Introduction
Of the gynecological cancers, ovarian cancer imposes
the highest mortality rate, even with surgery and adjuvant
chemotherapy. Even with good responsive to primary therapy, ∼80% of
patients who present with advanced cancers will experience
recurrence and succumb to the disease (1,2). In
the absence of screening, the need for novel treatments that
prevent disease progression following surgery, including
combination and intraperitoneal chemotherapies, becomes a matter of
some urgency. Compared with hematologic tumors and malignant
melanoma, ovarian cancer may be less vulnerable to immunotherapy
(3). Only recently have
tumor-specific antigens been identified that could serve as targets
for cytotoxic T-cell responses to ovarian cancer (4,5). New
strategies are needed to generate and enhance immune responses
against ovarian cancer, identify tumor-specific antigens and
modulate immune-suppressive activities.
The combination of platinum and taxane is used
currently as initial treatment for advanced epithelial ovarian
cancer. Patients treated with paclitaxel and cisplatin respond
better clinically and survive longer progression-free than with the
previous standard of care (cisplatin plus cyclophosphamide)
(6). Ovarian cancer is very
sensitive to paclitaxel and cisplatin combination. However,
acquired resistance to combined therapy with paclitaxel and a
platinum-based drug, which may develop by one of several pathways,
is a major reason for treatment failure and death in patients with
ovarian cancer (7,8). The mechanisms responsible for the
high resistance rate to paclitaxel are not well researched and
methods to prevent or regulate the resistance using another drug or
combination are not well studied either (9).
Toll-like receptors (TLRs) and ligand interaction
trigger immune cells (10). Mice
deficient in each TLR have demonstrated that each TLR has a
distinct function in terms of pathogen recognition and immune
responses (11). However, there is
significant amount of evidence for the involvement of TLRs in
disease largely from overexpression in multiple diseases, their
activation causing enhanced disease in models (12). Despite marked differences in
structure, paclitaxel and LPS share a receptor or signaling
molecule and paclitaxel was thereby identified as a TLR4 ligand in
murine macrophages (13,14). A previous study also reported
doxorubicin, chemical TLR2 ligand, may play a role in the
regulation of inflammatory and apoptotic mediators in the heart
after administration (15). TLR4
and MyD88 were detected in some human cancer cell lines including
ovarian cancer (16) and
paclitaxel activated MyD88-dependent pathway and recruited NF-κB,
leading anti-apoptotic molecule, to survive and resist drugs in
cancer cells (17,18).
In patients previously treated with platinum,
anthracyclines such as doxorubicin may be effective as single-agent
treatments (19). Addition of
doxorubicin to ovarian cancer regimens may significantly improve
survival compared to platinum-based combinations without
anthracyclines (20).
The use of single chemotherapeutic drug with high
dose has shown some limitations due to development of drug
resistance and high toxicity. However, attempts to delivery
chemotherapeutic drugs simultaneously have shown many difficulties,
such as different solubility in water. Following these results, we
focused on the effect of sequential hydrophobic paclitaxel and
hydrophilic doxorubicin treatment on survival of ovarian cancer
cells and antitumor immune responses induced by drug-treated cancer
cells concurrently in this study. Thus, tumor cells sequentially
treated with an anticancer drug combination may open a new route to
immune activation and disease management against advanced
cancer.
Materials and methods
Cells, antibodies, anticancer drugs and
mice
The MOSEC cell line was originally derived from
murine ovarian surface epithelial cells (21). Antibodies against mouse CD4
(557308) and IFN-γ (554411) were purchased from BD Pharmingen;
anti-CD40 (12-0401), CD80 (12-0801) and anti-CD86 (12-0862) were
purchased from eBioscience. The polyclonal antibodies to cleaved
caspase-3 were from Cell Signaling Technology; and female C57BL/6
mice, from the Chung-Ang Laboratory Animal Service (Seoul, Korea).
Cisplatin (P4394), paclitaxel (T7191) and doxorubicin-HCl (D1515)
were purchased from Sigma-Aldrich. Oregon Green 488-conjugated
paclitaxel (P22310, Molecular Probes) were reconstituted with DMSO
prior to use and diluted with RPMI-1640 to the required
concentrations. The ethics committee of the College of Medicine,
Chung-Ang University and College of Medicine, Inje University
approved all protocols and procedures used in this study.
Drug uptake and the effect of anticancer
drugs on MOSECs in vitro
On day 1, MOSECs (5.0×106/ml) were
cultured in the presence of paclitaxel (50 μg/ml) at 37°C
for 2 h and then cultured in the absence of the drug for 24 h.
MOSECs pre-exposed to paclitaxel were harvested and incubated with
doxorubicin and cisplatin at low concentrations (10 μg/ml)
for 2 h. An MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, a
yellow tetrazole) assay with Vybrant MTT cell assay kit (V-13154,
Molecular Probes) for cell viability was performed with the
drug-treated MOSECs after 24 and 48 h of incubation. Apoptosis was
also measured using an Annexin V-FITC detection kit (556570, BD
Pharmingen) according to the manufacturer’s protocol. MOSECs were
also cultured in the presence of Oregon green 488-conjugated
paclitaxel (50 μg/ml) and/or doxorubicin (10 μg/ml)
at 37°C for 2 h to determine the drug uptake according to a
previous study (22). Immunoblot
assay of cleaved caspase-3 expression was performed in
paclitaxel-treated MOSECs post-treated with doxorubicin or
cisplatin and harvested at 3 and 20 h after last drug treatment.
Primary antibodies used were; anti-cleaved caspase-3 (9662S, Cell
Signaling) (1:1,000) and anti-β-actin (4967L, Cell Signaling)
(1:2,000). Primary antibodies were detected using horseradish
peroxidase-conjugated goat anti-rabbit (7074S, Cell Signaling)
(1:2,000).
Characterization of immune responses to
the drug-treated tumor cells and the in vivo antitumor
responses
Prior to inoculation, all groups of tumor cells
(1.0×106/mouse) treated with single or sequential
anticancer drug regimens were irradiated at 100,000 cGy/10 min.
Mice were injected via the intraperitoneal route and boosted one
week later with the same dose of cells treated by the same regimen.
Immune responses were tested at one week or at day 50 after the
last treatment. To harvest and collect the peritoneal exudate cells
(PECs), 10 ml of cold sterile PBS was injected into the peritoneal
cavity. After de-contamination to remove the RBC, the PECs were
co-cultured with irradiated MOSECs for 16 h with complete medium in
the presence of GolgiPlug (555028, BD Pharmingen). Cells stained
with phycoerythrin-conjugated monoclonal rat anti-mouse CD4
antibody and cells were subjected to intracellular cytokine
staining using the Cytofix/ Cytoperm kit (554714, BD Pharmingen).
FITC-conjugated anti-IFN-γ was used for intracellular cytokine
staining. The numbers of CD4+ IFN-γ+
double-positive T cells in 1.0×105 PECs are calculated.
To translate immune responses to antitumor effects, female C57BL/6
mice were challenged intraperitoneally with 1.0×106
MOSECs per mouse. At day 3, tumor-bearing mice from each group (6–7
mice/group) were vaccinated twice at weekly intervals with the same
dose of cells treated by the same regimen. At 40–50 days after
tumor challenge, the general condition and weights of the mice were
monitored twice weekly to assess the tumor burden and ascites
accumulation resulting from progressive peritoneal carcinomatosis
(23). The moribund animals were
euthanized.
DC maturation and detection of cytokine
secretion with RT-PCR and ELISA
Dendritic cells were generated from murine bone
marrow cells as previously described with modifications (24). At day 6, MOSECs
(1.0×106/mouse) pre-exposed to paclitaxel and then
treated with doxorubicin or cisplatin were co-cultured with BMDCs
at a ratio of 1:1 (MOSEC:DCs) in a 24-well plate. After 24 h of
co-culture, cells were harvested and the BMDCs were isolated using
anti-CD11c antibody according to the manufacturer’s protocol
(130-052-001, Miltenyi Biotec). Cells were stained with antibodies
to CD80, CD86 and CD40 for detecting BMDCs maturation and extracted
RNA using TRIzol (15596-018, Life Technologies) to determine the
mRNA level of IL-12p40, IL-6, TNF-α. Following primers were used
for amplification: IL-12p40 (sense, 5’-CACCTGCCCAACTGCCGAGG-3’; and
antisense, 5’-TAGCTCCCTGGCTCTGCGGG-3’)/IL-6 (sense, 5’-ATGC
TGGTGACAACCACGGCC-3’; and antisense, 5’-GGCATA
ACGCACTAGGTTTGCCGA-3’)/TNF-α (sense, 5’-AGCCC CCAGTCTGTATCCTT-3’;
and antisense, 5’-CTCCCTTTGC AGAACTCAGG-3’)/GAPDH (sense,
5’-GTGGAGTCTACT GGCGTCTT-3’; and antisense, 5’-GCCTGCTTCACCACC
TTCTT-3’). The numbers of cycles and temperatures were used as
previously determined (25).
Cycling conditions for IL-12p40 were 30 sec at 95°C, 60 sec at
60°C, and 1 min at 72°C for 35 cycles; conditions for IL-6 were 30
sec at 95°C, 60 sec at 60°C and 1 min at 72°C for 35 cycles;
conditions for TNF-α were 30 sec at 95°C, 60 sec at 53°C and 1 min
at 72°C for 35 cycles and conditions for GAPDH were 30 sec at 95°C,
60 sec at 50.5°C and 1 min at 72°C for 35 cycles. PCR products were
electrophoresed and analyzed. After 24 h of co-culture, culture
media was collected and kept in −70°C to detect IL-12 protein level
with ELISA. ELISA was conducted according to the manufacturer’s
instructions (900-M97, PeproTech).
Immunoblot analysis for MyD88 and
downstream target proteins
MOSECs (1.0×106/mouse) treated with each
condition of anticancer drug isolated according to the time
schedule. BMDCs co-cultured with the drug-treated MOSECs for 24 h,
cells were harvested and the BMDCs were isolated using anti-CD11c
antibody according to the manufacturer’s protocol (130-052-001,
Miltenyi Biotec). All samples were lysed in Mammalian Protein
Extraction Reagent (M-PER) (78501, Pierce). The protein
transferred-membranes for western blotting were probed with an
appropriate antibody. Primary antibodies used were: anti-mouse
MyD88 (sc-74532), tumor necrosis factor receptor associated factor
6 (TRAF6) (sc-8409) and nuclear factor-κB (NF-κB) (sc-71675) from
Santa Cruz Biotechnology (1:1,000). Primary antibodies were
detected using horseradish peroxidase-conjugated goat anti-mouse
(1:5,000–1:10,000). Enhanced chemiluminescence was performed with
ECL-Plus (RPN2132, GE Healthcare). The bands were then quantified
using Thermo Scion Image Analysis software (Scion Corp.).
Statistical analysis
All data are expressed as means ± standard error of
the mean (SEM) and each value is representative of at least two
different experiments. Comparisons between all individual data were
made by analysis of variance (one-way ANOVA). Statistical
significance was defined as p<0.05.
Results
The death rate of the
paclitaxel-resistant MOSECs increases after treatment with
doxorubicin and cisplatin through an apoptotic pathway
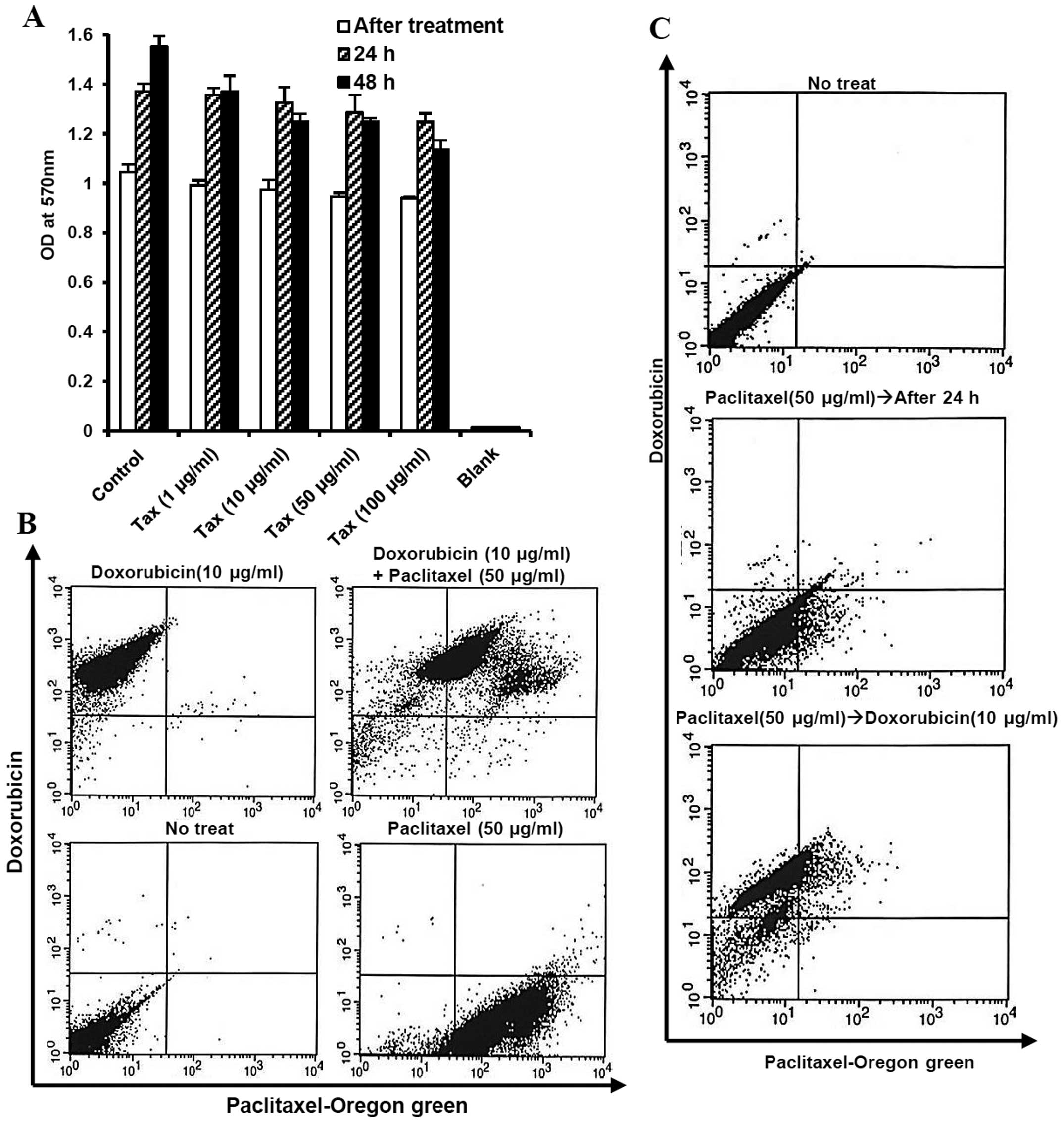
We first determined the single effect of paclitaxel,
first-line chemotherapy for ovarian cancer, on MOSECs exposed to
various doses of paclitaxel for a short time (2 h). However, the
high value on MTT assay showed a high level of viability in these
MOSECs at 24 and 48 h after short time treatment (Fig. 1). We observed that the paclitaxel
can be taken up by MOSECs and drug absorption rate did not affect
reciprocally hydrophobic paclitaxel and hydrophilic doxorubicin
after co-treatment. Most MOSEC cells co-expressed green
(paclitaxel) and red (doxorubicin) fluorescence after treatment
with paclitaxel and doxorubicin for 2 h (Fig. 1B). Doxorubicin was taken up by live
paclitaxel-exposed MOSECs for a short time (2 h) on day 1 (Fig. 1C).
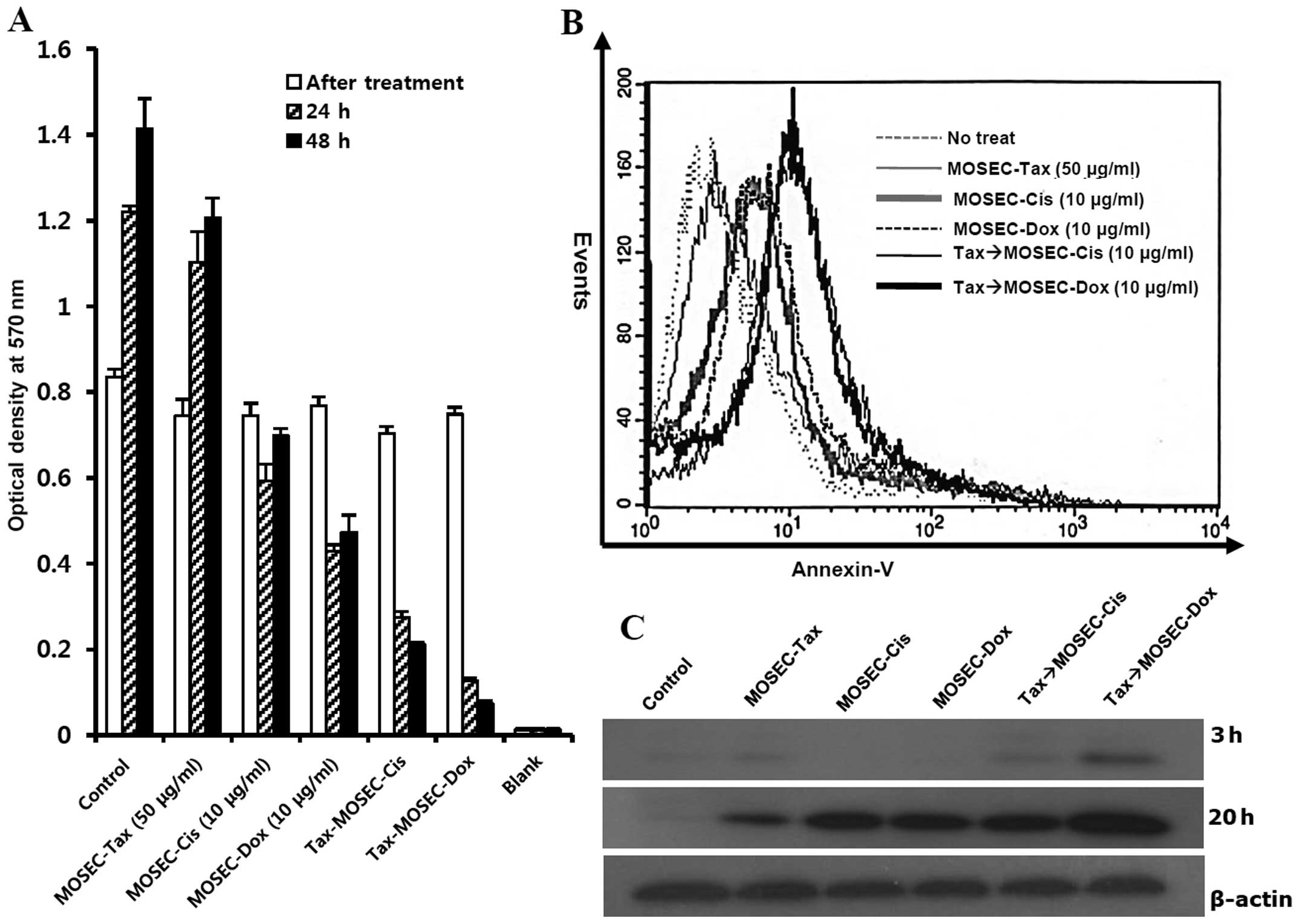
We next investigated the death rate of
paclitaxel-exposed MOSECs after post-treatment with doxorubicin (10
μg/ml) or cisplatin (10 μg/ml) for 2 h. In these
MOSECs, the MTT assay showed significant decreases in viability at
24 and 48 h after the second drug treatment (Fig. 2A). By cell-staining with
FITC-conjugated Annexin V after post-treatment with doxorubicin, we
showed that this decrease in viability occurred through apoptosis
(Fig. 2B). We determined the
extent of apoptotic death with immunoblot assay. Paclitaxel-exposed
MOSECs post-treated with doxorubicin for short time showed greater
caspase-3 activation than other groups at 20 h after finishing
treatment with last drug (Fig.
2C). Thus our results suggest that MOSECs can absorb the
paclitaxel after short incubation, but may be resistant. However,
brief exposure to low-dose doxorubicin or cisplatin activated
apoptosis in paclitaxel-exposed MOSECs.
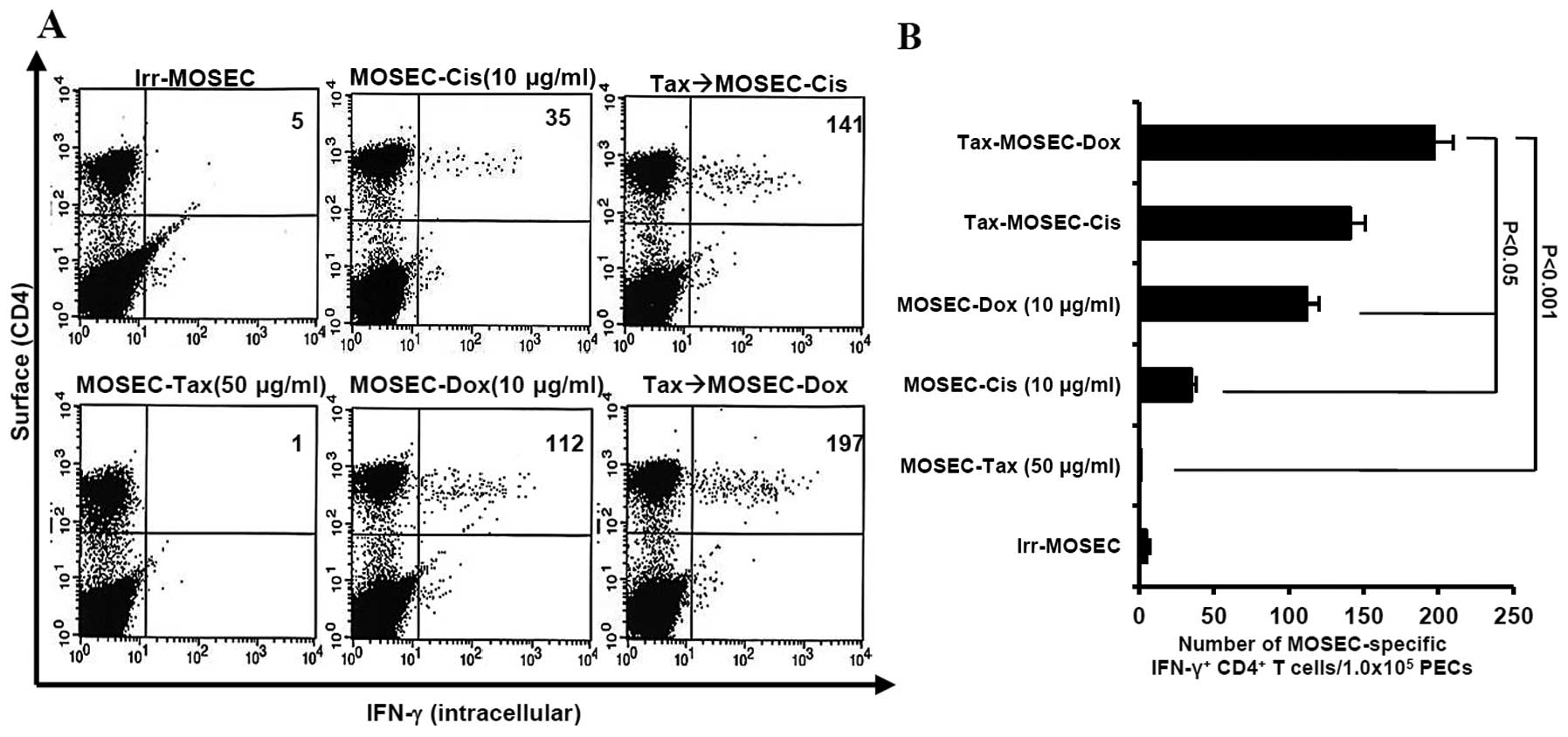
MOSECs sequentially treated with
paclitaxel and doxorubicin enhance MOSEC-specific CD4+
T-cell immune responses and prolong survival in vaccinated
mice
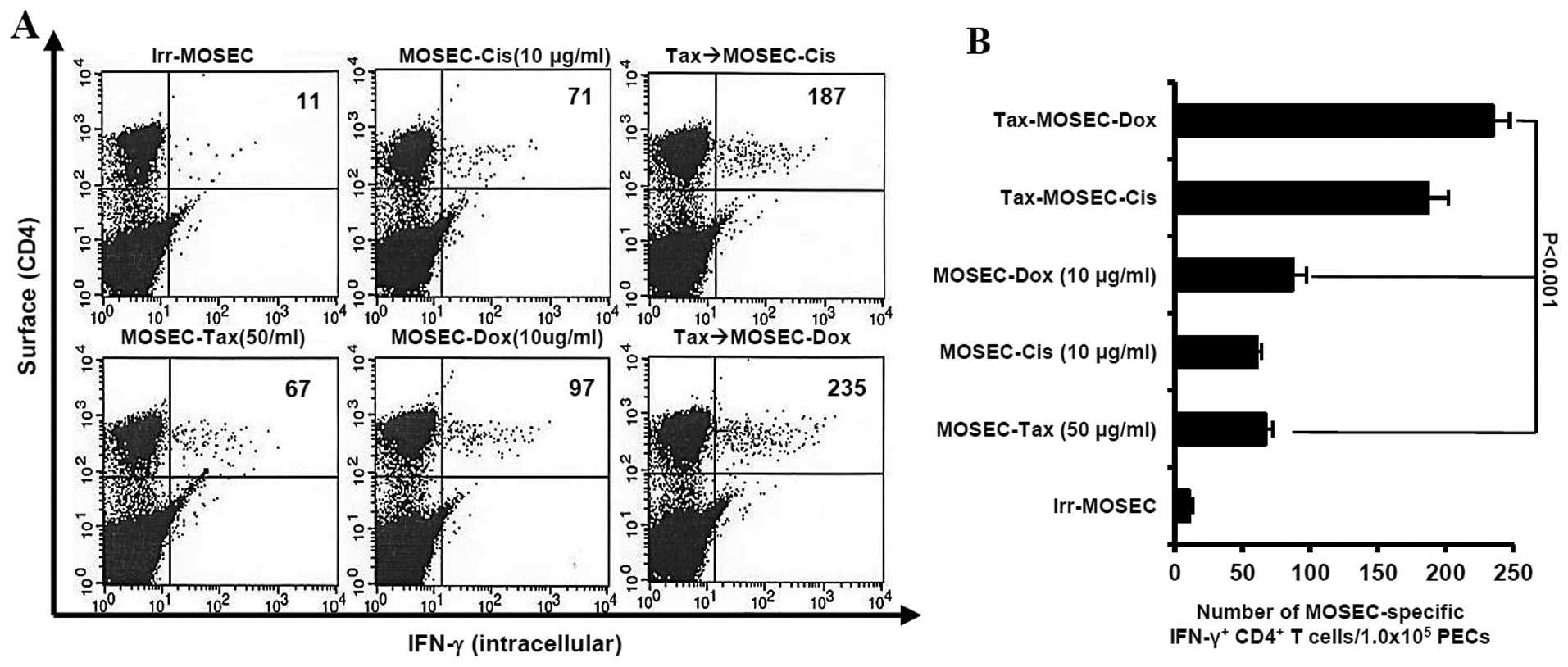
We determined whether paclitaxel-exposed MOSECs
treated with doxorubicin or cisplatin killed by apoptosis were
immunogenic in vivo. We observed that the mice vaccinated
with paclitaxel-exposed MOSECs after post-treatment with
doxorubicin induced highest MOSEC-specific CD4+ T-cell
immune responses (p<0.001) (Fig.
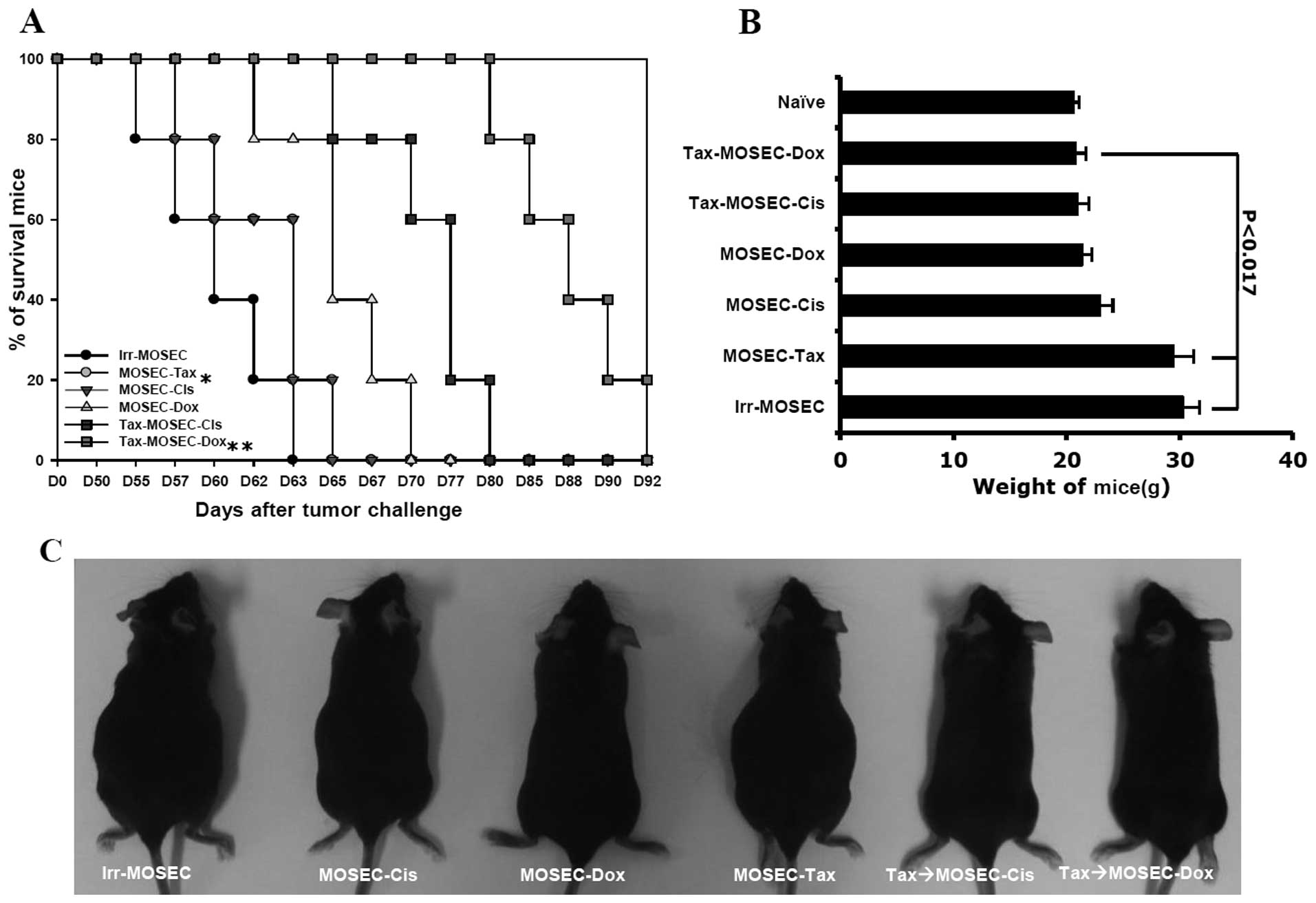
3). As a consequence of high immune responses, the survival
rate of the mice vaccinated with paclitaxel-exposed MOSECs after
post-treatment with doxorubicin was significantly higher at day 80
than the other groups (p<0.05) (Fig. 4A and B). Both the mice treated with
irradiated-only MOSECs and with paclitaxel-exposed MOSECs increased
in weight relatively early compared to those mice vaccinated with
doxorubicin-or cisplatin-treated MOSECs and paclitaxel-exposed
MOSECs post-treated with doxorubicin or cisplatin (p<0.017)
(Fig. 4B and C). We also
determined that the anticancer immune responses required
CD4+ T cells and NK cells based on antibody depletion
experiments in vivo (data not shown). Our results suggest
that MOSECs pre-exposed to paclitaxel and subsequently to
doxorubicin induces antitumor immune response and prolong survival
in tumor-bearing mice.
MOSECs pre-exposed to paclitaxel and
post-treated with doxorubicin and cisplatin induce specific
CD4+ long-lasting T cells in vaccinated mice
At day 50 after last vaccination, the mice
vaccinated with paclitaxel-exposed MOSECs post-treated with
doxorubicin or cisplatin induced MOSEC-specific CD4+
long-lasting T cells in greater numbers than the mice vaccinated
with MOSECs exposed to a single anticancer drug only (p<0.001,
Tax→MOSEC-Dox versus MOSEC-Tax; p<0.021, Tax→MOSEC-Dox versus
MOSEC-Dox). We also observed that the mice vaccinated with MOSECs
pre-exposed paclitaxel only did not generate MOSEC-specific
CD4+ long-lasting T cells (Fig. 5). Thus, our data suggest that
MOSECs pre-exposed to paclitaxel and treated briefly thereafter
with a different anticancer drug induces and sustains an effective
antitumor immune response against ovarian cancer.
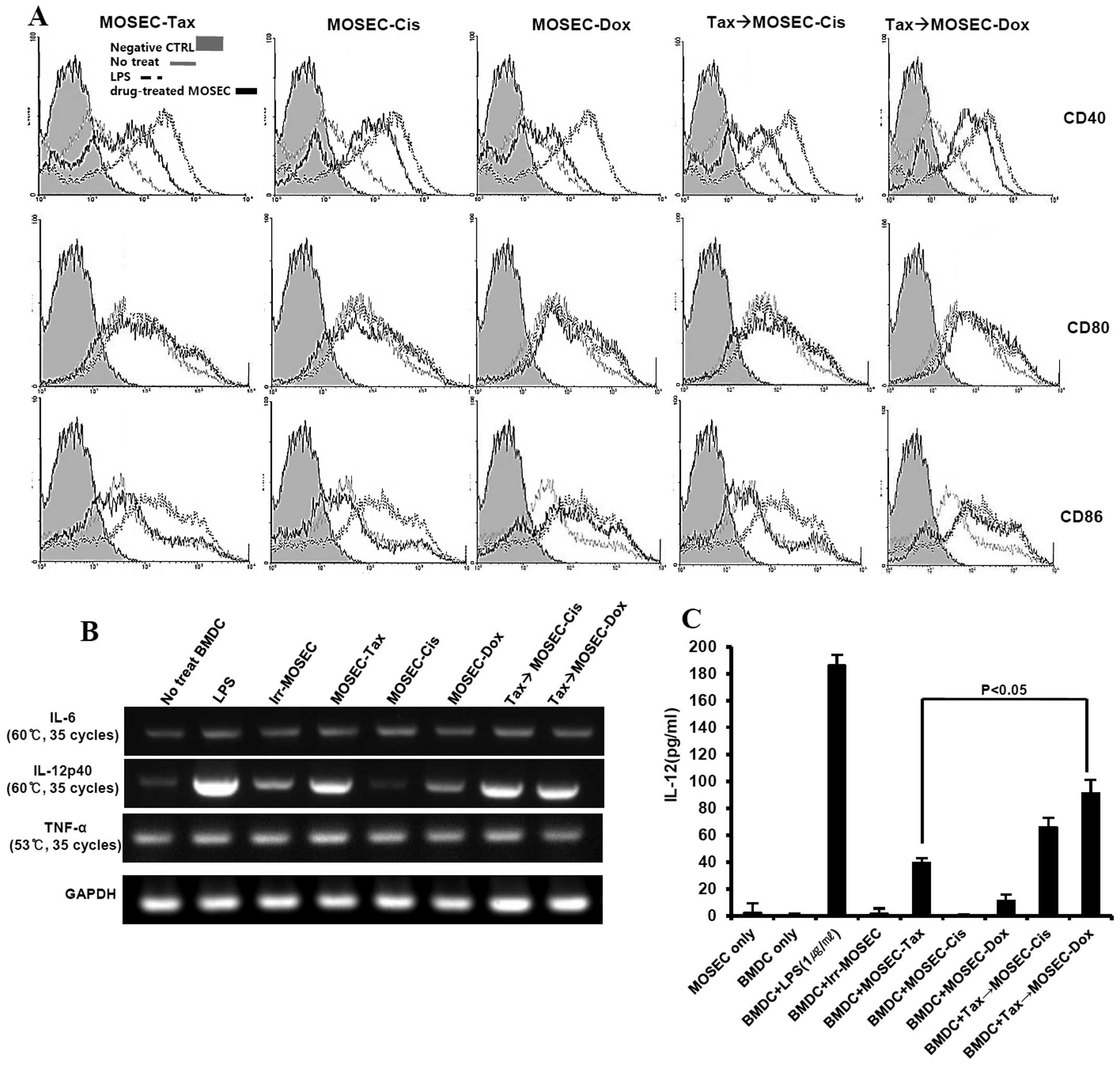
MOSECs pre-exposed to paclitaxel and then
treated with doxorubicin induce DC maturation and increase the
number of IL-12-producing DCs in vitro
We investigated whether the apoptotic MOSECs from
this treatment sequence could influence DC maturation. After
co-culture with drug-treated MOSECs and BMDCs for 24 h, BMDCs were
harvested and isolated using anti-CD11c antibody. Interestingly,
the expression of CD40 and CD86 in DCs co-cultured with
paclitaxel-exposed MOSECs post-treated with doxorubicin was higher
than in DCs co-cultured with paclitaxel-exposed MOSECs post-treated
with cisplatin or MOSECs treated with a single anticancer drug
(Fig. 6A). RT-PCR analysis was
performed with isolated BMDCs for cytokines that promote or inhibit
Th1 immune response. IL-12 (p40) mRNA levels were also
significantly upregulated in BMDCs co-cultured with
paclitaxel-exposed MOSECs post-treated with doxorubicin or
cisplatin as much as in BMDCs co-cultured with MOSEC exposed to
either LPS or paclitaxel alone. In contrast, IL-6 and TNF-α mRNA
levels did not change significantly compared with BMDCs co-cultured
with irradiated-only MOSECs as control (Fig. 6B). A significantly high level of
IL-12 concentration was also detected in culture media harvested
after paclitaxel-exposed MOSECs post-treated with doxorubicin or
cisplatin for 24 h compared to that of paclitaxel alone (Fig. 6C). Our results suggest that the
apoptotic MOSECs treated sequentially with paclitaxel and
doxorubicin stimulate BMDCs to mature and to secrete cytokine to
regulate Th1 cells.
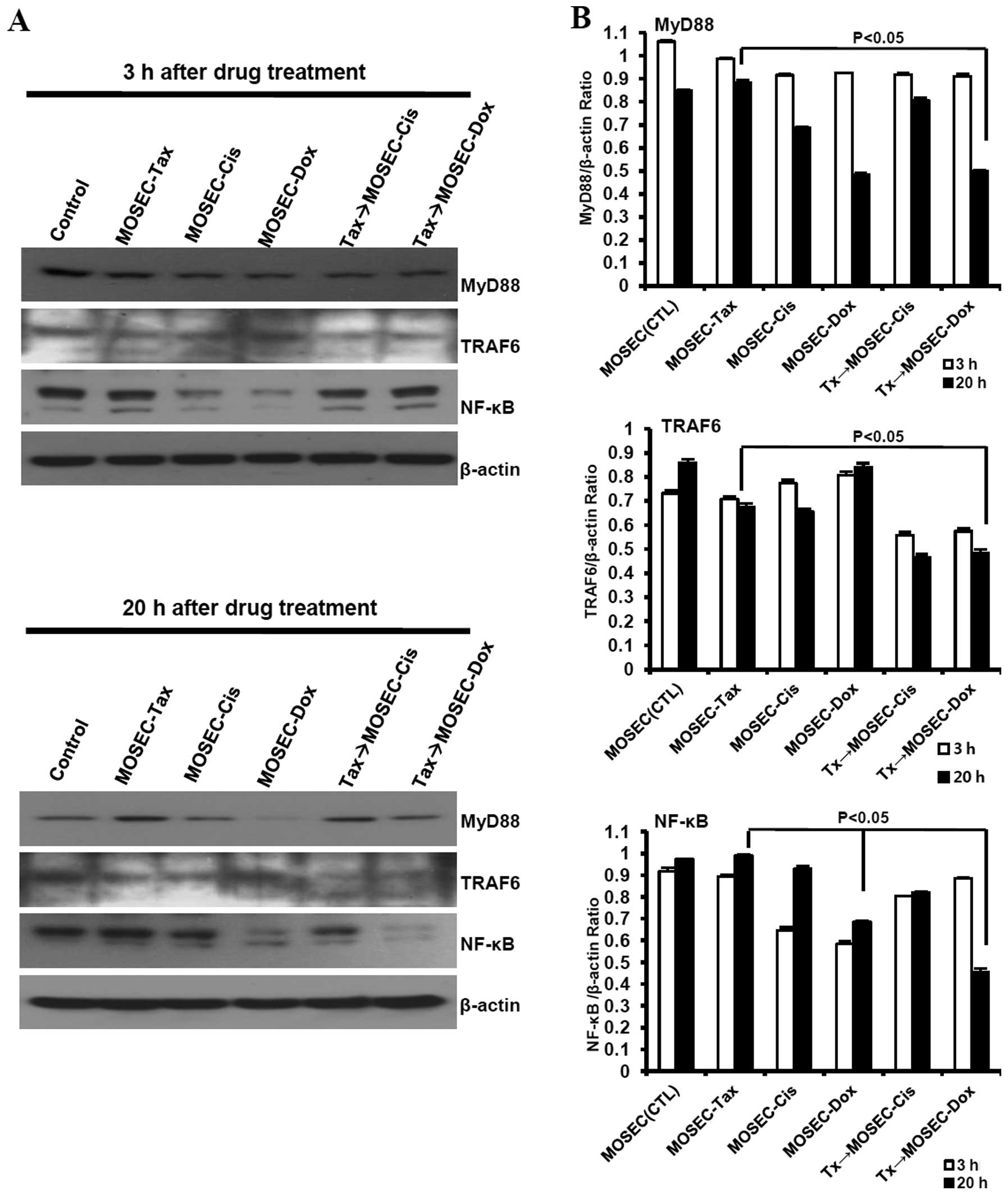
MOSECs exposed to paclitaxel and
doxorubicin in sequence downregulate MyD88 in cancer cells and
upregulate MyD88 in DCs
LPS, a ligand for Toll-like receptor 4 (TLR4),
shares with paclitaxel certain receptors and signaling molecules
for immune cell activation (10).
In patients with ovarian cancer, tumor expression of MyD88, adaptor
molecule for TLR4, may correlate negatively with survival (26). So, we first assessed the expression
levels of TRAF6 and NF-κB, downstream of MyD88 signaling, in
paclitaxel-exposed MOSECs post-treated with doxorubicin or
cisplatin to explore the mechanism of resistance or susceptibility
to the anticancer drug. We found that MOSECs constitutively
expressed MyD88 and TRAF6, even sustained or upregulated expression
of MyD88, TRAF6 and NF-κB at 20 h after exposure to paclitaxel.
NF-κB was downregulated in MOSECs treated with doxorubicin or
cisplatin only at 3 h after the end of treatment. The expression
level of MyD88, TRAF6 and NF-κB was significantly decreased in
paclitaxel-exposed MOSECs treated with doxorubicin compared to
MOSECs pre-exposed paclitaxel only at 20 h after the end of
treatment. Interestingly, we also observed a slight downregulation
of TRAF6 expression, but not MyD88 and NF-κB, in paclitaxel-exposed
MOSECs post-treated with cisplatin compared to MOSECs pre-exposed
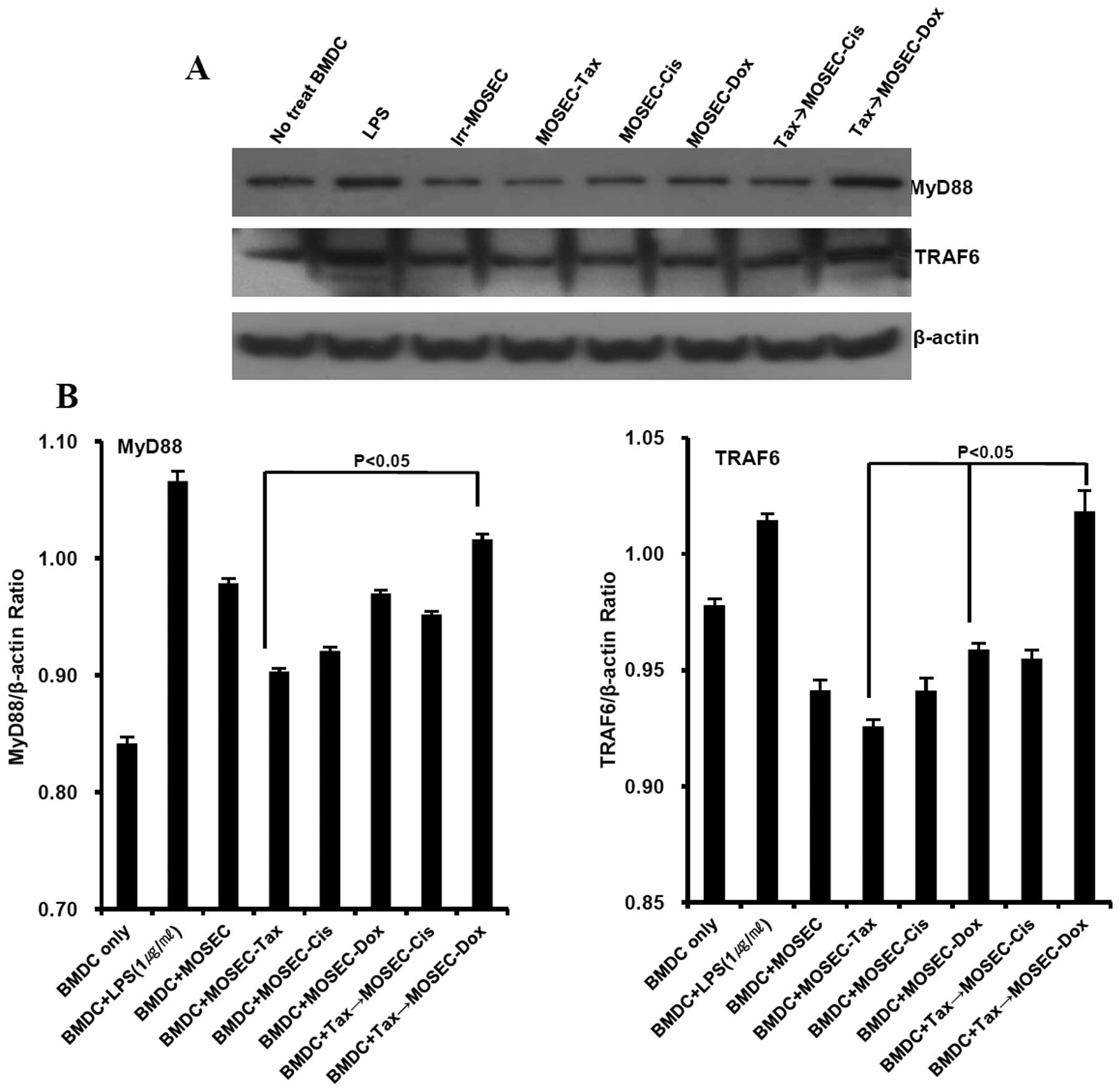
paclitaxel only at 20 h after the end of treatment (Fig. 7). Next, we examined the effect of
paclitaxel-exposed MOSECs post-treated with doxorubicin on MyD88
signaling in BMDCs because previous data showed that BMDCs
co-cultured with paclitaxel-exposed MOSECs post-treated with
doxorubicin produced significant amount of IL-12. In BMDCs
co-cultured with paclitaxel-exposed MOSECs post-treated with
doxorubicin, the MyD88 and TRAF6 expression increased; this
increase did not occur when the MOSECs were post-treated with
cisplatin or were treated with paclitaxel only (Fig. 8). These findings suggest that MyD88
downregulation in paclitaxel-exposed MOSECs post-treated with
doxorubicin increased susceptibility of these cells to apoptosis
and that doxorubicin post-treatment of the MOSECs enabled them to
induce MyD88-dependent BMDC maturation and IL-12 secretion to
generate immune responses.
Discussion
Whole tumor cell vaccines may be easily prepared and
administered directly by a physician without technical guidance.
Early forms of whole cell vaccines usually consisted of killed
tumor cells or lysates mixed with bacterial adjuvants (27,28).
However, the mechanisms of bacterial adjuvants are not understood
very well, and their use may result in side-effects and
inconsistent outcomes. In the present study we demonstrated an
increase in the immunogenicity of paclitaxel-exposed ovarian cancer
cells following brief exposure of the cells to a low dose of second
anticancer drug (10 μg/ml), doxorubicin. From these results,
we propose that tumor cells treated in this way may be used to
produce a whole cell vaccine against ovarian cancer.
Chemotherapy for cancer is severely
immune-suppressive, and is therefore very difficult to combine with
immunotherapy. Acquired resistance to combined therapy with
paclitaxel and cisplatin is also major reason for poor prognosis.
Paclitaxel is reported to be a ligand to TLR4 (10). Paclitaxel-induced signaling
activates NF-κB, leading anti-apoptotic molecule in cancer cells,
through mediation by adaptor protein MyD88, which links with the
cytoplasmic portion of TLR4. In mice, but not in humans,
paclitaxel-induced NF-κB activation occurs through an LPS-mimetic
pathway that also involves TLR4 (10). After receptor activation, a number
of adaptor proteins which are involved MyD88 downstream signaling
are recruited, such as IL-1 receptor-associated kinases (IRAK4),
tumor necrosis factor receptor-associated factor 6 (TRAF6) and
NF-κB. The kinase activity of IRAK-4 has also been shown to be
essential for signaling, as overexpression of the kinase-dead form
of IRAK-4 resulted in a reduction in LPS-induced NF-κB activation
(29). Recent studies also showed
that TLR4/MyD88 signaling via TRAF6 and IRAK4 enhances invasiveness
of human lung cancer cells through NF-κB and p38 MAPK pathway
(30). We observed that MOSECs
constitutively expressed MyD88 and TRAF6, even sustained or
upregulated expression of MyD88, TRAF6 and NF-κB at 20 h after
exposure to paclitaxel. However, our results also showed that
MyD88, TARF6 and NF-κB expression in paclitaxel-exposed MOSECs
post-treated with doxorubicin were all down-regulated compared to
MOSECs pre-exposed paclitaxel only. These results suggest that
sequential drug combination might be one choice to overcome
paclitaxel resistance in cancer cells. Another study also suggests
that paclitaxel promotes cell survival by upregulation of the
anti-apoptotic protein X-linked inhibitor of apoptosis (XIAP) and
of Akt phosphorylation (pAkt is inactive) through TLR4 ligation
(26). The essential role of MyD88
in this sequence is supported by observation that tumor expression
of MyD88 correlates negatively with patient survival in some
studies (26).
TLR4/MyD88 signaling generates immune responses
against cancer. Anthracycline drugs including doxorubicin induce
rapid, pre-apoptotic translocation of calreticulin (CRT) to the
cell surface and result in improved processing of tumor cells by
dendritic cells (31,32). However, synergistic effect of
paclitaxel plus doxorubicin on enhancement the antitumor
immunotherapy through an immune-modulatory action is not well
investigated. Our results showed that paclitaxel-exposed MOSECs
post-treated with doxorubicin induced CD4+ T-cell immune
responses without the immune-suppression associated with
chemotherapeutic drug treatment. Based on these results, paclitaxel
and doxorubicin might also be especially effective as first-line
chemotherapeutic drugs for MyD88-positive cancer in a situation
where chemo- and immunotherapy are combined. Further investigation
is needed, however, to fully understand the relationships between
TLR4-MyD88 signaling and other immune-suppressive pathways, which
may involve, for example, Stat3.
Previous studies show that MyD88−/− BMDCs
fail to upregulate IL-12 and IFN-α and -γ in response to viral
particles and thus fail to induce Th1 immune responses and that DC
activation by TLR4 ligands requires MyD88 (33,34).
IL-12 produced by DC augments the cytotoxicity of T cells and NK
cells and regulates IFN-γ production (35). Recently, another study also
suggested that TLR4 and TLR2 play different roles in inflammation
in a heart model (36). However,
our results showed that BMDCs co-cultured with sequential
paclitaxel and doxorubicin treatment activated MyD88 and TRAF6
signaling and result in generating significant IL-12p40 mRNA and
IL-12 protein compared to other groups. Although MOSECs treated
with paclitaxel only for a short time (2 h) also produced some
IL-12, it might not be useful for clinical application because they
showed resistance to paclitaxel. We also expect that little amount
of paclitaxel and doxorubicin brought by MOSECs might play an
important role to mature and activate DCs via TLR4 and TLR2
signaling. From these results, we need to further investigate and
confirm the sequential combination with other cancer drugs.
Several recent studies showed that CD4+ T
cells could eliminate tumors, even when the tumors expressed MHC
class I, but not MHC class II, and this suggested that the
CD4+ T cell responses could outperform the
CD8+ CTLs in mediating an antitumor effector function
(37,38). It has been reported that the
CD4+ T-cell functions against cancer is maximized in the
presence of NK cells (39). We
might expect NK cells help to sustain the CD4 response through the
activation of DCs or NK-DC interaction from the results that DCs
produced the IL-12 after co-culture with paclitaxel-exposed MOSECs
post-treated with doxorubicin. However, further investigation is
required to find the specific bridge between NK cells and
cancer-specific CD4+ T cells in antitumor immunotherapy.
We also recognize the need to discover additional tumor-specific
antigens expressed only on cancer cells that will induce CTLs
against ovarian cancer, and also to optimize the strategies and
conditions for using immunotherapy.
Taken together, our results using sequential
treatment of ovarian cancer cells with paclitaxel and doxorubicin
suggest a new model for overcoming cancer drug resistance and
generating antitumor immune responses. However, we do not know
exactly how much of each drug was delivered and the effects on the
cancer cells. Further investigations may reveal other potentially
effective combinations of drugs and through optimization of drug
dosages and immunization schedules may lead to new clinical
applications.
Abbreviations:
|
MOSECs
|
murine ovarian surface epithelial
cells;
|
|
BMDCs
|
bone marrow-derived dendritic
cells;
|
|
MyD88
|
myeloid differentiation primary
response gene 88;
|
|
TLR
|
Toll-like receptor;
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide;
|
|
PECs
|
peritoneal exudate cells;
|
|
TRAF6
|
tumor necrosis factor receptor
associated factor 6;
|
|
NF-κB
|
nuclear factor-κB
|
Acknowledgements
This study was supported by the Basic
Science Research Program through the National Research Foundation
of Korea (NRF) funded by the Ministry of Education, Science and
Technology (NRF-2009-0065166, NRF-2012- R1A1A3-013468).
References
|
1.
|
Yap TA, Carden CP and Kaye SB: Beyond
chemotherapy: targeted therapies in ovarian cancer. Nat Rev Cancer.
9:167–181. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J,
Murray T and Thun MJ: Cancer statistics, 2008. CA Cancer J Clin.
58:71–96. 2008. View Article : Google Scholar
|
|
3.
|
Dougan M and Dranoff G: Immune therapy for
cancer. Annu Rev Immunol. 27:83–117. 2009. View Article : Google Scholar
|
|
4.
|
Berchuck A, Iversen ES, Luo J, Clarke JP,
Horne H, Levine DA, Boyd J, Alonso MA, Secord AA, Bernardini MQ,
Barnett JC, Boren T, Murphy SK, Dressman HK, Marks JR and Lancaster
JM: Microarray analysis of early stage serous ovarian cancers shows
profiles predictive of favorable outcome. Clin Cancer Res.
15:2448–2455. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Maw MK, Fujimoto J and Tamaya T:
Overexpression of inhibitor of DNA-binding (ID)-1 protein related
to angiogenesis in tumor advancement of ovarian cancers. BMC
Cancer. 9:4302009. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Piccart MJ, Bertelsen K, James K, Cassidy
J, Mangioni C, Simonsen E, Stuart G, Kaye S, Vergote I, Blom R,
Grimshaw R, Atkinson RJ, Swenerton KD, Trope C, Nardi M, Kaern J,
Tumolo S, Timmers P, Roy JA, Lhoas F, Lindvall B, Bacon M, Birt A,
Andersen JE, Zee B, Paul J, Baron B and Pecorelli S: Randomized
intergroup trial of cisplatin-paclitaxel versus
cisplatin-cyclophosphamide in women with advanced epithelial
ovarian cancer: three-year results. J Natl Cancer Inst. 92:699–708.
2000.
|
|
7.
|
Fu Y, Hu D, Qiu J, Xie X, Ye F and Lu WG:
Overexpression of glycogen synthase kinase-3 in ovarian carcinoma
cells with acquired paclitaxel resistance. Int J Gynecol Cancer.
21:439–444. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Kobayashi Y, Seino K, Hosonuma S, Ohara T,
Itamochi H, Isonishi S, Kita T, Wada H, Kojo S and Kiguchi K: Side
population is increased in paclitaxel-resistant ovarian cancer cell
lines regardless of resistance to cisplatin. Gynecol Oncol.
121:390–394. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Duan Z, Duan Y, Lamendola DE, Yusuf RZ,
Naeem R, Penson RT and Seiden MV: Overexpression of MAGE/GAGE genes
in paclitaxel/doxorubicin-resistant human cancer cell lines. Clin
Cancer Res. 9:2778–2785. 2003.PubMed/NCBI
|
|
10.
|
Kawai T and Akira S: The role of
pattern-recognition receptors in innate immunity: update on
Toll-like receptors. Nat Immunol. 11:373–384. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Akira S, Uematsu S and Takeuchi O:
Pathogen recognition and innate immunity. Cell. 124:783–801. 2006.
View Article : Google Scholar
|
|
12.
|
Cook DN, Pisetsky DS and Schwartz DA:
Toll-like receptors in the pathogenesis of human disease. Nat
Immunol. 5:975–979. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Kawasaki K, Akashi S, Shimazu R, Yoshida
T, Miyake K and Nishijima M: Mouse toll-like receptor 4.MD-2
complex mediates lipopolysaccharide-mimetic signal transduction by
Taxol. J Biol Chem. 275:2251–2254. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Takeda K, Kaisho T and Akira S: Toll-like
receptors. Annu Rev Immunol. 21:335–376. 2003. View Article : Google Scholar
|
|
15.
|
Nozaki N, Shishido T, Takeishi Y and
Kubota I: Modulation of doxorubicin-induced cardiac dysfunction in
toll-like receptor-2-knockout mice. Circulation. 110:2869–2874.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Szajnik M, Szczepanski MJ, Czystowska M,
Elishaev E, Mandapathil M, Nowak-Markwitz E, Spaczynski M and
Whiteside TL: TLR4 signaling induced by lipopolysaccharide or
paclitaxel regulates tumor survival and chemoresistance in ovarian
cancer. Oncogene. 28:4353–4363. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Kreuz S, Siegmund D, Rumpf JJ, Samel D,
Leverkus M, Janssen O, Häcker G, Dittrich-Breiholz O, Kracht M,
Scheurich P and Wajant H: NFkappaB activation by Fas is mediated
through FADD, caspase-8, and RIP and is inhibited by FLIP. J Cell
Biol. 166:369–380. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Tanimura N, Saitoh S, Matsumoto F,
Akashi-Takamura S and Miyake K: Roles for LPS-dependent interaction
and relocation of TLR4 and TRAM in TRIF-signaling. Biochem Biophys
Res Commun. 368:94–99. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
No authors listed: Cyclophosphamide plus
cisplatin versus cyclophosphamide, doxorubicin, and cisplatin
chemotherapy of ovarian carcinoma: a meta-analysis. The Ovarian
Cancer Meta-Analysis Project. J Clin Oncol. 9:1668–1674. 1991.
|
|
20.
|
A’Hern RP and Gore ME: Impact of
doxorubicin on survival in advanced ovarian cancer. J Clin Oncol.
13:726–732. 1995.PubMed/NCBI
|
|
21.
|
Roby KF, Taylor CC, Sweetwood JP, Cheng Y,
Pace JL, Tawfik O, Persons DL, Smith PG and Terranova PF:
Development of a syngeneic mouse model for events related to
ovarian cancer. Carcinogenesis. 21:585–591. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Kim D, Hoory T, Monie A, Wu A, Hsueh WT,
Pai SI and Hung CF: Delivery of chemotherapeutic agents using
drug-loaded irradiated tumor cells to treat murine ovarian tumors.
J Biomed Sci. 17:612010. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Fewell JG, Matar MM, Rice JS, Brunhoeber
E, Slobodkin G, Pence C, Worker M, Lewis DH and Anwer K: Treatment
of disseminated ovarian cancer using nonviral interleukin-12 gene
therapy delivered intraperitoneally. J Gene Med. 11:718–728. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Jang MJ, Kim JE, Chung YH, Lee WB, Shin
YK, Lee JS, Kim D and Park YM: Dendritic cells stimulated with
outer membrane protein A (OmpA) of Salmonella typhimurium
generate effective anti-tumor immunity. Vaccine. 29:2400–2410.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Kim JE, Jang MJ, Lee JI, Chung YH, Jeong
JH, Hung CF and Kim D: Cancer cells containing nanoscale
chemotherapeutic drugs generate antiovarian cancer-specific
CD4+ T cells in peritoneal space. J Immunother. 35:1–13.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Kelly MG, Alvero AB, Chen R, Silasi DA,
Abrahams VM, Chan S, Visintin I, Rutherford T and Mor G: TLR-4
signaling promotes tumor growth and paclitaxel chemoresistance in
ovarian cancer. Cancer Res. 66:3859–3868. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Zbar B, Bernstein I, Tanaka T and Rapp HJ:
Tumor immunity produced by the intradermal inoculation of living
tumor cells and living Mycobacterium bovis (strain BCG).
Science. 170:1217–1218. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Baum H and Baum M:
Methyl-cholanthrene-induced sarcomata in mice after immunisation
with Corynebacterium parvum plus syngeneic subcellular membrane
fractions. Lancet. 2:1397–1398. 1974. View Article : Google Scholar
|
|
29.
|
Jiang Z, Ninomiya-Tsuji J, Qian Y,
Matsumoto K and Li X: Interleukin-1 (IL-1) receptor-associated
kinase-dependent IL-1-induced signaling complexes phosphorylate
TAK1 and TAB2 at the plasma membrane and activate TAK1 in the
cytosol. Mol Cell Biol. 22:7158–7167. 2002. View Article : Google Scholar
|
|
30.
|
Xu Z, Ren T, Xiao C, Li H and Wu T: Nickel
promotes the invasive potential of human lung cancer cells via
TLR4/MyD88 signaling. Toxicology. 285:25–30. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Coppolino MG, Woodside MJ, Demaurex N,
Grinstein S, St-Arnaud R and Dedhar S: Calreticulin is essential
for integrin-mediated calcium signalling and cell adhesion. Nature.
386:843–847. 1997. View Article : Google Scholar
|
|
32.
|
Obeid M, Tesniere A, Ghiringhelli F, Fimia
GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T,
Casares N, Métivier D, Larochette N, van Endert P, Ciccosanti F,
Piacentini M, Zitvogel L and Kroemer G: Calreticulin exposure
dictates the immunogenicity of cancer cell death. Nat Med.
13:54–61. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Yang R, Murillo FM, Cui H, Blosser R,
Uematsu S, Takeda K, Akira S, Viscidi RP and Roden RB:
Papillomavirus-like particles stimulate murine bone marrow-derived
dendritic cells to produce alpha interferon and Th1 immune
responses via MyD88. J Virol. 78:11152–11160. 2004. View Article : Google Scholar
|
|
34.
|
Sun J, Walsh M, Villarino AV, Cervi L,
Hunter CA, Choi Y and Pearce EJ: TLR ligands can activate dendritic
cells to provide a MyD88-dependent negative signal for Th2 cell
development. J Immunol. 174:742–751. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Kayashima H, Toshima T, Okano S, Taketomi
A, Harada N, Yamashita Y, Tomita Y, Shirabe K and Maehara Y:
Intratumoral neoadjuvant immunotherapy using IL-12 and dendritic
cells is an effective strategy to control recurrence of murine
hepatocellular carcinoma in immunosuppressed mice. J Immunol.
185:698–708. 2010. View Article : Google Scholar
|
|
36.
|
Ma Y, Zhang X, Bao H, Mi S, Cai W, Yan H,
Wang Q, Wang Z, Yan J, Fan G, Lindsey ML and Hu Z: Toll-like
receptor (TLR) 2 and TLR4 differentially regulate doxorubicin
induced cardiomyopathy in mice. PLoS One. 7:e407632012. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Kennedy R and Celis E: Multiple roles for
CD4+ T cells in anti-tumor immune responses. Immunol
Rev. 222:129–144. 2008.
|
|
38.
|
Hunder NN, Wallen H, Cao J, Hendricks DW,
Reilly JZ, Rodmyre R, Jungbluth A, Gnjatic S, Thompson JA and Yee
C: Treatment of metastatic melanoma with autologous CD4+
T cells against NY-ESO-1. N Engl J Med. 358:2698–2703. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Perez-Diez A, Joncker NT, Choi K, Chan WF,
Anderson CC, Lantz O and Matzinger P: CD4 cells can be more
efficient at tumor rejection than CD8 cells. Blood. 109:5346–5354.
2007. View Article : Google Scholar : PubMed/NCBI
|