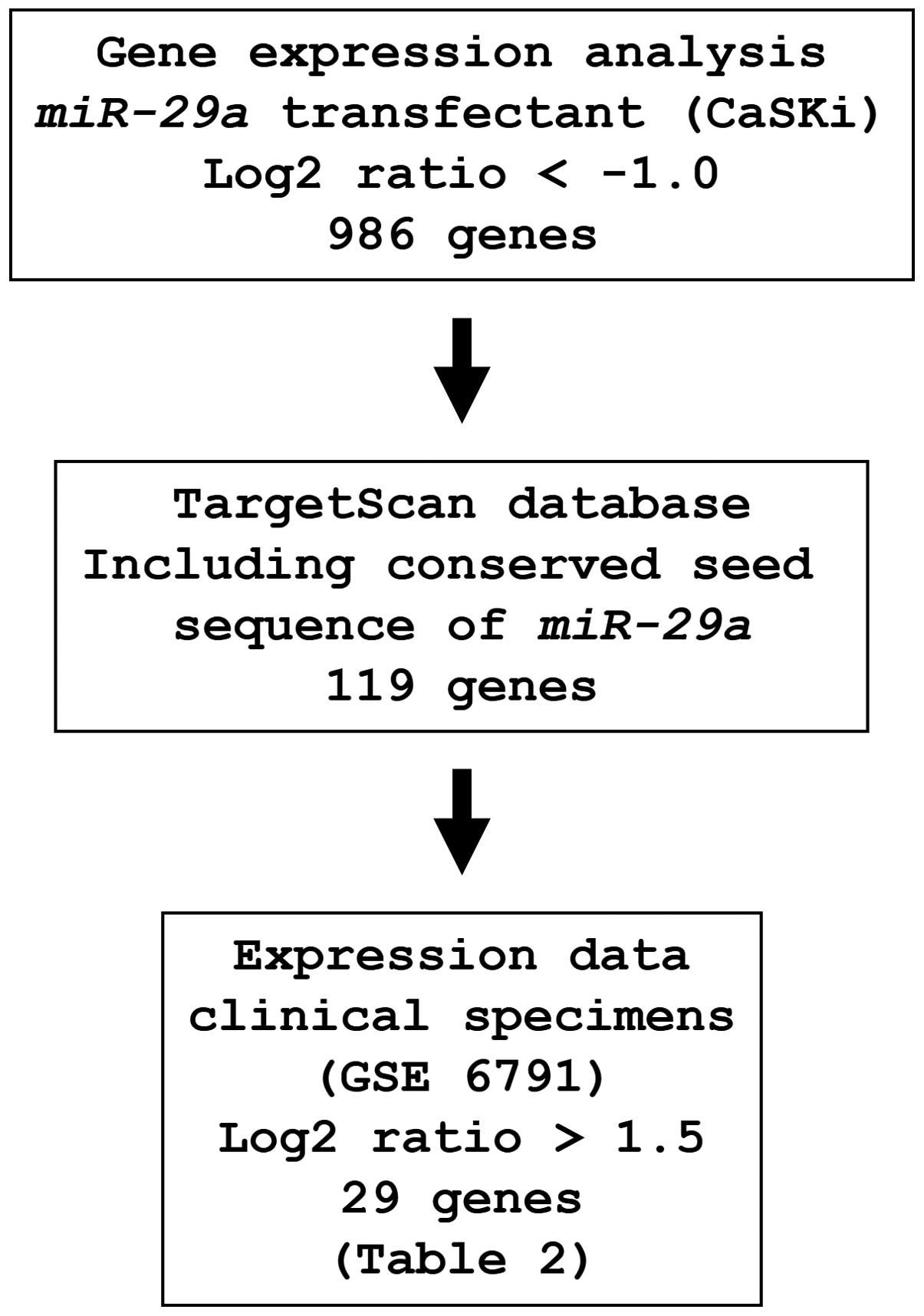
Identification of miR-29a-regulated
putative target genes
To identify genes regulated by miR-29a, we
used in silico and genome-wide gene expression analyses. The
strategy for the selection of miR-29a-target genes is shown
in Fig. 3. First, to gain further
insight into which genes were affected by miR-29a, we
performed genome-wide gene expression analysis using
miR-29a-transfected CaSKi cells; 986 genes were identified
as downregulated (log2 ratio <-1.0) by miR-29a
transfection. Next, we used the TargetScan database to examine
whether these genes contained miR-29a binding sequences in
their 3′UTRs. Finally, the gene set was analyzed with a publicly
available gene expression data set in the GEO (accession no.
GSE6791), and genes upregulated (log2 ratio >1.5) in
cervical SCC were chosen. A total of 29 genes were candidate
miR-29a-regulated oncogenic genes in cervical SCC (Table II).
| Table II.Candidate target genes regulated by
miR-29a. |
Table II.
Candidate target genes regulated by
miR-29a.
Expression
(log2 ratio)
| | | |
|---|
| CSCC clinical
specimen | miR-29a
transfectant | Entrez gene ID | Symbol | Gene name |
|---|
| 7.32 | −3.37 | 871 | HSP47 | Heat shock protein
47 |
| 4.75 | −3.04 | 4678 | NASP | Nuclear
autoantigenic sperm protein |
| 4.69 | −1.67 | 10951 | CBX1 | Chromobox homolog
1 |
| 3.29 | −1.68 | 144455 | E2F7 | E2F transcription
factor 7 |
| 3.08 | −2.25 | 4291 | MLF1 | Myeloid leukemia
factor 1 |
| 3.07 | −4.27 | 55920 | RCC2 | Regulator of
chromosome condensation 2 |
| 2.91 | −1.50 | 23186 | RCOR1 | REST corepressor
1 |
| 2.77 | −4.11 | 3300 | DNAJB2 | DnaJ (Hsp40)
homolog, subfamily B, member 2 |
| 2.64 | −2.66 | 3655 | ITGA6 | Integrin, α6 |
| 2.63 | −2.97 | 79017 | GGCT |
γ-glutamylcyclotransferase |
| 2.47 | −1.31 | 8936 | WASF1 | WAS protein family,
member 1 |
| 2.26 | −1.26 | 4140 | MARK3 | MAP/microtubule
affinity-regulating kinase 3 |
| 2.10 | −1.15 | 54851 | ANKRD49 | Ankyrin repeat
domain 49 |
| 1.91 | −1.56 | 9949 | AMMECR1 | Alport syndrome,
mental retardation, midface hypoplasia and elliptocytosis
chromosomal region gene 1 |
| 1.87 | −1.85 | 22877 | MLXIP | MLX interacting
protein |
| 1.85 | −1.49 | 8894 | EIF2S2 | Eukaryotic
translation initiation factor 2, subunit 2β, 38 kDa |
| 1.85 | −1.40 | 3927 | LASP1 | LIM and SH3 protein
1 |
| 1.84 | −1.42 | 54107 | POLE3 | Polymerase (DNA
directed), ε3 accessory subunit |
| 1.80 | −1.24 | 4361 | MRE11A | MRE11 meiotic
recombination 11 homolog A (S. cerevisiae) |
| 1.79 | −1.56 | 7328 | UBE2H |
Ubiquitin-conjugating enzyme E2H |
| 1.75 | −4.69 | 3915 | LAMC1 | Laminin, γl
(formerly LAMB2) |
| 1.67 | −1.57 | 80829 | ZFP91 | Zinc finger
protein |
| 1.65 | −2.31 | 8527 | DGKD | Diacylglycerol
kinase, δ 130 kDa |
| 1.61 | −3.07 | 4232 | MEST | Mesoderm specific
transcript |
| 1.60 | −2.75 | 7168 | TPM1 | Tropomyosin 1
(α) |
| 1.59 | −1.62 | 9618 | TRAF4 | TNF
receptor-associated factor 4 |
| 1.56 | −1.28 | 23380 | SRGAP2 | SLIT-ROBO Rho
GTPase activating protein 2 |
As a result of our selection strategy, we identified
HSP47 as one of the most highly upregulated genes in
cervical SCC tissues and one of the most significantly
downregulated genes in miR-29a-transfected cells.
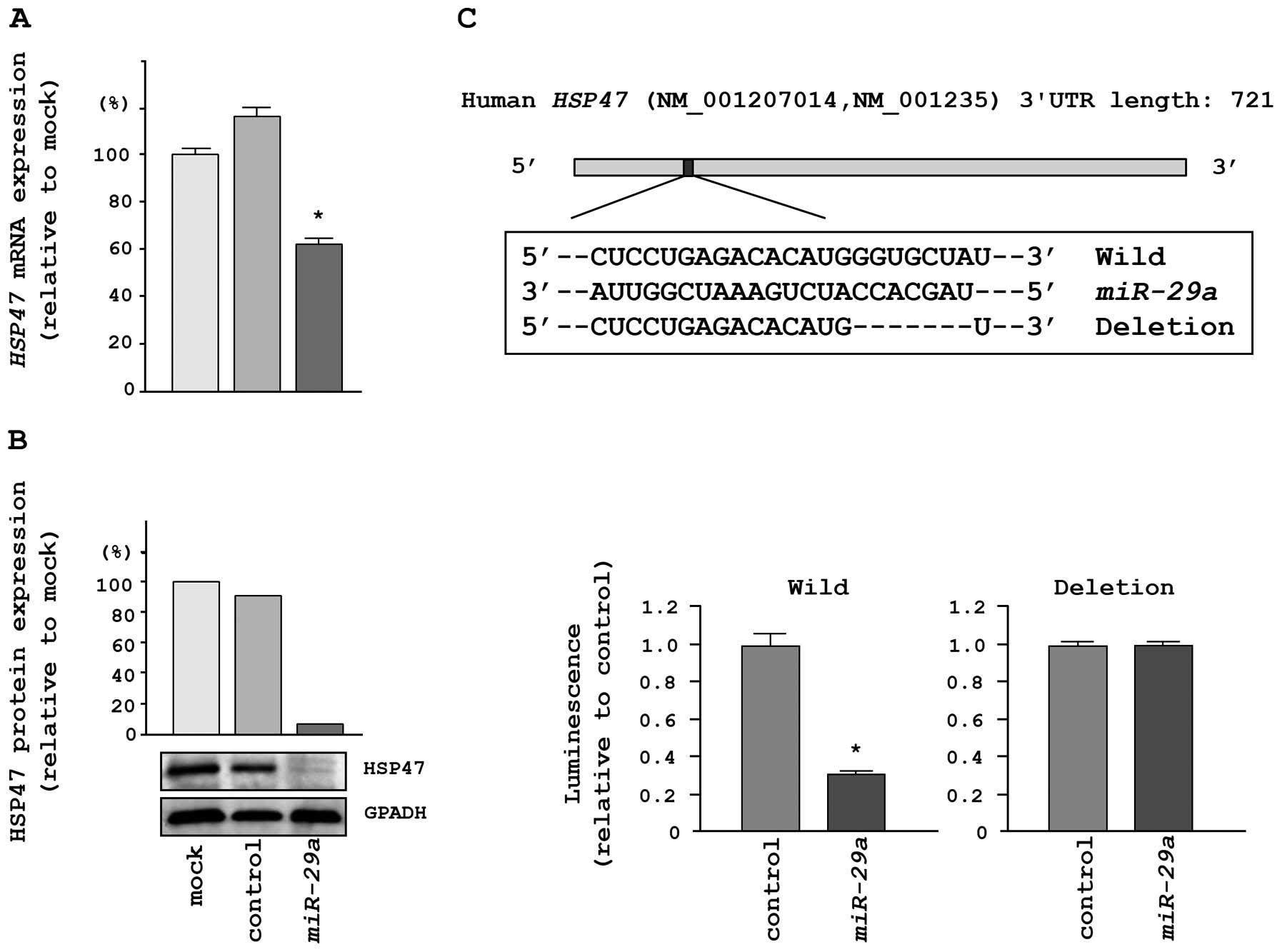
HSP47 was directly regulated by
miR-29a
We performed qRT-PCR and western blotting in HeLa
cells to investigate whether HSP47 expression was reduced by
restoration of miR-29a. The mRNA and protein expression
levels of HSP47 were significantly repressed in
miR-29a transfectants in comparison with mock- or
miR-control-transfected cells (Fig. 4A
and B).
To determine whether the 3′UTR of HSP47 mRNA
had an actual target site for miR-29a, we performed
luciferase reporter assays using a vector encoding the 3′UTR of
HSP47 mRNA. We found that the luminescence intensity was
significantly reduced by transfection with miR-29a and the
vector carrying the wild-type 3′UTR of HSP47, whereas
transfection with a deletion vector blocked the decrease in
luminescence (Fig. 4C).
Effects of silencing HSP47 on cell
proliferation, migration, and invasion in cervical SCC cell
lines
To investigate the functional role of HSP47, we
performed loss-of-function studies using si-T/SWZ-transfected CaSKi
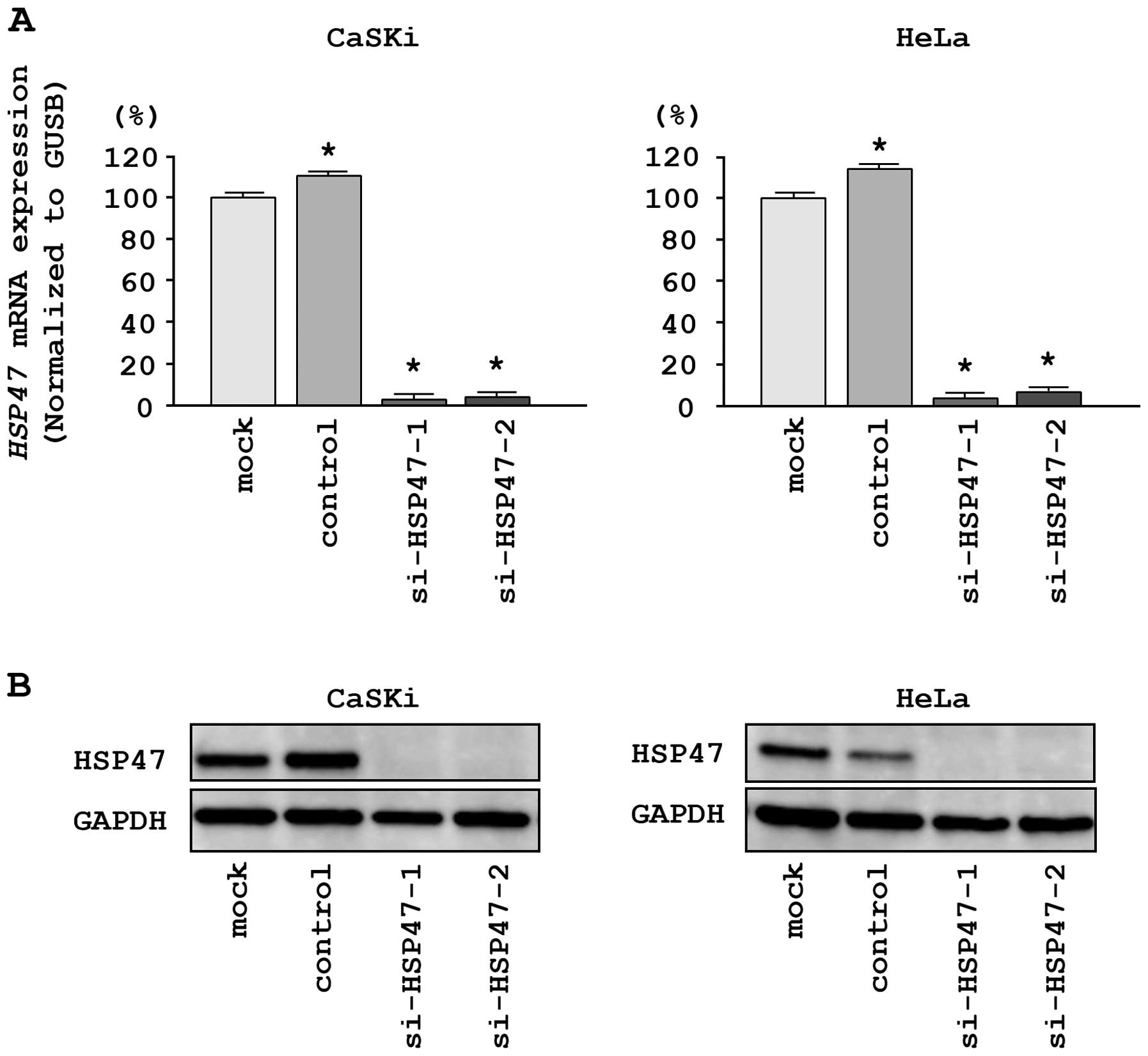
and HeLa cells. First, we evaluated the knockdown efficiency of
si-HSP47 transfection. The expression of HSP47 mRNA
was repressed in two si-HSP47 transfectants as compared with
mock and si-control transfectants (P<0.0001; Fig. 5A). Moreover, the expression of
HSP47 protein was repressed in si-HSP471-1 and
si-HSP47-2 transfectants as compared with mock and
si-control transfectants (Fig.
5B). These results showed that the two siRNAs were effective
for loss-of-function assays in this study.
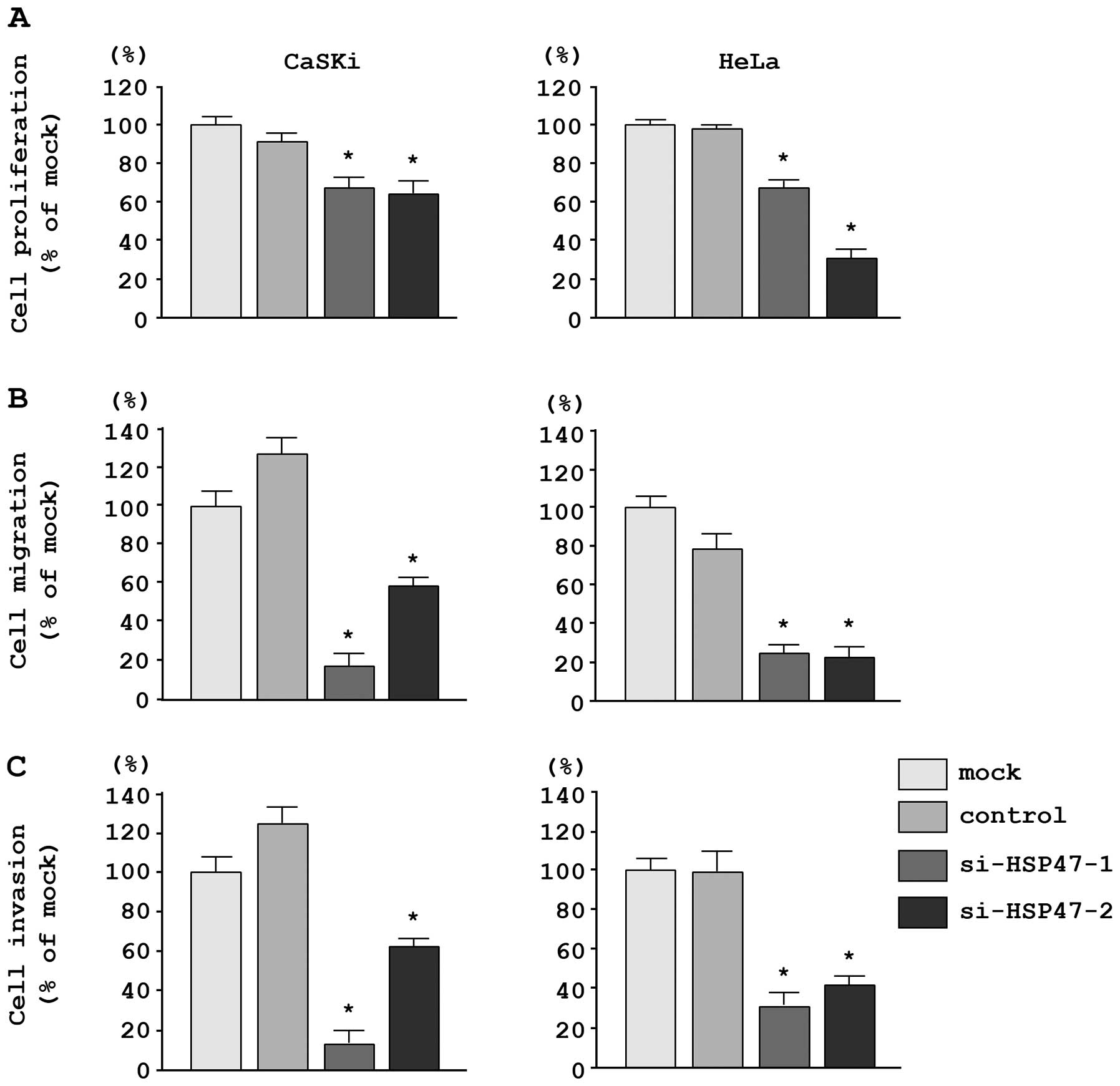
In CaSKi and HeLa cells, XTT assays revealed
significant inhibition of cell proliferation following transfection
with the two different siRNAs targeting HSP47 as compared
with the growth of mock- and si-control-transfected cells. The
following changes in growth were observed, expressed as a
percentage of control proliferation: i) CaSKi, mock, 100.0±4.7%;
miR-control, 87.0±5.7%; si-HSP47-1, 66.4±5.5%;
si-HSP47-2, 63.1±7.8%; ii) HeLa, mock, 100.0±8.5%;
miR-control, 100.7±8.7%; si-HSP47-1, 65.9±7.4%;
si-HSP47-2, 33.0±8.7% (Fig.
6A).
Moreover, migration assays demonstrated that cell
migration was significantly inhibited in CaSKi and HeLa cells
transfected with the two different si-HSP47 constructs. The
following changes in migration were observed, expressed as a
percentage of control migration: i) CaSKi, mock, 100.0±13.7%;
miR-control, 129.8±9.9%; si-HSP47-1, 17.1±7.4%;
si-HSP47-2, 57.7±5.3%; ii) HeLa, mock, 100.0±8.1%;
miR-control, 74.3±9.8%; si-HSP47-1, 24.5±3.5%;
si-HSP47-2, 22.9±4.7% (Fig.
6B).
Matrigel invasion assays demonstrated that cell
invasion was significantly inhibited in CaSKi and HeLa cells
transfected with the two different si-HSP47 constructs. The
following changes in invasion were observed, expressed as a
percentage of control invasion: i) CaSKi, mock, 100.0±24.5%;
miR-control, 128.0±18.9%; si-HSP47-1, 13.3+13.8%;
si-HSP47-2, 63.3±24.9%; ii) HeLa, mock, 100.0±13.1%;
miR-control, 97.8±17.4%; si-HSP47-1, 32.3±9.7%;
si-HSP47-2, 43.2±5.2% (Fig.
6C).
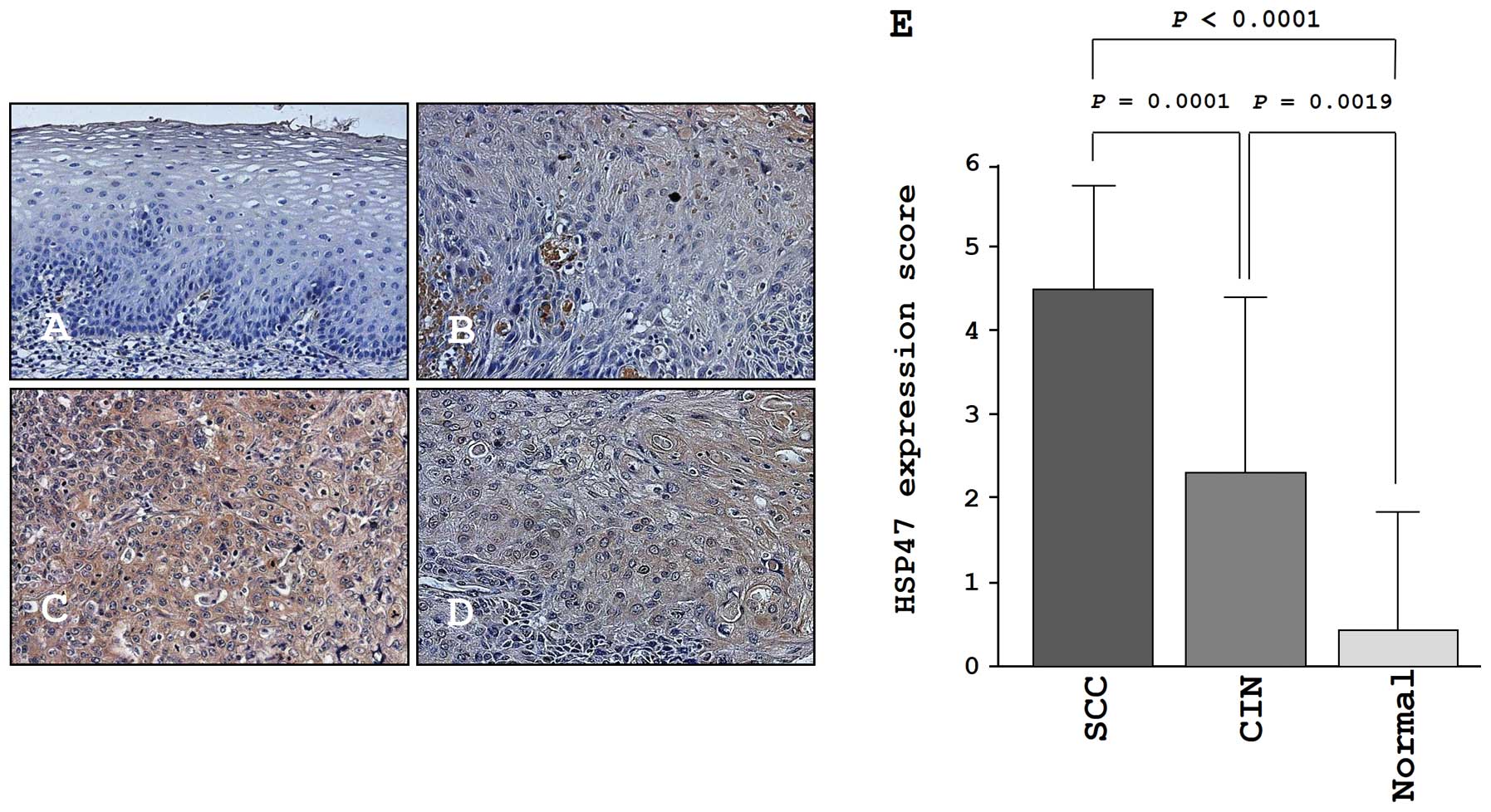
Immunohistochemistry of HSP47 in a tissue
microarray
We confirmed the expression levels of HSP47 in
normal cervical tissues, CIN tissues, and cancer tissues by
immunohistochemical staining. Very low expression of HSP47 was
observed in normal tissues (Fig.
7A). In CIN, weak expression of HSP47 was observed
(Fig. 7B). In contrast, HSP47 was
more strongly expressed in several tumor lesions compared to normal
tissues and CIN tissues (Fig. 7C and
D). The expression score of HSP47 in cervical SCC was
significantly higher than that in CIN (P=0.0001) and normal tissues
(P<0.0001; Fig. 7E).
Discussion
Aberrant expression of the miR-29 family
miRNAs has been reported in several types of human cancers;
however, the expression status varies according to the cancer type.
Decreased expression of the miR-29 family has been observed
in cholangiocarcinoma, nasopharyngeal cancer, non-small cell lung
cancer, hepatocellular carcinoma, malignant peripheral nerve sheath
tumors and mantle cell lymphoma. In contrast, upregulation of the
miR-29 family was reported in breast cancer, colon cancer
and acute myeloid leukemia (16).
In cervical cancer, it was reported that miR-29 targets the
HPV-related gene (17). In this
study, our data demonstrated that miR-29a was significantly
downregulated in cervical SCC clinical specimens and cell lines,
regardless of the type of HPV infection. Furthermore, restoration
of miR-29a in cervical cancer cells inhibited cancer cell
migration and invasion, suggesting that miR-29 a functions
as a tumor suppressor and may contribute to metastasis in cervical
SCC.
The molecular mechanism through which miR-29a
is silenced in cervical SCC is still unknown. Analysis of the
promoter region of miR-29 family miRNAs in the human genome
has revealed that the miR-29b-1/miR-29a promoter region
contains two putative E-box sites (MYC-binding sites), a
Gli-binding site and four NF-KB-binding sites (18). Moreover, increased expression of
the MYC oncogene silences miR-29b-1/miR-29a
expression and NF-κB signaling, which is known to be activated in
inflammation-related cancers, and directly represses
miR-29b-1/miR-29a promoter activity (19). Thus, it will be necessary to
identify the transcription factors that contribute to the silencing
of the miR-29 family in cervical SCC. Although the
miR-29b-1/miR-29a and miR-29b-2/miR-29c formed
cluster miRNAs are located within the same chromosomal regions, and
share transcriptional units, the expression of miR-29b was
not reduced in cancer tissues compared to normal tissues in this
study. The reason for this is not yet clear, and further
elucidation of the molecular mechanisms controlling the expression
of miR-29 family miRNAs in cancer cells is necessary.
MiRNAs are unique in their ability to regulate many
protein coding genes. Bioinformatic predictions have indicated that
miRNAs regulate >30% of protein coding genes (20). Aberrant expression of miRNAs causes
destruction of tightly regulated miRNA/protein-coding RNA networks
in human cancer cells. Therefore, identification of aberrantly
expressed miRNA-mediated cancer pathways and target genes is the
first step in elucidating the role of miRNAs in human cancers.
According to gene expression data and in
silico database analysis, a total of 29 genes were selected as
candidate miR-29 a targets. Previous reports have indicated
that the miR-29 family plays a dominant role in the
regulation of extracellular matrix (ECM) genes. Indeed, luciferase
reporter assays demonstrated that miR-29a directly targeted
HSP47, a collagen-binding, heat-inducible glycoprotein. This
is the first report demonstrating that HSP47 was regulated
by tumor-suppressive miR-29a in cervical SCC.
HSP47, a 47-kDa heat-shock protein, was first
identified in fibroblasts (21)
and is located within the human chromosome 11ql3.5 region, which is
frequently amplified in several types of human cancers, including
cervical SCC (22). Moreover,
HSP47 is localized in the endoplasmic reticulum, a cellular
organelle involved in the intercellular processing and secreting of
procollagens (23). Many studies
have demonstrated that HSP47 is overexpressed in fibrotic diseases,
including kidney fibrosis, pulmonary fibrosis, cardiac fibrosis,
and liver cirrhosis (24).
Fibrosis is a common disease of organ dysfunction and is closely
associated with ECM proteins, such as collagens, actins and
fibronectins (25). Interestingly,
members of the miR-29 family have been shown to be involved
in regulating ECM proteins and multiple studies have indicated that
aberrant expression of miR-29 family members contributes
substantially to the development of disease (26). Thus, down-regulation of the
miR-29 family and dysregulation of HSP47 and ECM components
are key events contributing to the pathogenesis of diseases,
suggesting that these molecules are potential therapeutic
targets.
Overexpression of HSP47 has been reported in
pancreatic cancer, gastric cancer, and head and neck squamous cell
carcinoma (27–29). Our present data also support
previous reports, suggesting that silencing of miR-29a
caused overexpression of HSP47 and was an important event in
the pathogenesis of cervical SCC, contributing to cancer cell
migration and invasion in particular. The epithelial-to-mesenchymal
transition (EMT) is a fundamental biological process whereby
epithelial cells lose their polarity and undergo a transition to a
mesenchymal phenotype (30).
Initiation of the EMT requires external signals, such as epidermal
growth factor (EGF), fibroblast growth factor (FGF), hepatocyte
growth factor (HGF), and transforming growth factor-β (TGF-β)
(31). The TGF-β pathway is a
prominent inducer of the EMT and expression of the miR-29
family has been shown to have an inverse relationship with the
TGF-β pathway. Restoration of miR-29 family members directly
suppresses TGF-β1 and TGF-β2 and disrupts the expression of ECM
proteins (32). Furthermore, ECM
molecules, including collagen type I, promote the EMT through
integrin and discoidin domain receptor-1 signaling (33,34).
Thus, the understanding of molecular pathways and targets regulated
by the tumor-suppressive miR-29 family may provide new
insights into the EMT process in cervical SCC and facilitate the
development of more effective strategies for future therapeutic
interventions for this disease.
In conclusion, downregulation of miR-29 a is
a frequent event in cervical SCC. Moreover, tumor-suppressive
miR-29a directly regulates HSP47, a molecular
chaperone involved in the maturation of collagen molecules.
Restoration of miR-29a or silencing of HSP47
inhibited cancer cell migration and invasion, suggesting that the
miR-29a-HSP47 pathway contributes to the metastasis of
cervical SCC. Identification of molecular targets regulated by
tumor-suppressive miRNAs will provide insights into the potential
mechanisms of cervical SCC oncogenesis and metastasis, facilitating
the development of novel therapeutic strategies for the treatment
of this disease.
Acknowledgements
This study was supported by the
KAKENHI (C), 24592590.
References
|
1.
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2.
|
Walboomers JM, Jacobs MV, Manos MM, et al:
Human papillomavirus is a necessary cause of invasive cervical
cancer worldwide. J Pathol. 189:12–19. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Munoz N, Bosch FX, de Sanjose S, et al:
Epidemiologic classification of human papillomavirus types
associated with cervical cancer. N Engl J Med. 348:518–527. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Clifford GM, Smith JS, Plummer M, Munoz N
and Franceschi S: Human papillomavirus types in invasive cervical
cancer worldwide: a meta-analysis. Br J Cancer. 88:63–73. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Munger K and Howley PM: Human
papillomavirus immortalization and transformation functions. Virus
Res. 89:213–228. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Filipowicz W, Bhattacharyya SN and
Sonenberg N: Mechanisms of post-transcriptional regulation by
microRNAs: are the answers in sight? Nat Rev Genet. 9:102–114.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Nelson KM and Weiss GJ: MicroRNAs and
cancer: past, present, and potential future. Mol Cancer Ther.
7:3655–3660. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Kano M, Seki N, Kikkawa N, et al: miR-145,
miR-133a and miR-133b: Tumor-suppressive miRNAs target FSCN1 in
esophageal squamous cell carcinoma. Int J Cancer. 127:2804–2814.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Moriya Y, Nohata N, Kinoshita T, et al:
Tumor suppressive microRNA-133a regulates novel molecular networks
in lung squamous cell carcinoma. J Hum Genet. 57:38–45. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Hidaka H, Seki N, Yoshino H, et al: Tumor
suppressive microRNA-1285 regulates novel molecular targets:
aberrant expression and functional significance in renal cell
carcinoma. Oncotarget. 3:44–57. 2012.
|
|
11.
|
Ichimi T, Enokida H, Okuno Y, et al:
Identification of novel microRNA targets based on microRNA
signatures in bladder cancer. Int J Cancer. 125:345–352. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Kojima S, Chiyomaru T, Kawakami K, et al:
Tumour suppressors miR-1 and miR-133a target the oncogenic function
of purine nucleoside phosphorylase (PNP) in prostate cancer. Br J
Cancer. 106:405–413. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Kikkawa N, Hanazawa T, Fujimura L, et al:
miR-489 is a tumour-suppressive miRNA target PTPN11 in
hypopharyngeal squamous cell carcinoma (HSCC). Br J Cancer.
103:877–884. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Nohata N, Hanazawa T, Kikkawa N, et al:
Tumour suppressive microRNA-874 regulates novel cancer networks in
maxillary sinus squamous cell carcinoma. Br J Cancer. 105:833–841.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Yamamoto N, Kinoshita T, Nohata N, et al:
Tumor suppressive microRNA-218 inhibits cancer cell migration and
invasion by targeting focal adhesion pathways in cervical squamous
cell carcinoma. Int J Oncol. 42:1523–1532. 2013.PubMed/NCBI
|
|
16.
|
Wang Y, Zhang X, Li H, Yu J and Ren X: The
role of miRNA-29 family in cancer. Eur J Cell Biol. 92:123–128.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Li Y, Wang F, Xu J, et al: Progressive
miRNA expression profiles in cervical carcinogenesis and
identification of HPV-related target genes for miR-29. J Pathol.
224:484–495. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Mott JL, Kurita S, Cazanave SC, Bronk SF,
Werneburg NW and Fernandez-Zapico ME: Transcriptional suppression
of miR-29b-l/miR-29a promoter by c-Myc, hedgehog, and NF-kappaB. J
Cell Biochem. 110:1155–1164. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Wang H, Garzon R, Sun H, et al:
NF-kappaB-YYl-miR-29 regulatory circuitry in skeletal myogenesis
and rhabdomyosarcoma. Cancer Cell. 14:369–381. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Nagata K, Saga S and Yamada KM:
Characterization of a novel transformation-sensitive heat-shock
protein (HSP47) that binds to collagen. Biochem Biophys Res Commun.
153:428–434. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Nagai N, Tetuya Y, Hosokawa N and Nagata
K: The human genome has only one functional hsp47 gene (CBP2) and a
pseudogene (pshsp47). Gene. 227:241–248. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Ishida Y and Nagata K: Hsp47 as a
collagen-specific molecular chaperone. Methods Enzymol.
499:167–182. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Sauk JJ, Nikitakis N and Siavash H: Hsp47
a novel collagen binding serpin chaperone, autoantigen and
therapeutic target. Front Biosci. 10:107–118. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Hubmacher D and Apte SS: The biology of
the extracellular matrix: novel insights. Curr Opin Rheumatol.
25:65–70. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Kriegel AJ, Liu Y, Fang Y, Ding X and
Liang M: The miR-29 family: genomics, cell biology, and relevance
to renal and cardiovascular injury. Physiol Genomics. 44:237–244.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Maitra A, Iacobuzio-Donahue C, Rahman A,
et al: Immunohistochemical validation of a novel epithelial and a
novel stromal marker of pancreatic ductal adenocarcinoma identified
by global expression microarrays: sea urchin fascin homolog and
heat shock protein 47. Am J Clin Pathol. 118:52–59. 2002.
View Article : Google Scholar
|
|
28.
|
Hirai K, Kikuchi S, Kurita A, et al:
Immunohistochemical distribution of heat shock protein 47 (HSP47)
in scirrhous carcinoma of the stomach. Anticancer Res. 26:71–78.
2006.PubMed/NCBI
|
|
29.
|
Lee SS, Tseng LH, Li YC, Tsai CH and Chang
YC: Heat shock protein 47 expression in oral squamous cell
carcinomas and upregulated by arecoline in human oral epithelial
cells. J Oral Pathol Med. 40:390–396. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Jing Y, Han Z, Zhang S, Liu Y and Wei L:
Epithelial-mesenchymal transition in tumor microenvironment. Cell
Biosci. 1:292011. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013.PubMed/NCBI
|
|
32.
|
Gebeshuber CA, Zatloukal K and Martinez J:
miR-29a suppresses tristetraprolin, which is a regulator of
epithelial polarity and metastasis. EMBO Rep. 10:400–405. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Shintani Y, Fukumoto Y, Chaika N, Svoboda
R, Wheelock MJ and Johnson KR: Collagen I-mediated up-regulation of
N-cadherin requires cooperative signals from integrins and
discoidin domain receptor 1. J Cell Biol. 180:1277–1289. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Shintani Y, Maeda M, Chaika N, Johnson KR
and Wheelock MJ: Collagen I promotes epithelial-to-mesenchymal
transition in lung cancer cells via transforming growth factor-beta
signaling. Am J Respir Cell Mol Biol. 38:95–104. 2008. View Article : Google Scholar : PubMed/NCBI
|