Introduction
Breast cancer is a sex steroid hormone-responsive
tumor; therefore sex steroid hormones and their receptors play a
pivotal role in the cell growth and progression of this type of
tumor. Since the 1950’s, there has been an enormous number of
reports on estrogen, estrogen receptor (ER), ER blockade,
aromatization inhibition and related topics that has been
accompanied by great progress in mainstream clinical management of
breast cancer (1). However,
androgen and androgen receptor (AR) have been clinically and
experimentally neglected and so less information is currently known
about the role of androgens and AR in breast cancer. To date
emerging evidence indicates androgen and AR have a complex and
significant function in breast cancer cell growth and tumor
progression. Studies have shown that the androgen signaling pathway
exerts inhibitory effects on the growth of normal mammary
epithelial cells and plays a protective role in the pathogenesis of
breast cancer (2–5). Emerging evidence indicates that AR is
expressed in breast cancer and may serve as a good prognostic
factor (6–10). Because AR expression has also been
reported in roughly 50% of patients with estrogen receptor-negative
and progesterone receptors-negative (ER−,
PR−) breast cancer (11,12),
identifying the underlying mechanisms of androgen and AR is
important in the design of appropriate therapies for
estrogen-insensitive neoplasms. Our previous study (13) showed that among 327 female Chinese
cases with invasive ductal breast carcinoma, 72.5% also had
detectable AR expression. AR was found in 53.2% of the
ER−, PR− breast cancers in this group.
Survival analysis suggested that the patients whose tumors
expressed AR had a more favorable prognosis than those whose tumors
did not. Similar to our results another study (14) showed that in a population-based
study, 77% of invasive breast cancers were AR-positive
(AR+) and AR expression was frequent even in molecular
subtypes of invasive cancer that are ER−. Further
exploration of the role of androgens and AR in breast cancer is
essential and will contribute to the development of new therapies
for AR+ tumors, especially ER−,
PR−, AR+ breast cancer patients who see
little or no benefit from anti-estrogen therapy.
Androgens act on target cells by binding to the
cognate receptor AR, a member of the nuclear receptor superfamily.
AR is a ligand-dependent transcription factor. Once bound to
androgen AR is activated. Activated AR can recognize the androgen
response elements (AREs) located at (or close to) the promoter and
enhancer region of androgen-dependent genes and then activate the
transcription machinery, including that for microRNA (miRNA)
transcription (15–17). Recently, several studies have been
published that suggest a role for the AR in the transcriptional
regulation of miRNA expression (18–20).
MiRNAs are evolutionally conserved approximately 22
nucleotide-long short non-coding RNA molecules that repress target
gene expression by binding to complementary sequences found in the
3′-untranslated region (UTRs) of target mRNAs. They participate in
diverse biological functions including development, cell
proliferation, differentiation, and apoptosis (21–24).
Accumulating evidence indicates that miRNA alterations are present
in various types of human cancer, including breast cancer, and play
a crucial role in the initiation and progression of human cancer
through their function as tumor suppressors or oncogenes (25).
The complex relationships among androgens, AR,
miRNAs and target mRNAs can be summed up as follows: androgens
activate AR by binding to AR and then activated AR can control
related miRNA transcription. Processed miRNA can suppress target
mRNA expression. In this study, we focused on a representative
miRNA in AR activated cells to explore the relationship among AR,
this miRNA and its targets in ER−, PR−,
AR+ breast cancer. To date, this type of question has
not been explored. The results presented here will contribute to
the understanding of ER−, PR−, AR+
breast cancer pathogenesis and help design new therapies for
estrogen-insensitive neoplasms.
Materials and methods
Cell culture and treatment
MCF-7, MDA-MB-453 and MDA-MB-231 human breast cancer
cell lines were chosen because MCF-7 cell line expresses high
levels of ER, PR and AR whereas MDA-MB-453 and MDA-MB-231 cell
lines express high levels of AR in the absence of ER and PR
(26–28). 5α-dihydrotestosterone (DHT,
Sigma-Aldrich, MO, USA) was used as it is a non-aromatisable
androgen and possesses the highest affinity for AR among natural
androgens. All cells were obtained from the American Type Culture
Collection (ATCC) and were maintained at 37°C in a humidified
atmosphere containing 5% CO2 in 75 cm2 flasks
containing minimal essential medium (MEM) supplemented with 10%
fetal bovine serum, 100 IU/ml penicillin and 100 mg/ml
streptomycin. Cells were passaged every 3–4 days when they reached
80% confluence and were harvested with 0.25% trypsin/EDTA. Before
each experiment, cells were grown in phenol red-free (PRF) DMEM,
containing 5% charcoal-treated fetal calf serum (PRF-CT), for 3
days and then serum starved in PRF DMEM for 24 h to synchronize the
cells. All experiments were performed in 2.5% PRF-CT. Cells were
treated with either DHT or vehicle alone at 10−8 M. DHT
was dissolved in 100% ethanol and added to media immediately prior
to use.
MiRNA microarray analysis
The MCF-7 and MDA-MB-453 cell samples were analyzed
by KangChen (KangChen Bio-tech Inc.) in miRNA microarray
experiments. Total RNA was extracted from MCF-7 and MDA-MB-453
cells that were treated with DHT or vehicle alone using TRIzol
reagent (Invitrogen, CA, USA) according to the manufacturer’s
instructions. RNA quantification and quality assurance were
assessed by NanoDrop ND-1000 and RNA integrity and DNA
contamination were assessed by denaturing agarose gel
electrophoresis. The total RNA was labeled with Cy3 or Cy5
fluorescent dyes by miRCURY™ Array Power Labeling kit (Cat
#208032-A, Exiqon) according to the manufacturer’s protocols. Each
miRCURY LNA microRNA array (v.11.0, Exiqon, Vedbaek, Denmark) was
hybridized with a single sample labeled with either Cy3 or Cy5.
Gene Pix 4000B scanner and GenePix Pro 6.0 software (Axon
Instruments, Union City, CA, USA) were used to scan images. Each
group was hybridized with three miRCURY LNA Arrays in triplicate
with independent samples for DHT-treated cells or vehicle-treated
cells. Background subtraction and normalization were performed. We
selected miRNAs whose expression intensities
(ForeGround:BackGround) reached at least 1000 and expression levels
differed by at least 2-fold between DHT-treated cells and
vehicle-treated cells.
Real-time reverse transcription PCR
(real-time RT-PCR)
ER−, PR−, AR+
MDA-MB-453 and MDA-MB-231 cells were analyzed. To detect the mature
miRNA let-7a level, a stem-loop RT-PCR assay was performed
(29). Briefly, 2 μg of
small RNA was reverse-transcribed to cDNA using M-MLV reverse
transcriptase (Promega) with the following primers: let-7a-RT,
5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAACTA-3′; and U6-RT,
5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAAAATAT GGAAC-3′,
which can fold to a stem-loop structure. The specific let-7a cDNA
fragment was amplified along with an endogenous control U6 snRNA
using the following primers: let-7a-Fwd,
5′-GCCGCTGAGGTAGTAGGTTGTA-3′; U6-Fwd, 5′-TGCGGGTGCTCGCTTCGGCAGC-3′,
and a universal downstream primer reverse,
5′-CCAGTGCAGGGTCCGAGGT-3′. PCR cycles were as follows: 94°C for 4
min, followed by 40 cycles of 94°C for 30 sec, 50°C for 30 sec,
72°C for 40 sec. SYBR Premix Ex Taq™ Kit (Takara) was used
following the manufacturer’s instructions, and the RT-PCR was
performed and analyzed by 7300 RT-PCR system (ABI). All primers
were synthesized by BGI Inc.
Chromatin immunoprecipitation (ChIP)
assay
ChIP assays were performed using MDA-MB-453 cells to
determine if activated AR directly upregulates the expression of
let-7a (30). Briefly, chromatin
DNA was extracted from harvested cells by sonication and precleaned
by incubating with normal rabbit serum (IgG) and protein
A/G-Sepharose beads. It was then immunoprecipitated with rabbit
anti-AR antibody (PG-21, Upstate). The following primers were used:
5′-CAAAGTTTCTAAACGGCTTC (forward) and 5′-AGGATATTTGGTACACTCTG
(reverse) for amplifying the -3922/-3707 fragment and
5′-TTTTACATTGGGCATAGCCG (forward) and 5′-TAGGCATTTGGAAGTTGGAC
(reverse) for the -3036/-2786 fragment.
Vector construction
To construct the pcDNA3.1/pri-let-7a expression
vector, we first amplified a 203-bp DNA fragment carrying
pri-let-7a from genomic DNA using the following PCR primers:
let-7a-sense, 5′-CGCGGATCCACTGTGGGATGAGGTAGTAGGT-3′ and
let-7a-antisense, 5′-CGCGAATTCTCCAGGCCATAAACAAATGC-3′. The
amplified fragment was inserted into the pcDNA3.1 (+) vector at the
BamHI and EcoRI sites.
Transfection
The vehicle-treated MDA-MB-453 and MDAMB-231 cells
were transfected with vectors and the DHT-treated MDA-MB-453 and
MDA-MB-231 cells were transfected with antisense oligonucleotide
(ASO) using Lipofectamine 2000 (Invitrogen) at 24 h after plating.
Transfection complexes were prepared according to the
manufacturer’s protocols. The final oligonucleotide concentration
was 10−9 M, and the final vector concentration was 0.5
mg/l. The transfection medium was replaced at 4 h
post-transfection. The oligonucleotides complementary to let-7a
were synthesized by IDT (Coralville, IA, USA) and their sequences
were as follows: let-7a ASO, 5′-AACTATACAACCTACTACCTCA-3′; and
control ASO, 5′-GTGGATATTGTTGCCATCA-3′.
Cell proliferation assay
Charcoal-stripped MDA-MB-453 and MDA-MB-231 cells
were plated in replicates of 6 at a density of 7000 cells/well in
96-well microtiter plates. At 24 h after seeding, cells were
treated with DHT or vehicle alone at 10−8 M and reagents
were replenished every 3 days. Cell viability and proliferation
were measured using the 3-(4,5 dimethylthiazol-2-yl)-2,5-diphenyl
tetrazolium bromide (MTT) colorimetric assay. At days 1–7, the
cells exposed to DHT or vehicle were added to MTT. Four hours
later, dimethyl sulfoxide (DMSO) was added to each well to dissolve
the resulting formazan crystals. After shaking for 20 min, the
absorbance at 570 nm was detected using a μQuant Universal
Microplate Spectrophotometer (Bio-Tek Instruments, Winooski, VT,
USA). MTT assay was also used to measure the viable proliferating
cells at days 1–7 after the MDA-MB-453 and MDA-MB-231 cells were
transfected.
Flow cytometry analysis
After 48 h, the charcoal-stripped MDA-MB-453 and
MDA-MB-231 cells exposed to DHT or vehicle were detached from the
plates using trypsin, rinsed with PBS and fixed in 70% (v/v)
ethanol. The cells were then rehydrated in PBS and incubated with
RNase (100 μg/ml) and propidium iodide (60 μg/ml)
(Sigma-Aldrich, MO, USA). Cells were analyzed using the FACS
Calibur System (BD Biosciences, San Jose, CA, USA), and the cell
cycle phase was determined by Cell Quest analysis. Proliferation
index (PI) was calculated as follows: PI = (S+G2/M)/G1 (S, G2/M and
G1 refer to the percentage of cells in S phase, G2/M phase and G1
phase, respectively) (31). Flow
cytometry analysis was also used to measure cell cycle phase at 48
h after the MDA-MB-453 and MDA-MB-231 cells were transfected.
Western blot analysis
After 48 h the charcoal-stripped MDAMB-453 and
MDA-MB-231 cells exposed to DHT or vehicle were lysed with RIPA
lysis buffer and proteins were harvested. All proteins were
resolved on 10% SDS denaturing polyacrylamide gel and then
transferred onto a nitrocellulose membrane. Membranes were
incubated with anti-KRAS, anti-CMYC or anti-GAPDH antibody (Saier
Biotech, Tianjin, P.R. China) with blotto overnight at 4°C. The
membranes were washed and incubated with horseradish peroxidase
(HRP) conjugated secondary antibody (Saier Biotech). Protein
expression was assessed by enhanced chemiluminescence and exposure
to chemiluminescent film. Lab Works™ Image Acquisition and Analysis
Software (UVP) were used to quantify band intensities.
Patients and tissue samples
Formalin-fixed and paraffin-embedded (FFPE) tissue
specimens were obtained from 24 female patients (mean age: 54.5
years, range from 41 to 70 years) who underwent surgical resection
for breast cancer from 2003 to 2004 at Tianjin Medical University
Cancer Institute and Hospital. All cases were ER− and
PR− invasive ductal carcinoma (IDC) and all the patients
had been treated according to modern guidelines, including the use
of adjuvant chemotherapy for IDC and irradiation for lymph node
metastasis. The study protocol was approved by the Hospital Human
Ethics Committee. Informed consent was obtained from all patients
before their surgery and the examination of the specimens.
In situ hybridization (ISH)
Locked nucleic acid (LNA) probes complementary to
mature let-7a (5′-AACTATACAACCTACTACCTCA-3′) and scrambled negative
control (5′-TTCACAATGCGTTATCGGATGT-3′) digoxigenin-labeled at the
5′-position were purchased from Exiqon. Detection of RNAs by ISH
utilizing oligonucleotide probes was performed. Briefly, human
tissues were deparaffinized, treated with protease (30 min in 2
mg/ml of pepsin), washed in sterile water, and then washed with
100% ethanol and air-dried. Hybridization was performed at 37°C
overnight followed by a low stringency wash in 0.2X SSC and 2%
bovine serum albumin at 4°C for 10 min. The probe-target complex
was visualized utilizing a digoxigenin antibody conjugated to
alkaline phosphatase acting on the chromogen nitro blue
tetrazolium/5-bromo-4-chloro-3-indolyl phosphate. Nuclear Fast Red
served as the counterstain. The slides were scored independently by
two pathologists and cases were considered positive if 10%
cytoplasmic and/or membranous staining was observed.
Immunohistochemical (IHC) staining
Deparaffinization; endogenous peroxidase
inactivation; antigen retrieval of FFPE clinical tissues; and
immunostaining with anti-ER (SP1, 1:200 dilution; ZETA), anti-PR
(SP2, 1:200 dilution; ZETA), anti-AR (AR441, 1:100 dilution;
LabVision), anti-CMYC (SRP00871, 1:200 dilution; Saierbio) and
anti-KRAS (SRP01436, 1:200 dilution; Saierbio) antibody were
performed as described previously (13). The immunostained sections were
evaluated independently by two pathologists in conjunction with the
H&E-stained sections from the same lesions. For each antibody,
the location of immunoreactivity, percentage of stained cells, and
intensity were determined. The evaluation of expression of each
protein was determined using the mean of the individual cases. AR,
ER, and PR stains were assessed using Allred scores (32). CMYC and KRAS stains were considered
positive if 10% of cells showed cytoplasmic and/or membranous
staining.
Statistical analysis
The data are reported as the mean ± SD of the values
from three independent determinations. Statistical analysis was
performed using Student’s t-test in comparison with corresponding
controls. Associations between ordinal variables were quantified by
Spearman rank correlation with Pearson’s χ2 test.
Probability values of <0.05 were considered statistically
significant. Analyses were run using SPSS17.0.
Results
Profile of miRNAs in DHT-treated and
vehicle-treated MCF-7 and MDA-MB-453 cells
To identify critical miRNAs related to
androgen-induced AR activating signal in breast cancer, we examined
global miRNA expression in DHT-treated and vehicle-treated MCF-7
and MDA-MB-453 cells using the microRNA array (v.11.0, Exiqon) that
consists of 847 capture probes for mature human miRNAs. In
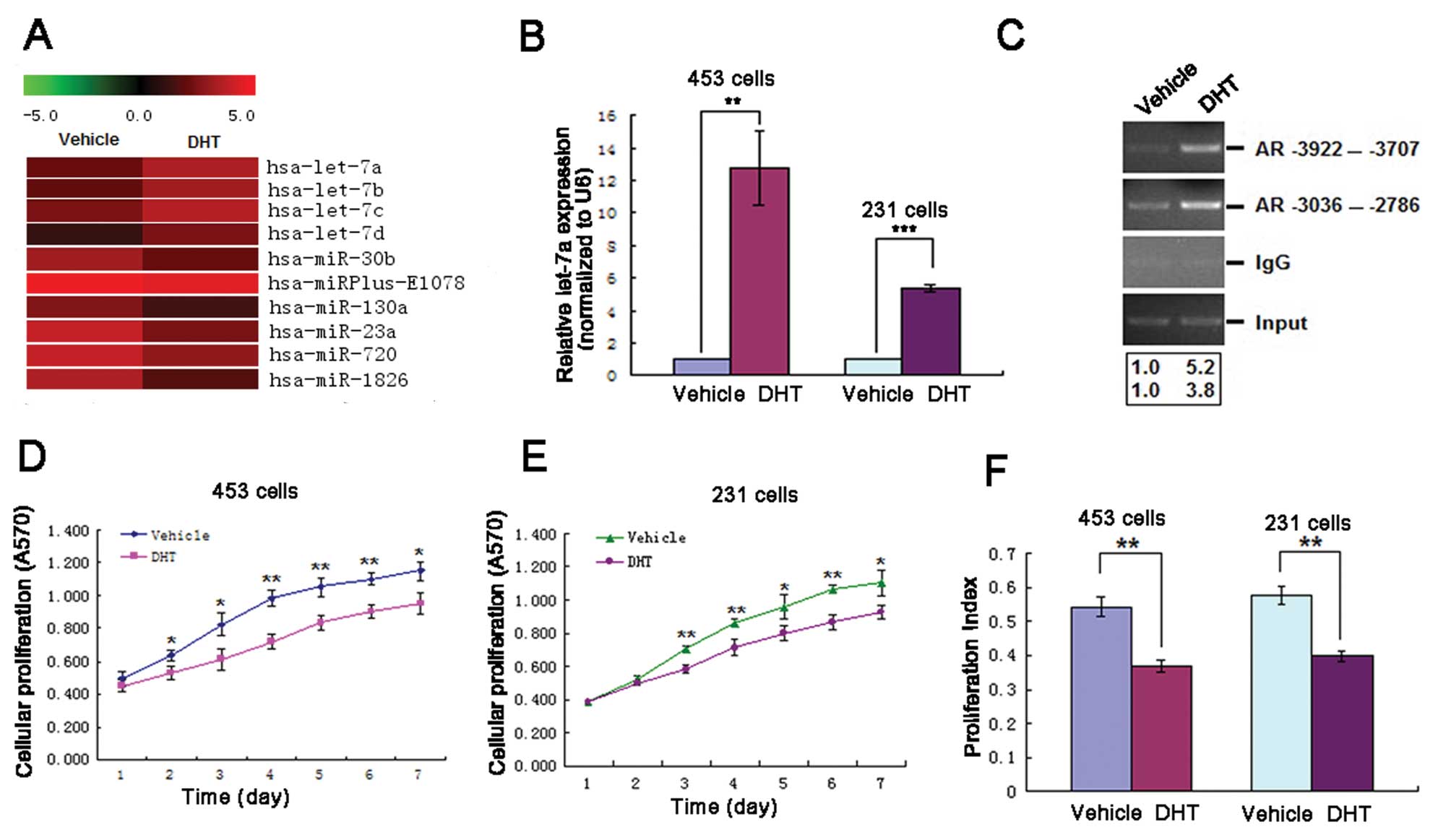
MDA-MB-453 cells a total number of 10 miRNAs were identified
(Fig. 1A) and in MCF-7 cells none
was identified using strict criteria that only miRNA undergoing
alterations at least 2-fold with expression intensities of at least
1000 were considered as differentially-expressed candidates. Among
these differentially-expressed miRNAs in MDA-MB-453 cells, 4
upregulated miRNAs - let-7a, b, c, d were found. Six downregulated
miRNAs were identified in the DHT-treated cells when compared with
the vehicle-treated cells (Table
I). Among them, let-7, as a tumor suppressor miRNA, is reported
to be downregulated in many types of solid tumors, including breast
cancer (33–35). In our experiments, only let-7a, b,
c, d were upregulated in MDA-MB-453 cells. This finding raises the
possibility that these upregulated let-7a, b, c, d miRNAs might
contribute to the pathogenesis of ER−, PR−,
AR+ breast cancer. We focused on let-7a and investigated
its involvement in ER−, PR−, AR+
breast cancer.
 | Table I.Differentially-expressed miRNAs in
MDA-MB-453 cells with at least 2-fold change and >1000
expression intensities. |
Table I.
Differentially-expressed miRNAs in
MDA-MB-453 cells with at least 2-fold change and >1000
expression intensities.
| miRNAa | Fold change | Expression
intensities (ForeGround-BackGround)
|
|---|
| DHTb-treated group | Vehicle-treated
group |
|---|
| hsa-let-7a | 2.505687288 | 2161 | 909.5 |
| hsa-let-7b | 2.297498628 | 1793 | 823 |
| hsa-let-7c | 2.168932179 | 2394 | 1164 |
| hsa-let-7d | 2.488421574 | 1105.5 | 468.5 |
| hsa-miR-30b | 0.457648129 | 1919.5 | 833 |
|
hsa-miRPlus-E1078 | 0.498143424 | 9110.5 | 4303.5 |
| hsa-miR-130a | 0.452923663 | 1248 | 536 |
| hsa-miR-29a | 0.503380318 | 3584.5 | 1711 |
| hsa-miR-23a | 0.36632922 | 3074.5 | 1068 |
| hsa-miR-720 | 0.4905976 | 3047 | 1417.5 |
| hsa-miR-1826 | 0.312695862 | 2281.5 | 676.5 |
DHT upregulates let-7a inhibiting cell
proliferation
In ER−, PR−, AR+
MDA-MB-453 and MDA-MB-231 cells the validity of let-7a ectopic
expression was confirmed by real-time RT-PCR, which revealed a
13-fold increase in let-7a expression in DHT-treated MDA-MB-453 and
a 5-fold increase in MDA-MB-231 cells over the vehicle-treated
groups (Fig. 1B). To determine if
the androgen-induced AR activating signal binds to the 5′-DNA
region of the let-7a locus to serve as a transcription factor, we
analyzed 4.0 kb in the 5′-region of the let-7a gene and identified
a typical TATA box. Using the PROMO 3.0 program 7 potential AREs
were identified in the 5′-region of let-7a. To determine AR
binding, ChIP was performed, and two primer pairs were used to
amplify the -3922/-3707 fragment (corresponding to ARE1) and the
-3036/-2786 fragment (corresponding to ARE3). Treatment of
MDA-MB-453 cells with 10−8 M DHT induced a 5.2-fold
increase in AR binding at ARE1 and a 3.8-fold increase at ARE3
(Fig. 1C). Taken together, the
results suggest that androgen-AR signaling directly mediates
regulation of let-7a. The effects of DHT on MDA-MB-453 and
MDA-MB-231 cell proliferation were measured by MTT assay using
cells exposed to 10−8 M DHT or vehicle for 1–7 days. DHT
treatment decreased cell proliferation when compared with vehicle
treatment (Fig. 1D and E). Flow
cytometric analysis was used to determine the cell cycle
distribution of MDA-MB-453 and MDA-MB-231 cells exposed to DHT or
vehicle for 48 h. The number of cells in the G1 phase was
significantly increased in both types of cells; G1-S arrest was
obvious and PI was decreased in the DHT-treated group compared with
the vehicle-treated group (Fig.
1F).
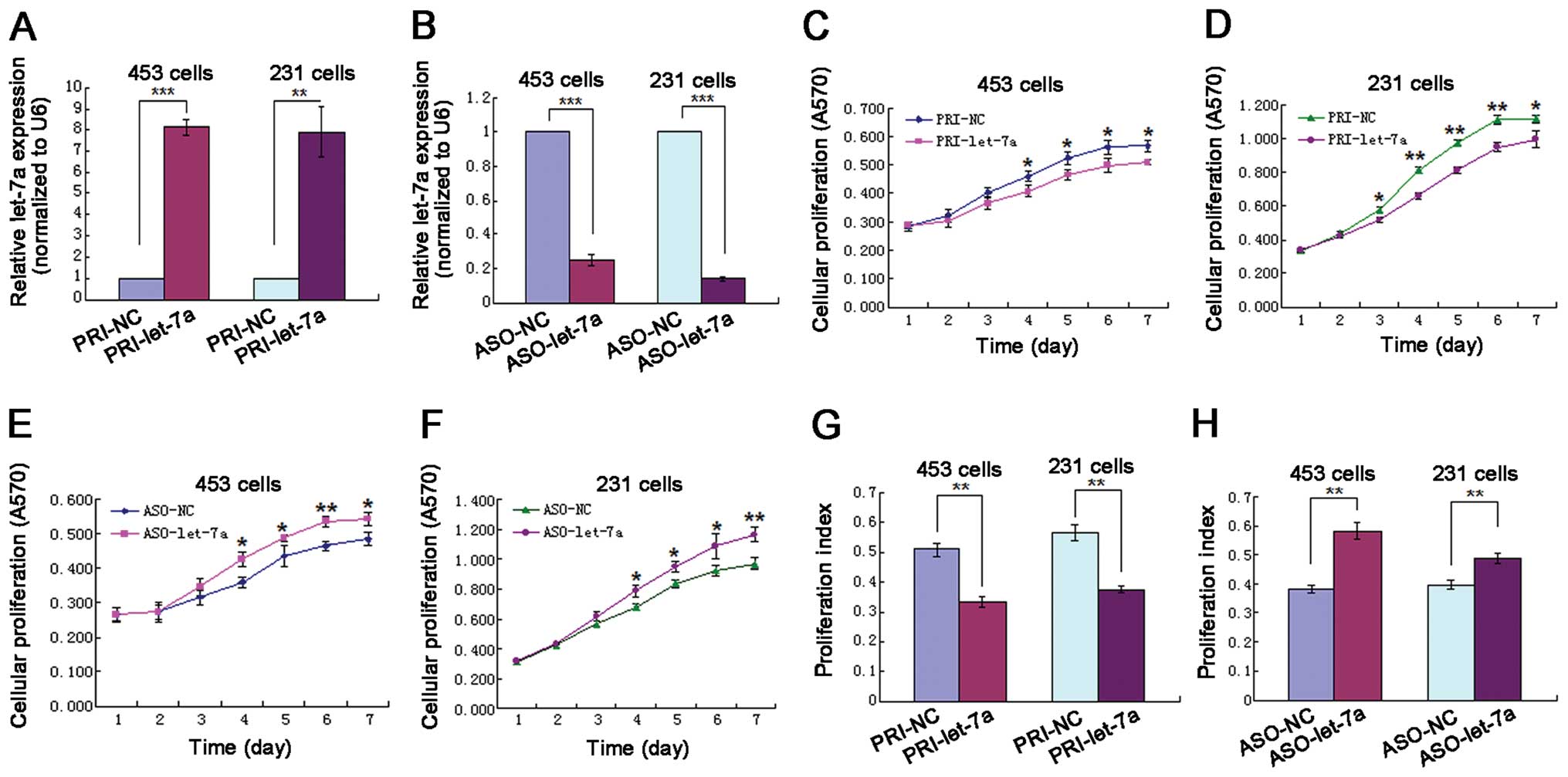
The effect of let-7a on the growth of
ER−, PR−, AR+ breast cancer
cells
In order to investigate the biological significance
of let-7a in MDA-MB-453 and MDA-MB-231 cells, we used a precursor
expression vector (pcDNA3.1/pri-let-7a) to enhance or let-7a ASO to
inhibit mature let-7a activity in vehicle-treated or DHT-treated
cells. Real-time RT-PCR was used to validate the alteration in
let-7a expression level. The let-7a level in the vector-treated
group was significantly increased compared with the control group
(Fig. 2A), whereas the let-7a
level in the let-7a ASO-treated group was significantly decreased
compared with the control group (Fig.
2B). These results suggest that the expression of let-7a was
successfully altered as designed in our experiments. MTT assay was
used to measure the viable proliferating cells at days 1–7 after
the MDA-MB-453 and MDA-MB-231 cells were transfected. The MTT data
suggests that when let-7a was overexpressed with vector, cell
growth was inhibited, an effect similar to that observed with
proliferative inhibition by DHT exposure (Fig. 2C and D). When let-7a was blocked
with let-7a ASO cell growth activity was elevated, an effect
directly opposite to that observed with let-7a overexpression
(Fig. 2E and F). Flow cytometry
analysis was used to assess the distribution of cells in the cell
cycle at 48 h after the MDA-MB-453 and MDA-MB-231 cells were
transfected. The data showed that when let-7a was overexpressed the
number of cells in the G1 phase was significantly increased, G1-S
arrest was obvious and PI was decreased (Fig. 2G), a pattern similar to that seen
in cells exposed to DHT. When let-7a was blocked the number of
cells in the S phase significantly increased and PI was increased,
an effect directly opposite to that observed in cells with let-7a
overexpressed (Fig. 2H).
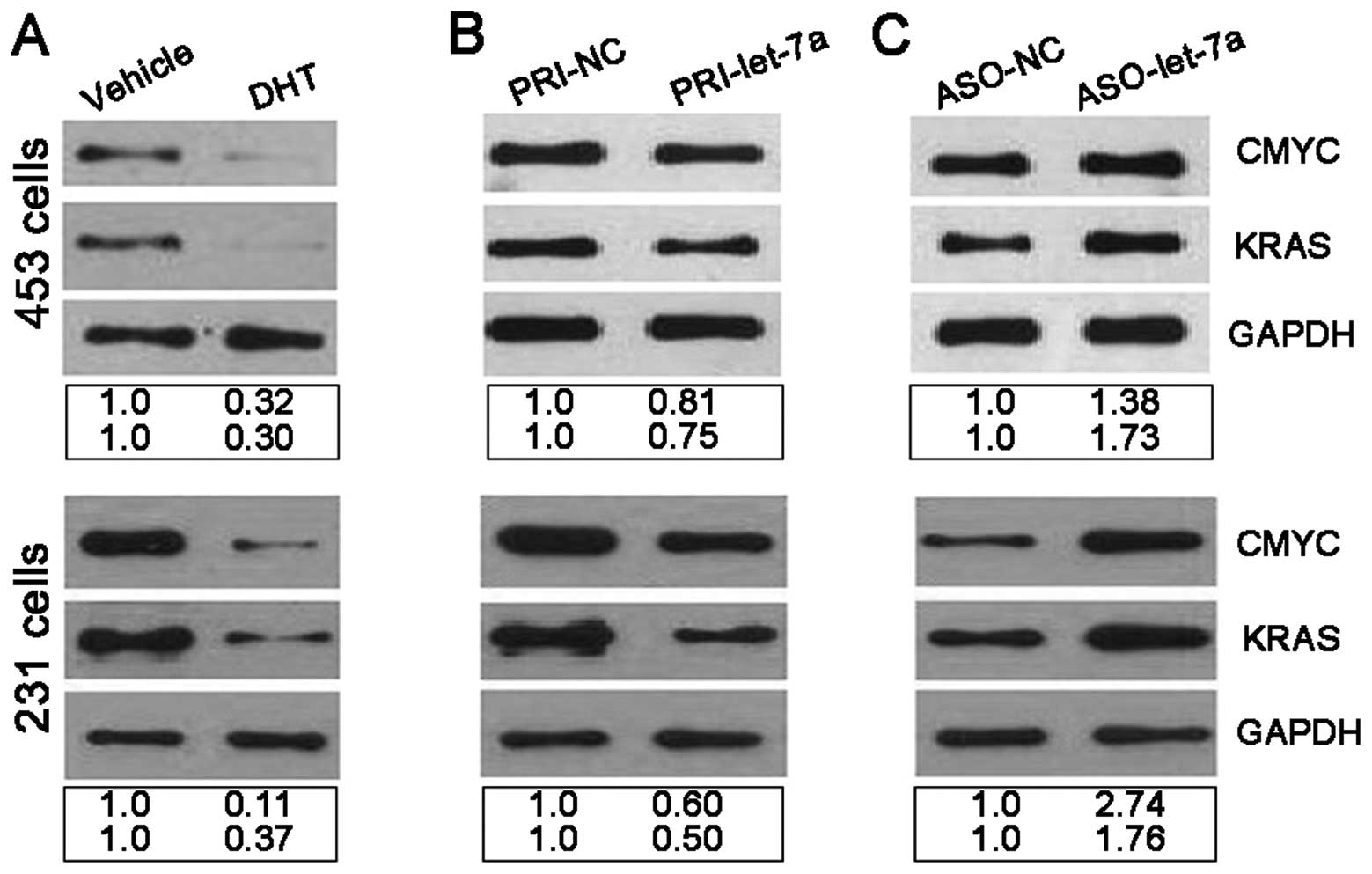
Let-7a negatively regulates CMYC and KRAS
at the post-transcriptional level
Translational repression is a major mechanism of
miRNA regulation of target gene expression (36). CMYC and KRAS, two oncogenes which
are involved in cell proliferation and cell cycle, are known target
genes of let-7a (37–40). To confirm the suppressing action of
let-7a on CMYC and KRAS the correlation between miRNA and target
protein expression was determined. The expression of CMYC and KRAS
protein was examined by western blot analysis using MDA-MB-453 and
MDA-MB-231 cells that were exposed to 10−8 M DHT or
vehicle for 48 h when let-7a was upregulated. CMYC and KRAS protein
were underexpressed when cells were exposed to 10−8 M
DHT (Fig. 3A). Next
vehicle-treated MDA-MB-453 and MDA-MB-231 cells were transfected
with vector and the expression of CMYC and KRAS protein was
examined by western blot analysis. CMYC and KRAS proteins were
underexpressed after let-7a was upregulated by vector (Fig. 3B). DHT-treated MDA-MB-453 and
MDA-MB-231 cells were then transfected with let-7a ASO and the
expression of CMYC and KRAS protein was examined by western blot
analysis. The amount of CMYC and KRAS protein was increased after
let-7a expression was blocked (Fig.
3C). In conclusion, the above data suggest that let-7a
negatively regulates endogenous CMYC and KRAS protein expression
through a translational repression mechanism, similar to previous
reports (37–40).
The relationship among let-7a, AR, CMYC
and KRAS in FFPE breast cancer tissue specimens
In FFPE breast cancer tissue specimens the staining
outcome of ISH for let-7a and IHC staining for AR, CMYC and KRAS
(Fig. 4; Table II) showed that let-7a expression
was negatively correlated with CMYC and KRAS expression. CMYC
expression was positively correlated with KRAS expression. No clear
correlation was observed between the staining pattern of AR and
let-7a, CMYC or KRAS (Table
III).
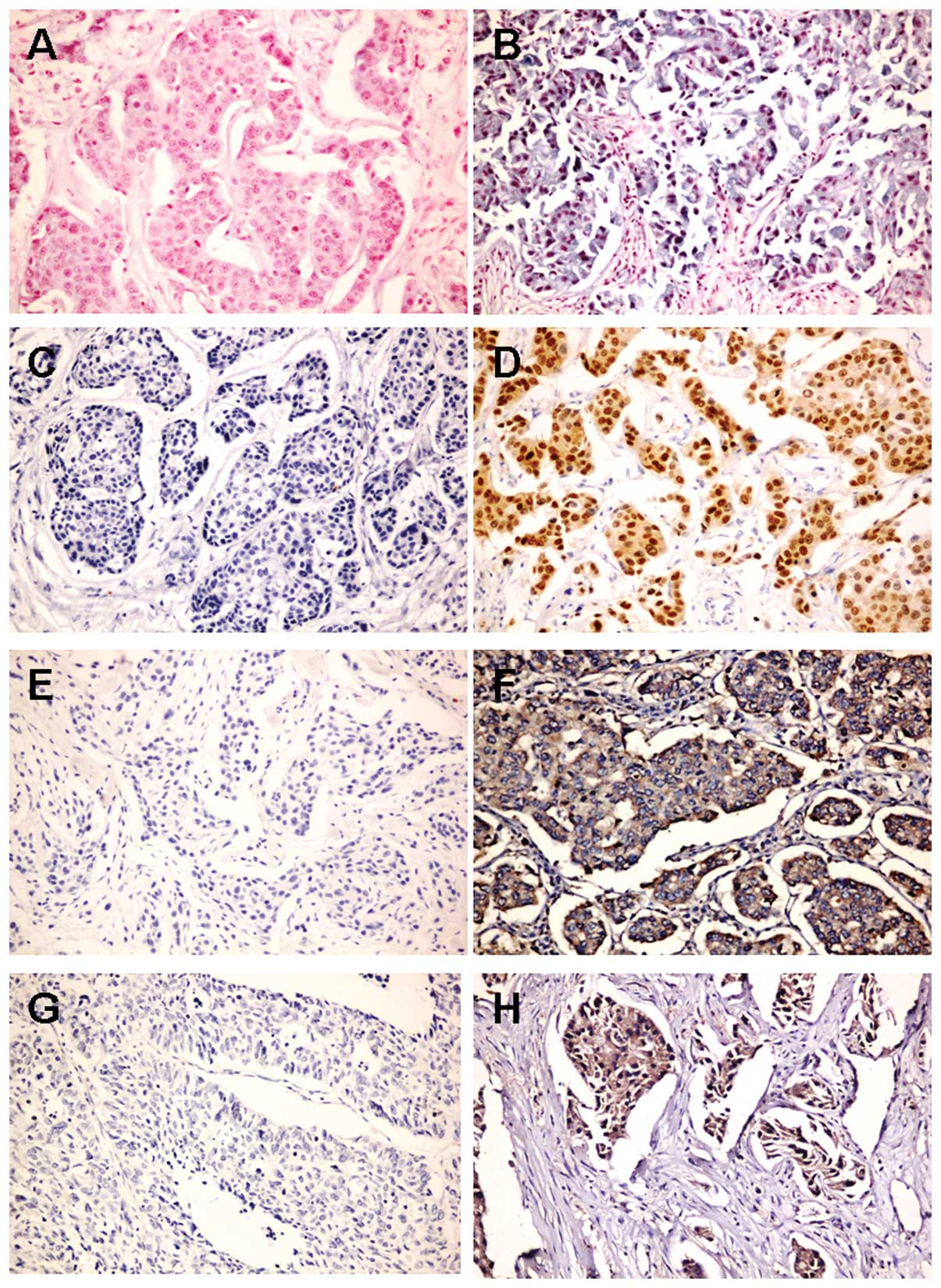 | Figure 4.Expression of let-7a by ISH and
expression of AR, CMYC and KRAS by IHC in ER−,
PR−, AR+ IDC. A, Negative expression of
let-7a (×200). B, Positive expression of let-7a (×200). C, Negative
expression of AR (×200). D, Positive expression of AR (×200). E,
Negative expression of CMYC (×200). F, Positive expression of CMYC
(×200). G, Negative expression of KRAS (×200). H, Positive
expression of KRAS (×200). |
| Table II.The results of the analysis of ISH
and IHC in 24 IDC cases with ER− and PR−. |
Table II.
The results of the analysis of ISH
and IHC in 24 IDC cases with ER− and PR−.
| Case | Age | Grade | Let-7a | AR | CMYC | KRAS |
|---|
| 1 | 43 | 2 | + | − | + | + |
| 2 | 56 | 2–3 | − | − | + | + |
| 3 | 54 | 2 | − | − | + | + |
| 4 | 51 | 2 | + | − | + | + |
| 5 | 57 | 2 | − | − | + | + |
| 6 | 50 | 2 | + | + | − | + |
| 7 | 43 | 2 | + | − | + | + |
| 8 | 63 | 1–2 | + | + | + | − |
| 9 | 62 | 2–3 | + | + | + | − |
| 10 | 41 | 3 | − | − | + | + |
| 11 | 61 | 2 | − | − | + | + |
| 12 | 44 | 3 | + | − | − | − |
| 13 | 58 | 3 | + | − | − | + |
| 14 | 70 | 3 | + | − | + | + |
| 15 | 51 | 2 | − | − | + | + |
| 16 | 46 | 2 | − | + | + | + |
| 17 | 54 | 1–2 | + | + | − | − |
| 18 | 55 | 2 | − | + | + | + |
| 19 | 48 | 3 | − | − | + | + |
| 20 | 60 | 2 | − | − | + | + |
| 21 | 42 | 1–2 | + | − | + | − |
| 22 | 67 | 1–2 | − | + | + | + |
| 23 | 59 | 2 | + | + | − | − |
| 24 | 56 | 2–3 | + | + | + | + |
| Table III.Correlations between the expression
of let-7a, AR, CMYC, KRAS, and two clinicopathological data,
expressed as Spearman’s ρ with Pearson’s χ2 test for
significance. |
Table III.
Correlations between the expression
of let-7a, AR, CMYC, KRAS, and two clinicopathological data,
expressed as Spearman’s ρ with Pearson’s χ2 test for
significance.
| Age | Grade | Let-7a | AR | CMYC | KRAS |
|---|
| Age | ρ=1.00 | | | | | |
| Grade | ρ=−0.068 | ρ=1.00 | | | | |
| P=0.754 | | | | | |
| Let-7a | ρ=−0.036 | ρ=−0.007 | ρ=1.00 | | | |
| P=0.866 | P=0.976 | | | | |
| AR | ρ=0.305 | ρ=−0.362 | ρ=0.194 | ρ=1.00 | | |
| P=0.147 | P=0.082 | P=0.363 | | | |
| CMYC | ρ=0.045 | ρ=−0.080 | ρ=−0.472a | ρ=−0.238 | ρ=1.00 | |
| P=0.836 | P=0.711 | P=0.020 | P=0.262 | | |
| KRAS | ρ=−0.063 | ρ=0.255 | ρ=−0.531b | ρ=−0.348 | ρ=0.415a | ρ=1.00 |
| P=0.771 | P=0.230 | P=0.008 | P=0.096 | P=0.044 | |
Discussion
Breast cancer is an extraordinarily
hormone-dependent tumor. The role of ER and PR are important in
regulating cell proliferation and differentiation. Anti-estrogen
therapy has therefore been successfully used in treatment of some
ER+ and/or PR+ breast cancers, yet patients
with ER− and PR− tumors gain little or no
benefit from anti-estrogen therapy. Increasing number of reports
show that AR is expressed in a considerable proportion of cases
and, of particular interest, AR is also expressed in almost 50% of
ER− and/or PR− breast cancer (6–14).
Identifying the underlying mechanisms of AR are crucial in the
design of therapies for estrogen-insensitive neoplasms.
Androgens act on target cells by binding to the
cognate receptor AR, a ligand-dependent transcription factor. The
AR protein possesses various domains that mediate its different
functions (15,16). Unbound AR exists in the cytoplasm
as a complex containing several molecular chaperones including
Hsp90, Hsp70 and Hsp56. Once bound to a hormone, the AR molecule
undergoes a conformational change that results in the shedding of
cytosolic heat shock proteins and is translocated into the nucleus
where it forms homodimers that associate with nuclear chaperons and
coactivators. This homodimer and associated proteins constitute the
active form of the receptor that is able to recognize the AREs
located at (or close to) the promoter region of androgen-dependent
genes. Once bound to AREs the homodimers recruit additional
coactivators and activate the transcription machinery, thus
increasing specific gene transcription by one or two orders of
magnitude (17).
AR can regulate mRNA transcription as well as miRNA
biogenesis. Several recent studies describe a role for AR in the
transcriptional regulation of global miRNA expression (18–20)
based on the observation that androgen-AR signaling directly
regulates the expression of certain miRNAs. Recent data indicate
that three miRNAs on the miR-125b-2 locus, let-7c, miR-99a and
miR-125b-2, are putative AR-regulated targets in prostate cancer
cells based on chromatin immunoprecipitation on array analysis
(19). In our study, miRNA
microarray analysis showed that let-7a, b, c, d were significantly
upregulated and real-time RT-PCR analysis revealed a significant
increase in let-7a expression in the DHT-treated MDA-MB-453 cells
and MDA-MB-231 compared with the vehicle-treated cells. Moreover,
ChIP analysis suggests androgen-AR signaling directly upregulates
the expression of let-7a.
The discovery of miRNA in the early 1990’s has
opened a new era of understanding post-transcriptional regulation
of genes by small RNAs (41).
miRNAs are endogenous, non-coding small RNAs known to repress
target gene expression by binding to complementary sequences in the
3′-UTRs of target mRNAs. There are many miRNA changes in different
types of human tumors and these miRNAs play an important role in
the initiation and progression of tumor as oncogenes and tumor
suppressors (42). There are 14
different let-7 family members in mouse and 13 members in human. In
human, these different members are let-7a-1, 7a-2, 7a-3, 7b, 7c,
7d, 7e, f7-1, 7f-2, 7g, 7i, mir-98, and mir-202 (43,44).
Among these members, let-7a has an identical sequence across
various animal species from Caenorhabditis elegans to human.
Let-7a is widely viewed as a tumor suppressor miRNA because the
expression of let-7a is downregulated in many cancer types and
during tumor progression (33–35).
Sempere et al reported that let-7a was decreased in breast
cancer cells after performing an ISH assay (45). Yu et al found that let-7
regulates key features of breast tumor-initiating cells (BT-IC):
self renewal, multipotent differentiation in vitro, and the
ability to form tumors in NOD/SCID mice (46). Protein expression of the let-7
targets RAS and HMGA2 is high in BT-IC and silenced during
differentiation. In our study, 4 upregulated miRNAs: let-7a, b, c,
d were identified in the DHT-treated cells as
differentially-expressed when compared with the vehicle-treated
cells. Expression of let-7a showed a 13-fold increase in MDA-MB-453
cells and a 5-fold increase in MDA-MB-231 cells. To obtain insight
into the role of let-7a in the androgen-AR signaling of
ER−, PR−, AR+ breast cancer cells,
we performed a series of cell function experiments. MTT assay was
used to measure the viable, proliferating cells and flow cytometry
analysis was used to measure cell cycle phase distribution after
the MDA-MB-453 and MDA-MB-231 cells were transfected. When let-7a
was overexpressed with vector, cell growth was inhibited and an
obvious G1-S arrest was observed, similar to the effect seen in
cells exposed to DHT. On the contrary, when let-7a was blocked with
let-7a ASO cell growth was elevated and the number of cells in the
S phase significantly increased.
The oncogenes CMYC and KRAS are known target genes
of let-7a that are involved in cell proliferation and cell cycle
(34–37). To elucidate the role of let-7a
targeting CMYC and KRAS in the process of DHT acting on MDA-MB-453
and MDA-MB-231 breast cancer cells, CMYC and KRAS protein was
examined by western blot analysis. The results showed that let-7a
negatively regulates endogenous CMYC and KRAS protein expression in
both DHT-treated cells and transiently transfected cells.
In FFPE breast cancer tissue specimens the staining
outcome of ISH for let-7a and IHC staining for AR, CMYC and KRAS
showed that let-7a expression was negatively correlated with CMYC
and KRAS expression and that CMYC expression was positively
correlated with KRAS expression. The results confirmed that let-7a
is indeed a negative regulator of CMYC and KRAS in vivo.
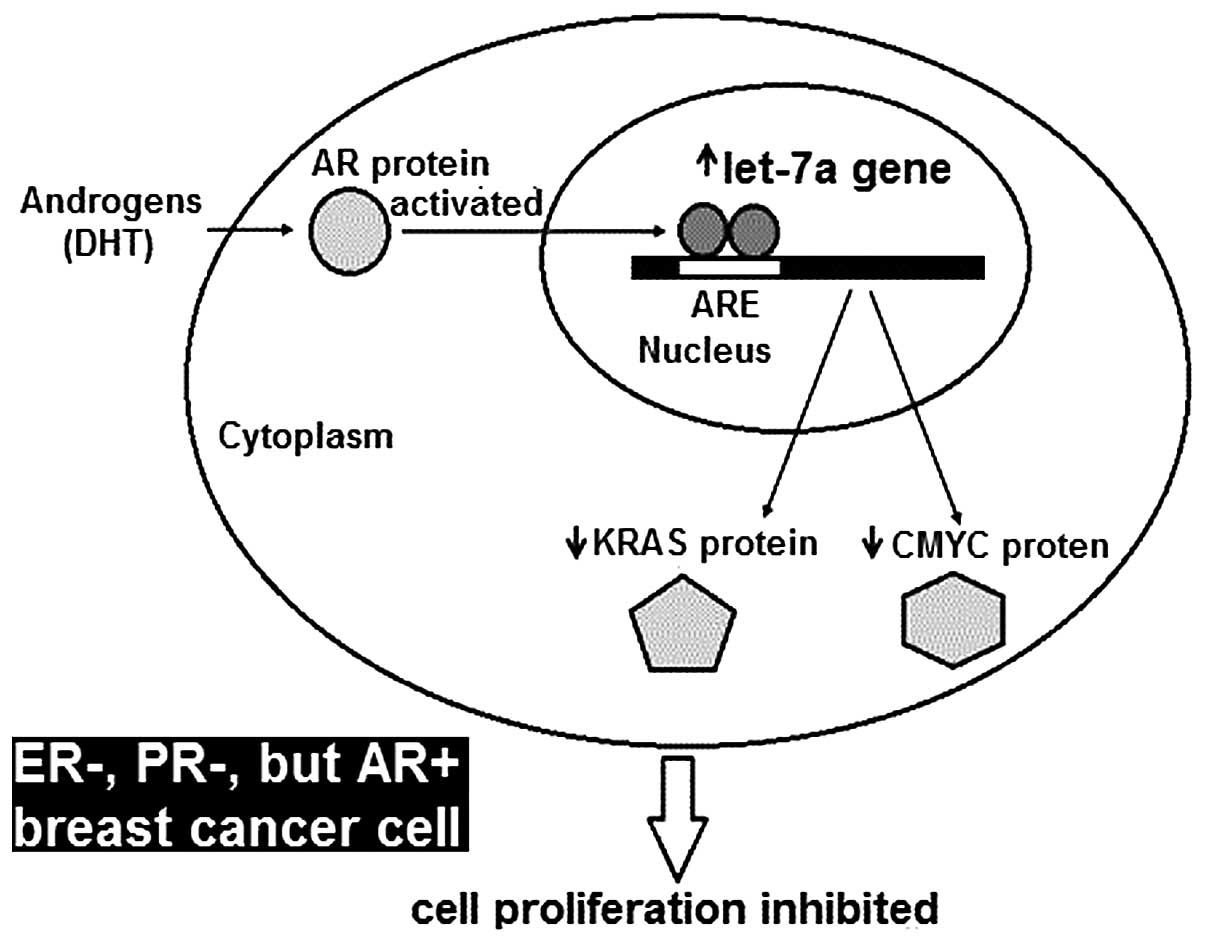
In conclusion, the androgen-induced AR activating
signal pathway directly upregulates let-7a miRNA expression. Let-7a
targets CMYC and KRAS and plays an important role in the process of
DHT inhibiting proliferation of ER−, PR−,
AR+ MDA-MB-453 and MDA-MB-231 breast cancer cells
(Fig. 5). In FFPE breast cancer
tissue, we further confirmed that let-7a is indeed a negative
regulator of CMYC and KRAS in vivo. These findings
contribute to the understanding of ER−, PR−,
AR+ breast cancer pathogenesis and will hopefully help
in the design of new therapies for estrogen-insensitive
neoplasms.
References
|
1.
|
Moe RE and Anderson BO: Androgens and
androgen receptors: a clinically neglected sector in breast cancer
biology. J Surg Oncol. 95:437–439. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Birrell SN, Hall RE and Tilley WD: Role of
the androgen receptor in human breast cancer. J Mammary Gland Biol
Neoplasia. 3:95–103. 1998.PubMed/NCBI
|
|
3.
|
Somboonporn W and Davis SR: Testosterone
effects on the breast: implications for testosterone therapy for
women. Endocr Rev. 25:374–388. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Labrie F, Luu-The V, Labrie C, Bélanger A,
Simard J, Lin SX and Pelletier G: Endocrine and intracrine sources
of androgens in women: inhibition of breast cancer and other roles
of androgens and their precursor dehydroepiandrosterone. Endocr
Rev. 24:152–182. 2003.
|
|
5.
|
Yeh S, Hu YC, Wang PH, Xie C, Xu Q, Tsai
MY, Dong Z, Wang RS, Lee TH and Chang C: Abnormal mammary gland
development and growth retardation in female mice and MCF7 breast
cancer cells lacking androgen receptor. J Exp Med. 198:1899–1908.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Agrawal AK, Jeleń M, Grzebieniak Z,
Zukrowski P, Rudnicki J and Nienartowicz E: Androgen receptors as a
prognostic and predictive factor in breast cancer. Folia Histochem
Cytobiol. 46:269–276. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Niemeier LA, Dabbs DJ, Beriwal S, Striebel
JM and Bhargava R: Androgen receptor in breast cancer: expression
in estrogen receptor-positive tumors and in estrogen
receptor-negative tumors with apocrine differentiation. Mod Pathol.
232:205–212. 2010. View Article : Google Scholar
|
|
8.
|
Lanzino M, Sisci D, Morelli C, Garofalo C,
Catalano S, Casaburi I, Capparelli C, Giordano C, Giordano F,
Maggiolini M and Andò S: Inhibition of cyclin D1 expression by
androgen receptor in breast cancer cells - identification of a
novel androgen response element. Nucleic Acids Res. 38:5351–5365.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Gonzalez LO, Corte MD, Vazquez J, Junquera
S, Sanchez R, Alvarez AC, Rodriguez JC, Lamelas ML and Vizoso FJ:
Androgen receptor expression in breast cancer: relationship with
clinicopathological characteristics of the tumors, prognosis, and
expression of metalloproteases and their inhibitors. BMC Cancer.
8:1492008. View Article : Google Scholar
|
|
10.
|
Castellano I, Allia E, Accortanzo V,
Vandone AM, Chiusa L, Arisio R, Durando A, Donadio M, Bussolati G,
Coates AS, Viale G and Sapino A: Androgen receptor expression is a
significant prognostic factor in estrogen receptor positive breast
cancers. Breast Cancer Res Treat. 124:607–617. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Agoff SN, Swanson PE, Linden H, Hawes SE
and Lawton TJ: Androgen receptor expression in estrogen
receptor-negative breast cancer. Immunohistochemical, clinical, and
prognostic associations. Am J Clin Pathol. 120:725–731. 2003.
View Article : Google Scholar
|
|
12.
|
Nahleh Z: Androgen receptor as a target
for the treatment of hormone receptor negative breast cancer: an
unchartered territory. Future Oncol. 4:15–21. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Yu Q, Niu Y, Liu N, Zhang JZ, Liu TJ,
Zhang RJ, Wang SL, Ding XM and Xiao XQ: Expression of androgen
receptor in breast cancer and its significance as a prognostic
factor. Ann Oncol. 226:1288–1294. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Collins LC, Cole KS, Marotti JD, Hu R,
Schnitt SJ and Tamimi RM: Androgen receptor expression in breast
cancer in relation to molecular phenotype: results from the Nurses’
Health Study. Mod Pathol. 24:924–931. 2011.PubMed/NCBI
|
|
15.
|
Baniahmad A: Nuclear hormone receptor
co-repressors. J Steroid Biochem Mol Biol. 93:89–97. 2005.
View Article : Google Scholar
|
|
16.
|
Dehm SM and Tindall DJ: Molecular
regulation of androgen action in prostate cancer. J Cell Biochem.
99:333–344. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Nicolás Díaz-Chico B, Germán Rodríguez F,
González A, Ramírez R, Bilbao C, Cabrera de León A, Aguirre Jaime
A, Chirino R, Navarro D and Díaz-Chico JC: Androgens and androgen
receptors in breast cancer. J Steroid Biochem Mol Biol. 105:1–15.
2007.
|
|
18.
|
Shi XB, Tepper CG and De Vere White RW:
Cancerous miRNAs and their regulation. Cell Cycle. 7:1529–1538.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Shi XB, Xue L, Yang J, Ma AH, Zhao J, Xu
M, Tepper CG, Evans CP, Kung HJ and De Vere White RW: An
androgen-regulated miRNA suppresses Bak1 expression and induces
androgen-independent growth of prostate cancer cells. Proc Natl
Acad Sci USA. 104:19983–19988. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Takayama K, Tsutsumi S, Katayama S,
Okayama T, Horie-Inoue K, Ikeda K, Urano T, Kawazu C, Hasegawa A,
Ikeo K, Gojyobori T, Ouchi Y, Hayashizaki Y, Aburatani H and Inoue
S: Integration of cap analysis of gene expression and chromatin
immunoprecipitation analysis on array reveals genome-wide androgen
receptor signaling in prostate cancer cells. Oncogene. 30:619–630.
2011. View Article : Google Scholar
|
|
21.
|
Jovanovic M and Hengartner MO: miRNAs and
apoptosis: RNAs to die for. Oncogene. 25:6176–6187. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Büssing I, Slack FJ and Grosshans H: Let-7
microRNAs in development, stem cells and cancer. Trends Mol Med.
14:400–409. 2008.
|
|
23.
|
Schickel R, Boyerinas B, Park SM and Peter
ME: MicroRNAs, key players in the immune system, differentiation,
tumorigenesis and cell death. Oncogene. 27:5959–5974. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Stefani G and Slack FJ: Small non-coding
RNAs in animal development. Nat Rev Mol Cell Biol. 9:219–230. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Cho WC: OncomiRs: the discovery and
progress of microRNAs in cancers. Mol Cancer. 6:602007. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Hall RE, Birrell SN, Tilley WD and
Sutherland RL: MDA-MB-453, an androgen-responsive human breast
carcinoma cell line with high level androgen receptor expression.
Eur J Cancer. 30A:484–490. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Yeap BB, Krueger RG and Leedman PJ:
Differential posttranscriptional regulation of androgen receptor
gene expression by androgen in prostate and breast cancer cells.
Endocrinology. 140:3282–3291. 1999.PubMed/NCBI
|
|
28.
|
Subik K, Lee JF, Baxter L, Strzepek T,
Costello D, Crowley P, Xing L, Hung MC, Bonfiglio T, Hicks DG and
Tang P: The expression patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67
and AR by immunohistochemical analysis in breast cancer cell lines.
Breast Cancer (Auckl). 4:35–41. 2010.PubMed/NCBI
|
|
29.
|
Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee
DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao
KQ, Livak KJ and Guegler KJ: Real-time quantification of microRNAs
by stem-loop RT-PCR. Nucleic Acids Res. 33:e1792005. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Louie MC, Yang HQ, Ma AH, Xu W, Zou JX,
Kung HJ and Chen HW: Androgen-induced recruitment of RNA polymerase
II to a nuclear receptor-p160 coactivator complex. Proc Natl Acad
Sci USA. 100:2226–2230. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Seifer DB, MacLaughlin DT, Penzias AS,
Behrman HR, Asmundson L, Donahoe PK, Haning RV Jr and Flynn SD:
Gonadotropin-releasing hormone agonist-induced differences in
granulosa cell cycle kinetics are associated with alterations in
follicular fluid müllerian-inhibiting substance and androgen
content. J Clin Endocrinol Metab. 76:711–714. 1993.PubMed/NCBI
|
|
32.
|
Harvey JM, Clark GM, Osborne CK and Allred
DC: Estrogen receptor status by immunohistochemistry is superior to
the ligand-binding assay for predicting response to adjuvant
endocrine therapy in breast cancer. J Clin Oncol. 17:1474–1481.
1999.
|
|
33.
|
Takamizawa J, Konishi H, Yanagisawa K,
Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y,
Mitsudomi T and Takahashi T: Reduced expression of the let-7
microRNAs in human lung cancers in association with shortened
postoperative survival. Cancer Res. 64:3753–3756. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Dahiya N, Sherman-Baust CA, Wang TL,
Davidson B, Shih IeM, Zhang Y, Wood W III, Becker KG and Morin PJ:
MicroRNA expression and identification of putative miRNA targets in
ovarian cancer. PLoS One. 3:e24362008. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
O’Hara AJ, Wang L, Dezube BJ, Harrington
WJ Jr, Damania B and Dittmer DP: Tumor suppressor microRNAs are
underrep-resented in primary effusion lymphoma and Kaposi sarcoma.
Blood. 113:5938–5941. 2009.PubMed/NCBI
|
|
36.
|
Engels BM and Hutvagner G: Principles and
effects of microRNA-mediated post-transcriptional gene regulation.
Oncogene. 25:6163–6169. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Sampson VB, Rong NH, Han J, Yang Q, Aris
V, Soteropoulos P, Petrelli NJ, Dunn SP and Krueger LJ: MicroRNA
let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt
lymphoma cells. Cancer Res. 67:9762–9770. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Johnson SM, Grosshans H, Shingara J, Byrom
M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D and Slack
FJ: RAS is regulated by the let-7 microRNA family. Cell.
120:635–647. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
He XY, Chen JX, Zhang Z, Li CL, Peng QL
and Peng HM: The let-7a microRNA protects from growth of lung
carcinomaby suppression of k-Ras and c-Myc in nude mice. J Cancer
Res Clin Oncol. 136:1023–1028. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Akao Y, Nakagawa Y and Naoe T: let-7
microRNA functions as a potential growth suppressor in human colon
cancer cells. Biol Pharm Bull. 29:903–906. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Lee RC, Feinbaum RL, Ambros V and The C:
Elegans heterochronic gene lin-4 encodes small RNAs with antisense
complementarity to lin-14. Cell. 75:843–854. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Cho WC: OncomiRs: the discovery and
progress of microRNAs in cancers. Mol Cancer. 6:602007. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Reinhart BJ, Slack FJ, Basson M,
Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR and Ruvkun G:
The 21-nucleotide let-7 RNA regulates developmental timing in
Caenorhabditis elegans. Nature. 403:901–906. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Ruby JG, Jan C, Player C, Axtell MJ, Lee
W, Nusbaum C, Ge H and Bartel DP: Large-scale sequencing reveals
21U-RNAs and additional microRNAs and endogenous siRNAs in C.
elegans. Cell. 127:1193–1207. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Sempere LF, Christensen M, Silahtaroglu A,
Bak M, Heath CV, Schwartz G, Wells W, Kauppinen S and Cole CN:
Altered microRNA expression confined to specific epithelial cell
subpopulations in breast cancer. Cancer Res. 67:11612–11620. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong
C, Huang Y, Hu X, Su F, Lieberman J and Song E: let-7 regulates
self renewal and tumorigenicity of breast cancer cells. Cell.
131:1109–1123. 2007. View Article : Google Scholar : PubMed/NCBI
|