Introduction
DNA damage is constantly caused by diverse sources
including endogenous metabolite and exogenous assaults (1). Of the mutagens, alkylating agents are
generally unavoidable owing to their abundant presence in the
environment and within living cells. By reacting with DNA bases,
alkylating agents generate covalent adducts and subsequently
trigger DNA strand breaks (2).
The maintenance of genome integrity and fidelity is
essential for cell growth, proliferation and cell survival. While
failure to repair DNA lesions may result in impairment of cellular
function and in occurrence of gene mutagenesis (3). When the injure is triggered by
alkylating agents, DNA damage response (DDR), as a major way to
eliminate lethal and tumorigenic mutations, is initiated (4), giving rise to transcriptional
activation, cell cycle arrest, DNA repair or apoptosis in cells
(5). Induction of DNA damage is
one of the primary mechanisms underlying the anticancer
chemotherapeutic drugs, for instance cisplatin and camptothecin
used in the clinic. Therefore, DNA damage response was considered
as double-edged sword, on one side it affects the sensitivity to
anticancer chemotherapy for cell, and on the other, perturbation of
DNA repair function leads to accumulation of DNA damage, thereby
resulting in genomic instability (6).
Forkhead transcription factors of the O class
(FOXOs) are characterized by a conserved forkhead box DNA-binding
domain, the mammalian orthologs of Caenorhabditis elegans
DAF-16. FOXO subfamily is composed of four members FOXO1, FOXO3a,
FOXO4, and FOXO6 (7). FOXO factors
regulate a variety of physiological and pathological processes and
are considered to be potential targets of tumor therapy (8). FOXO3a and FOXO4 have been shown to
function as a master switch initiating G1/G2 phase arrest and DNA
damage repair through upregulating p27Kip1 and growth
arrest and DNA damage inducible protein 45 (GADD45) expression
(9–11). As the first identified member of
the FOXO subfamily, FOXO1 has recently been demonstrated to
modulate gene expression, such as p27Kip1 and
p21cip1 involved in cell cycle arrest, GADD45 in DNA
damage repair, Bim and FasL in apoptosis (12). Therefore, FOXO1 is considered to
determine cell fate by a different mechanism after stress-induced
damage accroding to cell types and contexts. However, what role is
played in DDR by FOXO1 is not fully understood and needs to be
elucidated.
The transcriptional activity of FOXO1 is tightly
regulated by multiple posttranslational modifications such as
phosphorylation, acetylation and ubiquitination (13). Growth factor stimulates activation
of the phosphatidyl inositol 3-kinase (PI3K)-AKT pathway, leading
to phosphorylation of nuclear FOXO1 at specific sites (Thr24,
Ser256 and Ser319). Phosphorylated FOXO1 then binds to 14-3-3
chaperone proteins and becomes sequestered in the cytoplasm, which
is unable to influence its target gene expression (14). In addtion, AKT-mediated
phosphorylation of FOXO1 on S256 facilitates interaction with the
E3 ubiquitin ligase Skp2, leading to its subsequent ubiquitination
and proteasomal degradation (15).
To the contrary, the activation of c-Jun N-terminal kinase (JNK)
promotes transcriptional activity of FOXO in response to external
stimuli. JNK-dependent T447 and T451 phosphorylation enhances the
nuclear translocation thereby increasing transcriptional activity
of FOXO4 (16). However, it is
still unclear whether FOXO1 is a downstream effector of JNK and
whether this regulation is direct as well as positive.
In the present study, we investigated the protective
roles of FOXO1 in DNA damage caused by the alkylating agent in lung
cancer cells. N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) is a
biochemical tool used experimentally as a carcinogen and mutagen.
Since both FOXO and p53 transcription factors share the same target
genes, including p21cip1 and GADD45 (17), the p53-deficient cell line H1299
was selected to avoid p53 interference in the study. Our data show
that FOXO1 affects the output of DDR by facilitating nuclear import
and upregulation of target gene expression in H1299 cells.
FOXO1-dependent cell cycle arrest and apoptosis were observed
during MNNG-inflicted DNA damage. In addition, our results show
that FOXO1 formed a complex with JNK kinase during DNA damage in
H1299 cells. Furthermore, when using JNK inhibitor its activity was
suppressed, nuclear exclusion of FOXO1 was examined, and the
decrease in expression levels of FOXO1 target genes was measured.
The results confirmed that FOXO1 participates in the cellular
responses to genotoxic stimuli, and suggests that JNK make a
significant contribution to alteration in FOXO1 transcriptional
activity during DDR in H1299 cells.
Materials and methods
Cell culture and cytotoxicity assay
Human lung adenocarcinoma cancer cells H1299 and
human bronchial epithelial (HBE) cells were maintained in
high-glucose DMEM medium with 10% fetal bovine serum (Gibco, CA,
USA), penicillin (100 U/ml) and streptomycin (100 μg/ml). In
certain experiments, JNK inhibitor SP600125 (Sigma, St. Louis, MO,
USA), was added. All cells were incubated at 37°C in 5%
CO2.
For cell viability assay, H1299 cells were seeded in
96-well plates at 5×103 cells/well. Cells were allowed
to adhere for 24 h, and subconfluent cells were treated with MNNG
(0.5–10 μM) (Sigma) for 30 min. Then, the cells were washed
with phosphate-buffered saline (PBS) three times to remove the
alkylating agents. Cell growth inhibitory studies were carried out
following incubating for 24 or 48 h, and cells were tested by MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5- biphenyl tetrazolium bromide)
assay.
Alkaline comet assay
The alkaline comet assay was conducted according to
the Tice et al analyzing method (18). After exposure to 1–10 μM
MNNG for 30 min, H1299 cells were collected and resuspended in cold
PBS. The cells in the 0.5% low melting point agarose were placed on
a slide precoated with a layer of 1% regular agarose. These two
layers were solidified at 4°C, and immersed in a cold lysing
solution (100 mM Na2EDTA, 2.5 M NaCl, 10 mM 1% Triton
X-100, 10% DMSO, Tris-HCl, pH 10.5) for 1 h at 4°C. Subsequently,
the gel slides were dried and soaked in fresh electrophoresis
solution (1 mM Na2EDTA, 300 mM NaOH, pH 13.0) for 20
min. Electrophoresis was run at 300 mA, 25 V for 20 min at 4°C.
After staining with ethidium bromide (20 μg/ml) for 10 min,
the slides were neutralized with 0.4 M Tris-HCl (pH 7.5). The
slides were washed with water and observed under a fluorescent
microscope (BX51; Olympus, Tokyo, Japan). The parameters
representing DNA damage were obtained and recorded for 100 cells
per sample by CASP software.
Immunofluorescence
Cells grown on coverslips were fixed in 4%
paraformaldehyde for 15 min, and permeabilized with 0.5% Triton
X-100 for 30 min at room temperature. Subsequently, the cells were
blocked with 3% BSA for 30 min, and incubated with monoclonal
anti-FOXO1 antibody (1:100, Cell Signaling Technology, Boston, MA,
USA) overnight at 4°C. These cells was incubated with
tetramethylrhodamine (TRITC)-5-(and-6)-isothiocyanate)-conjugated
secondary antibody for 1 h. Finally, the slides were stained with
DAPI solution for 15 min at 37°C following rinsing with PBS.
Digital images were examined under a fluorescence microscope (BX61;
Olympus).
FACS analysis
For cell cycle and apoptosis analysis, subconfluent
cells were treated with 0.1–5 μM MNNG for 30 min, and
incubation was continued with growth medium for 24 h. In order to
analyze the cell cycle, harvested cells were fixed in 70% ethanol
overnight at 4°C, followed by rinsing with cold PBS. Resuspended in
PBS solution, cells were incubated with RNase I (10 g/l) for 30 min
at 37°C and stained with 50 mg/ml propidium iodide (PI) for 30 min
in the dark. The DNA content was evaluated by flow cytometry
(Becton-Dickinson, NJ, USA). For analysis of apoptosis, cells were
harvested and washed with PBS, then stained with Annexin V-FITC for
10 min and 50 mg/ml propidium iodide for 5 min successively.
Apoptotic cells were assessed by outer membrane phosphatidylserine
translocation as well as nuclear fluorescence.
Immunoblot and immunoprecipitation
analysis
Cell extracts were prepared with RIPA lysis buffer
adding 0.5% protease inhibitor cocktail and 1% phosphatase
inhibitors. Protein samples were separated on 10 or 12% SDS-PAGE
gel and transferred for 2.5 h to PVDF membranes. Blots were then
probed ordinally with primary antibody according to the respective
titers provided by their manufacturers, and with appropriate
secondary antibody, then visualized using enhanced
chemiluminescence reagent (Sigma). Membranes were blocked in 5%
non-fat dry milk and washed in TTBS. Anti-FOXO1 was purchased from
Cell Signaling Technology. Anti-AKT, anti-p-AKT Ser 473,
anti-p-FOXO1 Ser 256 and anti-FOXO4, anti-p-JNK, anti-GADD45,
anti-Skp2 and anti-normal rabbit IgG, anti-Bim, anti-p27,
anti-β-actin and anti-GAPDH were purchesed from Santa Cruz
Biotechnology (Santa Cruz, CA, USA).
For immunoprecipitation analysis, cell lysates were
precleared with Protein G Plus agarose for 30 min at 4°C. Primary
antibody or rabbit normal IgG and Protein G agarose beads (40
μl of 50% bead slurry, Santa Cruz Biotechnology) were then
added to 500 μl cell lysates for 2-h incubation at 4°C.
Following five washings with cold lysis buffer, immunoprecipitates
were analyzed by western blotting in 10% gel.
Results
MNNG inhibits cell growth by inducing DNA
damage in H1299 cells
In order to investigate the cytotoxic effect of MNNG
on cell viability, H1299 cells were incubated in the presence of
increasing concentrations of MNNG for 30 min, then maintained in
growth medium for 24 or 48 h. MTT assay demonstrated MNNG inhibited
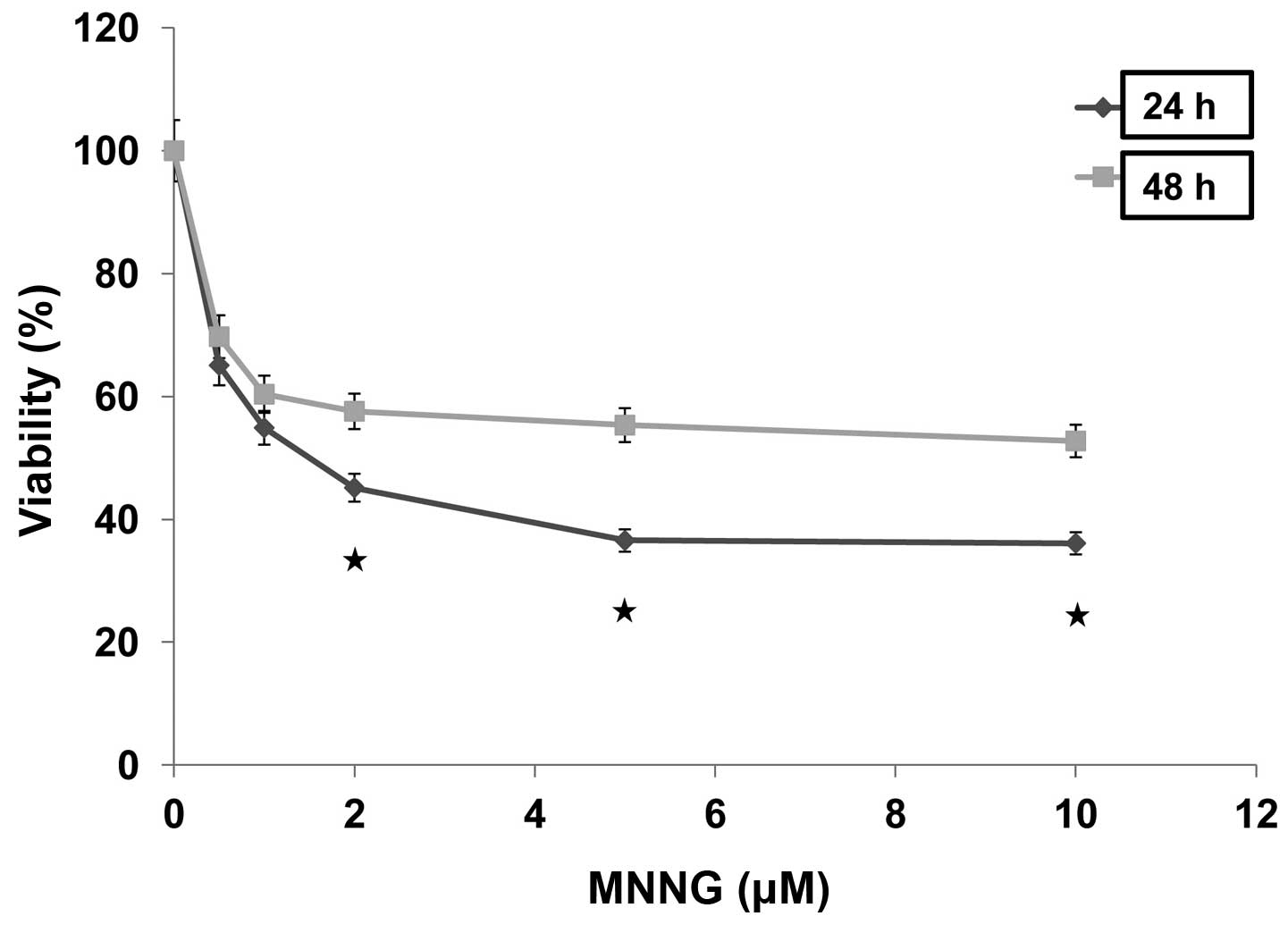
cell growth in a dose-dependent manner in H1299 cells (Fig. 1). The percentages of viable cells
cultured for 24 and 48 h after treatment with 0.5 μM MNNG
remarkably reduced to 65.1 and 69.7% compared with control,
respectively, and ultimately reduced to the plateau phases (36.6
and 55.3%) after treatment with 5 μM MNNG. In addition,
cells kept cultured for 24 h after treated with MNNG showed lower
viability than those cultured for 48 h after similar treatment.
MNNG was tested at a continuous cell exposure dose of 5 μM
with the experiments carried out at 0.5 to 10, and 5 μM was
selected for further analysis. These data indicated that cells may
have the ability to self-repair in response to MNNG-induced
damage.
MNNG is a wildly used alkylating agent considered to
cause methylation of DNA. To understand the mechanism underlying
MNNG-induced cytotoxicity in H1299 cells, we employed the alkaline
comet assay, because this assay is a sensitive and reliable
technique for evaluating the presence and level of DNA strand
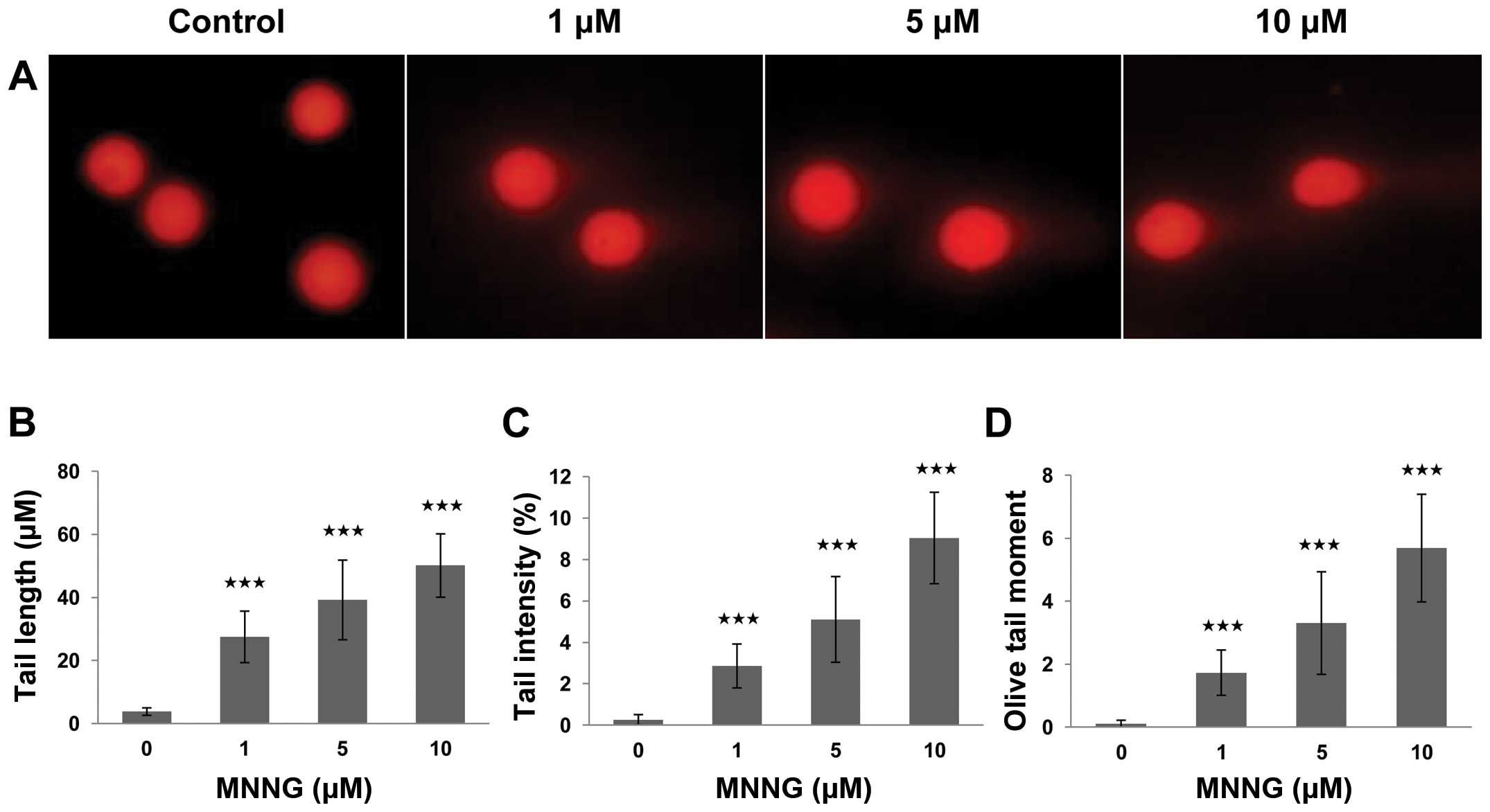
breaks (19). As shown by results
of the comet assay, fluorescence intense increased in the comet
tail, revealing the the fragments of DNA were excluded from their
nuclei, while the control cells displayed only fluorescent nuclei
without comet tails, after exposure to various concentrations of
MNNG (0, 1, 5 and 10 μM) for 30 min (Fig. 2). A statistically significant
difference was observed when these data from the exposed group was
compared to that from the control group. This difference was
manifested in the values of three comet parameters. The difference
of average value in tail length, tail intensity and tail moment
between cells of the exposed and the control group were 52.1±10.1
μm, 9.04±2.21% and 3.31±1.63 (p<0.001), respectively. The
data of MTT and comet assay revealed that MNNG inhibits cell growth
by inducing DNA strand breaks in a dose-dependent manner. Hence, we
speculated that the partial recovery of cell viability in p53-null
H1299 cells might be attributed to FOXO1-mediated DNA repair
response. Thus, it is essential to further define the function of
FOXO1 during DNA damage response.
DNA damage enhances the protein
expression and nuclear import of FOXO1
The expression levels and subcellular localization
of FOXO are two key factors influencing their transcriptional
activity. Increase in the protein level in the vicinity of the
nucleus contributes to the nuclear translocation of FOXO factors,
and intranuclear FOXOs can promote their target genes
transcription. Based on this, we first detected a change in its
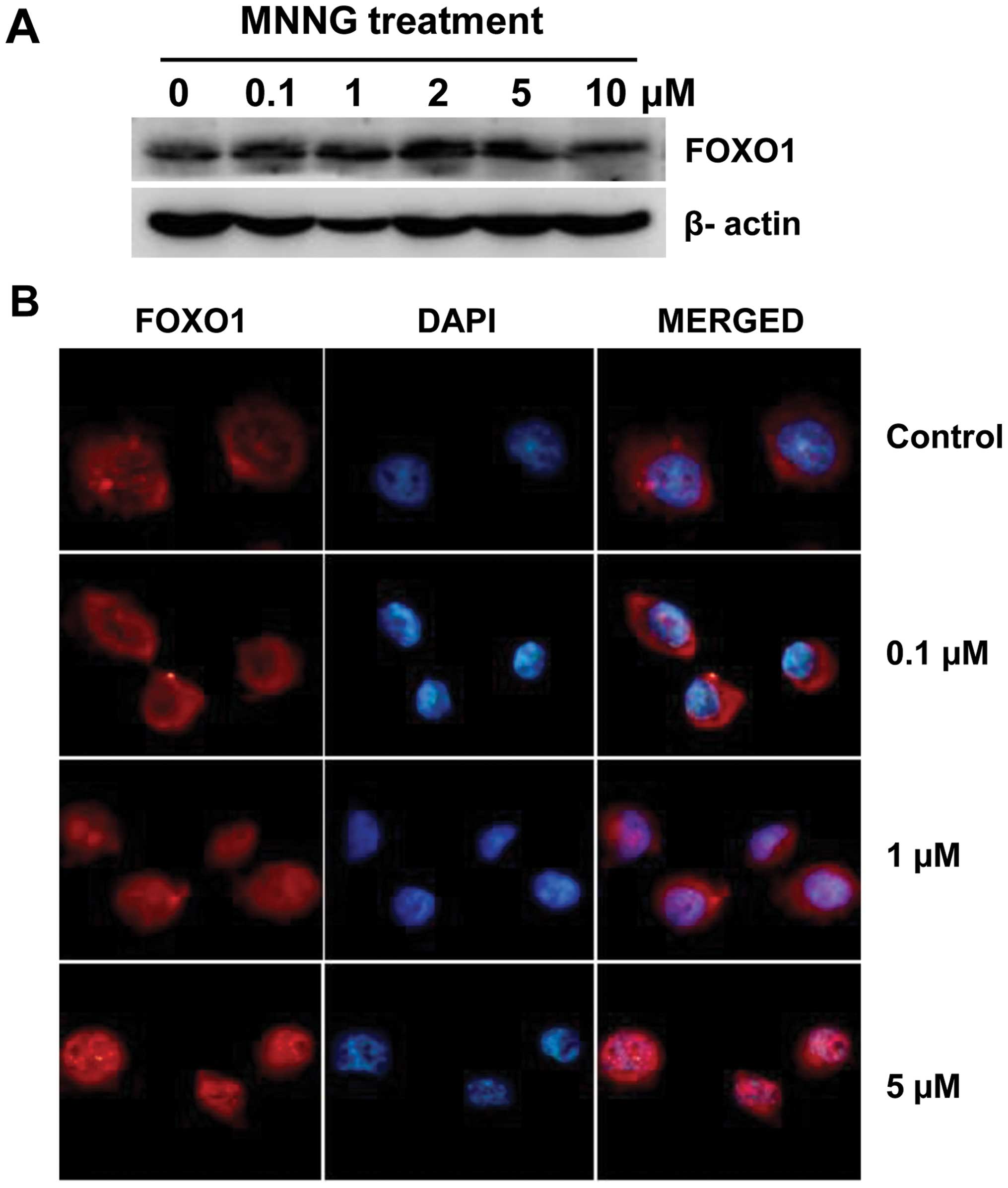
protein level after MNNG treatment, to see if FOXO1 is involved in
DNA damage repair in H1299 cells. Of note is the significant
increase in FOXO1 expression using western blotting was observed at
4 h after MNNG treatment, thus the analyses described below were
carried out at this time-point. Western blotting showed that
significant increase in the expression level of FOXO1 was found at
various concentrations but not at 10 μM (Fig. 3A). This result indicates that
tolerable DNA damage can induce FOXO1 expression, while excessive
cell injury suppresses FOXO1 expression.
Transcriptional factor FOXO1, when maintained in the
nucleus, is involved in negative regulation of cell cycle
progression. We were interested to see whether MNNG treatment give
rise to nuclear translocation of FOXO1 in H1299 cells. Therefore,
immunofluorescence investigation was carried out to visualize the
localization of FOXO1. Cells were subjected to 0–5 μM MNNG
treatment for 30 min and subsequent culture for 4 h prior to
staining and microscopic examination. As shown in Fig. 3B, the endogenous FOXO1 distributed
almost completely in the cytoplasm in untreated H1299 cells, and
nuclear staining was negligible. Compared with the control, nuclear
fluorescence was distinctly enhanced, indicating that FOXO1
proteins significantly accumulated in the nuclei, in H1299 cells
treated with increasing doses of MNNG. These results provide
evidence that nuclear translocation of FOXO1 takes place as a
response to MNNG. Results of western blotting and
immunofluorescence were in agreement, DNA damage promotes the
transcriptional activity of FOXO1 in H1299 cells.
FOXO1 is involved in DNA damage response
by enhancing target gene expression
To further investigate the role of FOXO1 in DNA
damage response, we also examined its target genes, such as
p27Kip1, GADD45 and Bim by western blotting after MNNG
treatment. Commonly, confronted with a variety of stresses
involving DNA damage, cells can rapidly take protective responses.
After treatment with 5 μM MNNG for 30 min, H1299 cells were
incubated in growth medium for different time-points (0, 2, 4, 6
and 8 h). As shown in Fig. 4A, the
transient increase in FOXO1 expression was observed at 4 h after
MNNG treatment, but the content of FOXO1 declined to the original
level 6 h later. Simultaneously, the expression of
p27Kip1, GADD45 and Bim displayed a continuous
upregulation from 4 to 8 h after MNNG treatment. Thus, the induced
p27Kip1, GADD45 and Bim proteins serve as regulators of
the cell cycle, DNA repair and apoptosis to execute their
functions. These data demonstrate further that FOXO1 is involved in
DNA damage response by modulating target gene expression in H1299
cells.
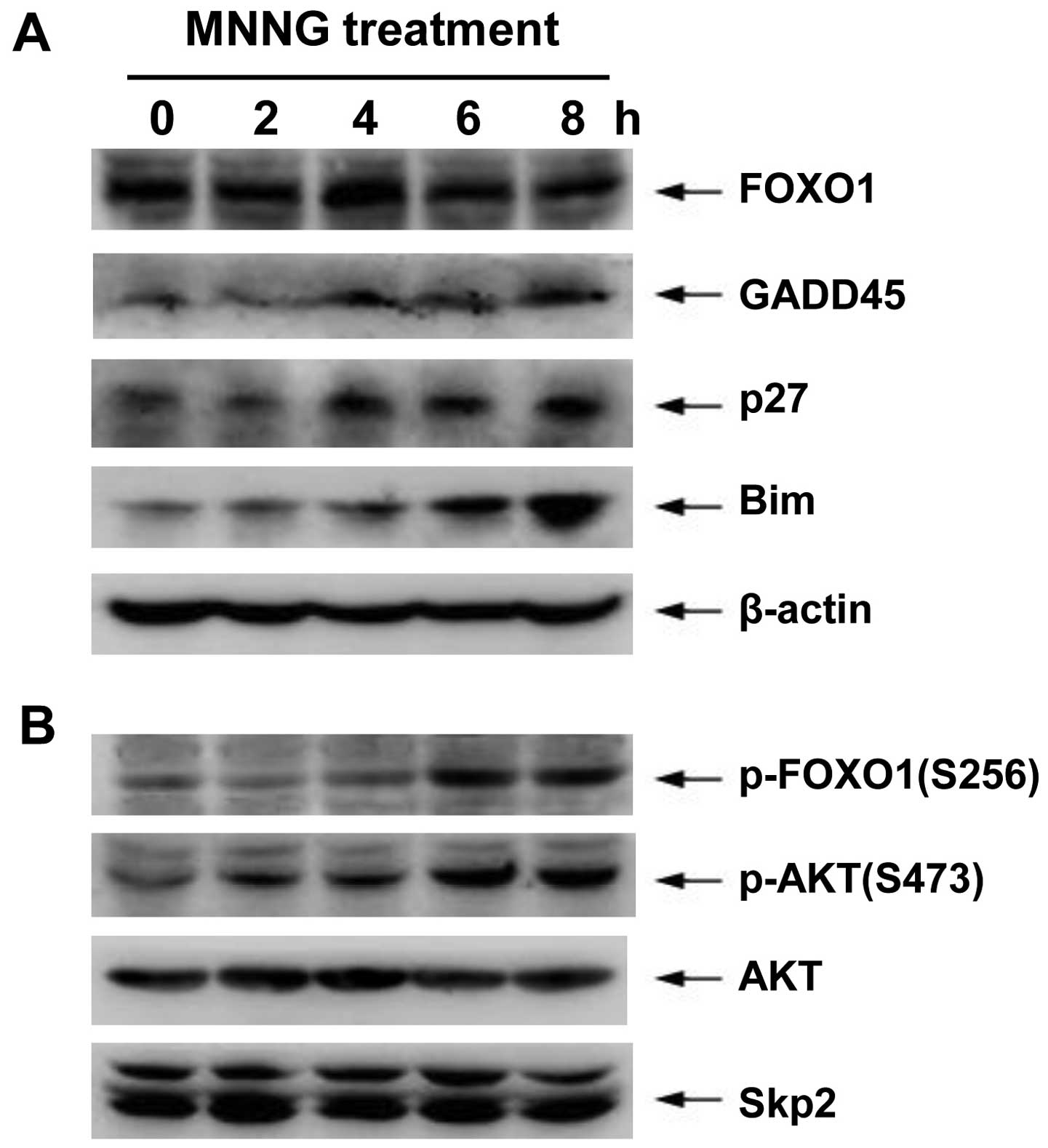 | Figure 4.Protein expression of FOXO1 target
genes and PI3K/AKT pathway during DNA damage. H1299 cells were
incubated with 5 μM MNNG for 30 min. Thereafter, cells were
incubated in growth medium for 0, 2, 4, 6 or 8 h before lysis.
FOXO1, p27Kip1, GADD45, Bim, p-FOXO1 (S256), p-AKT
(S473), AKT, Skp2 and β-actin protein levels were determined by
western blotting using the appropriate antibodies. Data are
representative of at least three independent experiments. |
It is known that AKT-dependent phosphorylation of
FOXO1 on S256 may lead to nuclear exclusion of FOXO1, and
subsequently Skp2 protein mediates FOXO1 degradation through
ubiquitin-proteasome system. Therefore, to study the effect on the
transcriptional activity of FOXO1 via the PI3K-AKT pathway, we
further determined the expression or activity of associated
proteins using western blotting during DNA damage response in H1299
cells. These results showed that both the phosphorylation levels of
AKT on S473 (AKT active form) and FOXO1 on S256 mediated by AKT
significantly increased at 6–8 h after MNNG treatment, coinciding
with the reduction of FOXO1 content. However, the MNNG exposure did
not cause changes in total proteins of AKT and Skp2 (Fig. 4B).
FOXO1-induced cell cycle arrest responds
to DNA damage
When DNA damage is induced, the cell cycle
progression will be blocked to provide time for repairing DNA
damage. Since FOXO1 increased the expression levels of
p27Kip1 and GADD45, cell cycle should be arrested,
thereby repairing DNA damage. In order to further examine the DNA
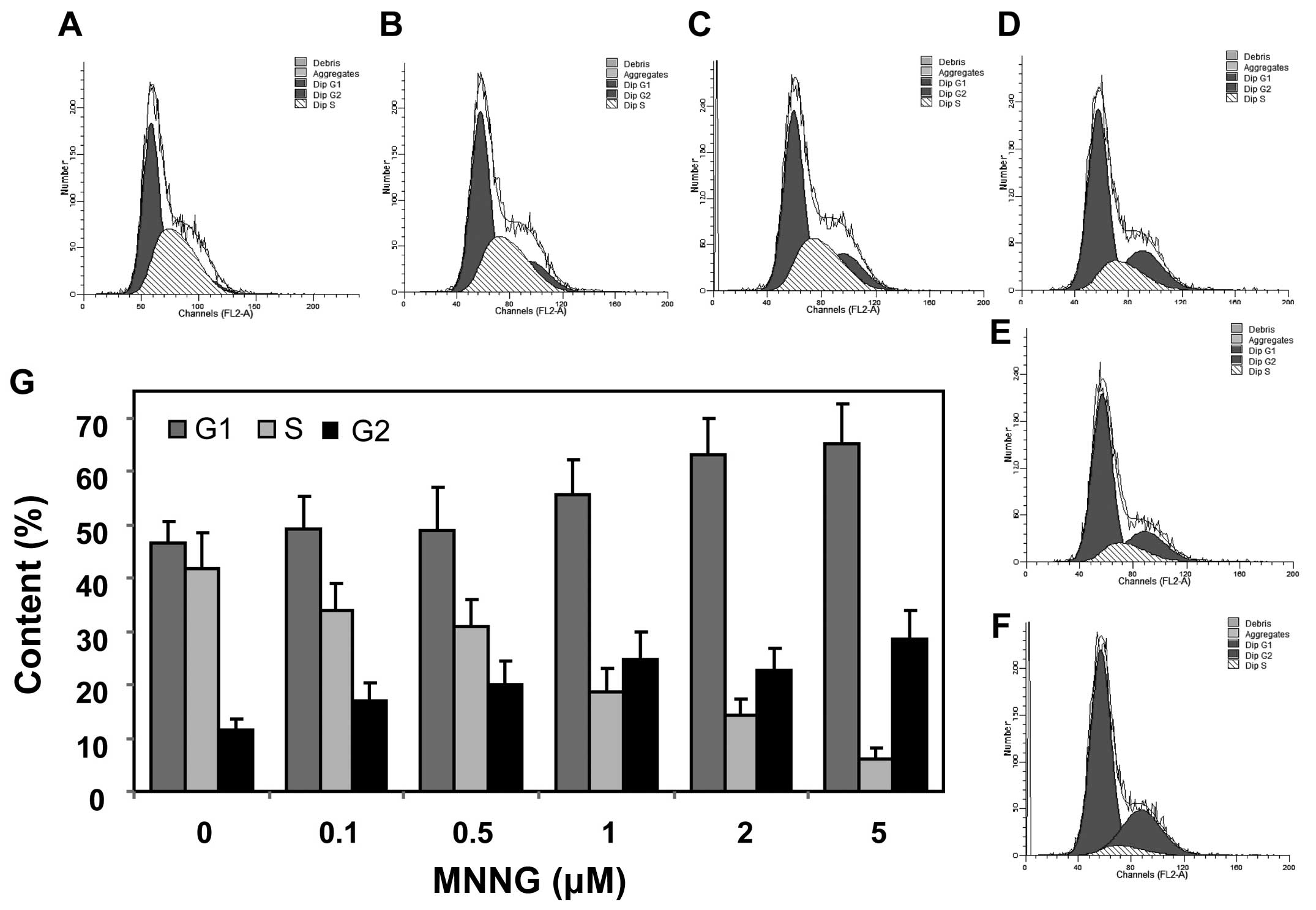
repair effect connected with FOXO1, we analyzed cell cycle after
DNA damage in H1299 cells. The results showed that the cells
arrested in G1 and G2 phase were significantly increased from 45.1
and 12.4% to 65.6 and 28.5%, respectively, after 5 μM MNNG
exposure (Fig. 5). Taken together,
these data display that FOXO1 negatively regulates cell cycle
progression, and facilitates DNA damage repair by modulating
expression of p27Kip1 and GADD45 in response to MNNG in
H1299 cells.
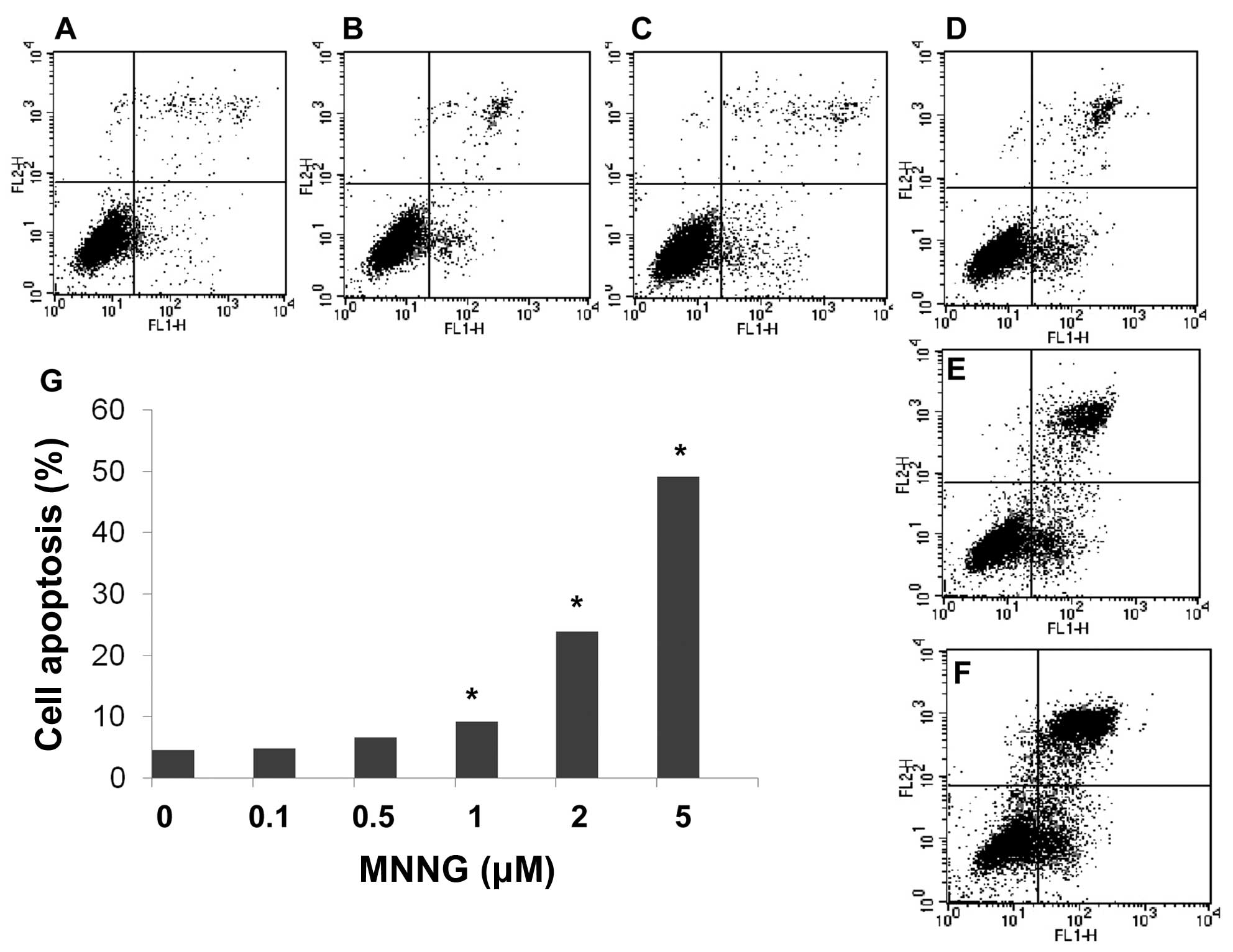
FOXO1 induces apoptosis during DNA
damage
Bim is a FOXO1 target gene serving as a proapoptotic
protein. We found that expression level of Bim increased in a
dose-dependent manner in H1299 cells. Moreover, flow cytometric
analysis using Annexin V and PI double staining further confirmed
apoptosis occurring after MNNG treatment. These results show that
the number of apoptotic cells significantly increased
dose-dependently after exposure to various doses of MNNG for 30 min
(Fig. 6). Thus, results of flow
cytometric analysis are consistent with detection using western
blotting.
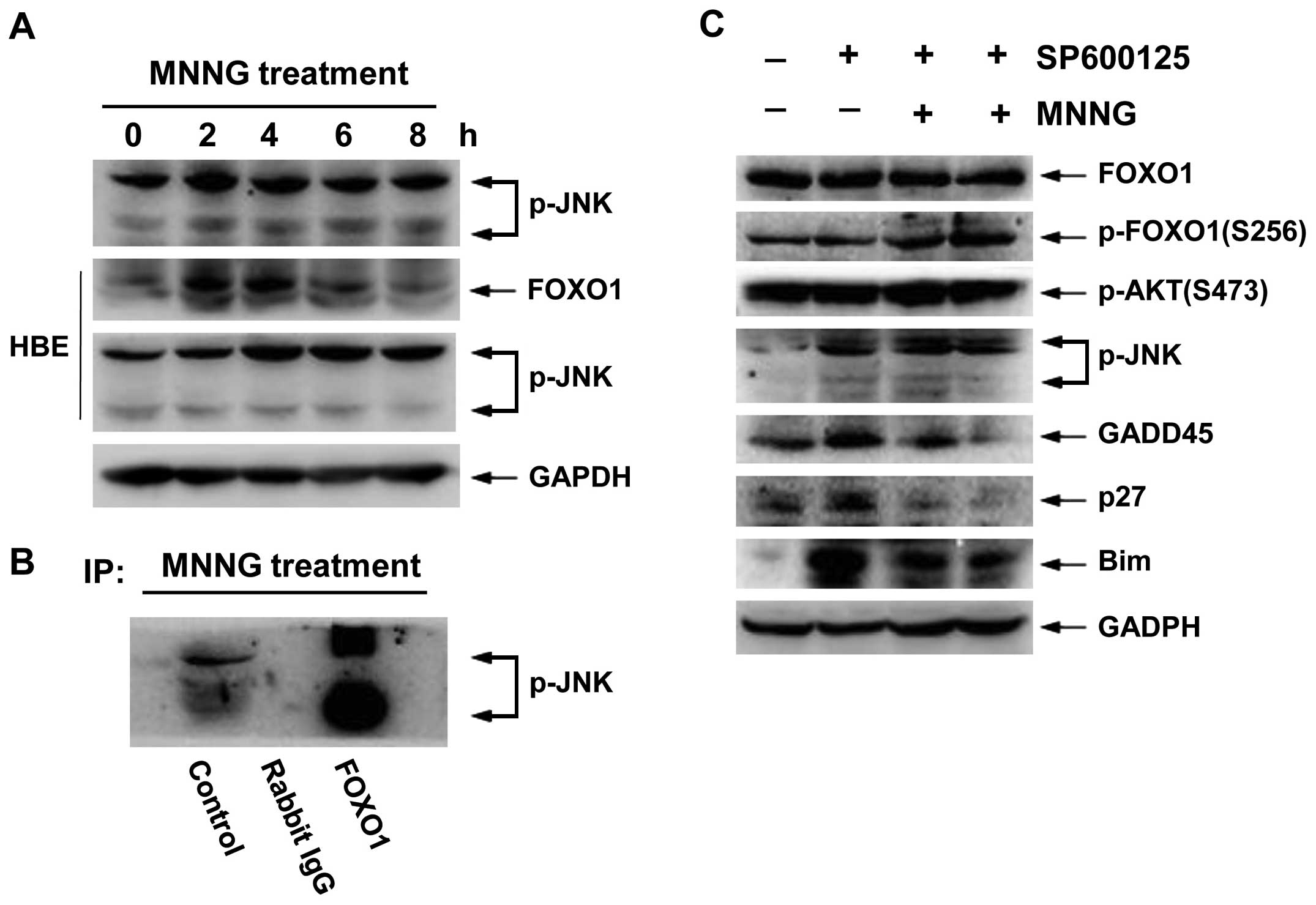
JNK improves the transcriptional activity
of FOXO1 by facilitating nuclear translocation
JNK, also known as stress-activated protein kinase
(SAPK), involves cellular response to a variety of external
stimuli. To define whether JNK pathway was also involved in the
response to MNNG-inflicted DNA damage in H1299 cells, we examined
the expression level of p-JNK (T183, Y185) at various time-points.
As shown in Fig. 7A,
phosphorylation status of JNK was transiently enhanced at 2 and 4 h
after MNNG treatment in H1299 cells, and high activity
phosphorylation status was maintained for 4 h in HBE cells.
Therefore, FOXO1 total protein level correspondingly increased at 4
h in H1299 cells and at 2 and 4 h in HBE cells (Figs. 2A and 7A). These data imply that activation of
JNK may be involved in regulation of transcriptional activity of
FOXO1. Thus, to determine the role of JNK in MNNG response, we
carried out immunoprecipitation analysis. The activated JNK
associated with FOXO1 (Fig. 7B),
suggesting FOXO1 also served as a substrate for JNK. Moreover,
inhibition of JNK activity by SP600125 (5 and 10 μM) reduced
the nuclear translocation of FOXO1 (Fig. 7D). In addition, we found that
SP600125 could suppress the expression levels of FOXO1 target
genes, such as GADD45, p27 and Bim. However, it showed no
significant change in the levels of either FOXO1 total protein or
p-AKT (S473) (Fig. 7C). These
results demonstrated that JNK can function as an important
regulator in MNNG response, promoting nuclear translocation and
target gene expression of FOXO1 by physical interaction with FOXO1
in H1299 cells.
Discussion
DNA damage can be caused by multiple stimuli from
the environment including chemical mutagens. Following genotoxic
stress, an inherent DNA damage response (DDR) is initiated to
eliminate DNA lesions. The DDR serves as a network of molecular
signalling events that modulate DNA repair, cell cycle arrest and
apoptosis (5). In this study, we
adopted MNNG as an alkylating agent that induces chemical DNA
damage, as the molecular mechanism of FOXO1-mediated DNA repair
response in p53-deficient cells is understood. We have found that
treatment of H1299 cells by short-term MNNG results in a
dose-dependent inhibition of proliferation. Interestingly,
viability of H1299 cells displayed a recovery at 48 h after MNNG
exposure in comparison with 24 h, suggesting that MNNG-inflicted
DNA damage might be defused gradually. It is known that alkylating
agent-induced genotoxicity is mainly due to triggering of DNA
single-strand or double-strand breaks. They react with the ring
nitrogens (N) and extracyclic oxygen (O) atoms of DNA bases to
generate a variety of covalent adducts (20). Therefore, we further analyzed the
levels of MNNG-caused DNA damage by assessing the content of DNA
strand breaks in H1299 cells using the comet assay. As cellular DNA
damage becomes serious, more DNA fragments migrate into the comet
tail region, and are quantified by the intensity of fluorescence
according to comet parameters including tail length, tail intensity
and Olive tail moment, representing the levels of DNA damage
(21). Our results exhibited
significant increase in these three parameters following MNNG
treatment, revealing that MNNG gave rise to DNA strand breaks in a
dose-dependent manner. This confirmed that a decline in cell
viability attributed to DNA damage occurred after MNNG
treatment.
In response to DNA strand breaks, cells undergo
corresponding DNA repair process such as homologous recombination
repair (HRR) or the non-homologous end-joining (NHEJ) (22,23).
It is well known that ATM and ATR play critical roles in DNA damage
response by phosphorylating CHK1, CHK2 and p53 (24,25).
FOXO proteins as transcription factors improve expression of a
number of genes that are involved in cell cycle, DNA repair and
cell death. The recovery of cell viability implies that
MNNG-induced DNA damage might be partly repaired in H1299 cells.
Therefore, we assumed that FOXO1-dependent DNA repair may be
subsequently induced after MNNG treatment. In this study, H1299
lung adenocarcinoma cells deficient in p53 gene were utilized,
thereby preventing p53 interference, and is thus appropriate for
investigating the role of FOXO1 in DNA damage repair. We show for
the first time that FOXO1 protein mediates DNA repair response by
facilitating its protein expression and nuclear translocation, and
then promoting expression of target genes in H1299 cells. Indeed,
p27Kip1 and GADD45 expression can block the cell cycle
and subsequently initiate DNA repair, respectively.
p27Kip1, a well-known cyclin-dependent kinase inhibitor
(CKI), can induce G1 phase arrest by inhibiting CDK-mediated
phosphorylation of Rb protein (26). Growth arrest and DNA damage
inducible protein 45 (GADD45) is involved in DNA repair and G2
phase arrest (27). Our results
demonstrate that MNNG treatment of H1299 cells induced cell cycle
arrest in either G1 or G2 phase, this coincides with the inducing
of p27Kip1 and GADD45. These results are consistent with
the literature on FOXO3a which describe G1 and G2 arrest induced by
DNA damage in cancer cells (9,10).
In addition, we observed an increase in Bim expression after MNNG
treatment. Bim is a proapoptotic member of the Bcl-2 family, and
ectopic expression of Bim is sufficient for triggering apoptosis in
a variety of cell types (28).
Apoptosis is induced after MNNG treatment, suggesting that FOXO1 is
involved in cell apoptosis in response to MNNG-mediated DNA damage
in H1299 cells. Our results are in agreement with the report of
Nakamura and Sakamoto, showing that FOXO1 plays an essential role
in reactive oxygen species-induced apoptosis by enhancing
expression of Bim in HeLa cells (29). Cell cycle arrest and apoptosis
response appear to be two critical mechanisms for eliminating DNA
lesions, the former provides time for repairing DNA damage, and the
latter prevents cells from passing on errant genetic codes that can
lead to disease. Based on the above, it is suggested that FOXO1 can
determine cell fate from survival to death by modulating expression
of different target genes based on the cell context.
The activity of FOXO1 is mainly in control of signal
pathway-dependent posttranslational modification, particularly
phosphorylation. Although the role of FOXO1 in multiple cellular
events such as proliferation, cell cycle arrest, apoptosis and
oxidative stress has been well studied (30), the regulatory mechanism of FOXO1 in
response to DNA damage is relatively unclear. Therefore, we further
investigated the relevant signal pathways involved in
FOXO1-dependent DNA damage repair.
Since FOXO1 plays an important role in cell cycle
arrest, DNA repair and apoptosis during MNNG response in H1299
cells, it is necessary to understand the regulatory mechanism of
FOXO1. In addition to expression level, the transcriptional
activity of FOXO1 is mainly regulated by posttranslational
modification, especially signal pathway-dependent phosphorylation.
It has been reported that PI3K-AKT signaling pathway is activated
and contributes to abolish FOXO-dependent cell cycle arrest in
hematopoietic cells during DNA damage (12). Besides, insulin treatment decreases
endogenous FOXO1 proteins via proteasomal degradation in HepG2
cells, and the degradation is mediated by the PI3K-AKT pathway
(31,32), indicating that PI3K-AKT pathway may
be a negative regulator of FOXO1 in response to DNA damage.
Therefore, we examined the levels of AKT-phosphorylated FOXO1 on
S256 and relevant proteins of PI3K/AKT pathway in H1299 cells. As
shown in the results, phosphorylation levels of both FOXO1 and AKT
were improved during MNNG-generated DNA damage, but the total FOXO1
expression was at its former level at the same time. These
observations suggest that PI3K/AKT pathway might restrain DNA
repair by causing the degradation of FOXO1.
In contrast, c-Jun-N-terminal kinase (JNK) pathway
appears to play a key role in response to environmental stresses
including DNA damage (33). It is
established that JNK could directly phosphorylate DAF-16 and FOXO4,
thereby regulating their transcriptional activity, however,
association between JNK and FOXO1 is as yet unidentified (18,34).
Here, we observed an increase in level of activated JNK (pT183,
pY185) by western blotting after MNNG treatment in both H1299 and
HBE cells (Fig. 7A). Furthermore,
the result by immunoprecipitation analysis showed that JNK could
directly interact with FOXO1 (Fig.
7B), suggesting FOXO1 may be a phosphorylated substrate for
JNK. In Drosophila, JNK has been demonstrated to promote the
nuclear translocation and target gene expression of FoxO1 (35). Using inhibitor of JNK, we observed
that inhibition of JNK suppressed the nuclear translocation of
FOXO1 preventing its target gene expression. These data suggest
that FOXO1 serving as a substrate, may be involved in JNK
pathway-mediated DNA repair response. Thus, FOXO1-dependent DNA
damage repair is regulated by JNK.
In conclusion, these findings suggest for the first
time that FOXO1 is a promising candidate substrate for JNK, and
FOXO1-dependent of DNA damage repair may be regulated by JNK
pathway positively in lung cancer H1299 cells.
References
|
1.
|
Hoeijmakers JH: DNA damage, aging, and
cancer. N Engl J Med. 361:1475–1485. 2009. View Article : Google Scholar
|
|
2.
|
Sedgwick B, Bates PA, Paik J, Jacobs SC
and Lindahl T: Repair of alkylated DNA: recent advances. DNA
Repair. 6:429–442. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Maher RL, Branagan AM and Morrical SW:
Coordination of DNA replication and recombination activities in the
maintenance of genome stability. J Cell Biochem. 112:2672–2682.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Fu D, Calvo JA and Samson LD: Balancing
repair and tolerance of DNA damage caused by alkylating agents. Nat
Rev Cancer. 12:104–120. 2012.
|
|
5.
|
Zhou BB and Elledge SJ: The DNA damage
response: putting checkpoints in perspective. Nature. 408:433–439.
2012.PubMed/NCBI
|
|
6.
|
Lam M, Carmichael AR and Griffiths HR: An
aqueous extract of Fagonia cretica induces DNA damage, cell
cycle arrest and apoptosis in breast cancer cells via FOXO3a and
p53 expression. PLoS One. 7:e401522012.PubMed/NCBI
|
|
7.
|
Greer EL and Brunet A: FOXO transcription
factors at the interface between longevity and tumor suppression.
Oncogene. 24:7410–7425. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Maiese K, Chong ZZ, Shang YC and Hou J:
Clever cancer strategies with FoxO transcription factors. Cell
Cycle. 7:3829–3839. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Tran H, Brunet A, Grenier JM, Datta SR,
Fornace AJ Jr, DiStefano PS, Chiang LW and Greenberg ME: DNA repair
pathway stimulated by the Forkhead transcription factor FOXO3a
through the Gadd45 protein. Science. 296:530–534. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Lei H and Quelle FW: FOXO transcription
factors enforce cell cycle checkpoints and promote survival of
hematopoietic cells after DNA damage. Mol Cancer Res. 7:1294–1303.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
de Keizer PL, Packer LM, Szypowska AA,
Riedl-Polderman PE, van den Broek NJ, de Bruin A, Dansen TB, Marais
R, Brenkman AB and Burgering BM: Activation of forkhead box O
transcription factors by oncogenic BRAF promotes p21cip1-dependent
senescence. Cancer Res. 70:8526–8536. 2010.PubMed/NCBI
|
|
12.
|
Huang H and Tindall DJ: Dynamic FoxO
transcription factors. J Cell Sci. 120:2479–2487. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Yang JY and Hung MC: A new fork for
clinical application: targeting forkhead transcription factors in
cancer. Clin Cancer Res. 15:752–757. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Vogt PK, Jiang H and Aoki M: Triple layer
control: phosphorylation, acetylation and ubiquitination of FOXO
proteins. Cell Cycle. 4:908–913. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Rena G, Prescott AR, Guo S, Cohen P and
Unterman TG: Roles of the forkhead in rhabdomyosarcoma (FKHR)
phosphorylation sites in regulating 14-3-3 binding, transactivation
and nuclear targeting. Biochem J. 354:605–612. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Essers MA, Weijzen S, Vries-Smits AM,
Saarloos I, de Ruiter ND, Bos JL and Burgering BM: FOXO
transcription factor activation by oxidative stress mediated by the
small GTPase Ral and JNK. EMBO J. 23:4802–4812. 2004. View Article : Google Scholar
|
|
17.
|
Fu Z and Tindall DJ: FOXOs, cancer and
regulation of apoptosis. Oncogene. 27:2312–2319. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Tice RR, Agurell E, Anderson D, Burlinson
B, Hartmann A, Kobayashi H, Miyamae Y, Rojas E, Ryu JC and Sasaki
YF: Single cell gel/comet assay: guidelines for in vitro and in
vivo genetic toxicology testing. Environ Mol Mutagen. 35:206–221.
2000. View Article : Google Scholar
|
|
19.
|
Dhawan A, Bajpayee M and Parmar D: Comet
assay: a reliable tool for the assessment of DNA damage in
different models. Cell Biol Toxicol. 25:5–32. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Drabløs F, Feyzi E, Aas PA, Vaagbø CB,
Kavli B, Bratlie MS, Peña-Diaz J, Otterlei M, Slupphaug G and
Krokan HE: Alkylation damage in DNA and RNA - repair mechanisms and
medical significance. DNA Repair. 3:1389–1407. 2004.PubMed/NCBI
|
|
21.
|
Cok I, Ulutaş OK, Okuşluk O, Durmaz E and
Demir N: Evaluation of DNA damage in common carp (Cyprinus
carpio L.) by comet assay for determination of possible
pollution in Lake Mogan (Ankara). Sci World J. 11:1455–1461.
2011.PubMed/NCBI
|
|
22.
|
Schroering AG and Williams KJ: Rapid
induction of chromatin-associated DNA mismatch repair proteins
after MNNG treatment. DNA Repair. 7:951–969. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Jasin M: Homologous repair of DNA damage
and tumori genesis: the BRCA connection. Oncogene. 21:8981–8993.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Abraham RT: Cell cycle checkpoint
signaling through the ATM and ATR kinases. Genes Dev. 15:2177–2196.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Matsuoka S, Ballif BA, Smogorzewska A,
McDonald ER III, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini
N, Lerenthal Y, Shiloh Y, Gygi SP and Elledge SJ: ATM and ATR
substrate analysis reveals extensive protein networks responsive to
DNA damage. Science. 316:1160–1166. 2007. View Article : Google Scholar
|
|
26.
|
Hirano M, Hirano K, Nishimura J and
Kanaide H: Transcriptional up-regulation of p27 (Kip1) during
contact-induced growth arrest in vascular endothelial cells. Exp
Cell Res. 271:356–367. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Hildesheim J and Fornace AJ Jr: Gadd45a:
an elusive yet attractive candidate gene in pancreatic cancer. Clin
Cancer Res. 8:2475–2479. 2002.PubMed/NCBI
|
|
28.
|
Stahl M, Dijkers PF, Kops GJ, Lens SM,
Coffer PJ, Burgering BM and Medema RH: The forkhead transcription
factor FoxO regulates transcription of p27Kip1 and Bim
in response to IL-2. J Immunol. 168:5024–5031. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Nakamura T and Sakamoto K: Forkhead
transcription factor FOXO subfamily is essential for reactive
oxygen species-induced apoptosis. Mol Cell Endocrinol. 281:47–55.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Maiese K, Chong ZZ, Hou J and Shang YC:
The ‘O’ class: crafting clinical care with FoxO transcription
factors. Adv Exp Med Biol. 665:242–260. 2009.
|
|
31.
|
Matsuzaki H, Daitoku H, Hatta M, Tanaka K
and Fukamizu A: Insulin-induced phosphorylation of FKHR (Foxo1)
targets to proteasomal degradation. Proc Natl Acad Sci USA.
100:11285–11290. 2003. View Article : Google Scholar
|
|
32.
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Lüpertz R, Chovolou Y, Unfried K,
Kampkötter A, Wätjen W and Kahl R: The forkhead transcription
factor FOXO4 sensitizes cancer cells to doxorubicin-mediated
cytotoxicity. Carcinogenesis. 29:2045–2052. 2008.PubMed/NCBI
|
|
34.
|
Wang MC, Bohmann D and Jasper H: JNK
extends life span and limits growth by antagonizing cellular and
organism-wide responses to insulin signaling. Cell. 121:115–125.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Hong YK, Lee S, Park SH, Lee JH, Han SY,
Kim ST, Kim YK, Jeon S, Koo BS and Cho KS: Inhibition of JNK/dFOXO
pathway and caspases rescues neurological impairments in Drosophila
Alzheimer’s disease model. Biochem Biophys Res Commun. 419:49–53.
2012.PubMed/NCBI
|