Introduction
Colorectal cancer is a major health issue and over
50,000 US residents alone are expected to die from this disease
this year (1). A crucial defect in
the vast majority of colorectal tumors is the overexpression of the
oncoprotein β-catenin. This is primarily due to the loss of the
tumor suppressor adenomatous polyposis coli (APC), which normally
directs the intracellular destruction of β-catenin, or activating
mutations in β-catenin itself (2).
Unfortunately, knowledge on the devastating impact of these genetic
mutations has not yet translated into improved therapy.
Aside from genetic mutations, epigenetic changes are
an underlying cause of tumorigenesis. Most prominently, such
epigenetic changes involve the methylation of DNA on cytosine
residues and the modification of histones by acetylation and
methylation (3,4). In contrast to DNA methylation and
histone acetylation, the methylation of histones was only recently
validated as a major epigenetic mechanism. Methylation occurs on
lysine and arginine residues at multiple sites on histones and the
tight regulation of the histone methylation status is absolutely
required for normal cell physiology and safeguards against aberrant
cell growth. Thus, enzymes affecting histone methylation play
seminal roles in cellular homeostasis and, accordingly,
dysregulation of both histone methyltransferases as well as the
opposing demethylases is thought to be capable of inducing cancer
(5,6). However, the roles of the enzymes
determining histone methylation in colorectal tumors are largely
unexplored.
The family of human Jumonji C domain containing
proteins comprises 30 members, many of which have been shown to
function as histone demethylases (7). These include the four related
lysine-specific demethylase 4 (KDM4) proteins, KDM4A-D (8). They can demethylate histone H3 on
lysines 9 and 36 as well as histone H1.4 on lysine 26 (9–15).
Notably, KDM4A, B and C are overexpressed in human breast tumors
and promote proliferation of breast tumor cells, in part due to
their ability to function as cofactors of estrogen receptor α
(16–21). Likewise, all four KDM4 proteins
were shown to coactivate the androgen receptor and may thereby
contribute to prostate tumor formation (22–24).
Despite these examples of overlapping function, KDM4 proteins can
also behave differently from each other (8). For instance, KDM4B seems to be less
catalytically active (9,12) and is the only KDM4 protein that is
robustly overexpressed under hypoxia (25,26).
Here, we explored the role of KDM4B in colorectal cancer.
Materials and methods
Lentivirus mediated KDM4B downregulation
with shRNA
To generate an inducible miRshRNA entry vector,
annealed oligonucleotides KDM4B-shRNA sense
(5′-AGCGAGCGCTGACACTGTATTCTTATTAGTGAAGCCACAGATGTAATAAGAATACAGTGTCAGCGC-3′)
and KDM4B-shRNA antisense
(5′-GGCAGGCGCTGACACTGTATTCTTATTACATCTGTGGCTTCACTAATAAGAATACAGTGTCAGCGCT-3′)
were cloned into pEN_TTRmiRc2 (Addgene). To transfer sequences into
the lentiviral destination vector, the miRshKDM4B entry vector was
incubated together with pSLIK Hygro (Addgene) and Clonase 2
(Invitrogen) per company instructions. The resulting miRshKDM4B
lentiviral expression vector was cotransfected along with packaging
plasmids pMD2.G (Addgene), pMDL/RRE g/p (Addgene) and pRSV-Rev
(Addgene) into 293T cells. Cells were seeded at ∼60% confluence and
DNA was delivered using the polyethylenimine transfection method
overnight. Polyethylenimine to DNA ratio was 3:1. Forty-eight and
72 h post-transfection, supernatant was harvested and filtered
through a 0.45-μm membrane. Cleared viral supernatant was
concentrated using poly(ethylene glycol)-8000 and used to infect
HT-29 cells (27).
Cell proliferation assay
HT-29 cells that inducibly expressed shRNA targeting
KDM4B were seeded into 96-wells and grown in DMEM medium plus 10%
fetal calf serum. One day thereafter, the cell number was
determined for the first time and defined as number of cells at day
0. Then, cells were mock treated or with 500 ng/ml doxycycline and
cell numbers measured at the indicated days thereafter with the
TACS (Trevigen) MTT
(3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) kit
(28). Averages with standard
errors of triplicate experiments were determined.
Clonogenic assay
Cells were seeded at 5,000 cells per well.
Thereafter, cells were mock treated or with 500 ng/ml doxycycline
and grown in DMEM medium plus 10% fetal calf serum. Media without
and with doxycycline were replenished every four days for twelve
days. The cells were then fixed in 3.7% formaldehyde for 10 min and
stained with 1% crystal violet blue for 30 min. After rinsing with
distilled water, cells were photographed.
Coimmunoprecipitation assays
Human embryonic kidney 293T cells were grown in a
humidified atmosphere in 10% CO2 at 37°C (29). Cells were seeded into 6-cm dishes
that were coated with poly-L-lysine 12–24 h before transfection
(30). Cells were then transiently
transfected by the calcium phosphate coprecipitation method
(31) and lysed 36 h after
transfection with 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 50 mM NaF,
0.25 mM Na3VO4, 0.5% Igepal CA-630, 10
μg/ml leupeptin, 2 μg/ml aprotinin, 1 μg/ml
pepstatin A, 0.5 mM PMSF, 0.2 mM DTT. Immunoprecipitation was
performed with anti-Flag M2 or anti-Myc 9E10 monoclonal antibodies
as described (32). Coprecipitated
proteins were detected by western blotting employing enhanced
chemiluminescence (33).
Glutathione S-transferase (GST)
pull-downs
GST fusion proteins were produced in Escherichia
coli and purified as previously described (34). Cell extract containing Myc-tagged
β-catenin protein was prepared from transiently transfected 293T
cells (35). This extract was then
incubated for 3 h at 4°C with GST fusion proteins bound to
glutathione agarose beads utilizing 20 mM HEPES (pH 7.4), 50 mM
NaCl, 1 mM DTT, 0.01% Tween-20, 0.5 mM PMSF as a binding buffer.
After three washes in the same buffer, any bound Myc-tagged
β-catenin was revealed by western blotting employing anti-Myc 9E10
monoclonal antibodies.
Reporter gene assay
HT-29 cells inducibly expressing KDM4B shRNA were
infected with pBARLS lentivirus (kind gift from Dr Randall Moon)
that encodes 12 TCF4 binding elements upstream of luciferase cDNA.
Equal numbers of cells were then split into two groups of
triplicates and treated without and with 500 ng/ml doxycycline for
72 h. Then, cells were lysed as described before (36) and luciferase activities derived
from pBARLS were measured in a Berthold LB9507 luminometer
(37).
Reverse transcription-polymerase chain
reaction (RT-PCR)
RNA was isolated employing TRIzol (Invitrogen) and
utilized in the Access RT-PCR kit (Promega) as described before
(38). Oligonucleotides used were
5′-GTGACCGCGACTTTTCAAAGC-3′ and 5′-CGTTGCTGGACTG GATTATCAG-3′ for
JUN, 5′-TGAGGAGACACCGCCCAC-3′ and 5′-CAACATCGATTTCTTCCTCATCTTC-3′
for MYC, 5′-AAGGCGGAGGAGACCTGCGCG-3′ and
5′-ATCGTGCGGGGTCATTGCGGC-3′ for Cyclin D1, and
5′-GAGCCACATCGCTCAGACACC-3′ and 5′-TGACAAGCTTCCGCTTCTCAGC-3′ for
GAPDH. Resulting PCR products were separated on agarose gels and
stained with ethidium bromide (39).
Chromatin immunoprecipitations
These were performed essentially as described before
(40) with rabbit KDM4B antibodies
either from Bethyl (A301-478A) in case of the JUN promoter or from
Novus Biologicals (NBP1-67802) in case of the Cyclin D1 promoter.
For promoter fragment amplification, nested PCR was employed using
the temperature program 98°C for 2 min; 8 cycles of 98°C for 30
sec, 65°C (−1°C per cycle) for 30 sec, 72°C for 25 sec; 18 cycles
(in the first PCR) or 15 cycles (in the second PCR) of 98°C for 30
sec, 57°C for 30 sec, 72°C for 25 sec (+ 1 sec per cycle), followed
by a final 4-min extension at 72°C (41). The iProof high fidelity DNA
polymerase (Bio-Rad) was used in the first PCR and GoTaq polymerase
with 5X Green buffer (Promega) in the second PCR. The primers for
the first PCR were: Jun-ChIP-2805-for
(5′-GGCAGCCACCGTCACTAGACAGTC-3′) and Jun-ChIP-3184-rev
(5′-GCCACACTCAGTGCAACTCTGAGC-3′), or D1-pro-for6
(5′-GTAACGTCACACGGACTACAGG-3′) and D1-pro-rev5
(5′-GCACACATTTGAAGTAGGACACC-3′). The primers for the second PCR
were: Jun-ChIP-2834-for (5′-CCAAGACGTCAGCCCACAATGCACC-3′) and
Jun-ChIP-3145-rev (5′-GCTCAACACTTATCTGCTACCAGTC-3′), or
D1-ChIP-2726-for (5′-GTTGCAAAGTCCTGGAGCCTCCAG-3′) and
D1-ChIP-2982-rev (5′-CGGTCGTTGAGGAGGTTGGCATCG-3′). The resultant
312 bp JUN promoter and 257 bp Cyclin D1 promoter fragments were
revealed by agarose gel electrophoresis (42).
Results
Overexpression of KDM4B in colorectal
tumors
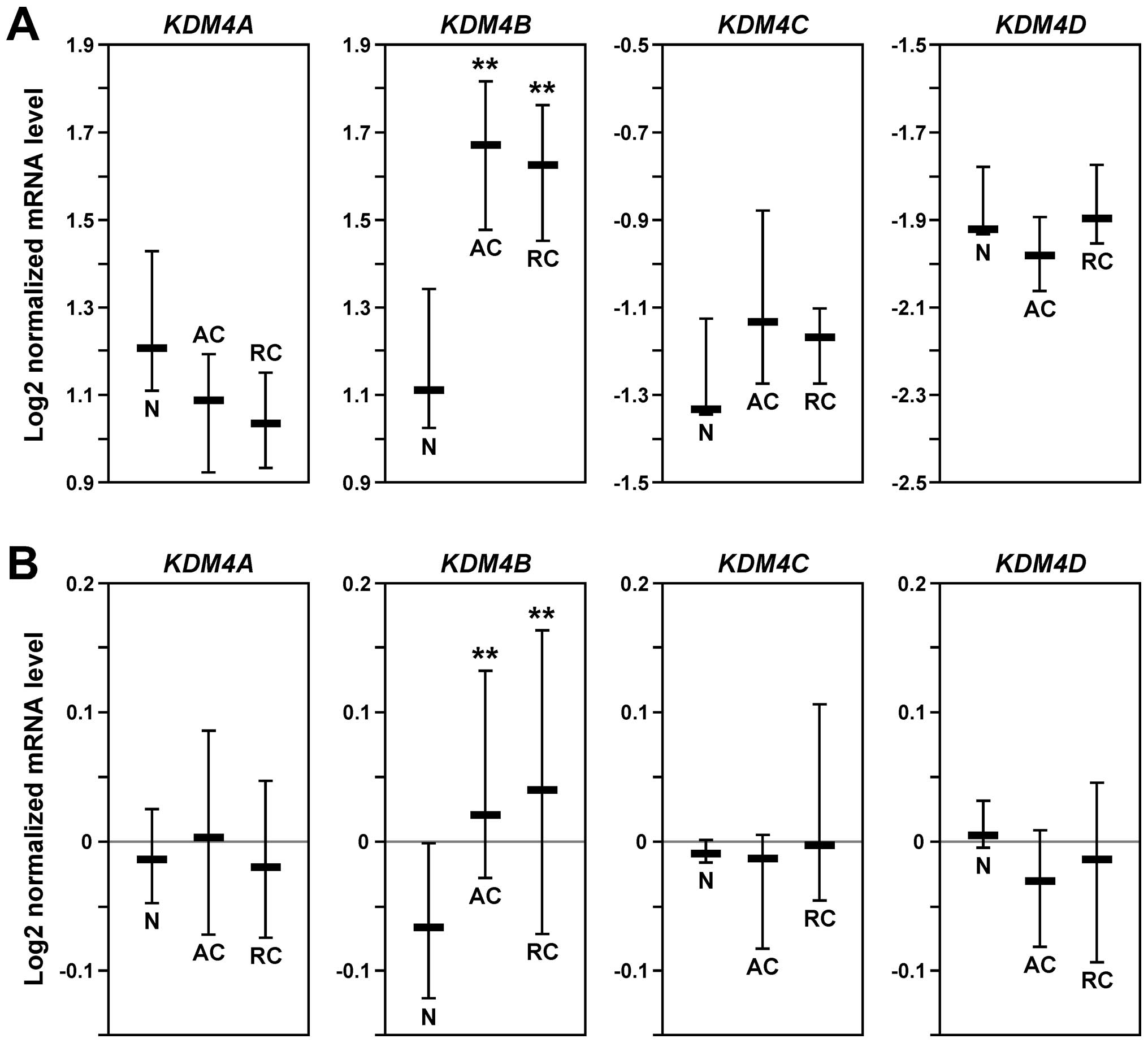
To comprehensively assess the expression pattern of
KDM4 genes in human colorectal tumors, we performed in
silico analyses comparing normal and cancer tissues with the
help of Oncomine (www.oncomine.org). In two independent
data sets (43,44), KDM4B mRNA was significantly
upregulated in both colon and rectal adenocarcinomas compared to
normal colon tissue, whereas no relevant upregulation of KDM4A,
KDM4C or KDM4D was observable (Fig.
1). These data implicate that elevated KDM4B levels, but not
overexpression of any of the other three KDM4 family members, play
a role in the development of colorectal tumors.
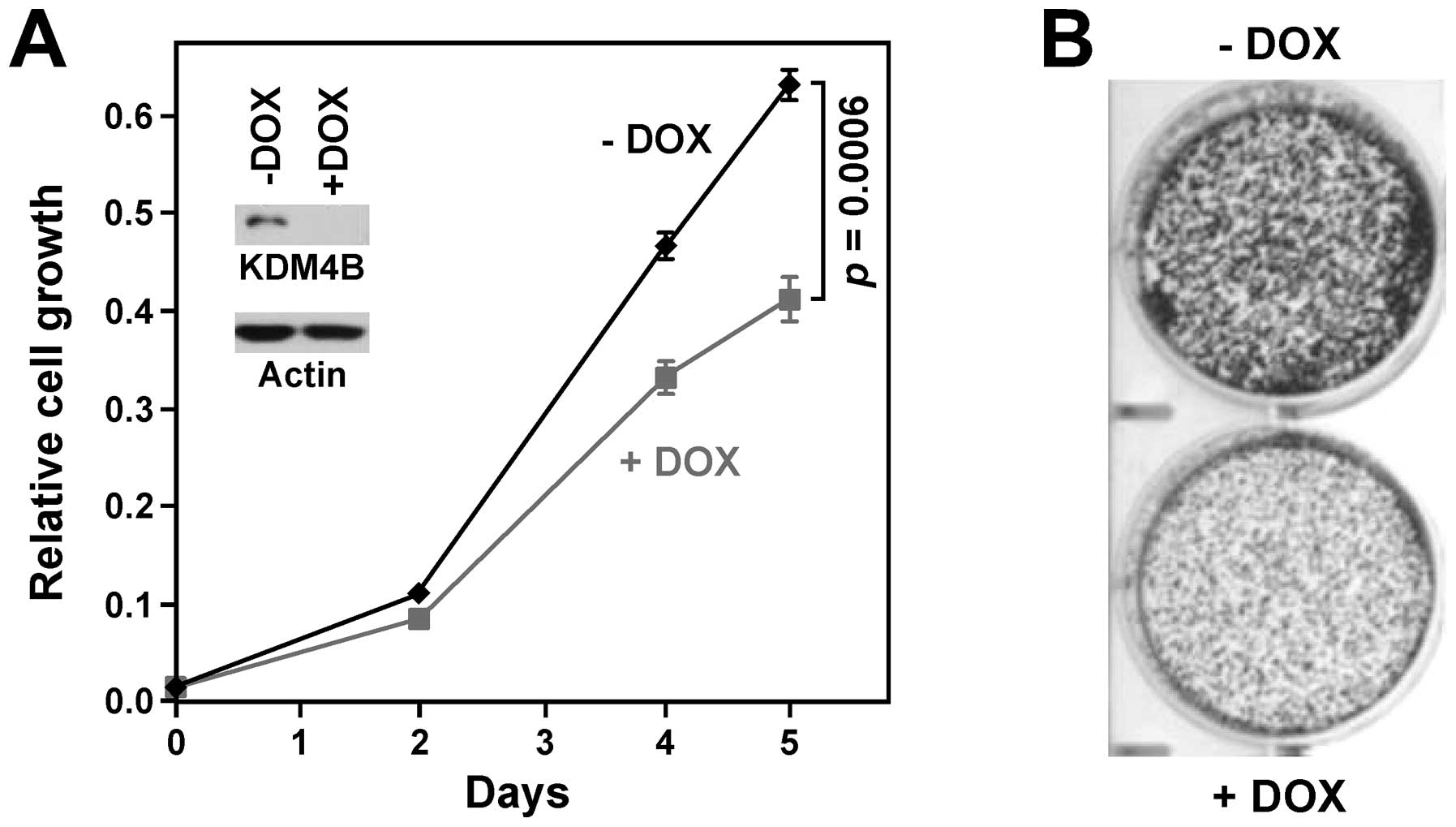
Growth deficit of colon cancer cells
following loss of KDM4B
To test the impact of KDM4B on cell physiology, we
infected human HT-29 colon cancer cells with lentivirus that
doxycycline-inducibly expressed KDM4B shRNA. After selection for
viral integration, these cells were treated with and without
doxycycline, which resulted in efficient downregulation of KDM4B
(Fig. 2A). Importantly,
doxycycline treatment significantly reduced HT-29 cell growth
(Fig. 2A), suggesting that KDM4B
has a pro-growth effect. We also determined the importance of KDM4B
for growing colonies from single cells, another litmus test for
oncogenic activity. When treated with doxycycline to ablate KDM4B
expression, HT-29 cells displayed a markedly reduced ability to
establish colonies (Fig. 2B).
Altogether, these results support the notion that KDM4B promotes
neoplastic growth of colon cells.
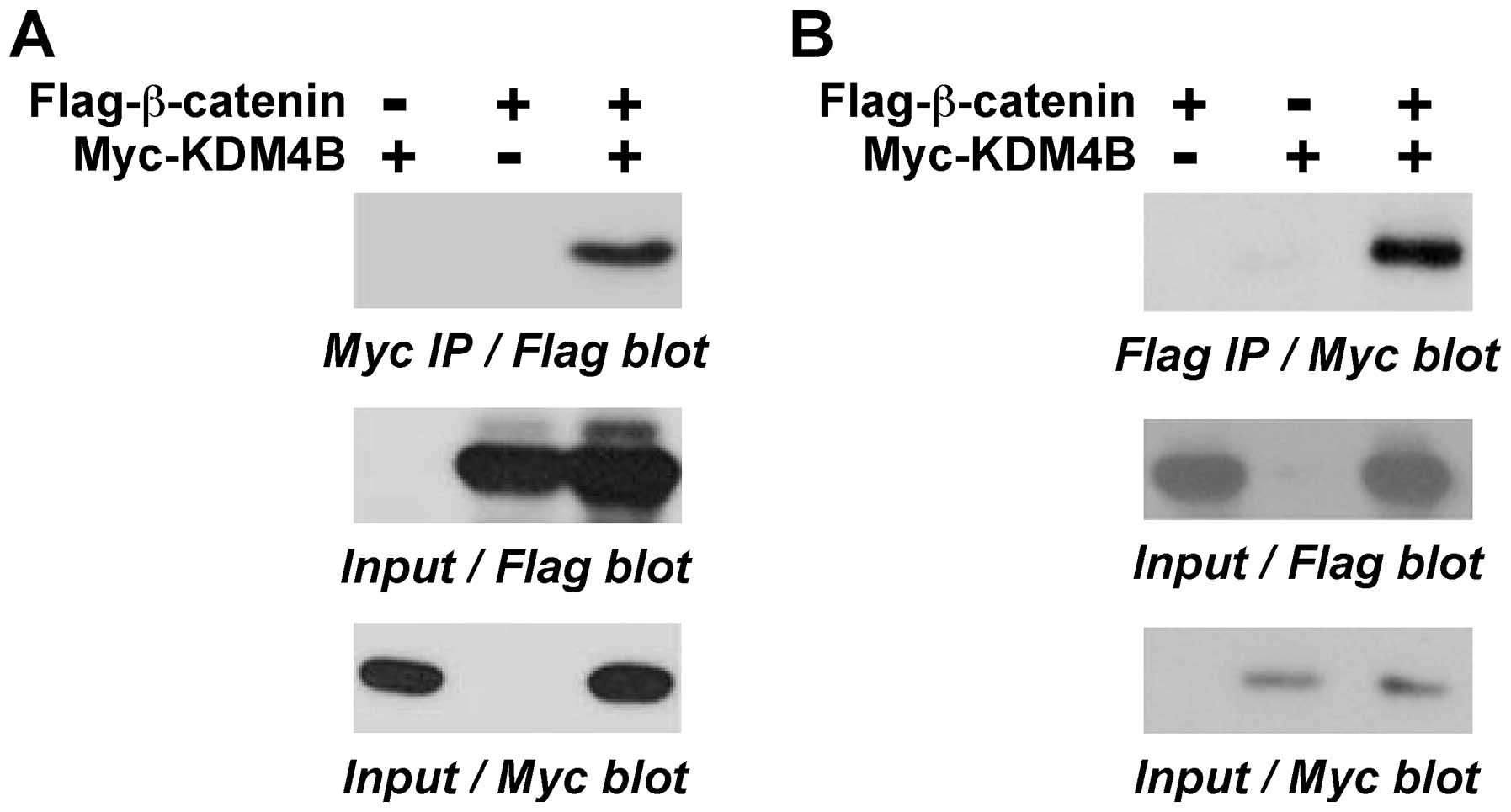
Complex formation of KDM4B with
β-catenin
Overexpression of the β-catenin oncoprotein, the
linchpin of colon tumorigenesis, is observed in >80% of sporadic
colorectal tumors (2). Like KDM4B,
β-catenin is a transcriptional cofactor (45). Therefore, we tested whether KDM4B
might interact with β-catenin. To this end, we coexpressed
Flag-tagged β-catenin with Myc-tagged KDM4B in 293T cells and
performed coimmunoprecipitation experiments. We found that
β-catenin coprecipitated with KDM4B (Fig. 3A). Similarly, in a reverse order
coimmunoprecipitation experiment, KDM4B coprecipitated with
β-catenin (Fig. 3B). These data
demonstrate that KDM4B and β-catenin form complexes in
vivo.
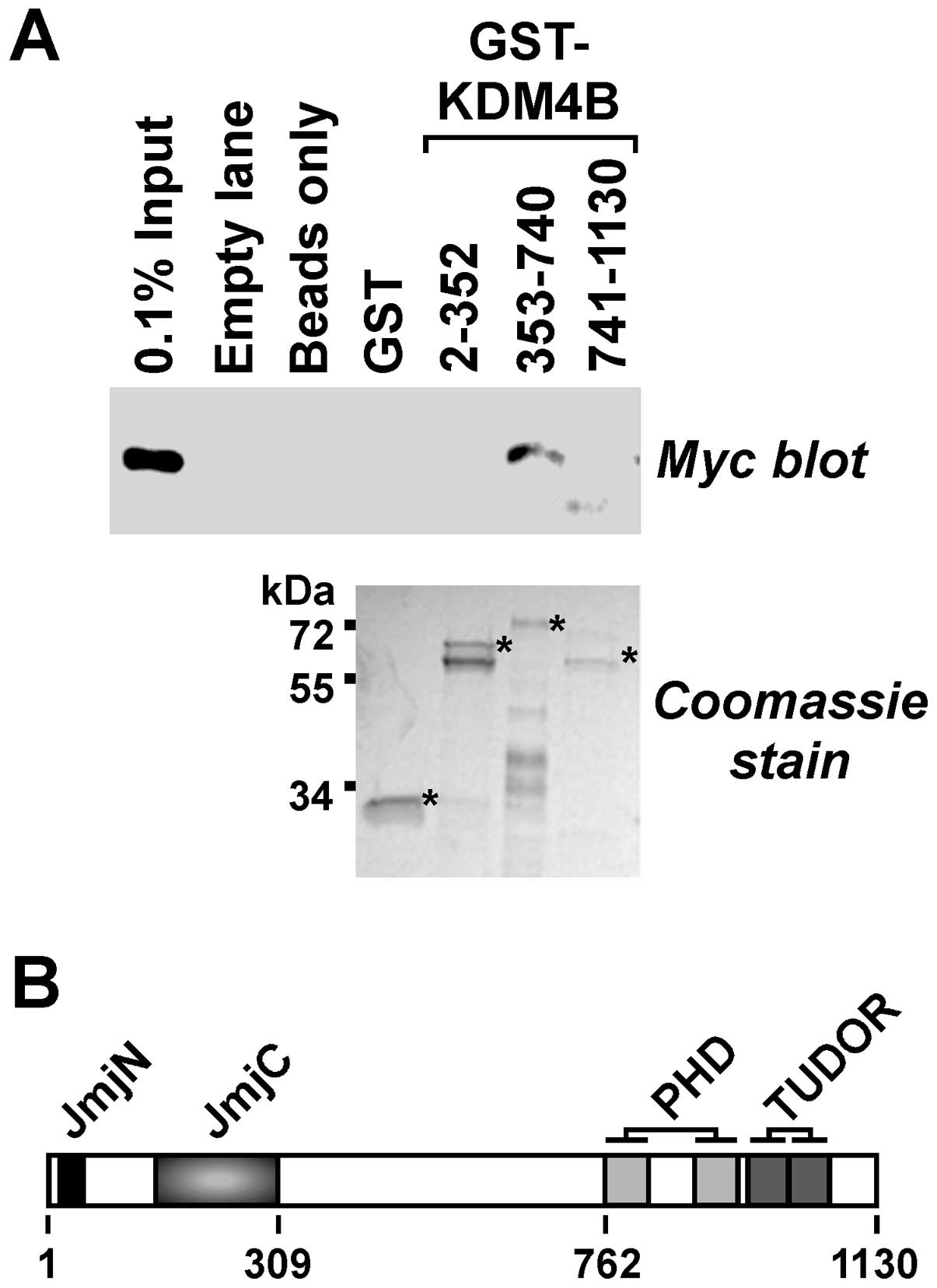
Next, we explored whether β-catenin would bind to
KDM4B in vitro and which domains of KDM4B would be involved.
To this end, we divided KDM4B into three parts and purified
respective GST fusion proteins. These were then bound to
glutathione agarose beads, which were subsequently incubated with a
cell extract containing Myc-tagged β-catenin. No binding of
β-catenin to the GST moiety itself was detectable, but it
interacted with KDM4B amino acids 353–740 (Fig. 4A). These amino acids are devoid of
any known structural motifs (Fig.
4B). In contrast, β-catenin did not appreciably bind to the
N-terminal KDM4B amino acids 2–352 encompassing the catalytic JmjC
and JmjN domains or the C-terminal amino acids 741–1130, which
contain the double PHD and TUDOR domains that are involved in
recognizing methylated histone residues (8,46).
Collectively, our data reveal that KDM4B and β-catenin interact
both in vitro and in vivo.
Coactivation of gene transcription by
KDM4B
The β-catenin protein itself does not directly bind
to DNA. Rather, it is primarily recruited to chromatin in the
intestine by the DNA-binding transcription factor TCF4 (47). Therefore, we reasoned that not only
β-catenin, but also TCF4 might form a complex with KDM4B. To test
this hypothesis, we coexpressed Flag-tagged KDM4B and Myc-tagged
TCF4 in 293T cells, immunoprecipitated with Flag antibodies and
then tested for coprecipitated TCF4 by anti-Myc western blotting.
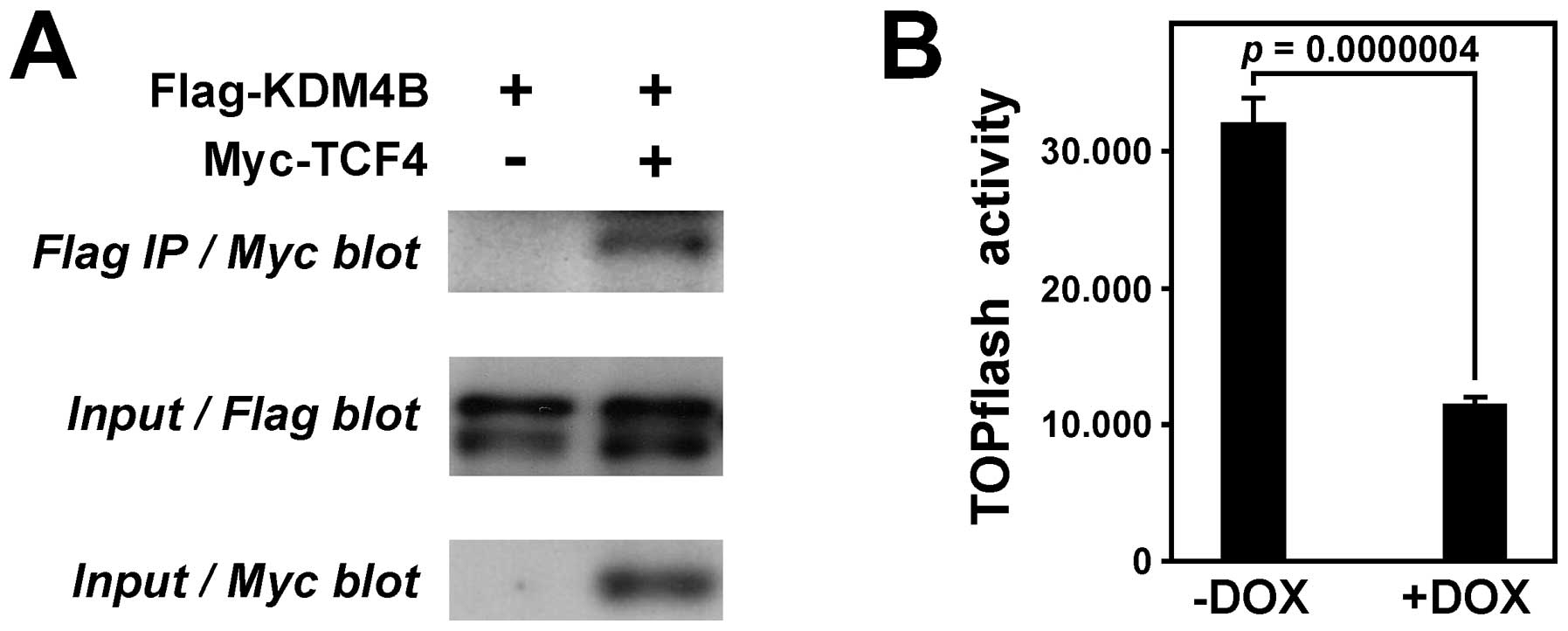
Indeed, TCF4 coprecipitated with KDM4B (Fig. 5A), implicating that a tripartite
complex of TCF4, β-catenin and KDM4B can be formed.
To determine whether KDM4B stimulates or represses
β-catenin/TCF4-dependent transcription, we employed a TOPflash
luciferase reporter system that specifically measures
β-catenin/TCF4-dependent transcription in colon cells (48). A lentiviral vector was employed to
stably introduce the luciferase reporter gene into our HT-29 colon
cancer cells that doxycycline-inducibly expressed KDM4B shRNA. In
the absence of doxycycline, high luciferase activity was expectedly
observable (Fig. 5B). However,
upon depletion of KDM4B after doxycyline treatment, luciferase
activity was ∼3-fold reduced, indicating that KDM4B can stimulate
β-catenin/TCF4-dependent gene transcription.
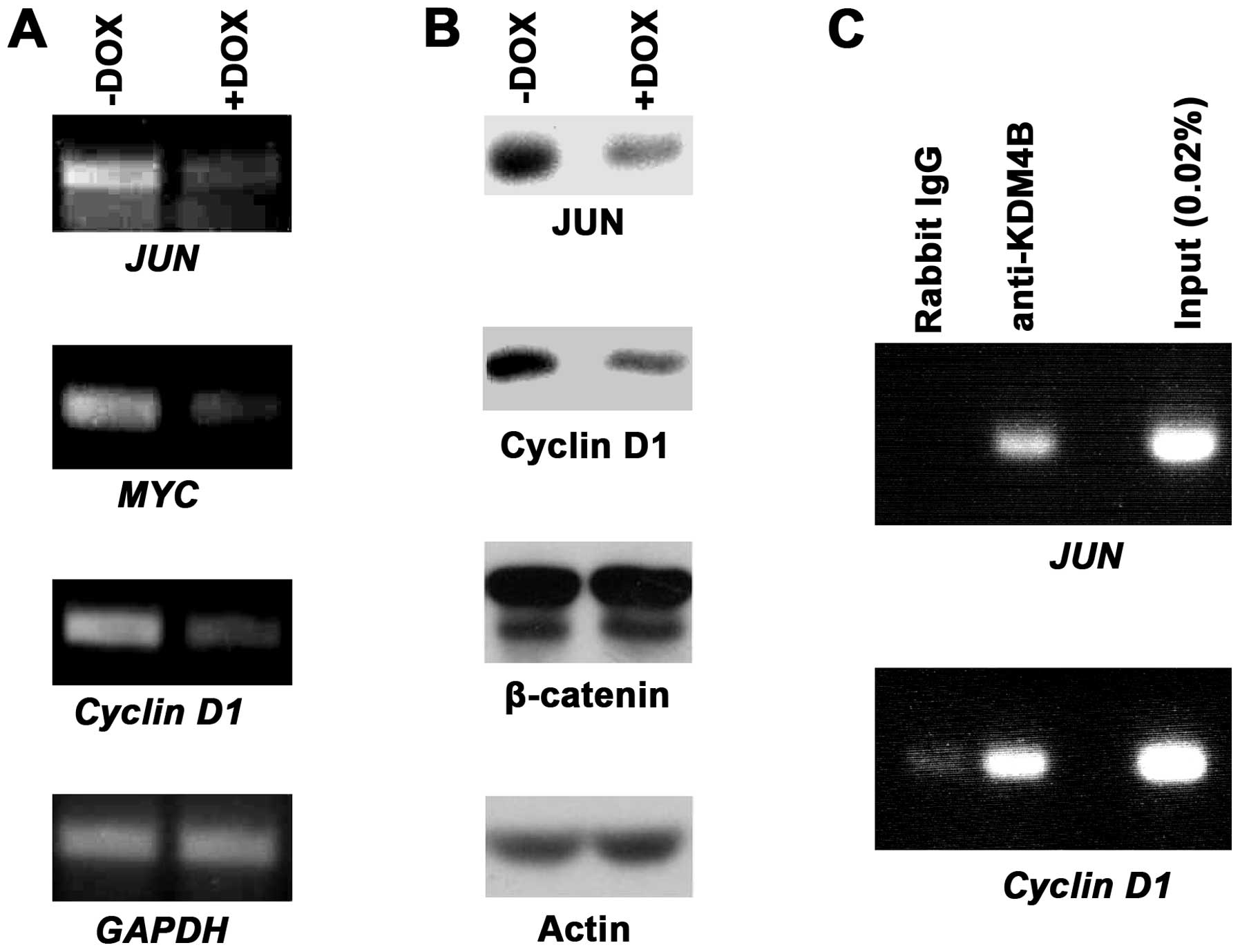
We next sought to assess how endogenous target genes
of β-catenin/TCF4 would be affected by KDM4B downregulation.
Specifically, we studied three oncogenes encoding the JUN or MYC
transcription factor or the cell cycle regulator Cyclin D1, all of
which are bona fide targets of β-catenin/TCF4 (48–51).
When utilizing our doxycycline-inducible KDM4B shRNA expressing
HT-29 cells, we observed that JUN, MYC and Cyclin D1 transcription
became reduced upon KDM4B downregulation, whereas the control GAPDH
was unaffected (Fig. 6A). In
addition, western blotting showed that this also held true at the
protein level for JUN and Cyclin D1 (Fig. 6B), whereas MYC protein expression
was below our detection level (not shown); as controls, neither
β-catenin nor actin protein levels were affected by KDM4B
downregulation (Fig. 6B).
Finally, we assessed whether KDM4B would bind to the
JUN or Cyclin D1 gene promoter. To this end, we performed chromatin
immunoprecipitation experiments. While control IgG antibodies
barely or not at all led to precipitation of promoter fragments,
KDM4B antibodies were able to robustly precipitate both JUN and
Cyclin D1 promoter fragments (Fig.
6C). This indicates that JUN and Cyclin D1 are directly
regulated by KDM4B. Altogether, our data provide evidence that
KDM4B coactivates β-catenin/TCF4-dependent gene transcription.
Discussion
The data described in this report provide novel
information. First, we discovered that the KDM4B histone
demethylase is appreciably upregulated at the mRNA level in
colorectal tumors, whereas none of the other three KDM4 family
members is. Second, we showed that growth and colony formation
ability of HT-29 colon cancer cells is stimulated by KDM4B, and
third, we revealed that KDM4B is capable of forming complexes with
β-catenin and TCF4 and can thereby enhance transcription of
oncogenes such as JUN, MYC and Cyclin D1. Altogether, these data
strongly suggest that KDM4B has oncogenic properties in colorectal
cells.
KDM4B is a histone demethylase that especially
demethylates trimethylated H3K9, H3K36 and H1.4K26 (9,12,14).
In general, both trimethylated H3K9 and H1.4K26 at a gene promoter
are associated with a transcriptionally repressed status of
chromatin (52,53), providing an explanation how KDM4B
may stimulate gene transcription by removing these repressive
marks. On the other hand, trimethylated H3K36 has a variety of
functions: it may promote transcription elongation but suppress
transcription initiation at the promoter (54). Thus, the net effect on gene
expression upon removal of H3K36 trimethylation by KDM4B is
debatable. However, since KDM4 proteins appear to more efficiently
target H3K9 than H3K36 (8), it is
likely that the effect of KDM4B on gene transcription is primarily
governed by its ability to demethylate trimethylated H3K9 (and
H1.4K26) and accordingly KDM4B overexpression should result in
enhanced transcription of target genes like JUN, MYC and Cyclin
D1.
MYC and JUN encode for DNA-binding transcription
factors and are prominent oncogenes (55,56).
They are also β-catenin target genes (48,49)
and upregulated in human colorectal tumors (57,58).
Interestingly, JUN can form complexes with β-catenin and TCF4,
which stabilizes the β-catenin/TCF4 complex and enhances β-catenin
dependent transcription (59,60).
Furthermore, a dominant-negative version of JUN was able to
suppress the tumorigenic potential of HT-29 colon cancer cells and
JUN inactivation also reduced gastrointestinal tumor formation in
the APCMin mouse model, in which inactivation of APC
leads to β-catenin overexpression (59,61).
Likewise, the cell cycle regulator Cyclin D1 is upregulated in
colorectal tumors (62) and its
ablation suppressed tumor formation in APCMin mice
(63). Thus, stimulation of JUN,
MYC and Cyclin D1 expression by KDM4B is predicted to promote
colorectal tumor formation.
Cooperation with β-catenin may not be the only
mechanism by which KDM4B contributes to the causation of colorectal
cancer. Recently, it was reported that KDM4B is involved in the
stimulation of hypoxia-inducible genes in colon cancer (64). Possibly, this may involve complex
formation of KDM4B with the hypoxia-inducible factor 1α, the master
mediator of the hypoxic response, since a relative of KDM4B, the
KDM4C protein, can actually bind to hypoxia-inducible factor 1α
(65). Moreover, KDM4A is capable
of binding to the p53 tumor suppressor in colon cancer cells and
inhibit p53-dependent transcription (66), raising the possibility that also
KDM4B might do so.
Apart from transcription regulation, KDM4B is also
involved in DNA repair. Its TUDOR domains can bind to dimethylated
H4K20, which normally recruits 53BP1 to sites of DNA damage and
thereby facilitates repair. Notably, this activity of KDM4B is
independent of its enzymatic activity (67). Accordingly, KDM4B overexpression
could impair DNA repair by preventing the recruitment of 53BP1 and
thereby induce genomic instability, another mechanism by which
KDM4B could contribute to tumor formation. However, a recent report
posits that KDM4B enhances DNA repair in a manner dependent on its
enzymatic activity and thus promotes cell survival (68). This may be relevant during therapy,
when KDM4B overexpression could help cancer cells to repair the DNA
damage caused by radiotherapy and many chemotherapeutic agents.
In conclusion, our study highlighted the pro-growth
function of KDM4B in colorectal tumors and provided a mechanism by
which KDM4B may attain this through interaction with
β-catenin/TCF4. Given that β-catenin is involved in many other
types of cancer (2,45), including breast and bladder cancer
where KDM4B is also overexpressed (18,69,70),
the oncogenic functions of KDM4B may be of widespread importance.
Thus, inhibition of KDM4B enzymatic activity or preventing its
interaction with β-catenin by small molecule drugs, similar to the
inhibition of chromatin recruitment of the BRD4 epigenetic
regulator in acute myeloid leukemia (71,72),
may hold promise in cancer therapy.
References
|
1.
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar
|
|
2.
|
Clevers H: Wnt/beta-catenin signaling in
development and disease. Cell. 127:469–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Chi P, Allis CD and Wang GG: Covalent
histone modifications--miswritten, misinterpreted and mis-erased in
human cancers. Nat Rev Cancer. 10:457–469. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Kulis M and Esteller M: DNA methylation
and cancer. Adv Genet. 70:27–56. 2010. View Article : Google Scholar
|
|
5.
|
Dawson MA and Kouzarides T: Cancer
epigenetics: from mechanism to therapy. Cell. 150:12–27. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Black JC, Van Rechem C and Whetstine JR:
Histone lysine methylation dynamics: establishment, regulation, and
biological impact. Mol Cell. 48:491–507. 2012. View Article : Google Scholar
|
|
7.
|
Kooistra SM and Helin K: Molecular
mechanisms and potential functions of histone demethylases. Nat Rev
Mol Cell Biol. 13:297–311. 2012.PubMed/NCBI
|
|
8.
|
Berry WL and Janknecht R: KDM4/JMJD2
histone demethylases: epigenetic regulators in cancer cells. Cancer
Res. 73:2936–2942. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Whetstine JR, Nottke A, Lan F, Huarte M,
Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M and Shi
Y: Reversal of histone lysine trimethylation by the JMJD2 family of
histone demethylases. Cell. 125:467–481. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Cloos PA, Christensen J, Agger K, Maiolica
A, Rappsilber J, Antal T, Hansen KH and Helin K: The putative
oncogene GASC1 demethylates tri- and dimethylated lysine 9 on
histone H3. Nature. 442:307–311. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Klose RJ, Yamane K, Bae Y, Zhang D,
Erdjument-Bromage H, Tempst P, Wong J and Zhang Y: The
transcriptional repressor JHDM3A demethylates trimethyl histone H3
lysine 9 and lysine 36. Nature. 442:312–316. 2006. View Article : Google Scholar
|
|
12.
|
Fodor BD, Kubicek S, Yonezawa M,
O’Sullivan RJ, Sengupta R, Perez-Burgos L, Opravil S, Mechtler K,
Schotta G and Jenuwein T: Jmjd2b antagonizes H3K9 trimethylation at
pericentric heterochromatin in mammalian cells. Genes Dev.
20:1557–1562. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Shin S and Janknecht R: Diversity within
the JMJD2 histone demethylase family. Biochem Biophys Res Commun.
353:973–977. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Trojer P, Zhang J, Yonezawa M, Schmidt A,
Zheng H, Jenuwein T and Reinberg D: Dynamic histone H1 isotype 4
methylation and demethylation by histone lysine methyltransferase
G9a/KMT1C and the Jumonji domain-containing JMJD2/KDM4 proteins. J
Biol Chem. 284:8395–8405. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Weiss T, Hergeth S, Zeissler U, Izzo A,
Tropberger P, Zee BM, Dundr M, Garcia BA, Daujat S and Schneider R:
Histone H1 variant-specific lysine methylation by G9a/KMT1C and
Glp1/KMT1D. Epigenetics Chromatin. 3:72010. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Liu G, Bollig-Fischer A, Kreike B, van de
Vijver MJ, Abrams J, Ethier SP and Yang ZQ: Genomic amplification
and oncogenic properties of the GASC1 histone demethylase gene in
breast cancer. Oncogene. 28:4491–4500. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Yang J, Jubb AM, Pike L, Buffa FM, Turley
H, Baban D, Leek R, Gatter KC, Ragoussis J and Harris AL: The
histone demethylase JMJD2B is regulated by estrogen receptor alpha
and hypoxia, and is a key mediator of estrogen induced growth.
Cancer Res. 70:6456–6466. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Kawazu M, Saso K, Tong KI, McQuire T, Goto
K, Son DO, Wakeham A, Miyagishi M, Mak TW and Okada H: Histone
demethylase JMJD2B functions as a co-factor of estrogen receptor in
breast cancer proliferation and mammary gland development. PLoS
One. 6:e178302011. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Shi L, Sun L, Li Q, Liang J, Yu W, Yi X,
Yang X, Li Y, Han X, Zhang Y, Xuan C, Yao Z and Shang Y: Histone
demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes
hormonally responsive breast carcinogenesis. Proc Natl Acad Sci
USA. 108:7541–7546. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Berry WL, Shin S, Lightfoot SA and
Janknecht R: Oncogenic features of the JMJD2A histone demethylase
in breast cancer. Int J Oncol. 41:1701–1706. 2012.
|
|
21.
|
Gaughan L, Stockley J, Coffey K, O’Neill
D, Jones DL, Wade M, Wright J, Moore M, Tse S, Rogerson L and
Robson CN: KDM4B is a master regulator of the estrogen receptor
signalling cascade. Nucleic Acids Res. 41:6892–6904. 2013.
View Article : Google Scholar
|
|
22.
|
Wissmann M, Yin N, Muller JM, Greschik H,
Fodor BD, Jenuwein T, Vogler C, Schneider R, Gunther T, Buettner R,
Metzger E and Schule R: Cooperative demethylation by JMJD2C and
LSD1 promotes androgen receptor-dependent gene expression. Nat Cell
Biol. 9:347–353. 2007. View
Article : Google Scholar
|
|
23.
|
Shin S and Janknecht R: Activation of
androgen receptor by histone demethylases JMJD2A and JMJD2D.
Biochem Biophys Res Commun. 359:742–746. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Coffey K, Rogerson L, Ryan-Munden C,
Alkharaif D, Stockley J, Heer R, Sahadevan K, O’Neill D, Jones D,
Darby S, Staller P, Mantilla A, Gaughan L and Robson CN: The lysine
demethylase, KDM4B, is a key molecule in androgen receptor
signalling and turnover. Nucleic Acids Res. 41:4433–4446. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Pollard PJ, Loenarz C, Mole DR, McDonough
MA, Gleadle JM, Schofield CJ and Ratcliffe PJ: Regulation of
Jumonji-domain-containing histone demethylases by hypoxia-inducible
factor (HIF)-1alpha. Biochem J. 416:387–394. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Beyer S, Kristensen MM, Jensen KS,
Johansen JV and Staller P: The histone demethylases JMJD1A and
JMJD2B are transcriptional targets of hypoxia-inducible factor HIF.
J Biol Chem. 283:36542–36552. 2008. View Article : Google Scholar
|
|
27.
|
Kim TD, Oh S, Shin S and Janknecht R:
Regulation of tumor suppressor p53 and HCT116 cell physiology by
histone demethylase JMJD2D/KDM4D. PLoS One. 7:e346182012.
View Article : Google Scholar
|
|
28.
|
Oh S, Shin S, Lightfoot SA and Janknecht
R: 14-3-3 proteins modulate the ETS transcription factor ETV1 in
prostate cancer. Cancer Res. 73:5110–5119. 2013. View Article : Google Scholar
|
|
29.
|
Mooney SM, Grande JP, Salisbury JL and
Janknecht R: Sumoylation of p68 and p72 RNA helicases affects
protein stability and transactivation potential. Biochemistry.
49:1–10. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Janknecht R: Regulation of the ER81
transcription factor and its coactivators by mitogen- and
stress-activated protein kinase 1 (MSK1). Oncogene. 22:746–755.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Dowdy SC, Mariani A and Janknecht R:
HER2/Neu- and TAK1-mediated up-regulation of the transforming
growth factor beta inhibitor Smad7 via the ETS protein ER81. J Biol
Chem. 278:44377–44384. 2003. View Article : Google Scholar
|
|
32.
|
Goel A and Janknecht R: Concerted
activation of ETS protein ER81 by p160 coactivators, the
acetyltransferase p300 and the receptor tyrosine kinase HER2/Neu. J
Biol Chem. 279:14909–14916. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Papoutsopoulou S and Janknecht R:
Phosphorylation of ETS transcription factor ER81 in a complex with
its coactivators CREB-binding protein and p300. Mol Cell Biol.
20:7300–7310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Knebel J, De Haro L and Janknecht R:
Repression of transcription by TSGA/Jmjd1a, a novel interaction
partner of the ETS protein ER71. J Cell Biochem. 99:319–329. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Wu J and Janknecht R: Regulation of the
ETS transcription factor ER81 by the 90-kDa ribosomal S6 kinase 1
and protein kinase A. J Biol Chem. 277:42669–42679. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Rossow KL and Janknecht R: The Ewing’s
sarcoma gene product functions as a transcriptional activator.
Cancer Res. 61:2690–2695. 2001.
|
|
37.
|
Mooney SM, Goel A, D’Assoro AB, Salisbury
JL and Janknecht R: Pleiotropic effects of p300-mediated
acetylation on p68 and p72 RNA helicase. J Biol Chem.
285:30443–30452. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Goel A and Janknecht R:
Acetylation-mediated transcriptional activation of the ETS protein
ER81 by p300, P/CAF, and HER2/Neu. Mol Cell Biol. 23:6243–6254.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Oh S and Janknecht R: Histone demethylase
JMJD5 is essential for embryonic development. Biochem Biophys Res
Commun. 420:61–65. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Shin S, Oh S, An S and Janknecht R: ETS
variant 1 regulates matrix metalloproteinase-7 transcription in
LNCaP prostate cancer cells. Oncol Rep. 29:306–314. 2013.PubMed/NCBI
|
|
41.
|
DiTacchio L, Bowles J, Shin S, Lim DS,
Koopman P and Janknecht R: Transcription factors ER71/ETV2 and SOX9
participate in a positive feedback loop in fetal and adult mouse
testis. J Biol Chem. 287:23657–23666. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Shin S, Kim TD, Jin F, van Deursen JM,
Dehm SM, Tindall DJ, Grande JP, Munz JM, Vasmatzis G and Janknecht
R: Induction of prostatic intraepithelial neoplasia and modulation
of androgen receptor by ETS variant 1/ETS-related protein 81.
Cancer Res. 69:8102–8110. 2009. View Article : Google Scholar
|
|
43.
|
Kaiser S, Park YK, Franklin JL, Halberg
RB, Yu M, Jessen WJ, Freudenberg J, Chen X, Haigis K, Jegga AG,
Kong S, Sakthivel B, Xu H, Reichling T, Azhar M, Boivin GP, Roberts
RB, Bissahoyo AC, Gonzales F, Bloom GC, Eschrich S, Carter SL,
Aronow JE, Kleimeyer J, Kleimeyer M, Ramaswamy V, Settle SH, Boone
B, Levy S, Graff JM, Doetschman T, Groden J, Dove WF, Threadgill
DW, Yeatman TJ, Coffey RJ Jr and Aronow BJ: Transcriptional
recapitulation and subversion of embryonic colon development by
mouse colon tumor models and human colon cancer. Genome Biol.
8:R1312007. View Article : Google Scholar
|
|
44.
|
Kurashina K, Yamashita Y, Ueno T, Koinuma
K, Ohashi J, Horie H, Miyakura Y, Hamada T, Haruta H, Hatanaka H,
Soda M, Choi YL, Takada S, Yasuda Y, Nagai H and Mano H: Chromosome
copy number analysis in screening for prognosis-related genomic
regions in colorectal carcinoma. Cancer Sci. 99:1835–1840. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Clevers H and Nusse R: Wnt/beta-catenin
signaling and disease. Cell. 149:1192–1205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Yap KL and Zhou MM: Keeping it in the
family: diverse histone recognition by conserved structural folds.
Crit Rev Biochem Mol Biol. 45:488–505. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Mosimann C, Hausmann G and Basler K:
Beta-catenin hits chromatin: regulation of Wnt target gene
activation. Nat Rev Mol Cell Biol. 10:276–286. 2009. View Article : Google Scholar
|
|
48.
|
He TC, Sparks AB, Rago C, Hermeking H,
Zawel L, da Costa LT, Morin PJ, Vogelstein B and Kinzler KW:
Identification of c-MYC as a target of the APC pathway. Science.
281:1509–1512. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Mann B, Gelos M, Siedow A, Hanski ML,
Gratchev A, Ilyas M, Bodmer WF, Moyer MP, Riecken EO, Buhr HJ and
Hanski C: Target genes of beta-catenin-T
cell-factor/lymphoid-enhancer-factor signaling in human colorectal
carcinomas. Proc Natl Acad Sci USA. 96:1603–1608. 1999. View Article : Google Scholar
|
|
50.
|
Shtutman M, Zhurinsky J, Simcha I,
Albanese C, D’Amico M, Pestell R and Ben-Ze’ev A: The Cyclin D1
gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad
Sci USA. 96:5522–5527. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Tetsu O and McCormick F: Beta-catenin
regulates expression of Cyclin D1 in colon carcinoma cells. Nature.
398:422–426. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Kouzarides T: Chromatin modifications and
their function. Cell. 128:693–705. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Daujat S, Zeissler U, Waldmann T, Happel N
and Schneider R: HP1 binds specifically to Lys26-methylated histone
H1.4, whereas simultaneous Ser27 phosphorylation blocks HP1
binding. J Biol Chem. 280:38090–38095. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Wagner EJ and Carpenter PB: Understanding
the language of Lys36 methylation at histone H3. Nat Rev Mol Cell
Biol. 13:115–126. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Eilers M and Eisenman RN: Myc’s broad
reach. Genes Dev. 22:2755–2766. 2008.
|
|
56.
|
Eferl R and Wagner EF: AP-1: a
double-edged sword in tumorigenesis. Nat Rev Cancer. 3:859–868.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
57.
|
Smith DR, Myint T and Goh HS:
Over-expression of the c-myc proto-oncogene in colorectal
carcinoma. Br J Cancer. 68:407–413. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
58.
|
Magrisso IJ, Richmond RE, Carter JH, Pross
CB, Gilfillen RA and Carter HW: Immunohistochemical detection of
RAS, JUN, FOS, and p53 oncoprotein expression in human colorectal
adenomas and carcinomas. Lab Invest. 69:674–681. 1993.PubMed/NCBI
|
|
59.
|
Nateri AS, Spencer-Dene B and Behrens A:
Interaction of phosphorylated c-Jun with TCF4 regulates intestinal
cancer development. Nature. 437:281–285. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
60.
|
Gan XQ, Wang JY, Xi Y, Wu ZL, Li YP and Li
L: Nuclear Dvl, c-Jun, beta-catenin, and TCF form a complex leading
to stabilization of beta-catenin-TCF interaction. J Cell Biol.
180:1087–1100. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
61.
|
Suto R, Tominaga K, Mizuguchi H, Sasaki E,
Higuchi K, Kim S, Iwao H and Arakawa T: Dominant-negative mutant of
c-Jun gene transfer: a novel therapeutic strategy for colorectal
cancer. Gene Ther. 11:187–193. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
62.
|
Bartkova J, Lukas J, Strauss M and Bartek
J: The PRAD-1/Cyclin D1 oncogene product accumulates aberrantly in
a subset of colorectal carcinomas. Int J Cancer. 58:568–573. 1994.
View Article : Google Scholar
|
|
63.
|
Hulit J, Wang C, Li Z, Albanese C, Rao M,
Di Vizio D, Shah S, Byers SW, Mahmood R, Augenlicht LH, Russell R
and Pestell RG: Cyclin D1 genetic heterozygosity regulates colonic
epithelial cell differentiation and tumor number in ApcMin mice.
Mol Cell Biol. 24:7598–7611. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
64.
|
Fu L, Chen L, Yang J, Ye T, Chen Y and
Fang J: HIF-1alpha-induced histone demethylase JMJD2B contributes
to the malignant phenotype of colorectal cancer cells via an
epigenetic mechanism. Carcinogenesis. 33:1664–1673. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
65.
|
Luo W, Chang R, Zhong J, Pandey A and
Semenza GL: Histone demethylase JMJD2C is a coactivator for
hypoxia-inducible factor 1 that is required for breast cancer
progression. Proc Natl Acad Sci USA. 109:E3367–E3376. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
66.
|
Kim TD, Shin S, Berry WL, Oh S and
Janknecht R: The JMJD2A demethylase regulates apoptosis and
proliferation in colon cancer cells. J Cell Biochem. 113:1368–1376.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
67.
|
Mallette FA, Mattiroli F, Cui G, Young LC,
Hendzel MJ, Mer G, Sixma TK and Richard S: RNF8- and
RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1
recruitment to DNA damage sites. EMBO J. 31:1865–1878. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
68.
|
Young LC, McDonald DW and Hendzel MJ:
Kdm4b histone demethylase is a DNA damage response protein and
confers a survival advantage following gamma-irradiation. J Biol
Chem. 288:21376–21388. 2013. View Article : Google Scholar
|
|
69.
|
Slee RB, Steiner CM, Herbert BS, Vance GH,
Hickey RJ, Schwarz T, Christan S, Radovich M, Schneider BP,
Schindelhauer D and Grimes BR: Cancer-associated alteration of
pericentromeric heterochromatin may contribute to chromosome
instability. Oncogene. 31:3244–3253. 2012. View Article : Google Scholar
|
|
70.
|
Toyokawa G, Cho HS, Iwai Y, Yoshimatsu M,
Takawa M, Hayami S, Maejima K, Shimizu N, Tanaka H, Tsunoda T,
Field HI, Kelly JD, Neal DE, Ponder BA, Maehara Y, Nakamura Y and
Hamamoto R: The histone demethylase JMJD2B plays an essential role
in human carcinogenesis through positive regulation of
cyclin-dependent kinase 6. Cancer Prev Res. 4:2051–2061. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
71.
|
Dawson MA, Prinjha RK, Dittmann A,
Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C,
Savitski MM, Huthmacher C, Gudgin E, Lugo D, Beinke S, Chapman TD,
Roberts EJ, Soden PE, Auger KR, Mirguet O, Doehner K, Delwel R,
Burnett AK, Jeffrey P, Drewes G, Lee K, Huntly BJ and Kouzarides T:
Inhibition of BET recruitment to chromatin as an effective
treatment for MLL-fusion leukaemia. Nature. 478:529–533. 2011.
View Article : Google Scholar
|
|
72.
|
Zuber J, Shi J, Wang E, Rappaport AR,
Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, Taylor
MJ, Johns C, Chicas A, Mulloy JC, Kogan SC, Brown P, Valent P,
Bradner JE, Lowe SW and Vakoc CR: RNAi screen identifies Brd4 as a
therapeutic target in acute myeloid leukaemia. Nature. 478:524–528.
2011. View Article : Google Scholar
|