Introduction
Breast cancer is the most commonly occurring cancer
in women. Each year approximately one million of new cases occurred
in the world, 25–40% of patients developing metastasis and dying
from cancer (1,2). Many problems remain in its clinical
management which is generally related to the malignancy types and
drug resistance mechanisms as well as metastasis development.
Additional investigations to understand the physiopathology of
breast cancer are urgently necessary to develop new therapies based
on new targets involved in cancer cell invasion and
proliferation.
In previous studies we have already demonstrated
that the upregulation and activation of the small GTPase RhoA or
RhoC contribute to cell invasion leading to breast cancer
metastasis (3,4). RhoA signaling pathways are implicated
in the activation of FAK, Akt/PI3K, p38MAPK and MLCK, which have
been shown to be responsible for cytoskeleton actin reorganization,
cell adhesion, motility, migration and invasion (5–9). In
many types of cancers, RhoA appears to be overexpressed and/or
constitutively activated (10,11),
and considered to be a negative clinical prognosis marker (10,12,13).
Recently, it has been shown that the voltage-gated
Na+ channels (VGSCs) could be an accelerating factor in
malignant cancers (14–17). VGSCs are membrane panning proteins
expressed in a wide variety of excitable and non-excitable cells,
as well as in many carcinomas including breast cancer (18,19).
VGSCs mainly mediate rapid and transient Na+ influx into
cells and are classically responsible for generation and
propagation of action potential. In cancer cells, overexpression of
VGSCs and/or their upregulation has been observed. In particular,
the Nav1.5 channel type correlates with cancer cell invasion
(20–22). Although it seems that the neonatal
isoform of Nav1.5 channel may be a potential target for cancer
therapy, so far the mechanisms regulating its expression and its
functional activity in cancer cells have not been clarified. A
previous report revealed that the expression of the neonatal
isoform of Nav1.5 was regulated by protein kinase A (23).
In the present study, the Nav1.5 channel expression
was analyzed in invasive (MDA-MB-231) or less (MCF-7) invasive
breast cancer cell lines. Due to the importance of RhoA in
signaling pathways in cancer cell invasion, the involvement of RhoA
in the expression and regulation of Nav1.5 was investigated using
the real-time RT-PCR and the electrophysiological patch-clamp
techniques.
Materials and methods
Cell culture
MDA-MB-231 cells were grown in RPMI-1640 medium with
10% fetal bovine serum (FBS) (Eurobio), 2 mM L-glutamine. MCF-7
cells were grown in H-DMEM medium with 10% FBS, 4 mM L-glutamine.
Both cell lines were obtained from ECACC. All cultures contained
100 IU/ml penicillin and 100 μg/ml of streptomycin (Eurobio)
and were incubated at 37°C in a humidified 5% CO2
atmosphere.
siRNA transfection
Specific siRNAs directed against human RhoA or
Nav1.5 was designed using the criteria established by Tuschl.
Candidate sequences were compared with cDNA sequences and their
specificity verified in the non-redundant human DNA database using
a BLAST algorithm [accession through NCBI]. The RhoA siRNA selected
was: sense 5′-GAC AUGCUUGCUCAUAGUC-3′, antisense 5′-CUGUACGAACG
AGUAUCAG-3′ and the Nav1.5 siRNA selected was: sense
5′-GGCACAUGAUGGACUUCUU-3′, antisense 5′-CCGU GUACCUGAAGAA-3′.
Eurogentec negative control siRNA was used as control. siRNAs (10
nM) were introduced into cells by INTERFERin™-mediated transfection
(Ozyme). In patch-clamp experiments, siRNAs were labeled with
tetramethyl-rhodamin, and only red cells detected under fluorescent
microscope were selected for recording.
Cell proliferation and viability
Cells were seeded (6×103/well) in 96-well
plates in growth medium complemented with 5% FBS. Cell
proliferation/viability was evaluated using a
[3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium,
inner salt] (MTS, Promega) assay at 24, 48 and 72 h after
transfection. Cells were incubated with MTS in culture medium at
37°C for 2 h. Optical density was read at 490 nm using a
PowerWavex spectrophotometer (Bio-Tek Instruments
Inc.).
Cancer cell invasion
Cells were cultured for 24 h in serum-free medium.
Cells (7.5×104) were seeded in the insert coated with
Matrigel. The lower chamber was filled with 0.75 ml of RPMI-1640
containing 10% FCS to induce chemotaxis. Twenty-four or 72 hours
later, the non-migrated cells in the upper chamber were gently
scraped away, and invasive cells were fixed with methanol, stained
with 1% toluidine/1% borax solution, and counted with Mercator
software (Explora Nova). The invasion by MDA-MB-231 was tested
after tranfection with control, RhoA or Nav1.5 siRNAs.
Quantitative polymerase chain reaction
(qPCR) assay (real-time RT-PCR)
The transfected cells were harvested and total RNA
was prepared by SV total RNA isolation system kit (Promega). The
purity of total RNA was checked by a ratio of A260/A280 (>1.9).
Total RNA (50 ng) was used to synthesize cDNA in 20 μl
reaction solution using iScript™ cDNA Synthesis kit (Bio-Rad). Then
2 μl of cDNA was used for qPCR assay in duplicates with SYBR
Green gene expression assay method. The primers for RhoA (forward
primer: 5′-CGC TTTTGGGTACATGGAGT-3′, reverse primer: 5′-GAGCAGC
TCTCGTAGCCATT-3′), Nav1.5 (forward primer: 5′-CGCCTA
CGTGATGAGTGAGA-3′, reverse primer: 5′-TAGGAGGG TGGGAAGGAAGT-3′) and
GAPDH (forward primer: 5′-TGC ACCACCAACTGCTTAGC-3′, reverse primer:
5′-GGCATG GACTGTGGTCATGAG-3′) were purchased from Eurogentec,
Belgium. The qPCR was performed using Quantifast™ SYBR®
Green PCR kit (Qiagen) by 10 min of initial denaturation and 44
cycles of 15 sec at 95°C, 60 sec at 60°C in a Master Cycler System
(Eppendorf). The ratio of interest, mRNA and GAPDH mRNA, was used
for analyzing qPCR results.
Western blot analysis
For protein extractions, 2×106 cells were
seeded into 75-cm2 flasks. Forty-eight hours after
treatment, proteins were extracted by RIPA buffer complemented with
protease and phosphatase inhibitor cocktail, concentration of
protein was measured by BCA Protein Assay kit (Pierce). Protein
fractions were separated by SDS-PAGE, then transferred onto
polyvinylidene difluoride membranes (Amersham) using a dry transfer
system (Invitrogen). Membranes were blocked with skim milk, and
probed using anti-RhoA (Santa Cruz) and anti-GAPDH (Sigma-Aldrich)
primary antibodies. GAPDH is used as control protein and for
protein normalization. The detection was done using a secondary
peroxidase-conjugated antibody (Dako). After washing the bound
antibody was detected with Immobilon western chemiluminescente HRP
substrate (Millipore). Then chemiluminescent emission was captured
on Kodak XAR film. Images were analyzed by ImageJ software.
Electrophysiology
Whole-cell currents of MDA-MB-231 human breast
cancer cells were recorded using a conventional patch-clamp
technique. Prior to each experiment, the growth medium was replaced
with an external bath solution containing (in mM): NaCl 144, KCI
5.4, MgCl2 1, CaCl2 2.5, D-glucose 5.6, and
HEPES 5, adjusted to pH 7.3 with 1 M NaOH. Soft glass patch
pipettes of resistance of 5–7 MΩ were filled with a solution
containing (in mM): NaCl 5, CsCl 145, MgCl2 2,
CaCl2 1, HEPES 10 and EGTA 11, adjusted to pH 7.4 with 1
M CsOH, to block outward K+ currents. A holding
potential of −100 mV was applied, unless indicated otherwise.
Standard voltage-clamp protocols were used to study the
electrophysiological properties of the voltage-gated sodium
channels on MDA-MB-231 cells. Whole-cell currents of isolated cells
were recorded at room temperature (23–25°C).
Data acquisition and analysis
All current signals were recorded with Axopatch 700B
amplifier (Axon Instruments, Union City, CA, USA) in the
voltage-clamp mode. Analogue signals were filtered at 3 kHz using a
lowpass Bessel filter and series resistance was compensated by
∼70%. Currents were corrected on-line for leak and residual
capacitance transients by a P/4 protocol. Electrophysiological
signals were sampled at 10 kHz and digitised using Digidata 1440A
(Axon Instruments). Data acquisition and analysis of membrane
currents were performed with pClamp 10.2 software (Axon
Instruments) and/or Origin 8.0 (Origin Lab, Northampton, MA, USA).
Values are given as mean ± SEM with n as the number of cells
tested. Statistical analysis was performed using the Student’s
t-test with paired comparisons where it was relevant. When multiple
comparisons were made, data were analyzed by a one-way ANOVA test,
followed by Tukey test when significant differences were
observed.
Statistical analysis
The Dunnett test was used for quantitative
comparisons between treatments. All experiments were reproduced at
least 3 times on different days unless specified otherwise.
Results
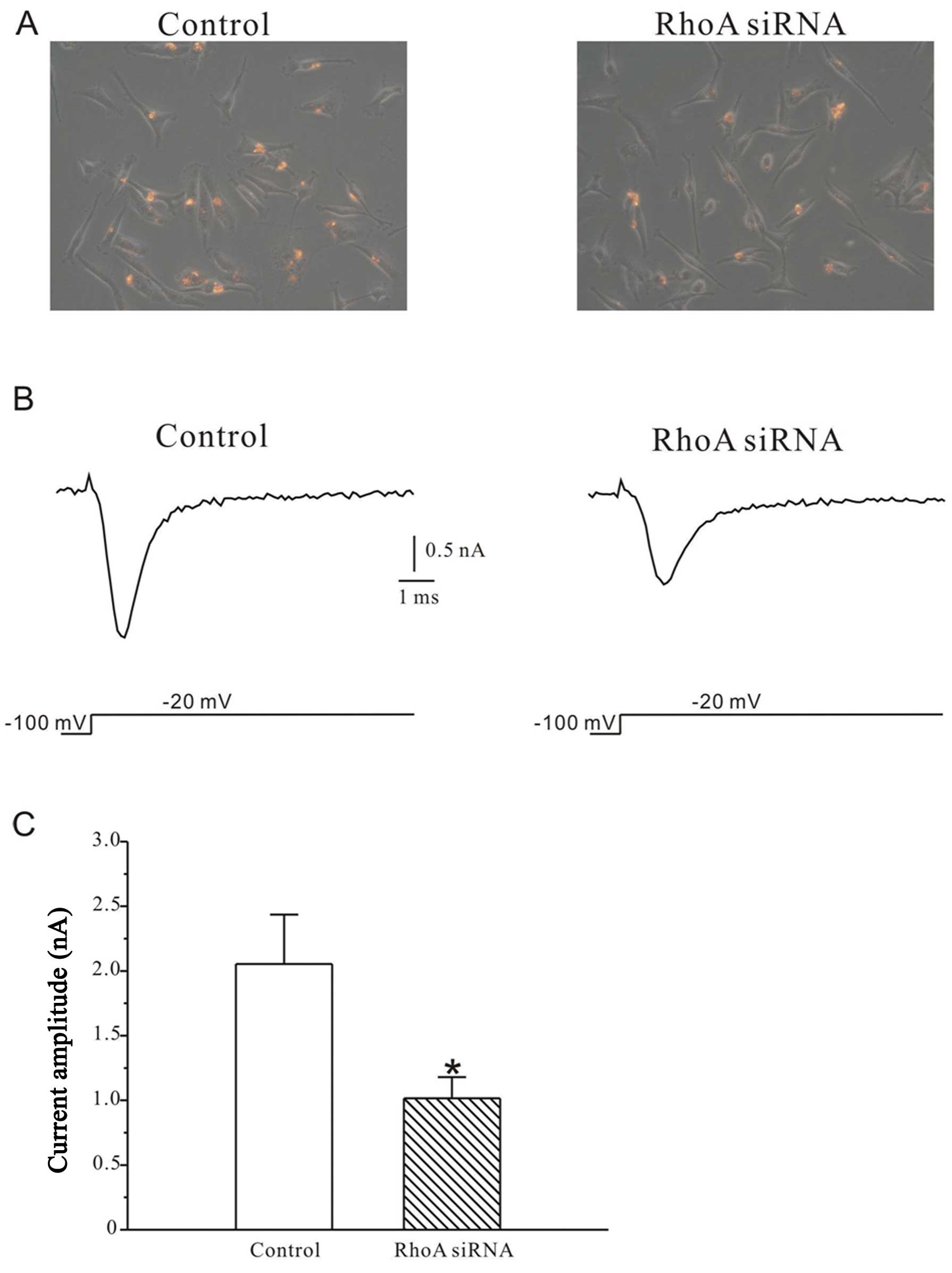
Silencing of RhoA reduces sodium
current
Whole-cell patch clamp recordings were used to
measure sodium current (INa) in MDA-MB-231 human
breast cells. Only the cells with red signals indicating successful
transfection were chosen (Fig.
1A). Small interference RNAs targeting RhoA (RhoA siRNAs)
dramatically reduced INa evoked by depolarizing
pulses to −20 mV from a holding potential of −100 mV. As shown in
Fig. 1B and C, the peak current in
RhoA siRNA-treated cells (1016.1±163.2 pA, n=20) was decreased by
50.5% (p<0.05) compared to the control cells (2054.7±380.5 pA,
n=16).
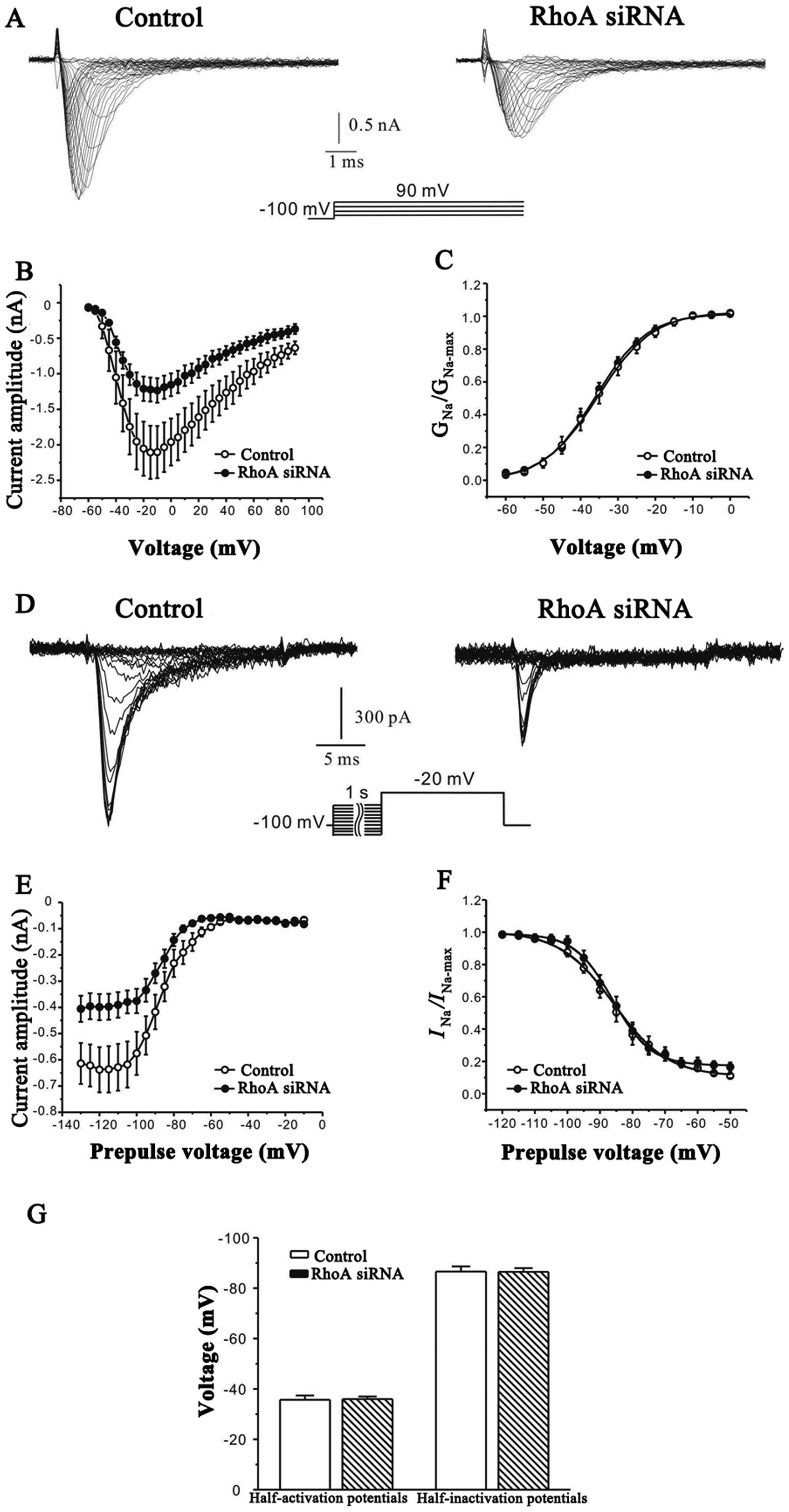
In order to clarify whether the decrease of the peak
current may result from changes in functional properties or the
number of channels expressed on the cell membrane, the effect of
RhoA siRNAs on the steady-state activation and inactivation
properties of INa was further investigated. A
protocol of 20-msec depolarizing pulses from a holding potential of
−100 mV to between −60 and 90 mV, with 5 mV steps at intervals of 5
sec, was used. As shown in Fig. 2A and
B, the evoked sodium current families were markedly reduced
after RhoA silencing. However, RhoA silencing did not significantly
modify (p<0.05) the steady-state activation property of sodium
channels. In all treated and untreated cells (16 cells for each
group), the half-activation potential was −35.7±1.7 and −35.9±1.0,
respectively (Fig. 2C and G).
The effect of RhoA siRNAs on the voltage dependence
of steady-state inactivation of sodium channels was studied by
inducing INa using 1-sec conditioning pre-pulses
ranging from −130 to −10 mV, in steps of 5 mV prior to a −20 mV
test pulse. Typical current traces and curves illustrating the
relationship between the peak current and the pre-pulse potential
is illustrated in Fig. 2D and E.
The steady-state inactivation curves were then fitted using the
Boltzmann equation of INa/INamax=1/
{1+exp [(Vm−Vm1/2)/k]}
+A (Fig. 2F). In all
treated (n=16) and untreated (n=13) cells, Vm 1/2
was −85.8±0.6 mV and −86.1±0.4 mV, respectively without any
significant (p<0.05) changes (Fig.
2G).
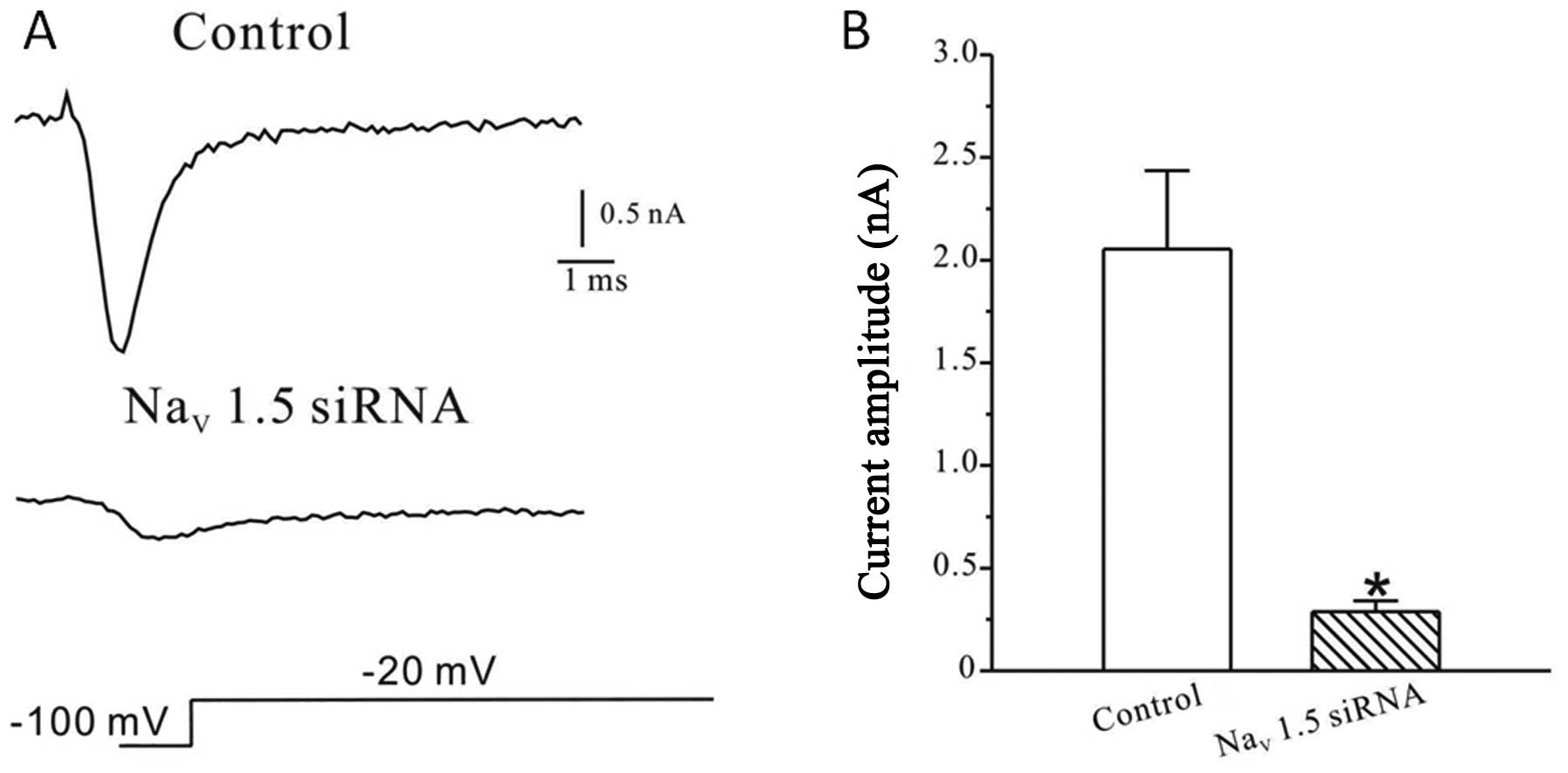
Among the different subtypes of VGSCs expressed in
MDA-MB-231 human breast cancer cells, the Nav1.5 channel is
suggested to be the major functional sodium channel subtype
expressed in these cells. To further investigate the expression
level of Nav1.5 channels, whole-cell patch clamp recordings were
performed in cells treated with the small interference RNAs
targeting Nav1.5 channels. As shown in Fig. 3A, transfection of Nav1.5 siRNA
strongly reduced the Na current. Compared to control (2054.7±380.5
pA, n=16), the peak current amplitude in transfected cells was
reduced to 288.0±53.5 pA (n=25) (Fig.
3B).
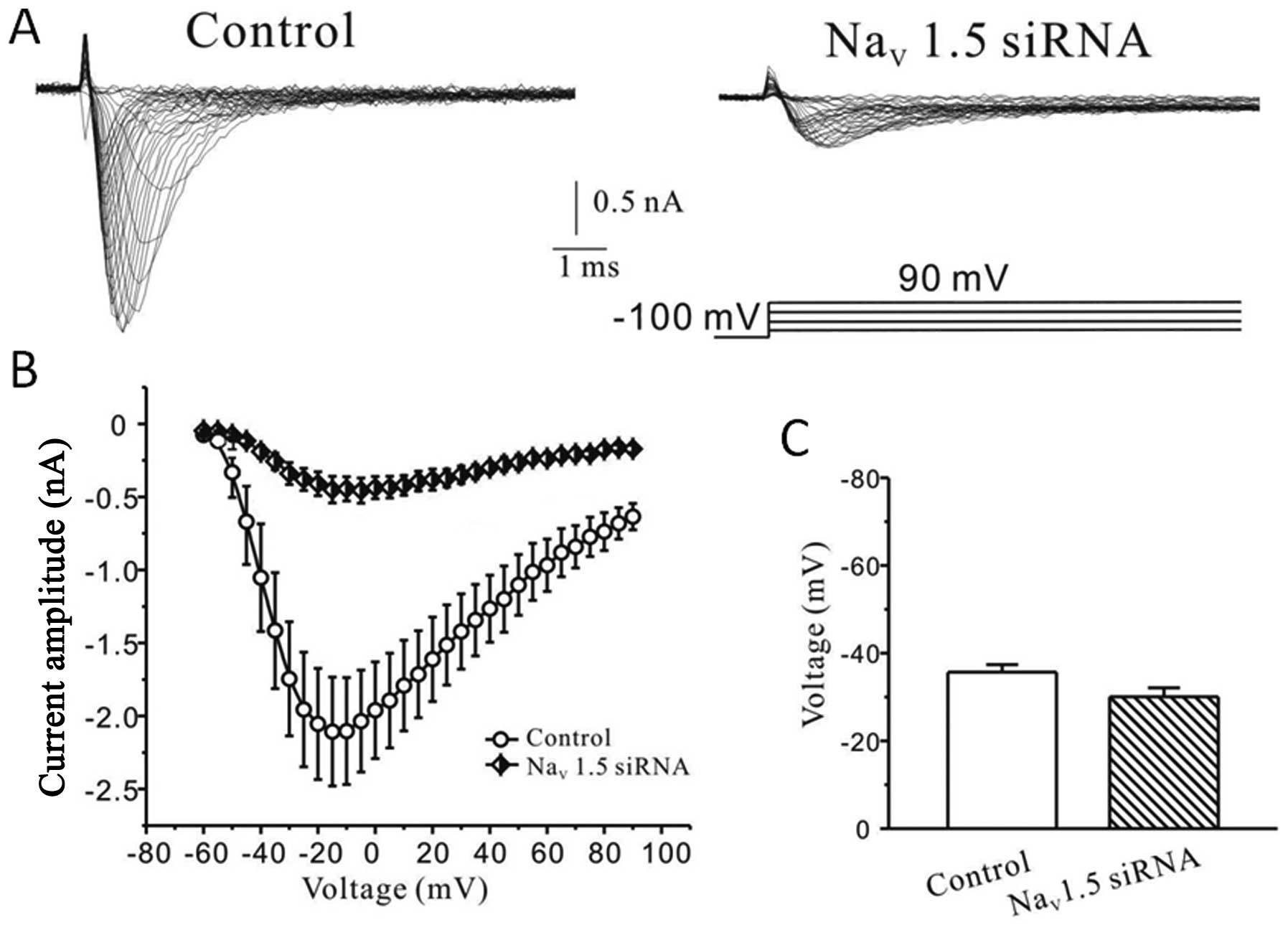
We also examined the steady-state activation
property of sodium current after interference with Nav1.5 siRNA.
Fig. 4A shows representative
whole-cell current families recorded from control or Nav1.5 siRNA
treated cells. As observed in Fig.
4B the mean I/V curves obtained from control (n=7) or treated
(n=6) cells showed that silencing the Nav1.5 sodium channel subtype
significantly reduced the sodium peak current elicited by each
depolarizing pulse tested. Fig. 4C
illustrates that the evoked currents were half-activated at
−30.0±2.0 mV after silencing Nav1.5 channels without any
significant difference (p>0.05) compared to the control
(−35.7±1.7 mV).
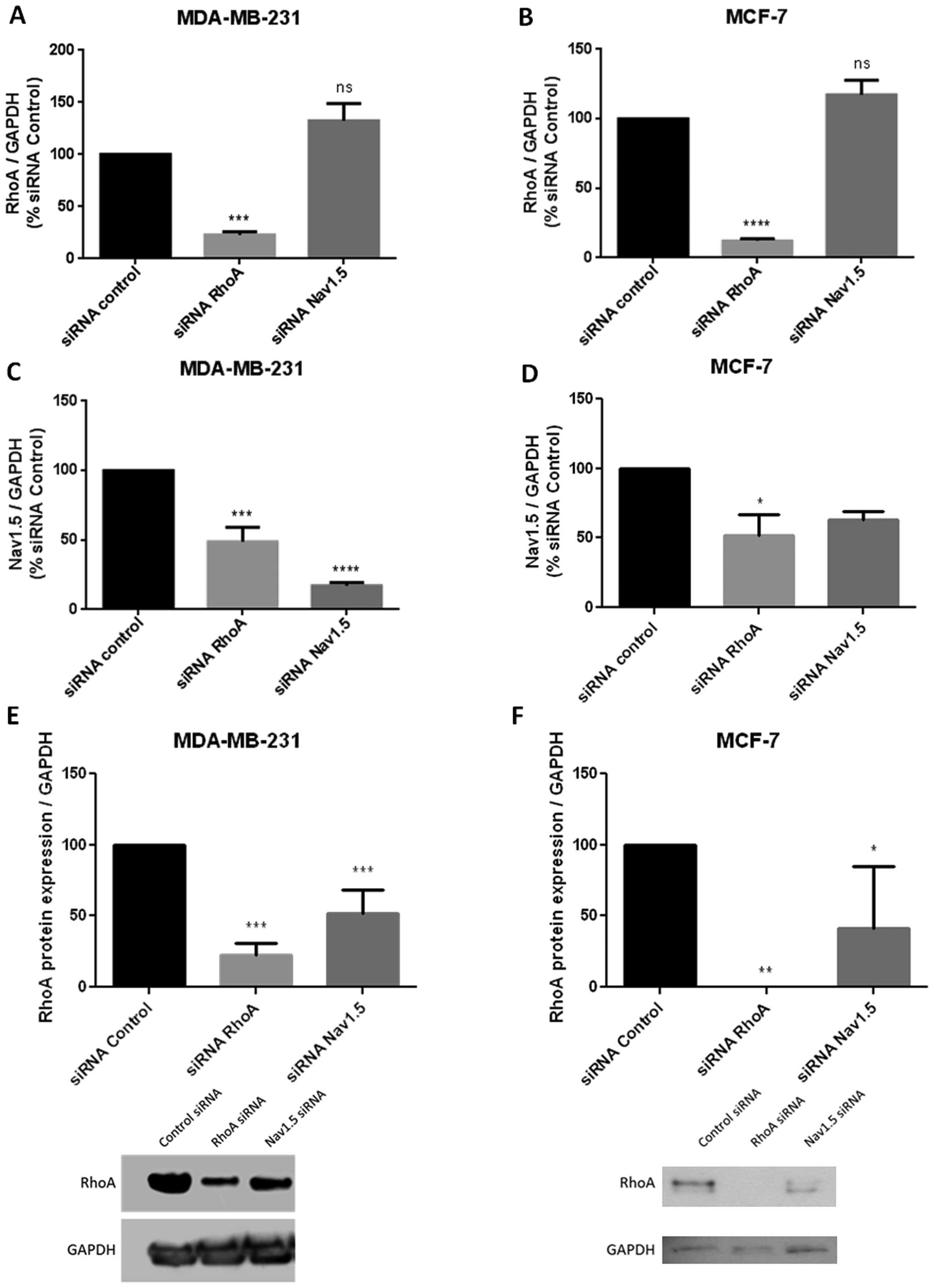
Inhibition of RhoA expression
downregulates Nav1.5 channels at transcription level
To further investigate the regulation mechanisms of
RhoA on Nav1.5 channels, we compared the expression level of the
channel molecules in the presence of RhoA or after RhoA silencing.
In the present set of experiments, the small interference RNA
anti-RhoA or anti-Nav1.5 were used in both aggressive MDA-MB-231
and less aggressive MCF-7 breast cancer cells. Real-time RT-PCR was
applied to evaluate the transcriptional level of RhoA or Nav1.5
expression. The histograms of Fig. 5A
and B revealed that siRNA anti-RhoA markedly and significantly
inhibited RhoA expression in both types of tumor cells (MDA-MB-231
cells: p<0.001, n=7; MCF-7 cells: p<0.0001, n=5) while siRNA
anti-Nav1.5 had no effect on RhoA transcriptional level. In
contrast, interestingly, as observed in Fig. 5C and D, after siRNA anti-RhoA
treatment, the transcriptional level of Nav1.5 channels appeared to
be significantly decreased (MDA-MB-231 cells: p<0.001, n=4;
MCF-7 cells: p<0.05, n=3) with a similar range of inhibition in
both types of breast cancer cells. It has to be noted that the
inhibition level of Nav1.5 expression by siRNA anti-Nav1.5 was
stronger in MDA-MB-231 than in MCF-7 cells.
Inhibition of Nav1.5 expression
downregulates the RhoA protein level
We investigated a potent effect of Nav1.5 channels
on RhoA expression. Nav1.5 silencing did not modify the
transcriptional level of RhoA (Fig. 5A
and B) while it decreased its protein level in both aggressive
(MDA-MB-231) and less aggressive (MCF-7) cells (Fig. 5E and F). The western blots shown in
Fig. 5 clearly revealed that after
a 48-h transfection, siRNA anti-Nav1.5 reduced by >50% the RhoA
protein level (MDA-MB-231 cells: p<0.01, n=4; MCF-7 cells:
n=2).
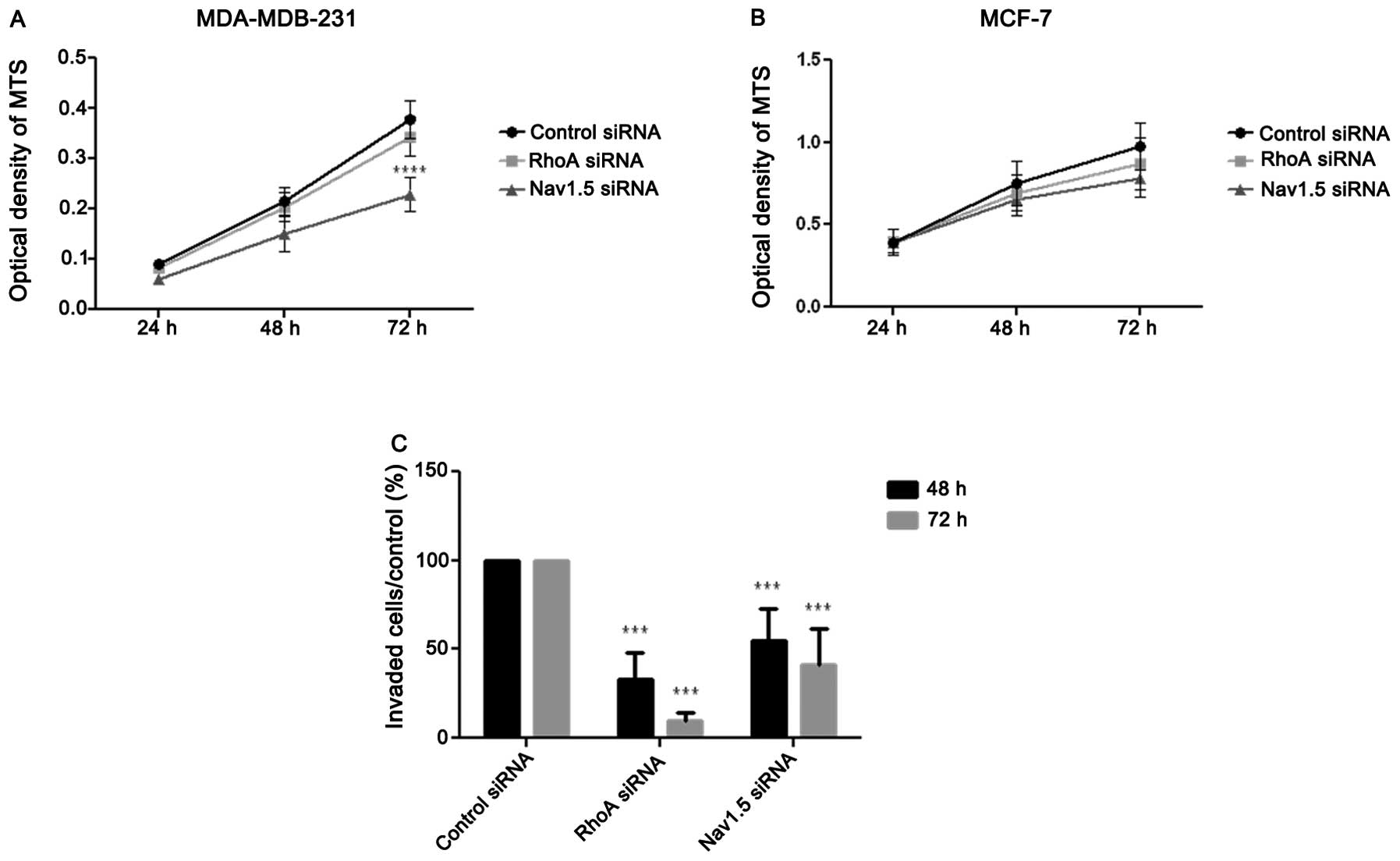
siRNA anti-Nav1.5 inhibits cell
proliferation of MDA-MB-231 but not of MCF-7
Additional experiments were conducted on cell
proliferation. Results illustrated in Fig. 5A and B indicated that only siRNA
anti-Nav1.5 inhibited cell proliferation in MDA-MB-231 cell line
(p<0.0001; n=7 after 72-h treatment) while RhoA silencing failed
to exert any effect. In contrast, in MCF-7 cells no significant
effect was detected after treatment with siRNA anti-RhoA or
anti-Nav1.5 (n=8).
RhoA and Nav1.5 promote tumor cell
invasion
In order to establish the relationship between RhoA
or Nav1.5 and tumor cell invasion, Boyden chamber assays were
performed on MDA-MB-231 cells (MCF-7 cells have no invasive
activity in this condition) after treatments with siRNA anti-RhoA
or anti-Nav1.5. As shown in Fig.
6C, each siRNA tested significantly (p<0.0001, n=6 after
48-h treatment; p<0.001, n=4 after 72 h) inhibited cell
invasion. It was noted that the RhoA silencing appeared to have a
more pronounced effect than that of Nav1.5.
Discussion
In previous studies, it was suggested that the
upregulation and activation of the small GTPase RhoA contribute to
cell invasion leading to metastasis (10,13).
Numerous data have elucidated that high RhoA expression and
activation levels may be a negative prognostic marker in cancer
treatment (10,12,13).
We have also demonstrated that in the aggressive breast cancer
cells, the proliferation and invasiveness were considerably reduced
when the small GTPase RhoA was inhibited (24,25).
Thus, we have developed a strategy of molecular therapy using
anti-RhoA siRNAs. The efficacy of i.v.-administered encapsulated
anti-RhoA siRNA in chitosan-coated polyiso-hexylcyanoacrylate
(PIHCA) nanoparticles in xenografted aggressive breast cancer
MDA-MB-231 was demonstrated (3,4).
To understand the physiopathology of breast cancer,
other reports suggested that upregulation of voltage-gated
Na+ channels could be an accelerating factor in
metastatic disease (26). The Na
channel expression level appeared to be closely linked to the
metastatic development in human breast cancer (19). As MDA-MB-231 cells are known to
highly express Nav1.5 channels on the cytoplasmic membrane, we
investigated the relationship between the expression of RhoA and
that of Na channels, particularly the Nav1.5 channels.
The present study was carried out on both invasive
(MDA-MB-231) and less-invasive (MCF-7) cells. The expression level
of Nav1.5 channels in both cell lines showed the expected results.
The expression level appeared to be higher in MDA-MB-231 than in
MCF-7 cells, in correlation with RhoA expression and activation
pattern.
We found that RhoA controls the expression of Nav1.5
that is recognized as the main type of functional VGSCs. The
silencing of RhoA dramatically reduced the expression of Nav1.5 at
its transcriptional level. Sustained (>48 h) treatment with
siRNA anti-RhoA markedly depressed sodium current. In addition, we
found that RhoA silencing exerted a much stronger inhibitory effect
on Nav1.5 than Nav1.7 (data not shown). These results indicated
that RhoA plays a key role in sodium current, most probably via
modulating the level of Nav1.5 channels. So far, this effect of
RhoA on VGSC expression and function has not been reported in
cancer cells or in non-tumor cells. Investigations in different
types of normal and tumor cells remain to be done to clarify
whether this effect of RhoA corresponds to physiological ubiquitous
or tumor specific mechanism. Since RhoA is implicated in the
formation of actin fibers, it would be of interest to know whether
the suppression of RhoA by siRNA may lead to an interrupted
integration of sodium channels into the cell membrane.
We noted that anti-Nav1.5 siRNA, not only markedly
decreased sodium current by 70%, but also inhibited cell invasion
and migration by 60–80%. Such results strongly support the notion
that Nav1.5 is the main VGSC implicated in MDA-MB-231 cell
invasiveness as suggested previously by Brackenbury et al
and Gao et al (20,27).
Interestingly, a positive feed-back of Nav1.5 on
RhoA was observed in this study. We found that inhibition by
anti-Nav1.5 siRNA led to a decrease in RhoA protein level in the
cells. Surprisingly, the result of RT-PCR showed no change in the
RhoA mRNA level. It suggests that Nav1.5 might affect the stability
of the RhoA protein. Many post-transcriptional and
post-translational events occur in cells such as phosphorylation,
transglutamination, palmitoylation, AMPylation, isoprenylation, and
ubiquitylation, and these modifications determine the distribution
and life cycle of RhoA (28,29).
Described mechanisms governing the stability of Rho proteins
include phosphorylation of serine 188 on RhoA that protect against
ubiquitin-mediated proteasomal degradation and geranylgeranylation
which facilitates proteasomal degradation of Rho (30,31).
Nav1.5-regulated RhoA protein level might be
partially explained by a regulatory mechanism which involves sodium
currents and intracellular calcium gradient. As a rule,
Nav1.5-mediated sodium currents influence the electrochemical
gradient of calcium because sodium influx activates
voltage-dependent calcium channels and induces calcium entry.
Indeed, the RhoA level was regulated by cytosolic calcium
concentration. Rao et al demonstrated that the reduction of
calcium concentration led to a decrease in Rho expression and the
decrease in cytosolic calcium markedly destabilized the RhoA
protein and accelerates its degradation (32). This allows assuming a likely
accelerated RhoA degradation when blocking Nav1.5 function.
Therefore, a positive feed-back between RhoA and
VGSCs would represent a crucial mechanism in oncogenesis. As RhoA
is overexpressed and constitutively activated in various cancers
including breast cancer (10,11),
RhoA-mediated cancer invasiveness and metastasis could be partially
via VGSC upregulation. Effectively it has been shown that in
prostate cancer cells a significant increase in voltage-dependent
channels was observed and found relative to the stimulation by EGF,
a growth factor known for activating RhoA (33). Therefore, targeting RhoA signaling
may be an interesting approach in cancer therapy.
Recently, the dominant VGSC was found to be an
embryonic/neonatal splice variant (nNav1.5) consistent with the
gene expression being ‘oncofoetal’. The molecular differences
between the adult and neonatal isoforms of the VGSC/Nav1.5 are 31
nucleotide differences, resulting in 7 amino acid differences
(26). It was proposed that
nNav1.5 is a novel marker with significant clinical potential for
management of metastatic breast cancer. However, the mechanisms
responsible for the expression of VGSC especially the neonatal
isoforms in metastatic cancers are not well elucidated. Therefore,
further investigation of nNav1.5 expression and RhoA regulation
seems to be of interest to better understand the regulation
mechanisms between RhoA signaling and VGSC function and their
implication in cancer malignancy.
Acknowledgements
C. Dulong was surpported finacially by
La Région Normandie. Grant sponsor: Groupement des Entreprises
Francaises dans la Lutte contre le Cancer de Rouen (GEFLUC of
Rouen), the Ligue Regionale de Haute Normandie de Lutte contre le
Cancer, Association Ti’Toine. We thank Catherine Bouquet and
Elisabeth Legrand for their technical assistance.
References
|
1.
|
Guarneri V and Conte P: Metastatic breast
cancer: therapeutic options according to molecular subtypes and
prior adjuvant therapy. Oncologist. 14:645–656. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Decensi A, Dunn BK, Puntoni M, Gennari A
and Ford LG: Exemestane for breast cancer prevention: a critical
shift? Cancer Discov. 2:25–40. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Pillé JY, Denoyelle C, Varet J, Bertrand
JR, Soria J, Opolon P, Lu H, Pritchard LL, Vannier JP, Malvy C,
Soria C and Li H: Anti-RhoA and anti-RhoC siRNAs inhibit the
proliferation and invasiveness of MDA-MB-231 breast cancer cells in
vitro and in vivo. Mol Ther. 11:267–274. 2005.PubMed/NCBI
|
|
4.
|
Pillé JY, Li H, Blot E, Bertrand JR,
Pritchard LL, Opolon P, Maksimenko A, Lu H, Vannier JP, Soria J,
Malvy C and Soria C: Intravenous delivery of anti-RhoA small
interfering RNA loaded in nanoparticles of chitosan in mice: safety
and efficacy in xenografted aggressive breast cancer. Hum Gene
Ther. 17:1019–1026. 2006.PubMed/NCBI
|
|
5.
|
Nobes CD and Hall A: Rho, rac, and cdc42
GTPases regulate the assembly of multimolecular focal complexes
associated with actin stress fibers, lamellipodia, and filopodia.
Cell. 81:53–62. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Schmitz AA, Govek EE, Böttner B and Van
Aelst L: Rho GTPases: signaling, migration, and invasion. Exp Cell
Res. 261:1–12. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Del Re DP, Miyamoto S and Brown JH: Focal
adhesion kinase as a RhoA-activable signaling scaffold mediating
Akt activation and cardiomyocyte protection. J Biol Chem.
283:35622–35629. 2008.PubMed/NCBI
|
|
8.
|
Cardone RA, Bagorda A, Bellizzi A, Busco
G, Guerra L, Paradiso A, Casavola V, Zaccolo M and Reshkin SJ:
Protein kinase A gating of a pseudopodial-located
RhoA/ROCK/p38/NHE1 signal module regulates invasion in breast
cancer cell lines. Mol Biol Cell. 16:3117–3127. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Gutjahr MC, Rossy J and Niggli V: Role of
Rho, Rac, and Rho-kinase in phosphorylation of myosin light chain,
development of polarity, and spontaneous migration of Walker 256
carcinosarcoma cells. Exp Cell Res. 308:422–438. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Gou L, Wang W, Tong A, Yao Y, Zhou Y, Yi C
and Yang J: Proteomic identification of RhoA as a potential
biomarker for proliferation and metastasis in hepatocellular
carcinoma. J Mol Med. 89:817–827. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Leve F and Morgado-Díaz JA: Rho GTPase
signaling in the development of colorectal cancer. J Cell Biochem.
113:2549–2559. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Horiuchi A, Kikuchi N, Osada R, Wang C,
Hayashi A, Nikaido T and Konishi I: Overexpression of RhoA enhances
peritoneal dissemination: RhoA suppression with Lovastatin may be
useful for ovarian cancer. Cancer Sci. 99:2532–2539. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Liu Y, Wang Y, Zhang Y, Miao Y, Zhao Y,
Zhang PX, Jiang GY, Zhang JY, Han Y, Lin XY, Yang LH, Li QC, Zhao C
and Wang EH: Abnormal expression of p120-catenin, E-cadherin, and
small GTPases is significantly associated with malignant phenotype
of human lung cancer. Lung Cancer. 63:375–382. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Balasuriya D, Stewart AP, Crottès D,
Borgese F, Soriani O and Edwardson JM: The sigma-1 receptor binds
to the Nav1.5 voltage-gated Na+ channel with 4-fold
symmetry. J Biol Chem. 287:37021–37029. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Gillet L, Roger S, Besson P, Lecaille F,
Gore J, Bougnoux P, Lalmanach G and Le Guennec JY: Voltage-gated
sodium channel activity promotes cysteine cathepsin-dependent
invasiveness and colony growth of human cancer cells. J Biol Chem.
284:8680–8691. 2009. View Article : Google Scholar
|
|
16.
|
House CD, Vaske CJ, Schwartz AM, Obias V,
Frank B, Luu T, Sarvazyan N, Irby R, Strausberg RL, Hales TG,
Stuart JM and Lee NH: Voltage-gated Na+ channel SCN5A is
a key regulator of a gene transcriptional network that controls
colon cancer invasion. Cancer Res. 70:6957–6967. 2010.PubMed/NCBI
|
|
17.
|
Hernandez-Plata E, Ortiz CS,
Marquina-Castillo B, Medina-Martinez I, Alfaro A, Berumen J, Rivera
M and Gomora JC: Overexpression of NaV 1.6 channels is associated
with the invasion capacity of human cervical cancer. Int J Cancer.
130:2013–2023. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Diss JK, Fraser SP and Djamgoz MB:
Voltage-gated Na+ channels: multiplicity of expression,
plasticity, functional implications and pathophysiological aspects.
Eur Biophys J. 33:180–193. 2004.
|
|
19.
|
Fraser SP, Diss JK, Chioni AM, Mycielska
ME, Pan H, Yamaci RF, Pani F, Siwy Z, Krasowska M, Grzywna Z,
Brackenbury WJ, Theodorou D, Koyutürk M, Kaya H, Battaloglu E, De
Bella MT, Slade MJ, Tolhurst R, Palmieri C, Jiang J, Latchman DS,
Coombes RC and Djamgoz MB: Voltage-gated sodium channel expression
and potentiation of human breast cancer metastasis. Clin Cancer
Res. 11:5381–5389. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Brackenbury WJ, Chioni AM, Diss JK and
Djamgoz MB: The neonatal splice variant of Nav1.5 potentiates in
vitro invasive behaviour of MDA-MB-231 human breast cancer cells.
Breast Cancer Res Treat. 101:149–160. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Brisson L, Gillet L, Calaghan S, Besson P,
Le Guennec JY, Roger S and Gore J: Na(V)1.5 enhances breast cancer
cell invasiveness by increasing NHE1-dependent H(+) efflux in
caveolae. Oncogene. 30:2070–2076. 2011.PubMed/NCBI
|
|
22.
|
Yang M, Kozminski DJ, Wold LA, Modak R,
Calhoun JD, Isom LL and Brackenbury WJ: Therapeutic potential for
phenytoin: targeting Na(v)1.5 sodium channels to reduce migration
and invasion in metastatic breast cancer. Breast Cancer Res Treat.
134:603–615. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Chioni AM, Shao D, Grose R and Djamgoz MB:
Protein kinase A and regulation of neonatal Nav1.5 expression in
human breast cancer cells: activity-dependent positive feedback and
cellular migration. Int J Biochem Cell Biol. 42:346–358. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Denoyelle C, Vasse M, Körner M, Mishal Z,
Ganné F, Vannier JP, Soria J and Soria C: Cerivastatin, an
inhibitor of HMG-CoA reductase, inhibits the signaling pathways
involved in the invasiveness and metastatic properties of highly
invasive breast cancer cell lines: an in vitro study.
Carcinogenesis. 22:1139–1148. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Vincent L, Chen W, Hong L, Mirshahi F,
Mishal Z, Mirshahi-Khorassani T, Vannier JP, Soria J and Soria C:
Inhibition of endothelial cell migration by cerivastatin, an
HMG-CoA reductase inhibitor: contribution to its anti-angiogenic
effect. FEBS Lett. 495:159–166. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Onkal R and Djamgoz MB: Molecular
pharmacology of voltage-gated sodium channel expression in
metastatic disease: clinical potential of neonatal Nav1.5 in breast
cancer. Eur J Pharmacol. 625:206–219. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Gao R, Wang J, Shen Y, Lei M and Wang Z:
Functional expression of voltage-gated sodium channels Nav1.5 in
human breast cancer cell line MDA-MB-231. J Huazhong Univ Sci
Technolog Med Sci. 29:64–67. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Liu M, Bi F, Zhou X and Zheng Y: Rho
GTPase regulation by miRNAs and covalent modifications. Trends Cell
Biol. 22:365–373. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
David M, Petit D and Bertoglio J: Cell
cycle regulation of Rho signaling pathways. Cell Cycle.
11:3003–3010. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Rolli-Derkinderen M, Sauzeau V, Boyer L,
Lemichez E, Baron C, Henrion D, Loirand G and Pacaud P:
Phosphorylation of serine 188 protects RhoA from
ubiquitin/proteasome-mediated degradation in vascular smooth muscle
cells. Circ Res. 96:1152–1160. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Von Zee CL and Stubbs EB Jr:
Geranylgeranylation facilitates proteasomal degradation of rho
G-proteins in human trabecular meshwork cells. Invest Ophthalmol
Vis Sci. 52:1676–1683. 2011.PubMed/NCBI
|
|
32.
|
Rao JN, Li L, Golovina VA, Platoshyn O,
Strauch ED, Yuan JX and Wang JY: Ca2+-RhoA signaling
pathway required for polyamine-dependent intestinal epithelial cell
migration. Am J Physiol Cell Physiol. 280:993–1007. 2001.
|
|
33.
|
Uysal-Onganer P and Djamgoz MB: Epidermal
growth factor potentiates in vitro metastatic behaviour of human
prostate cancer PC-3M cells: involvement of voltage-gated sodium
channel. Mol Cancer. 6:762007. View Article : Google Scholar
|