Introduction
Pancreatic ductal adenocarcinoma (PDA) is the fourth
leading cause of cancer-related deaths in the United States and is
almost uniformly lethal with an estimated annual incidence of
43,140 new cases approximating 36,800 annual deaths and a 5-year
survival rate of <5% (1–3).
Late initial diagnosis, aggressive metastatic behavior and
resistance to chemoradiotherapy render pancreatic cancer one of the
most difficult to treat malignant diseases. Surgical resection is
curative; however, nearly 80% of the patients are diagnosed with
locally advanced metastatic disease, precluding surgical
intervention. Gemcitabine, the current standard of care for
advanced pancreatic cancer, provides only short-term symptomatic
improvement with minor impact on survival (4,5). The
integration of multiple modalities also has not improved survival.
Thus, there is the dire need for more active agents and novel
strategies for the treatment of pancreatic cancer.
Herbal remedies are used in traditional medicine to
treat and prevent human diseases including cancer. Numerous plant
derived flavonoids and phenolic/polyphenolic compounds with
antioxidant and anti-inflammatory activities are currently used by
cancer patients as dietary supplements to complement chemotherapy.
In fact, isolation and identification of bioactive components from
medicinal plants have led to the synthesis and development of
potent anticancer drugs, including Vinca alkaloids, taxol,
camptothecan, etoposide and retinoids. Triterpenoids are members of
a larger family of structurally related compounds known as
cyclosqualenoids that are widely distributed in nature. Pristimerin
(PM) is a quinonemethide triterpenoid present in various plant
species in the Celastraceae and Hippocrateaceae
families (6,7). PM and related compounds have shown
anti-inflammatory, antioxidant and antimalarial activities
(8–10). Recent studies have shown potent
antiproliferative and apoptosis-inducing activity of PM in glioma,
leukemia, breast, lung and prostate cancer cell lines (11–14).
Induction of apoptosis by PM involved activation of caspases,
mitochondrial dysfunction, inhibition of anti-apoptotic nuclear
factor-κB (NF-κB) and Akt (15–17).
Recent evidence also shows participation of reactive oxygen species
(ROS) in the antitumor activity of PM (18). In addition, PM also inhibited
proteasome activity, tumor cell migration and angiogenesis
(13,19,20).
To the best of our knowledge there has been only one
published report showing the antitumor activity of PM against
pancreatic cancer cells through the inhibition of cell cycle
progression and induction of apoptosis (21). Since this report, there has been no
additional report on the anticancer activity and mechanism(s) of
action of PM in pancreatic cancer cells. In the present study, we
demonstrate that PM inhibits proliferation and induces apoptosis in
pancreatic cancer cells by inhibiting the pro-survival Akt, NF-κB
and mTOR signaling proteins and their downstream mediators as well
as anti-apoptotic Bcl-2. The inhibition of Bcl-2 by PM was not
through the proteolytic degradation but resulted from the
inhibition of Bcl-2 gene transcription.
Materials and methods
Reagents
Pristimerin was purchased from Sigma Chemicals (St.
Louis, MO). Anti-caspase-3 and caspase-9 antibodies were purchased
from BD Pharmingen (San Diego, CA). Anti-PARP-1, anti-Bcl-2,
anti-Bcl-xL and anti-survivin antibodies were purchased from Santa
Cruz Biotechnology (Santa Cruz, CA). 96 AQueous One Solution
Proliferation Assay System was from Promega (Madison, WI). Annexin
V-FITC apoptosis detection kit was purchased from BD Pharmingen and
mitochondrial potential sensor JC-1 was obtained from Molecular
Probes, Invitrogen (San Diego, CA). Caspase-3 activity kit was from
Clontech Laboratories (Mountain View, CA). Stock solution of PM
(100 mM) was prepared in DMSO and all test concentrations were
prepared by diluting stock solution in tissue culture medium.
Cell lines
MiaPaCa-2 and Panc-1 PDA cell lines were obtained
from the American Type Culture Collection (ATCC, Rockville, MD).
Both cell lines were grown in DMEM tissue culture medium
(Gibco-BRL, Rockville, MD) supplemented with 10% fetal bovine
serum, 1% penicillin/streptomycin, and 25 mM HEPES buffer. Cells
were incubated at 37°C in a humidified atmosphere consisting of 5%
CO2, 95% air and maintained by splitting cultures twice
a week.
Measurement of cell viability (MTS
assay)
Tumor cells (1×104) in 100 μl of
tissue culture medium were seeded into each well of a 96-well
plate. After 24-h incubation to allow cells to adhere, cells were
treated with PM at concentrations ranging from 0 to 5 μM.
Cultures were incubated for additional 72 h and cell viability was
then determined by the colorimetric MTS assay using CellTiter 96
AQueous One Solution Proliferation Assay System from Promega. This
assay measures the bioreduction of the tetrazolium compound MTS by
intracellular dehydrogenases in the presence of electron-coupling
reagent phenazine methosulfate. MTS and phenazine methosulfate were
added to the culture wells, and cultures were incubated for 2 h at
37°C. The absorbance, which is directly proportional to the number
of viable cells in the cultures, was measured at 490 nm using a
microplate reader.
Annexin V-FITC binding
Induction of apoptosis was assessed by the binding
of Annexin V-FITC to the cells. Briefly, MiaPaCa-2 and Panc-1 cells
treated with PM (0 to 5 μM) for 20 h were incubated with 5
μl of Annexin V-FITC and 5 μl of PI for 30 min at
room temperature in the dark. Stained cells were analyzed by flow
cytometry.
Western blot analysis
Cell lysates were prepared by detergent lysis [1%
Triton X-100 (v/v), 10 mM Tris-HCl (pH 7.5), 5 mM EDTA, 150 mM
NaCl, 10% glycerol, 2 mM sodium vanadate, 5 μg/ml leupeptin,
1 μg/ml aprotinin, 1 μg/ml pepstatin A and 10
μg/ml 4-2-aminoethyl-benzenesulfinyl fluoride]. Lysates were
clarified by centrifugation at 14,000 × g for 10 min at 4°C, and
protein concentrations were determined by Bradford assay. Samples
(50 μg) were boiled in an equal volume of sample buffer [20%
glycerol, 4% SDS, 0.2% bromophenol blue, 125 mM Tris-HCl (pH 7.5),
and 640 mM 2-mercaptoethanol] and separated on 10%
SDS-polyacrylamide gels. Proteins resolved on the gels were
transferred to nitrocellulose membranes and probed with antibodies
to procaspases -3 and -9 (1:1,000), Akt, NF-κB, mTOR, Bcl-2,
Bcl-xL, survivin, cytochrome c, Cox IV or β-actin (all at
1:500 dilution) followed by HRP-conjugated secondary antibody.
Immune complexes were visualized by chemiluminescence. Protein
bands were imaged and band densities analyzed using the NIH/Scion
image analysis software. The protein band densities were normalized
to the corresponding β-actin band densities and relative change in
signal strength was calculated.
Measurement of mitochondrial
depolarization and cytocyto-chrome c release
The loss of mitochondrial potential was determined
using mitochondrial potential sensor JC-1 (Molecular Probes,
Invitrogen). Control or cells treated with PM (1×106)
were loaded with mitochondrial sensor JC-1 dye (10 μg/ml)
for 10 min at 22°C. Cells were then analyzed by flow cytometry. In
normal cells, dye is aggregated in mitochondria, fluoresces red,
and is detected in the FL2 channel. In cells with altered
mitochondrial potential, the dye fails to accumulate in the
mitochondria, remains as monomers in the cytoplasm, fluoresces
green and is detected in the FL1 channel.
For effect on mitochondrial cytochrome c,
mitochondrial and cytosolic fractions of control and treated cells
were prepared using ApoAlert Cell Fractionation Kit (Clontech
Laboratories) and were separated on a 14% SDS-PAGE gel. After
transfer of proteins, membranes were probed with cytochrome
c antibody.
Measurement of Bcl-2 gene expression
The effect of PM on Bcl-2 gene expression was
measured by analyzing Bcl-2 mRNA by RT-PCR. Total cellular RNA was
extracted with TRIzol reagent (Gibco-BRL) and 1 μg of RNA
was reverse transcribed by oligo-dt primer and high fidelity
reverse transcriptase (Boehringer, Mannheim, Germany) to generate
cDNAs. cDNA was used (1 μl) of as the template for
polymerase chain reaction (PCR) using Bcl-2 primers: upper, 5′-CGA
CTT CGC CGA GAT GTC CAG CCA G-3′; and lower, 5′-ACT TGT GGC CCA GAT
AGG CAC CCA G-3′; and GAPDH primers: upper, 5′-TCC CTC AAG, ATT,
GTC AGC AA-3′; and lower, 5′-AGA TCC ACA ACG GAT ACA TT-3′. The PCR
reaction conditions used were: a first cycle of 10 min at 95°C, 45
sec at 65°C and 1 min at 72°C followed by 45 sec at 95°C, 45 sec at
65°C and 1 min at 72°C for 36 cycles. The PCR products were
separated on 2% agarose gel and visualized by ethidium bromide
staining. The size of DNA fragments amplified by Bcl-2 primers
GAPDH primers was 388 and 173 bp, respectively.
Statistical analysis
Data are presented as the means ± S.D. The
differences between control and treatment groups were analyzed
using Dunnett’s multiple comparison test and differences with
p<0.05 were considered statistically significant.
Results
Pristimerin inhibits proliferation of
pancreatic cancer cells
To measure the effect of PM on proliferation of
pancreatic cancer cells, MiaPaCa-2 and Panc-1 cells were treated
with PM for 72 h at concentrations ranging from 0.625 to 5
μM. At the end of the treatment, viability of cultures was
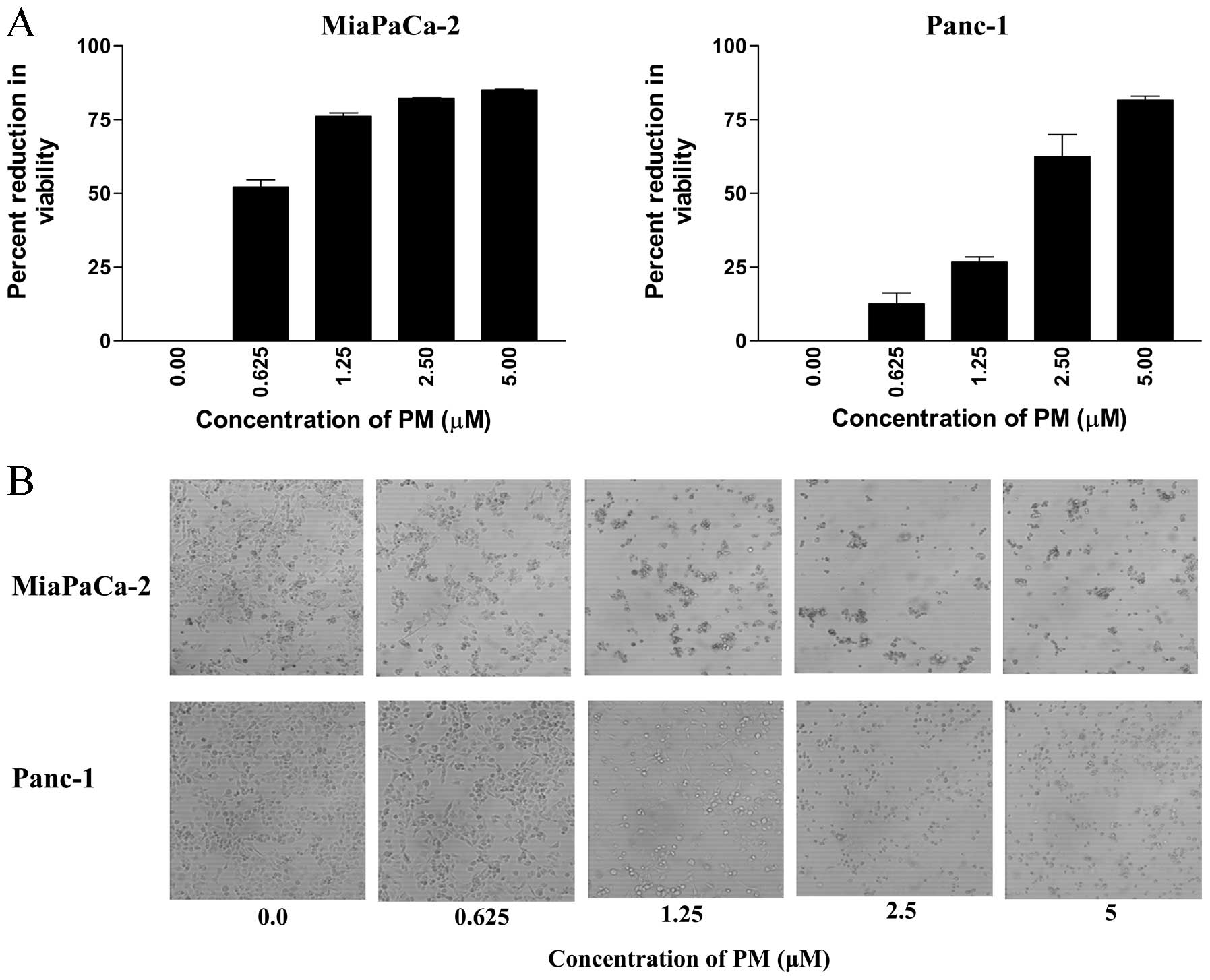
determined by MTS assay. As shown in Fig. 1A, PM significantly reduced the
proliferation of both cell lines (measured from the loss of
viability of cultures) at concentrations of 0.625 to 5 μM
(MiaPaCa-2, 52 to 85% reduction; Panc-1, 13 to 81% reduction,
p<0.05).
The antiproliferative effect of PM in MTS assay
correlated with the morphological changes in cell cultures treated
with PM. Microscopic examination of MiaPaCa-2 and Panc-1 cell
cultures at 48 h after treatment with PM showed partial rounding of
cells at 0.625 μM. At higher concentrations of PM (1.25 to 5
μM), there was extensive cellular detachment, rounding,
shrinkage and clumping of cells in cultures of both cell lines in a
dose-dependent manner (Fig. 1B).
Together, MTS and morphological data indicated strong
antiproliferative effect of PM on pancreatic cancer cells.
Pristimerin induces apoptosis in
pancreatic cancer cells
To determine whether PM induces apoptosis in
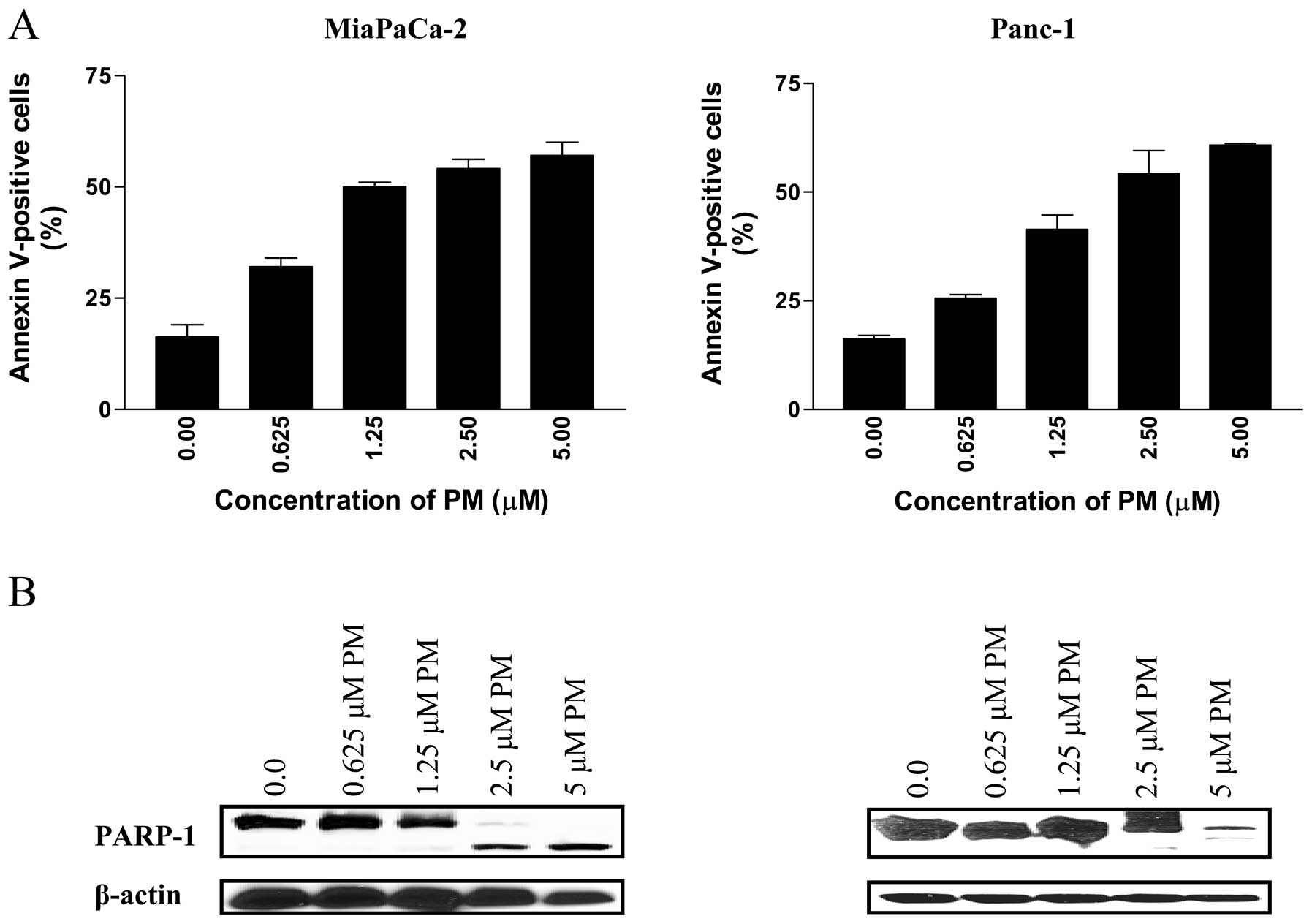
pancreatic cancer cells, we first measured the binding of Annexin
V-FITC to MiaPaCa-2 and Panc-1 cells treated with PM by flow
cytometry. As shown in Fig. 2A,
treatment with PM (0.625 to 5 μM) for 20 h significantly
increased the percentage of Annexin V-FITC binding cells in both
cell lines. In MiaPaCa-2 cells, the percentage of Annexin
V-FITC-binding cells increased from 32% at 0.625 μM to 61%
at 5 μM PM (p<0.05). Similarly, the percentage of Annexin
V-FITC-binding Panc-1 cells increased from 25% at 0.625 μM
to 57% at 5 μM PM (p<0.05).
The induction of apoptosis by PM was confirmed by
the cleavage of PARP-1 by western blot analysis. As shown in
Fig. 2B, PARP-1 was cleaved in
both cell lines at PM concentrations of 2.5 to 5 μM.
Pristimerin causes cleavage of
procaspases in pancreatic cancer cells
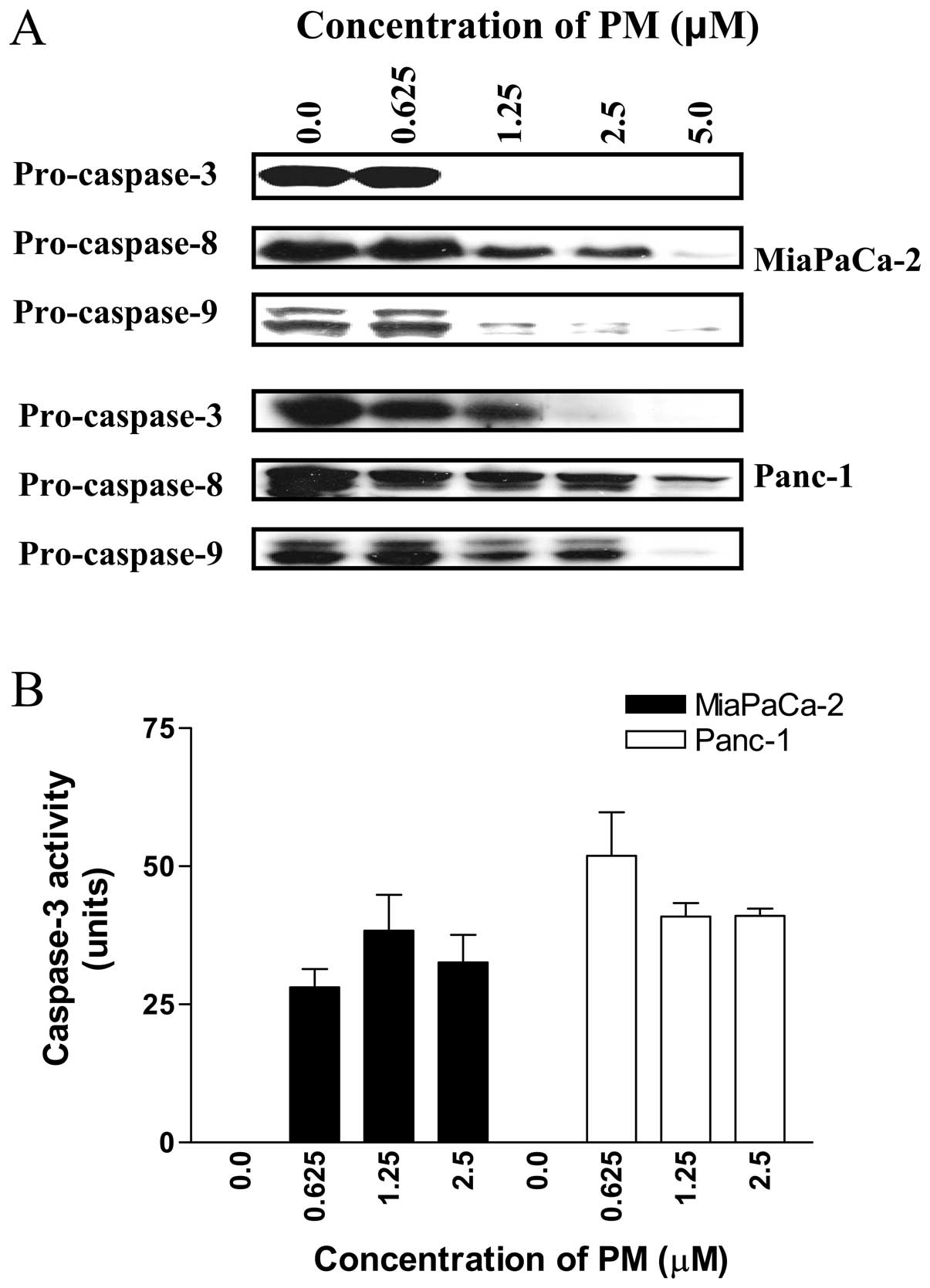
For further evidence that PM induces apoptosis in
pancreatic cancer cells we also examined the effect of PM on the
activation of procaspases-3, -8 and -9. Western blot analysis of
cell lysates prepared from cells treated with PM showed partial to
complete processing of procaspases-3, -8 and -9 in MiaPaCa-2 cells
at 2.5 to 5 μM PM (Fig.
3A). In Panc-1 cells, significant to complete processing of
procaspases-3, -8 and -9 was evident at PM concentrations of 1.25
to 5 μM.
We also measured caspase-3 in cells treated with PM
using ApoAlertR caspase-3 assay kit from Clontech Laboratories.
MiaPaCa-2 and Panc-1 cells treated with PM (0 to 2.5 μM) for
20 h were processed according to the manufacturer’s protocol for
the colorimetric measurement of caspase-3 activity. As shown in
Fig. 3B, PM significantly induced
caspase-3 activity at concentrations of 0.625 to 2.5 μM in
both cell lines compared to control cells (p<0.05).
Taken together, increase in Annexin V-FITC-binding
cells, cleavage of PARP-1 and procaspases-3, -8, -9 and increase in
caspase-3 activity indicated induction of apoptosis by PM.
Pristimerin induces mitochondrial
depolarization and release of cytochrome c
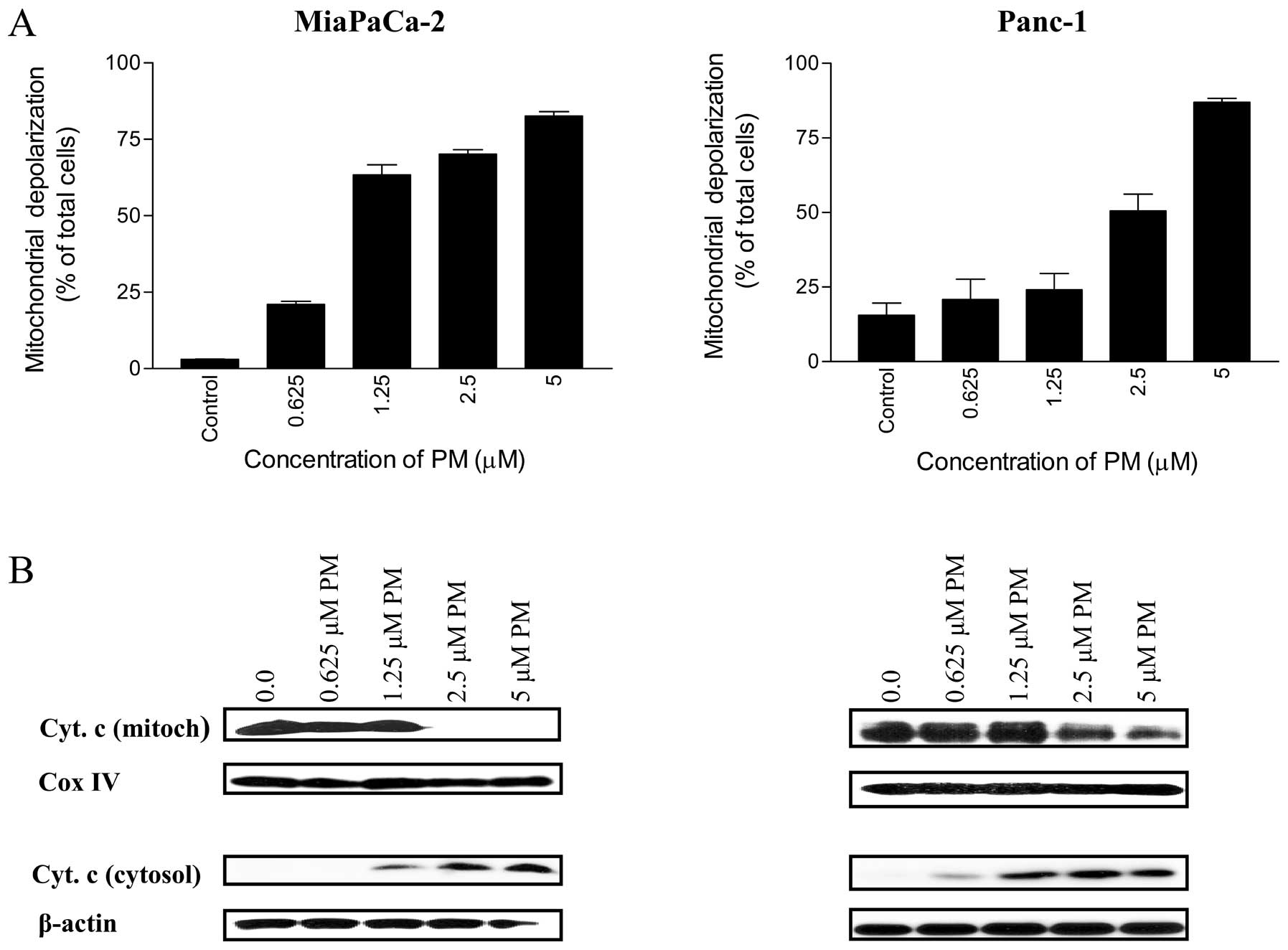
Whether PM utilizes mitochondrial ‘intrinsic’
pathway in apoptotic death of pancreatic cancer cells was
investigated next. For this, we evaluated the change in
mitochondrial membrane potential following the treatment of cells
with PM. Thus, MiaPaCa-2 and Panc-1 cells treated with PM (0.625–5
μM) for 20 h were loaded with mitochondrial
membrane-potential JC-1 probe and fluorescent shift in cells was
measured by flow cytometry. As shown in Fig. 4A, the percentage of MiaPaCa-2 cells
with green fluorescence changed from 3% at 0 μM PM to 21,
63, 70 and 82% at 0.625, 1.25, 2.5 and 5 μM PM,
respectively. The percentage of Panc-1 cells with green
fluorescence also increased significantly (p<0.05) after
treatment with PM (e.g., 15, 21, 24, 50 and 87% of cells with green
fluorescence at 0, 0.625, 1.25, 2.5 and 5 μM PM,
respectively).
To further determine the effect of PM on
mitochondrial integrity, the release of cytochrome c from
mitochondria in cells treated with PM was measured. Western blot
analysis of mitochondrial and cytosolic fractions of MiaPaCa-2 and
Panc-1 cells treated with PM (0.625–5 μM) demonstrated the
release of cytochrome c from the mitochondria in both cell
lines (Fig. 4B). Mitochondrial
cytochrome c levels were more than 90% decreased in
MiaPaCa-2 cells at 2.5 μM and were completely devoid of it
at 5 μM PM without significant change in Cox IV levels
(loading control). PM also induced the release of cytochrome
c from mitochondria in Panc-1 cells at 2.5 and 5 μM
PM. As expected, decrease in mitochondrial cytochrome c
correlated with the corresponding increase in cytosolic cytochrome
c in both cell lines (Fig.
4B). Together, these data demonstrated induction of
mitochondrial ‘intrinsic’ pathway of apoptosis by PM in the
pancreatic cancer cells.
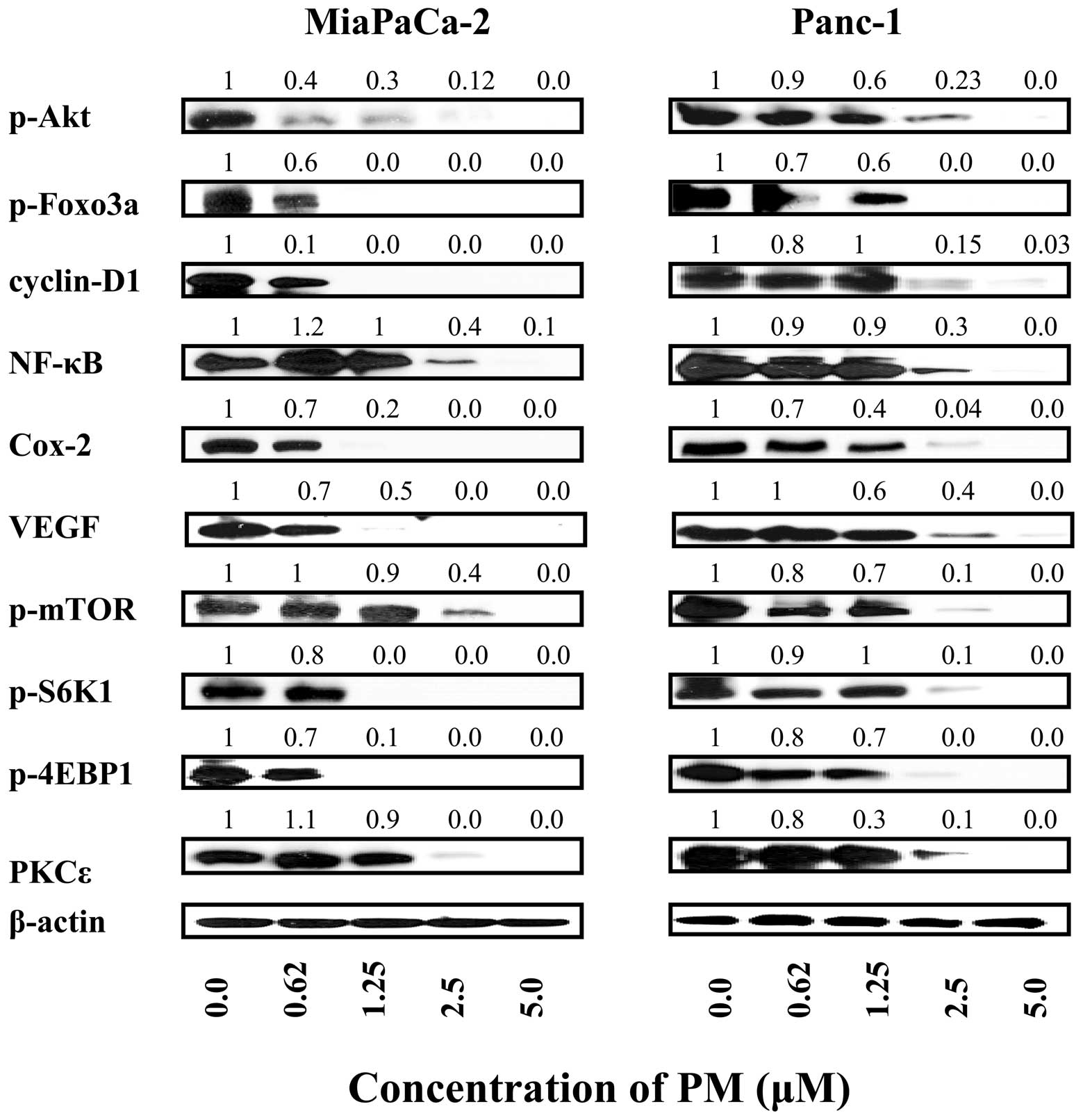
Pristimerin inhibits pro-survival
signaling proteins in pancreatic cancer cells
Akt, NF-κB and mTOR are pro-survival signaling
proteins that are constitutively active in a variety of human
cancers and confer survival advantage and resistance of cancer
cells to various forms of anticancer therapies. We investigated
whether induction of apoptosis in pancreatic cancer cells by PM
involved the inhibition of Akt, NF-κB, mTOR and downstream
mediators of these signaling molecules. Cellular lysates prepared
from MiaPaCa-2 and Panc-1 cells treated with PM (0 to 5 μM)
for 20 h were analyzed by western blot analysis for levels of p-Akt
and Akt regulated Foxo-3α and cyclin D1; NF-κB (p65) and
NF-κB-regulated Cox-2 and VEGF; p-mTOR and mTOR-regulated p-S6K1
and p-4E-BP1. p-Akt, NF-κB and p-mTOR were significantly to
completely inhibited in both cell lines by PM at concentrations of
1.25 to 5 μM (Fig. 5). PM
also inhibited the levels of various downstream mediators of these
signaling proteins at similar concentrations. Since PKCɛ has been
invoked in induction of apoptosis by anticancer agents we analyzed
the effect of PM on the levels of PKCɛ. PM dramatically reduced the
level of PKCɛ in both cell lines at concentrations of 2.5 to 5
μM, suggesting that PKCɛ-mediated apoptotic pathway is also
a target of PM. Together, these data indicated that induction of
apoptosis in pancreatic cancer cells by PM involves the inhibition
of pro-survival p-Akt, NF-κB and mTOR and their downstream
mediators as well as PKCɛ mediated pathway of apoptosis.
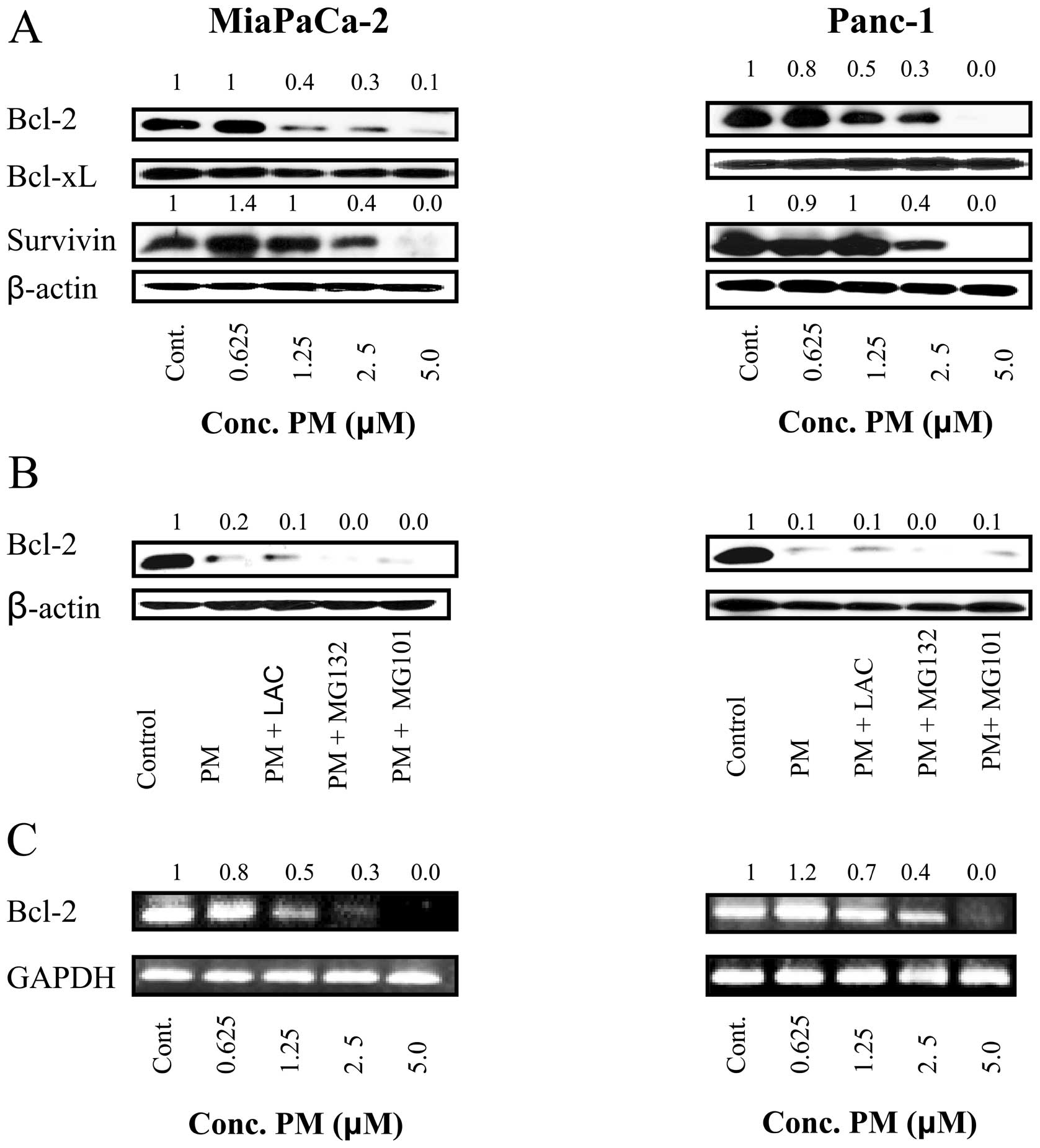
Pristimerin downregulates anti-apoptotic
Bcl-2 but not Bcl-xL expression in pancreatic cancer cells
Bcl-2 and Bcl-xL are anti-apoptotic proteins located
in mitochondrial wall that regulate mitochondrial or ‘intrinsic’
apoptosis pathway by controlling mitochondrial permeability. We
determined the effect of PM on expression of these proteins in
MiaPaCa-2 and Panc-1 cells. Lysates of cells treated with PM (0.625
to 5 μM) for 20 h were analyzed by western blot analysis for
the levels of Bcl-2 and Bcl-xL. As shown in Fig. 6A, PM partially to completely
inhibited Bcl-2 in both cell lines at 1.25 to 5 μM PM. In
contrast, the levels of Bcl-xL were not affected by PM in these
cell lines. Furthermore, survivin, which is an inhibitor of
apoptosis was also reduced by PM in both cell lines.
In order to investigate the mechanism by which PM
inhibits Bcl-2, we examined whether PM mediates proteasomal or
lysosomal degradation of Bcl-2. MiaPaCa-2 and Panc-1 cells were
treated with PM (5 μM) in the absence or presence of
proteasomal inhibitors lactacystin and MG132 or calpain inhibitor
MG101 for 20 h and cell lysates were analyzed for Bcl-2 by western
blot analysis. As shown in Fig.
6B, neither proteasomal nor calpain inhibitors prevented the
down-regulation of Bcl-2 by PM, indicating that PM does not induce
proteolytic degradation of Bcl-2.
Next we evaluated the effect of PM on Bcl-2 gene
expression by RT-PCR. Treatment with PM at 1.25 to 5 μM for
20 h inhibited Bcl-2 mRNA in both cell lines in a
concentration-related manner without affecting the expression of
GAPDH, a housekeeping gene (Fig.
6C). Thus, RT-PCR data suggested that PM downregulates Bcl-2 by
inhibiting Bcl-2 gene transcription.
Discussion
Pristimerin containing plant products have been used
in traditional medicine to treat a variety of diseases including
inflammation and cancer. However, the purported medicinal effects
of pristimerin have only recently come under scientific scrutiny
and the antiproliferative and apoptosis-inducing activity of
pristimerin has been validated against diverse types of tumor cell
lines in a small number of studies (11–18).
Although, these studies have provided some insights into the mode
of cell death by pristimerin; the molecular mechanisms of the
proapoptotic activity of pristimerin remain to be understood. In
the present study, we investigated the mechanism of the antitumor
activity of pristimerin in MiaPaCa-2 and Panc-1 pancreatic cancer
cell lines with a focus on the effect of pristimerin on
pro-survival (anti-apoptotic) cellular mechanisms. PM significantly
inhibited the proliferation of both cell lines at concentrations of
1.25 to 5 μM. The inhibition of cell proliferation by PM was
associated with induction of apoptosis as demonstrated by the
increased binding of Annexin V due to the externalization of
phosphatidylserine to the outer leaflet of the cell membrane and
cleavage of PARP-1, both well recognized markers of apoptosis.
These results indicated that induction of apoptosis is part of the
mechanism by which PM destroys pancreatic cancer cells and
corroborate the result of a previous study that also showed
induction of apoptosis in pancreatic cancer cells by PM (21).
Two major pathways of apoptotic cell death program
have been identified, namely receptor-mediated (extrinsic) and
mitochondrial (intrinsic) apoptotic cell death pathways (22). In both cases, caspases, a family of
cysteine proteases, play an important role in the apoptotic cell
death. In the extrinsic pathway, binding of the death ligands
(e.g., TNF-α, FasL, TRAIL) with their cognate receptors activates
initiator caspase-8 which then cleaves and activates effector
caspases -3, -6 and -7 leading to apoptosis (23). In mitochondrial or intrinsic
pathway, undefined signals induce release of cytochrome c
from mitochondria, which in conjunction with Apaf-1 causes the
activation of initiator caspase-9. Activated caspase-9, in turn,
activates effector caspases-3, -6 and -7 (22). Pristimerin induced the cleavage of
initiator procaspases-8 and -9 and effector caspase-3. The cleavage
of procaspase-9 indicated that PM activates the mitochondrial
(intrinsic) pathway of apoptosis in pancreatic cancer cells. The
activation of the mitochondrial pathway by PM is also supported by
the loss of mitochondrial membrane potential and release of
cytochrome c from mitochondria in cancer cells treated with
PM (Fig. 4). Thus, the cleavage of
procaspase-9 and release of cytochrome c from mitochondria
leading to the activation of effector caspase-3 indicated that the
mitochondrial apoptosis pathway plays a significant role in the
apoptotic cell death of pancreatic cancer cells by PM. The
processing of procaspase-8 by PM suggested that the extrinsic
pathway of apoptosis may also be involved in the apoptotic cell
death of pancreatic cancer cells by PM. Activated caspase-8 may
directly cleave and activate caspase-3 or it may facilitate
caspase-9 mediated apoptosis through the truncation of Bid, a
proapoptotic BH3 only Bcl-2 family member that links extrinsic
pathway with the intrinsic pathway of apoptosis (24,25).
Phosphatidylinositol-3 kinase/Akt (PI3K/Akt)) signal
transduction pathway which controls cell proliferation, survival,
apoptosis and malignant transformation is frequently activated in a
variety of malignancies including pancreatic cancer (26). p-Akt promotes cell growth and
survival by inactivating downstream substrates such as Bad,
procaspase-9, and forkhead transcription factors (27,28).
Anti-apoptotic NF-κB and progrowth mTOR signaling pathways are
downstream targets of activated Akt. NF-κB family of transcription
factors controls the expression of genes involved in immune and
inflammatory responses, cell proliferation, oncogenesis,
angiogenesis and Bcl-2 family members (29). NF-κB plays a critical role in
resistance of cancer cells to anticancer therapies by protecting
them from apoptosis (30). mTOR is
a 290 kDa serine-threonine kinase, which controls cell growth,
survival, division and motility is activated in human tumors
(31,32). PM inhibited p-Akt and its
downstream targets Foxo-3α and cyclin D1; NF-κB (p65) and
NF-κB-regulated Cox-2 and VEGF; p-mTOR and mTOR-regulated p-S6K1
and p-4E-BP1 in both pancreatic cancer cell lines. Thus, each of
the three major pro-survival (anti-apoptotic) signaling pathways
related proteins were inhibited by PM, suggesting that targeting
multiple pro-survival signaling pathways may be critical for the
antiproliferative and apoptosis-inducing activity of PM.
The intrinsic (mitochondrial) pathway of apoptosis
is regulated by members of the Bcl-2 family of proteins and
inhibitors of apoptosis (33,34).
Bcl-2 and Bcl-xL are major anti-apoptotic members of Bcl-2 family
that inhibit apoptosis by preventing the activation of inner
mitochondrial permeability transition pore and release of
proapotogenic mitochondrial contents including cytochrome c
(33). PM inhibited Bcl-2 but had
no effect on the expression of Bcl-xL. In addition to Bcl-2, PM
also reduced the levels of anti-apoptic survivin. Thus, inhibition
of these anti-apoptotic proteins seems essential for induction of
apoptosis by PM. These data also imply that inhibition of Bcl-2 by
PM is sufficient to facilitate Bax and Bak mediated mitochondrial
permeability transition and release of cytochrome c without
the inhibition of Bcl-xL.
The stability of Bcl-2 is regulated through
post-translational modifications, such as dephosphorylation,
ubiquitination and proteasomal degradation (35). However, PM-induced downregulation
of Bcl-2 was not mediated through the proteolytic degradation,
since treatment of tumor cells with PM in the presence of
proteasomal and calpain inhibitors failed to prevent downregulation
of Bcl-2. On the other hand, RT-PCR analysis revealed that the
inhibition of Bcl-2 by PM is mediated through the inhibition of
Bcl-2 gene transcription. Collectively, results of the present
study suggest that a better understanding of the mechanism of the
proapoptotic activity of PM can potentially facilitate the clinical
development of pristimerin for the treatment of pancreatic
cancer.
Acknowledgements
This study was supported by NIH grant
1R01 CA130948 from the National Cancer Institute to S.C.G.
References
|
1.
|
Pancreatic Cancer-National Cancer
Institute, U.S National Institutes of Health: Cancer.
Gov.http:www.cancer.gov/cancer-topics/types/pancreaticRetrieved
06-042010.
|
|
2.
|
Maitra A and Hruban RH: Pancreatic cancer.
Annu Rev Pathol. 3:157–188. 2008. View Article : Google Scholar
|
|
3.
|
Li D, Xie K, Wolff R and Abbruzzese JL:
Pancreatic cancer. Lancet. 363:1049–1057. 2004. View Article : Google Scholar
|
|
4.
|
Mulcahy MF, Wahl AO and Small W Jr: The
current status of combined radiotherapy and chemotherapy for
locally advanced or resected pancreas cancer. J Natl Compr Canc
Netw. 3:637–642. 2005.PubMed/NCBI
|
|
5.
|
Pino SM, Xiong HQ, McConkey D and
Abbruzzese JL: Novel therapies for pancreatic adenocarcinoma. Curr
Oncol Rep. 6:199–206. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Buffa Filho W, Corsino J, Bolzani da SV,
Furlan M, Pereira AM and Franca SC: Quantitative determination for
cytotoxic Friedo-nor-oleanane derivatives from five morphological
types of Maytenus ilicifolia (Celastraceae) by reverse-phase
high-performance liquid chromatography. Phytochem Anal. 13:75–78.
2002.PubMed/NCBI
|
|
7.
|
Chang FR, Hayashi K, Chen IH, et al:
Antitumor agents. 228. Five new agarofurans, Reissantins A-E, and
cytotoxic principles from Reissantia buchananii. J Nat Prod.
66:1416–1420. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Sassa H, Kogure K, Takaishi Y and Terada
H: Structural basis of potent antiperoxidation activity of the
triterpene celastrol in mitochondria: effect of negative membrane
surface charge on lipid peroxidation. Free Radic Biol Med.
17:201–207. 1994. View Article : Google Scholar
|
|
9.
|
Dirsch VM, Kiemer AK, Wagner H and Vollmar
AM: The triterpenoid quinonemethide pristimerin inhibits induction
of inducible nitric oxide synthase in murine macrophages. Eur J
Pharmacol. 336:211–217. 1997. View Article : Google Scholar
|
|
10.
|
Figueiredo JN, Raz B and Sequin U: Novel
quinone methides from Salacia kraussii with in vitro
antimalarial activity. J Nat Prod. 61:718–723. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Brinker AM, Ma J, Lipsky PE and Raskin I:
Medicinal chemistry and pharmacology of genus Tripterygium
(Celastraceae). Phytochemistry. 68:732–766. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Costa PM, Ferreira PM, Bolzani Vda S, et
al: Antiproliferative activity of pristimerin isolated from
Maytenus ilicifolia (Celastraceae) in human HL-60 cells.
Toxicol In Vitro. 22:854–863. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Yang H, Landis-Piwowar KR, Lu D, et al:
Pristimerin induces apoptosis by targeting the proteasome in
prostate cancer cells. J Cell Biochem. 103:234–244. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Yan YY, Bai JP, Xie Y, Yu JZ and Ma CG:
The triterpenoid pristimerin induces U87 glioma cell apoptosis
through reactive oxygen species-mediated mitochondrial dysfunction.
Oncol Lett. 5:242–248. 2013.
|
|
15.
|
Lee JS, Yoon IS, Lee MS, et al: Anticancer
activity of pristimerin in epidermal growth factor receptor
2-positive SKBR3 human breast cancer cells. Biol Pharm Bull.
36:316–325. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Wu CC, Chan ML, Chen WY, Tsai CY, Chang FR
and Wu YC: Pristimerin induces caspase-dependent apoptosis in
MDA-MB-231 cells via direct effects on mitochondria. Mol Cancer
Ther. 4:1277–1285. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Lu Z, Jin Y, Chen C, Li J, Cao Q and Pan
J: Pristimerin induces apoptosis in imatinib-resistant chronic
myelogenous leukemia cells harboring T315I mutation by blocking
NF-kappaB signaling and depleting Bcr-Abl. Mol Cancer. 9:1122010.
View Article : Google Scholar
|
|
18.
|
Byun JY, Kim MJ, Eum DY, et al: Reactive
oxygen species-dependent activation of Bax and poly(ADP-ribose)
polymerase-1 is required for mitochondrial cell death induced by
triterpenoid pristimerin in human cervical cancer cells. Mol
Pharmacol. 76:734–744. 2009. View Article : Google Scholar
|
|
19.
|
Mu XM, Shi W, Sun LX, et al: Pristimerin
inhibits breast cancer cell migration by up-regulating regulator of
G protein signaling 4 expression. Asian Pac J Cancer Prev.
13:1097–1104. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Mu X, Shi W, Sun L, Li H, Jiang Z and
Zhang L: Pristimerin, a triterpenoid, inhibits tumor angiogenesis
by targeting VEGFR2 activation. Molecules. 17:6854–6868. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Wang Y, Zhou Y, Zhou H, et al: Pristimerin
causes G1 arrest, induces apoptosis, and enhances the
chemosensitivity to gemcitabine in pancreatic cancer cells. PLoS
One. 7:e438262012. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Sun XM, MacFarlane M, Zhuang J, Wolf BB,
Green DR and Cohen GM: Distinct caspase cascades are initiated in
receptor-mediated and chemical-induced apoptosis. J Biol Chem.
274:5053–5060. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Ashkenazi A and Dixit VM: Death receptors:
signaling and modulation. Science. 281:1305–1308. 1998. View Article : Google Scholar
|
|
24.
|
Li H, Zhu H, Xu CJ and Yuan J: Cleavage of
BID by caspase 8 mediates the mitochondrial damage in the Fas
pathway of apoptosis. Cell. 94:491–501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Luo X, Budihardjo I, Zou H, Slaughter C
and Wang X: Bid, a Bcl2 interacting protein, mediates cytochrome c
release from mitochondria in response to activation of cell surface
death receptors. Cell. 94:481–490. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Altomare DA and Testa JR: Perturbations of
the AKT signaling pathway in human cancer. Oncogene. 24:7455–7464.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Datta SR, Dudek H, Tao X, et al: Akt
phosphorylation of BAD couples survival signals to the
cell-intrinsic death machinery. Cell. 91:231–241. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Brunet A, Bonni A, Zigmond MJ, et al: Akt
promotes cell survival by phosphorylating and inhibiting a Forkhead
transcription factor. Cell. 96:857–868. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Karin M, Cao Y, Greten FR and Li ZW:
NF-kappaB in cancer: from innocent bystander to major culprit. Nat
Rev Cancer. 2:301–310. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Mayo MW and Baldwin AS: The transcription
factor NF-kappaB: control of oncogenesis and cancer therapy
resistance. Biochim Biophys Acta. 1470:M55–M62. 2000.PubMed/NCBI
|
|
31.
|
Liu L, Li F, Cardelli JA, Martin KA,
Blenis J and Huang S: Rapamycin inhibits cell motility by
suppression of mTOR-mediated S6K1 and 4E-BP1 pathways. Oncogene.
25:7029–7040. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Bjornsti MA and Houghton PJ: The TOR
pathway: a target for cancer therapy. Nat Rev Cancer. 4:335–348.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar
|
|
34.
|
Deveraux QL and Reed JC: IAP family
proteins - suppressors of apoptosis. Genes Dev. 13:239–252. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Breitschopf K, Haendeler J, Malchow P,
Zeiher AM and Dimmeler S: Posttranslational modification of Bcl-2
facilitates its proteasome-dependent degradation: molecular
characterization of the involved signaling pathway. Mol Cell Biol.
20:1886–1896. 2000. View Article : Google Scholar
|