Introduction
Glioblastoma (GBM) is the most common and aggressive
type of primary brain cancer in adults (1). Despite multimodality treatment
consisting of maximally safe resection, adjuvant chemoradiation
with temozolomide, median survival remains dismal at 12–15 months
(2), where less than 2% of
patients survive 3 years post-diagnosis (3). Infiltrating cancer cells in the
surrounding brain that prevent complete resection and their
intrinsic resistance to chemoradiation treatment cause the poor
prognosis of the GBM patient (4,5). GBM
that arise de novo usually occurs as the result of
progression from lower grade astrocytomas. Several histological
changes occur during transition to GBM and these reflect a profound
alteration in the tumor vascular biology. Particularly, the main
change is represented by the appearance of hypoxic areas bounded by
regions with a high rate of cell proliferation (6,7). In
GBM hypoxia and its microenvironment are predominant features
associated with the tumor growth, progression and resistance to
chemo- and radiation therapies (8–10).
Furthermore, it has been shown that hypoxia favors the maintenance
of GBM stem cell population that is intrinsically resistant to
standard therapies (11,12). Thus, targeting the molecular
mechanism regulated by hypoxia could represent a valid alternative
strategy in order to chemoradio-sensitize GBM cells (13). One of the main early cellular
events evoked upon exposure to hypoxia is activation of HIF-1
transcription factor that, through the binding of
hypoxia-responsive elements (HREs), induces the expression of
several target genes involved in tumor angiogenesis, invasion, cell
survival and glucose metabolism (14). Very little is known about the
molecular mechanisms implicated in hypoxia-induced resistance to
therapies, although the central role of HIF-1α in promoting the
cancer cells resistance to therapies has been reported. Eukaryotic
cells respond to hypoxia through the modulation of several
downstream effector pathways, including intracellular signal
transduction cascades, which in turn interfere with gene expression
regulation. In particular, members of the family of mitogen
activated protein (MAP) kinases were shown to be involved in the
transduction of the hypoxic signals (15–17).
Even though the mitogenic Ras/Raf/MEK/ERK cascade signalling
pathway, which responds to growth factors and factors inducing
cellular differentiation, such as epidermal growth factor (EGF) and
platelet derived growth factor (PDGF), has been intensively studied
(18), little is known about its
relationship to hypoxia (19). It
has been shown that ERKs, activated during hypoxia, positively
regulate HIF-1α gene expression (20), phosphorylate HIF-1α protein
regulating its activity (21) and
are needed for its activity as a transcription factor (22). Resistance to chemo-and radiotherapy
may be caused primarily by DNA repair mechanisms (23). The main deleterious damage induced
by chemo- and radiotherapy is DNA-double-strand breaks (Dsbs) and
DNA repair remains one of the main responses through which cancer
cells guarantee their own survival thus contributing to tumor
chemo- and radioresistance. The major mechanism underlying the
repair of DNA-Dsbs in mammalian cells requires the DNA-PKcs, a
serine-threonine protein kinase that forms a complex of 450,000 kDa
catalytic subunit (DNA-PKcs), in a heterodimeric complex composed
of the proteins Ku70 (70,000 Da) and Ku80 (86,000 Da). Ku binds to
both ends of a double-strand break and recruits DNA-PKcs to the DNA
end (24,25). Functional interplay has been shown
between HIF-1α and DNA-PKcs, which could contribute to
radioresistance and chemoresistance in hypoxic tumor cells
(26). GBM is characterized by
several aberrantly activated signalling pathways such as EGF,
vascular endothelial growth factor receptor (VEGF) and PDGF
pathways (27,28). All these pathways converge on the
RAS-MEKs-ERKs signal transduction activation that plays a key role
in the regulation of tumor progression and treatments response
(29). Herein, the role of
RAS-MEKs-ERKs signal transduction pathway in controlling GBM
transformed phenotype and response to radiotherapy treatment was
investigated under hypoxic condition. Our results showed that
RAS/MAPK pathway, through the regulation of DNA-PKcs/HIF-1α
interplay, governs the GBM transformed phenotype and regulates the
hypoxia-mediated increase of GBM radioresistance. Particularly we
showed for the first time that DNA-PKcs is an upstream regulator of
HIF-1α protein expression, which is the determinant in sustaining
GBM refractoriness to radiation therapy.
Materials and methods
Cell cultures in conventional and hypoxic
conditions: treatments and radiation exposure
The human glioblastoma T98G and U138MG cell lines
were obtained from American Type Culture Collection (Manassas, VA,
USA). The human glioblastoma U87MG cell lines were from Life
Technologies (Frederick, MD, USA). The human glioblastoma U251MG
cell lines were obtained from Sigma-Aldrich (St. Louis, MO, USA).
The T98G U138MG and U251MG cell lines were cultured in Eagle’s
minimum essential medium containing fetal bovine serum to a final
concentration of 10%. The U87MG cell lines were cultured in
RPMI-1640 medium containing fetal bovine serum to a final
concentration of 10%. The cell lines are tested in our laboratory
every year for the expression of specific markers by western blot
analysis and were last tested in 2012. For hypoxia experiments,
cultures were maintained at 37°C in a humidified incubator in an
atmosphere of 20% O2, 5% CO2 and 75%
N2. The Xvivo Closed Incubation System (Xvivo system 300
C, BioSpherix, New York, NY, USA) was used in this study in order
to accurately maintain different oxygen tensions in different
chambers. After 24 h of cultivation in conventional cell culture
(allowing cells to attach onto the flasks), the cells were
transferred into different chambers with 0.1% O2, 5%
CO2 and 94.9% N2 for variable periods of time
before being harvested for additional analysis. Treatment with 10
μmol/l MEK/ERK inhibitor U0126
(1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene;
Promega, Madison, WI, USA) or 300 mM HIF-1α inhibitor FM19G11
[2-oxo-2-(p-tolyl)ethyl] 3-[(2,4-dinitrobenzoyl)amino]benzoate,
3-[(2,4-dinitrobenzoyl)amino]-benzoic acid
2-(4-methylphenyl)-2-oxoethyl ester were done for the times shown
in the figures and started before radiation, lasting for 24 h.
Radiation was delivered at room temperature using an x-6 MV photon
linear accelerator as already described (30). The total single dose of 400 cGy was
delivered with a dose rate of 2 Gy/min using a source-to-surface
distance (SSD) of 100 cm. Doses of 200 kV X-rays (Yxlon Y.TU 320;
Yxlon, Copenhagen, Denmark) filtered with 0.5 mm Cu. The absorbed
dose was measured using a Duplex dosimeter (PTW, Freiburg,
Germany). The dose-rate was approximately 1.3 Gy/min and applied
doses ranged from 0 to 600 cGy.
Immunoblot analysis
Immunoblot analysis was performed as described
(31). Briefly, cells were lysed
in 2% SDS containing phosphatase and protease inhibitors sonicated
for 30 sec. Proteins of whole cell lysates were assessed using the
method of Lowry et al (32), and equal amounts were separated on
SDS-PAGE. The proteins were transferred to a nitrocellulose
membrane (Schleicher & Schuell, BioScience GmbH, Dassel,
Germany) by electroblotting. Immunoblot analyses were performed
with the following antibodies: anti-c-Myc (9E10), anti-N-Myc
(C-19), anti-phospho ERK1/2 (E-4), anti-ERK2 (C-14 positive also
for ERK1), anti-cyclin-D1 (M-20), anti-HIF-1α (28b), anti-DNA-PKcs
(28b), α-tubulin (B-7) all from Santa Cruz Biotechnology (Santa
Cruz, CA, USA) and anti-Ku70/Ku80 (ab53126) from Abcam (Cambridge,
UK). Peroxidase-conjugate anti-mouse or anti-rabbit IgG
(Amersham-Pharmacia Biotech, Amersham, UK or Santa Cruz) were used
for enhanced chemiluminescence (ECL) detection.
Cell proliferation, soft agar assays and
FACS analysis
Cells from adherent and suspension culture were
counted using hemocytometer, and tested for exclusion of trypan
blue. Data are expressed as mean ± SE of experiments performed in
triplicate. Suspension culture proliferation assay was performed by
using polyHema assay as already described (36). Briefly, polyHEMA-coated 96-well
plates were used. A total of 50 μl of polyHEMA solution (5
mg/ml in 95% ethanol) was overlaid into wells and dried for 2 days
with lids in place. Cells were inoculated in a volume of 135
μl at a density of 5,000 cells per well. Cells were cultured
for 4 days in the presence or absence of inhibitors. At the end of
the treatment, 15 μl of
3-[4,5-dimethylthiazol-2yl]-2,5-diphenyltetrazolium bromide
solution (5 mg/ml in PBS) was added, and the mixture was further
incubated for 4 h. The resulting
3-[4,5-dimethylthiazol-2yl]-2,5-diphenyltetrazolium bromide
formazan was solubilized by addition of 100 μl of SDS
solution (20% in 10 mm HCl), and the absorbance was measured after
24 h at 570 nm and a reference wavelength of 690 nm using a
microplate reader (33).
Colony-forming in soft agar assays were based on standard methods.
Briefly, 2×104 cells were resuspended in 4 ml of 0.33%
special Noble agar (Difco, Detroit, MI, USA) and plated (6-cm
plate) in growth medium-containing 0.5% soft agar. Colonies were
photographed 14 days after plating. FACS analysis was performed as
described (34). Briefly, cells
were harvested by trypsin-EDTA and washed; pellets were resuspended
in 0.3 ml 50% FCS in PBS, additioned with 0.9 ml 70% ethanol and
left O/N in the dark at 4°C before FACS analysis (Coulter Epics XL
Flow Cytometer, Beckman Coulter, Brea, CA, USA).
Invasion and migration assays
Transwell membrane (Corning Costar Corporation,
Corning, NY, USA) was used. Cancer cells were trypsinized, washed
and kept suspended in the appropriate medium without FCS.
Migration-inducing medium (with 10% FCS) was added to the lower
wells of the chambers, while the upper wells were filled with
serum-free medium with cells (20,000 cells per well) in the
absence, as controls, or in the presence of the appropriate
treatments. After 8 h, filters were removed and fixed with methanol
and subsequently wiped on the cells on the upper side using the
Q-tip. Filters were stained with 20% Giemsa solution. Evaluation of
completed transmigration was performed under a microscope, and
random fields were scanned (four fields per filter) for the
presence of cells at the lower membrane side only. Invasion assays
were done in a similar manner as the migration assays described
above, unless the inserts were pre-coated with Matrigel (BD
Biosciences, Franklin Lakes, NJ, USA).
HIF-1α transcription factor assay
HIF-1α transcription factor activity was evaluated
by HIF-1α Transcription Factor Assay (ab133104) from
Abcam®. Transfection experiments were performed with
siRNA for ERK1 and ERK2 or DNA-PKcs (Sancta Cruz Biotechnology)
using Lipofectamine 2000 reagent (Invitrogen, San Giuliano
Milanese, Milan, Italy), according to the manufacturer’s
instructions. Briefly, cells were plated at 40–50% confluence and
transfected 24 h later with 100 nM siRNA, which we ascertained to
be sufficient to detect maximum fluorescence using
fluorescein-conjugated control siRNA.
Results
MEK/ERK inhibition blocks G0/G1 cell
cycle phase transition and reverts transformed phenotype of
glioblastoma cancer cells
Our experiments were started by verifying the role
of MEKs/ERKs pathway through controlling cell cycle progression of
growing GBM cancer cells in hypoxic conditions. For this purpose,
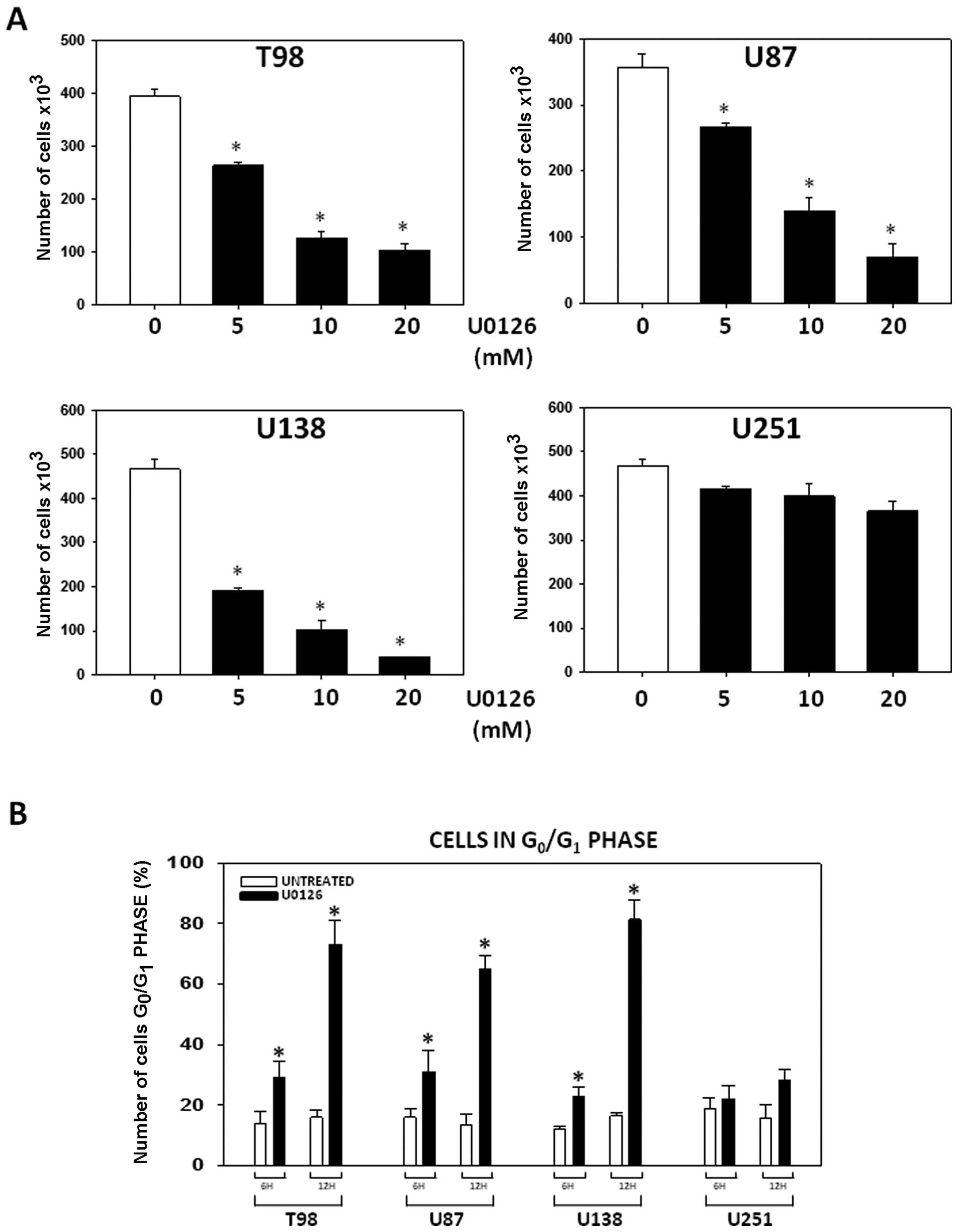
T98G, U87MG, U138MG and U251MG GBM cell lines maintained under
hypoxic conditions were treated with either U0126 (5, 10 or 20
μM) or vehicle and counted 4 after days. MEKs/ERKs
inhibition by U0126 arrested T98G, U87MG, U138MG in a concentration
dependent manner while barely affecting the U251MG cells growth
(Fig. 1A). FACS analysis showed
that U0126 (10 μM) induced accumulation of T98G, U87MG,
U138MG tumor cells in the G1 phase of cell cycle with weak effects
on U251MG cells (Fig. 1B).
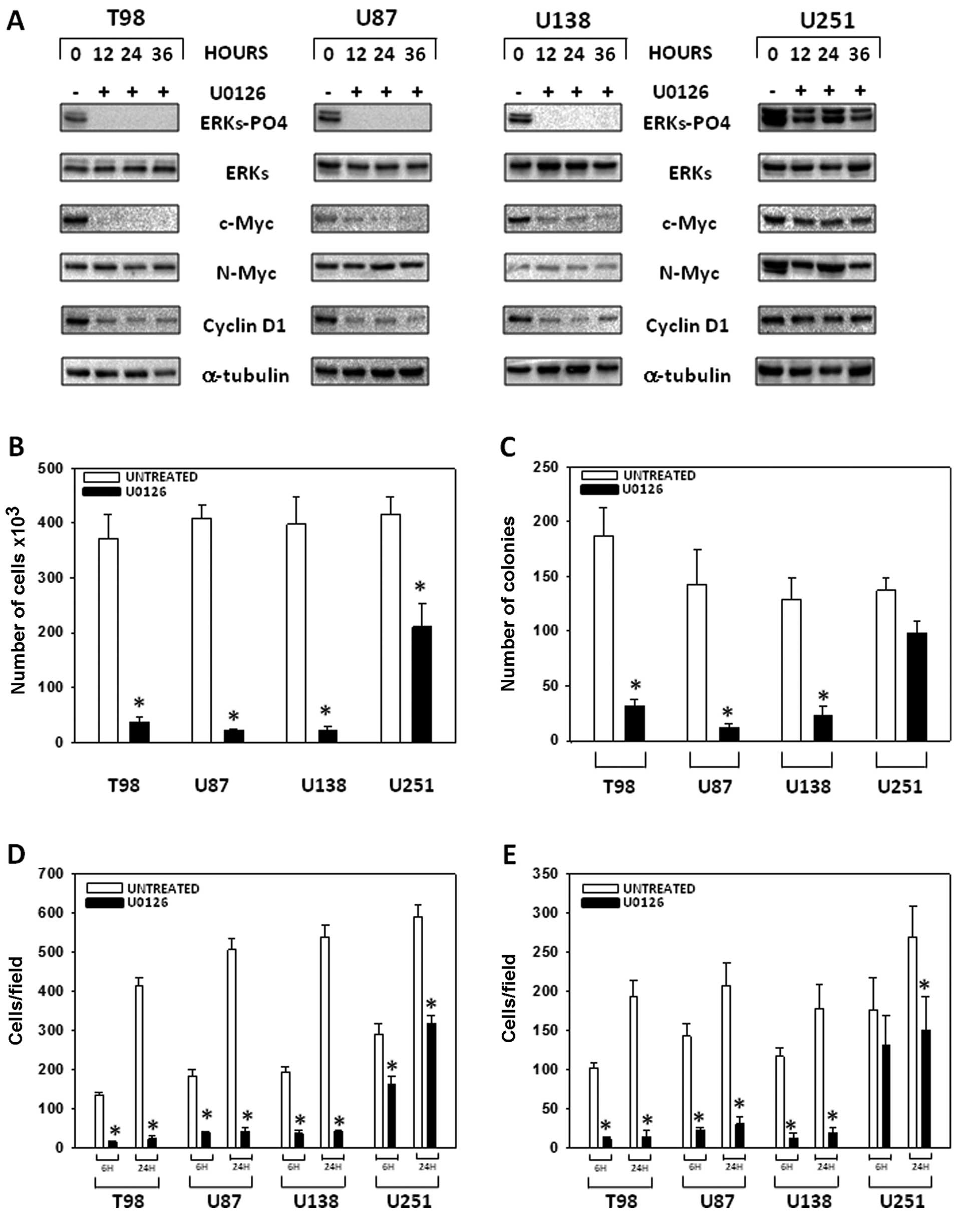
Analysis of phospho/active-ERKs status and markers of G1 arrest was
performed (Fig. 2A). As shown in
Fig. 2, U0126 induced a rapid (12
h), highly persistent (24 h) ERK de-phosphorylation/inactivation in
T98G, U87MG, U138MG cells while a much weaker phospho/active-ERK
inhibition was observed in U251MG cells. ERK inhibition was
concomitant to c-Myc and cyclin D1 downregulation while N-Myc
levels where not modified with the exception of U251 line which
expresses N-Myc at higher levels than the other cell lines. We
tested the effects of MEK/ERK inhibition assessing the ability of
GBM cells to grow, migrate and/or invade in an
anchorage-independent manner under hypoxic conditions. As show in
Fig. 2, U0126 (10 μM)
drastically reduced the ability of T98G, U87MG and U138MG to grow
in suspension (Fig. 2B) and form
colonies in soft agar (Fig. 2C),
while these effects were less evident in U251MG cancer cells.
Finally, U0126 (10 μM) significantly inhibited T98G, U87MG
and U138MG invasion and migration at both 3 and 24 h post-treatment
time points (Fig. 2D and E).
Therefore, we concluded that MEK/ERK inhibition by U0126 induces
growth arrest, which blocks the molecular mechanism responsible for
G1 progression and reduces the tumorigenic and metastatic potential
of GBM tumor cell growth under hypoxic conditions.
The hypoxia-induced increase of
radioresistance and HIF-1α activity are counteracted by
U0126-mediated MEK/ERK inhibition
It has been shown that cancer cells exhibit
different response to radiation therapy under hypoxia and normoxia.
Since GBM usually acquires the ability to progress in hypoxic
conditions, we tested whether U0126 affected radiosensitization
ability under these two conditions. Tumor cells were cultured in
the presence or absence of U0126 (10 μM) in normoxic or
hypoxic conditions for 12 h before the delivery of increasing doses
of ionizing radiation (0–6 Gy) (Fig.
3. left panels). After radiation treatment U0126 was removed
and a colony assay was then performed. All cell lines were
basically radioresistant under normoxic conditions (Fig. 3, left panels, normoxia+RT), while
the hypoxic conditions highly increased (Fig. 3, left panels, hypoxia+RT) the
intrinsic levels of radioresistance. U0126 increased the
radiosensitivity of T98G, U87MG and U138MG with effects evident
both in normoxic and hypoxic conditions (Fig. 3, left panels, normoxia+U0126+RT,
hypoxia+U0126+RT). No statistically significant differences were
observed in the U251MG cell line (Fig.
3, left panels). We analyzed the HIF-1α activity present in the
GBM cells after RT treatment (Fig.
3, right panels). HIF-1α basal activity increased under hypoxic
condition in T98G, U87MG and U138MG while it was drastically
reduced upon MEK/ERK inhibition combined treatment (Fig. 3, right panels) which, by contrast,
had no effect on HIF-1α activity status of U251MG cancer cells
(Fig. 3, right panels).
Noteworthy, RT treatment alone increased the HIF-1α basal activity
both in normoxic and hypoxic conditions (Fig. 3, right panels).
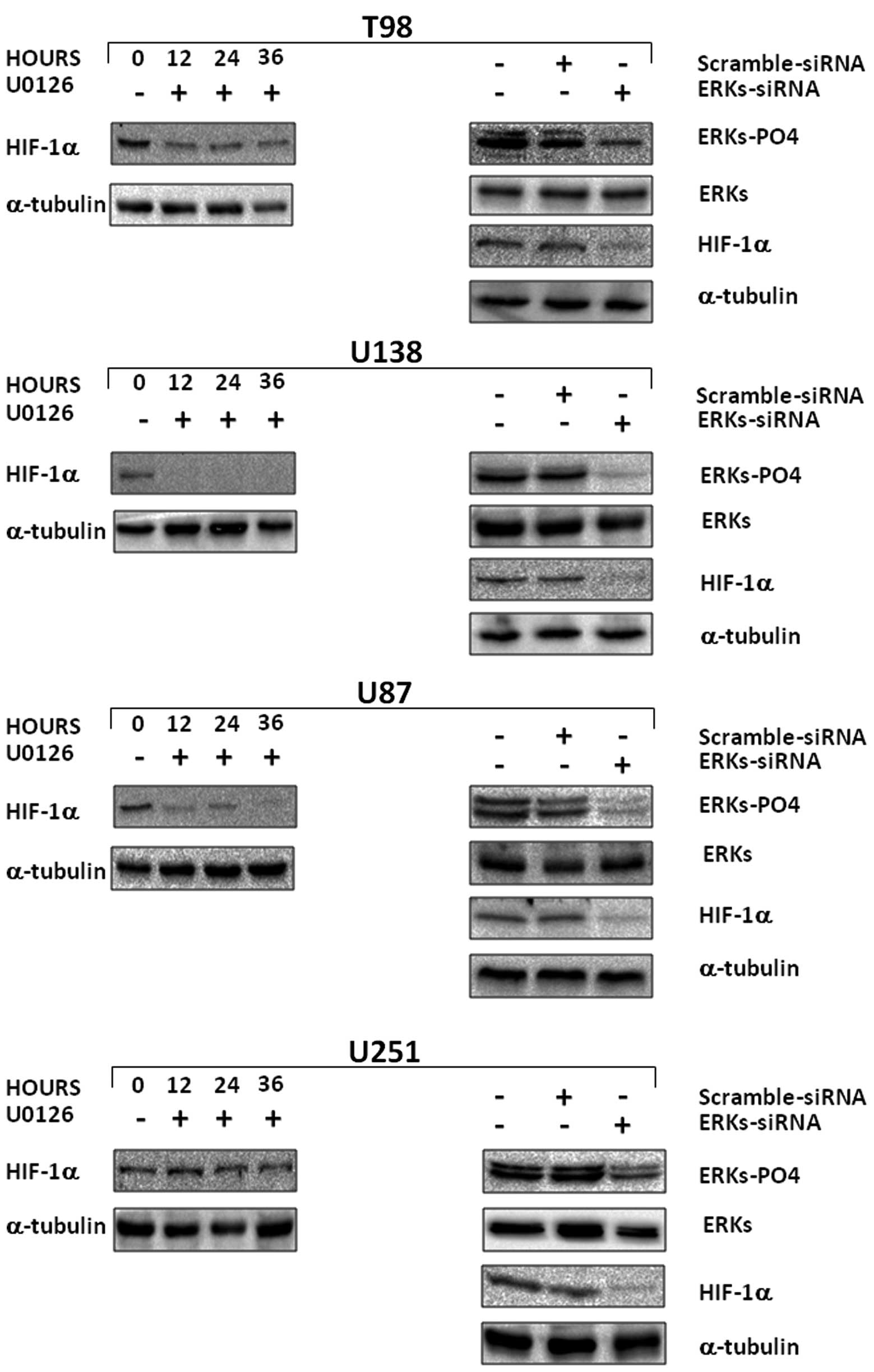
MEK/ERK pathway is an upstream regulator
of HIF-1α protein expression in GBM cancer cells
Since radiation increased HIF-1α activity in hypoxic
conditions and MEK/ERK inhibition combined with radiation affected
HIF-1α activity, we investigated whether MEK/ERK pathways regulate
HIF-1α protein expression in hypoxic conditions. GBM cell lines
were cultured with or without U0126 (10 μM) in hypoxic
conditions for 12, 24 or 36 h; analysis of HIF-1α protein
expression levels was performed (Fig.
4, right panels). MEK/ERK inhibition by U0126 dramatically and
persistently (12 to 36 h) reduced HIF-1α protein expression levels.
In T98G as well as in U87MG and in U138MG cell lines the protein
expression was completely abrogated (Fig. 4, left panels). U0126 did not affect
HIF-1α protein expression levels in U251MG GBM cancer cells.
MEK/ERK pathway functioning upstream of HIF-1α in GBM, as suggested
by U0126 experiments, was further demonstrated by RNA interference
experiment with ERK1/ERK2- and scramble-siRNA in transient
transfection. Three days after ERK1/ERK2 siRNA transfection, we
observed a downregulation of total ERKs and a drastic reduction of
HIF-1α in T98G, U87MG, U138MG and U251MG transfected cells
(Fig. 4, right panels).
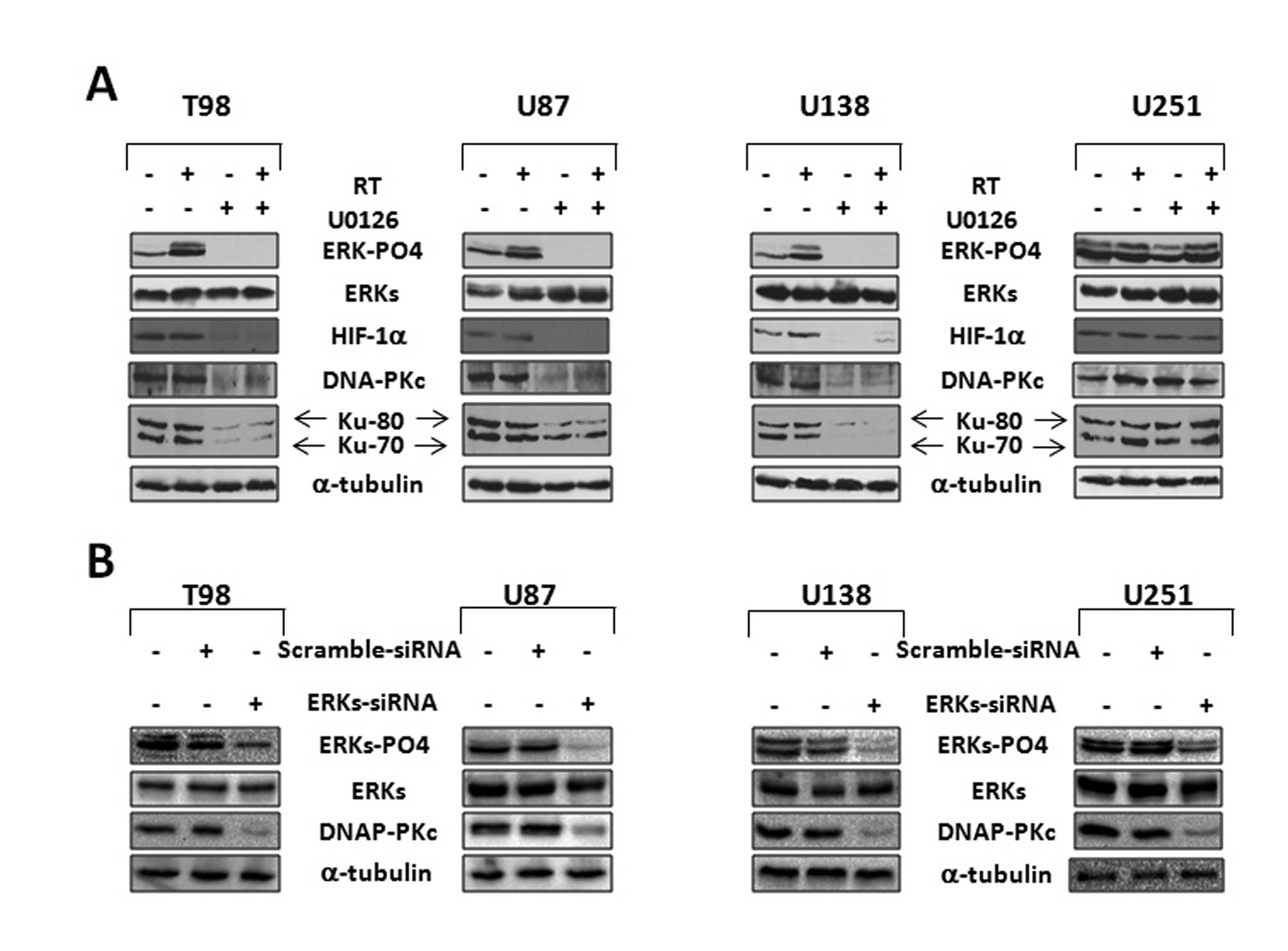
MEK/ERK inhibition by U0126
radiosensitizes GBM cancer cell lines by affecting the DNA repair
molecular mechanism
We next investigated the molecular mechanism
responsible of radiosensitization induced by MEK/ERK inhibition.
DNA-PKcs and Ku proteins are upregulated in various tumors and are
implicated in the radiation response. DNA-PKcs, Ku-80 and Ku-70
expression levels in response to U0126 were assessed by western
blot analysis (Fig. 5A). The 24 h
treatment with U0126 alone, and in combination with radiation
reduced DNA-PKcs, Ku-80 and Ku-70 protein expression levels
concomitantly with HIF-1α downregulation in the T98G, U87MG and
U138MG is shown in Fig. 5A. No
change was observed in U0126-treated U251MG cells. RT treatment
alone increased the level of phospho/active ERKs. RNA interference
experiment with ERK1/ERK2- or scramble-siRNA in transient
transfection confirmed that DNA-PKcs expression is under the
control of ERKs (Fig. 5B). Three
days after the transfection we observed a downregulation of total
ERKs and a drastic reduction of DNA-PKcs in ERK1/ERK2 siRNA T98G,
U87MG, U138MG and U251MG transfected cells (Fig. 5B).
MEK/ERK pathway regulates DNA-PKcs
protein expression levels and controls HIF-1α protein accumulation
and radioresistance of GBM cancer cells
Since MEK/ERK inhibition by U0126 or siRNA-mediated
silencing induced HIF-1α and DNA-PKcs downregulation, we
investigated if there was a functional correlation between DNA-PKcs
and HIF-1α. T98G, U87MG, U138MG and U251MG were transiently
transfected with either scramble control or DNA-PKcs-siRNA. Sixty
hours after transfection the cells were exposed to hypoxic
condition for 12 h. Finally, total lysates were processed for
western blot analysis (Fig. 6A)
and HIF-1α activity evaluation (Fig.
6B). DNA-PKcs silencing by siRNA reduced the HIF-1α protein
expression levels (Fig. 6A) and
activity (Fig. 6B) in T98G, U87MG,
U138MG as well as in U251MG cell line. Since HIF-1α seemed to be
the final regulator of the molecular mechanism governed by MEK/ERK
pathway and responsible of GBM radioresistance, we tested whether
the HIF-1α inhibition could radiosensitize GBM cells. For this
experiment T98G, U87MG, U138MG and U251MG cancer cell lines were
cultured in the presence or absence of FM19G11 (300 nM), a specific
HIF-1α inhibitor, in hypoxic conditions for 12 h before the
delivery of increasing doses of ionizing radiation (0–6 Gy). After
radiation treatment, FM19G11 was removed and colony assay was
performed (Fig. 6C). To test the
inhibitory efficiency of FM19G11, HIF-1α activity was measured
before the radiation delivery (Fig.
6D). HIF-1α inhibition by FM19G11 (Fig. 6D) reduced the radioresistance of
T98G, U87MG, U138MG and U251MG cancer cells (Fig. 6C).
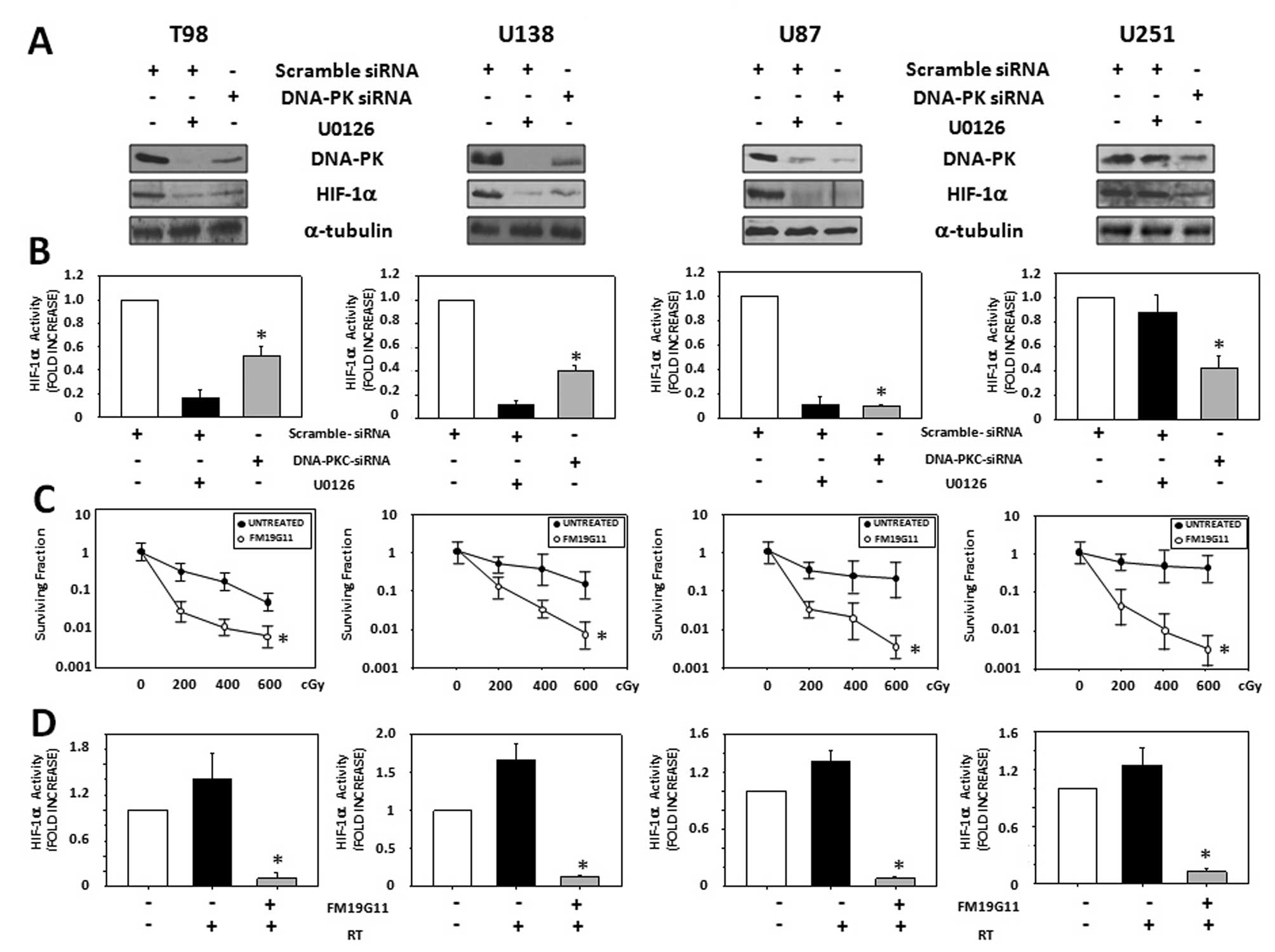 | Figure 6.DNA-PKcs regulates HIF-1α protein
accumulation and activity which inhibition affects GBM cancer cell
lines survival. (A) T98G, U87MG, U138MG and U251MG cells were
transfected with control (scramble) or DNA-PKcs siRNAs and cultured
for 3 days under hypoxia conditions. Immunoblot analyses of total
lysates were performed using specific antibodies recognizing the
indicated proteins. Similar results were obtained in three
experiments. (Right panels) (B) Analysis on HIF-1α activation
status were performed. The data shown are the mean ± SEM of
triplicates of a representative experiment. (C) T98G, U87MG, U138MG
and U251MG GBM cell lines in the exponential phase of growth under
hypoxic condition, were pre-treated for 24 h with FM19G11 (300 nM)
and then exposed to the indicated doses of γ-radiation. Clonogenic
survival was determined by counting the number of colonies
containing >50 cells after 2 weeks of growth. The surviving
fraction is shown in a semilogarithmic plot against radiation dose.
Points, means from triplicate flasks from two to three independent
experiments; bars, SE. (Right panels) T98G, U87MG, U138MG and
U251MG GBM cell lines in the exponential phase of growth under
hypoxic condition, were exposed to 400 cGy of γ-radiation. (D)
Analysis on HIF-1α activation status were performed after 1 h from
γ-radiation. Similar results were obtained in three experiments.
The data shown are the mean ± SEM of triplicates of a
representative experiment. |
Discussion
Hypoxia is a negative prognostic and predictive
factor, known to contribute to chemoresistance, radioresistance,
angiogenesis, vasculogenesis, invasiveness, metastasis, resistance
to cell death, altered metabolism and genomic instability. Given
its central role in tumor progression and resistance to therapy,
tumor hypoxia might well be considered the best validated target
that has yet to be explored in oncology (35). Hypoxic stress has been linked to
several phenotypic changes that are fundamental to GBM progression
and unresponsiveness of treatments (36). Even though Ras-MAPK pathway is
important in radioresistance, where activated oncogenic Ras
mediates resistance to ionizing radiation (37), the role of its inhibition under
hypoxic conditions and the downstream target in radiation response
has not yet been investigated in GBM. In the present study, we
addressed the issue on whether MEK/ERK inhibition, through
targeting HIF-1α, prevents the transformed phenotype expression and
radiation therapy resistance in several GBM cell lines grown under
hypoxic conditions. Experimental conditions chosen, were based on
data already present in the literature for which a near-maximal
HIF-1α expression occurs at 12 h of hypoxia at an oxygen
concentration of 0.1% O2, representing a level of
hypoxia that is frequently observed in solid tumors and is
radiobiologically relevant (38).
Our data show that MEK/ERK inhibition by U0126 reverts the
transformed phenotype of T98G, U87MG and U138MG GBM cell lines by
blocking the proliferation, migration and invasion under hypoxic
conditions. U251MG GBM cell line in which U0126 did not induce a
persistent MEK/ERK inhibition, was unresponsive to the treatment.
According to the literature (8–10),
in the clonogenic assay, hypoxia increased the intrinsic levels of
radioresistance in GBM cell lines that was counteracted by U0126 in
T98G, U87MG and U138MG but not in U251MG cells. It has been shown
that tumors deficient in the function or expression of HIF-1α
present an enhanced response to radiotherapy in vivo and
in vitro models (39–42).
In this study we show that MEK/ERK inhibition counteracted
hypoxia-induced increment of HIF-1α basal activity and reduced
HIF-1α protein expression levels in T98G, U87MG and U138MG GBM cell
lines. Furthermore, we note that RT treatment, independently of
oxygen concentration, increased MEK/ERK pathway activation and
HIF-1α activity. We speculate that GBM cancer cells respond to
radiation treatment by activating a pro-survival molecular
mechanism characterized by the MEK/ERK-HIF-1α interplay, which is
interrupted by U0126 treatment. Thus, the molecular mechanism
responsible of U0126 mediated radiosensitiziation was further
dissected. DNA-PKcs activity is known to be essential for the
repair of DSBs in the cell, and substantial scientific evidence has
shown that DNA-PKcs and Ku proteins strongly correlate with
radiosensitivity/resistance in several types of cancer (43,44).
Consistently, in T98G, U87MG and U138MG, but not in U251MG cell
line, the synergistic effect of MEK/ERK inhibitor on irradiation
results from the targeting of DNA-PKcs and Ku protein expression
levels, most likely compromising DNA-repair mechanism. Even though
functional interplay between HIF-1α and DNA-PK has been shown
(26), there is no evidence on the
molecular mechanism responsible for this relationship. Herein, for
the first time we show that DNA-PKcs positively regulate HIF-1α
protein expression levels and support its accumulation.
Interestingly, the selective inhibition of DNA-PKcs by siRNA
downregulates HIF-1α protein expression levels in all the GBM cell
line tested including the U251MG cell line, which are unresponsive
to U0126 treatment. Thus, HIF-1α appears to be a downstream target
of a complex molecular signal transduction pathway responsible of
GBM radioresistance and its inhibition by FM19G11, HIF-1α
inhibitor, drastically radiosensitizes GBM cells. Our data strongly
suggest that HIF-1α is an attractive target for overcoming
hypoxia-induced radioresistence. The selection of clinically
promising HIF-1α targeting agents represents an essential step
towards identifying new radiosensitive agents. In conclusion, we
demonstrated that through signal transduction-based chemotherapy it
is possible to radiosensitize GBM cancer cells by interrupting the
interplay between MEK/ERK and DNA-PKcs pathways and preventing
HIF-1α-mediated hypoxic cell survival and DNA-PKcs-mediated cancer
cells radiation escape. MEK/ERK or HIF-1α inhibitors combined with
radiation are an efficient antitumor treatment for GBM cell lines
suggesting a successful therapy to be tested in vivo and
translated to clinic.
References
|
1.
|
Schwartzbaum JA, Fisher JL, Aldape KD and
Wrensch M: Epidemiology and molecular pathology of glioma. Nat Clin
Pract Neurol. 2:494–503. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Stupp R, Hegi ME, Mason WP, Van Den Bent
MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B,
Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ,
Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M,
Lacombe D, Cairncross JG and Mirimanoff RO: European Organisation
for Research and Treatment of Cancer Brain Tumour and Radiation
Oncology Groups; National Cancer Institute of Canada Clinical
Trials Group: Effects of radiotherapy with concomitant and adjuvant
temozolomide versus radiotherapy alone on survival in glioblastoma
in a randomised phase III study: 5-year analysis of the EORTC-NCIC
trial. Lancet Oncol. 10:459–466. 2009.
|
|
3.
|
Senger D, Cairncross JG and Forsyth PA:
Long-term survivors of glioblastoma: Statistical aberration or
important unrecognized molecular subtype? Cancer J. 9:214–221.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q,
Hjelmeland AB, Dewhirst MW, Bigner DD and Rich JN: Glioma stem
cells promote radioresistance by preferential activation of the DNA
damage response. Nature. 444:756–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Liu G, Yuan X, Zeng Z, Tunici P, Ng H,
Abdulkadir IR, Lu L, Irvin D, Black KL and Yu JS: Analysis of gene
expression and chemoresistance of CD133+ cancer stem
cells in glioblastoma. Mol Cancer. 5:672006. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Brat DJ and Van Meir EG: Glomeruloid
microvascular proliferation orchestrated by VPF/VEGF: A new world
of angiogenesis research. Am J Pathol. 158:789–796. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Brat DJ and Van Meir EG: Vaso-occlusive
and prothrombotic mechanisms associated with tumor hypoxia,
necrosis, and accelerated growth in glioblastoma. Lab Invest.
84:397–405. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Yang L, Lin C, Wang L, Guo H and Wang X:
Hypoxia and hypoxia-inducible factors in glioblastoma multiforme
progression and therapeutic implications. Exp Cell Res.
318:2417–2426. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Harris AL: Hypoxia - a key regulatory
factor in tumor growth. Nat Rev Cancer. 2:38–47. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Wouters BG, Weppler SA, Koritzinsky M,
Landuyt W, Nuyts S, Theys J, Chiu RK and Lambin P: Hypoxia as a
target for combined modality treatments. Eur J Cancer. 38:240–257.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Nduom EK, Hadjipanayis CG and Van Meir EG:
Glioblastoma cancer stem-like cells: implications for pathogenesis
and treatment. Cancer J. 18:100–106. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Li P, Zhou C, Xu L and Xiao H: Hypoxia
enhances stemness of cancer stem cells in glioblastoma: an in vitro
study. Int J Med Sci. 10:399–407. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Meijer TW, Kaanders JH, Span PN and
Bussink J: Targeting hypoxia, HIF-1, and tumor glucose metabolism
to improve radiotherapy efficacy. Clin Cancer Res. 18:5585–5594.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Wang GL, Jiang BH, Rue EA and Semenza GL:
Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS
heterodimer regulated by cellular O2 tension. Proc Natl
Acad Sci USA. 92:5510–5514. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Laderoute KR and Webster KA:
Hypoxia/reoxygenation stimulates Jun kinase activity through redox
signaling in rat cardiac myocytes. Circ Res. 80:336–344. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Laderoute KR, Mendonca HL, Calaoagan JM,
Knapp AM, Giaccia AJ and Stork PJS: Mitogen-activated protein
kinase phosphatase-1 (MKP-1) expression is induced by low oxygen
conditions found in solid tumor microenvironment. J Biol Chem.
274:12890–12897. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Kunz M, Ibrahim S, Koczan D, Thiesen HJ,
Koehler HJ, Acker T, Plate KH, Ludwig S, Rapp UR and Broecker EB:
Activation of c-Jun NH2-terminal kinase/stress-activated protein
kinase (JNK/SAPK) is critical for hypoxia-induced apoptosis of
human malignant melanoma. Cell Growth Differ. 12:137–145.
2001.PubMed/NCBI
|
|
18.
|
Daum G, Eisenmann-Tappe I, Fries HW,
Troppmair J and Rapp UR: The ins and outs of Raf kinases. Trends
Biochem Sci. 19:474–480. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Kunz M and Ibrahim SM: Molecular responses
to hypoxia in tumor cells. Mol Cancer. 17:2–23. 2003.
|
|
20.
|
Lim JH, Lee ES, You HJ, Lee JW, Park JW
and Chun YS: Ras-dependent induction of HIF-1alpha785 via the
Raf/MEK/ERK pathway: a novel mechanism of Ras-mediated tumor
promotion. Oncogene. 23:9427–9431. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Minet E, Arnould T, Michel G, Roland I,
Mottet D, Raes M, Remacle J and Michiels C: ERK activation upon
hypoxia: involvement in HIF-1 activation. FEBS Lett. 468:53–58.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Berra E, Milanini J, Richard DE, Le Gall
M, Vinals F, Gothie E, Roux D, Pages G and Pouyssegur J: Signaling
angiogenesis via p42/p44 MAP kinase and hypoxia. Biochem Pharmacol.
60:1171–1178. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Eastman A and Barry MA: The origins of DNA
breaks: a consequence of DNA damage, DNA repair, or apoptosis?
Cancer Invest. 10:229–240. 1992. View Article : Google Scholar
|
|
24.
|
Chu G: Double strand break repair. J Biol
Chem. 272:24097–24100. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Kanaar R, Hoeijmakers JH and van Gent DC:
Molecular mechanism of DNA double-strand break repair. Trends Cell
Biol. 8:483–489. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Um JH, Kang CD, Bae JH, Shin GG, Kim DW,
Kim DW, Chung BS and Kim SH: Association of DNA-dependent protein
kinase with hypoxia inducible factor-1 and its implication in
resistance to anticancer drugs in hypoxic tumor cells. Exp Mol Med.
36:233–242. 2004. View Article : Google Scholar
|
|
27.
|
Sanson M, Thillet J and Hoang-Xuan K:
Molecular changes in gliomas. Curr Opin Oncol. 16:607–613. 2004.
View Article : Google Scholar
|
|
28.
|
De Groot JF and Gilbert MR: New molecular
targets in malignant gliomas. Curr Opin Neurol. 20:712–718.
2007.PubMed/NCBI
|
|
29.
|
Minniti G, Muni R, Lanzetta G, Marchetti P
and Enrici RM: Chemotherapy for glioblastoma: current treatment and
future perspectives for cytotoxic and targeted agents. Anticancer
Res. 29:5171–5184. 2009.PubMed/NCBI
|
|
30.
|
Marampon F, Gravina GL, Di Rocco A,
Bonfili P, Di Staso M, Fardella C, Polidoro L, Ciccarelli C,
Festuccia C, Popov VM, Pestell RG, Tombolini V and Zani BM: MEK/ERK
inhibitor U0126 increases the radiosensitivity of rhabdomyosarcoma
cells in vitro and in vivo by downregulating growth and DNA repair
signals. Mol Cancer Ther. 10:159–168. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Marampon F, Ciccarelli C and Zani BM:
Down-regulation of c-Myc following MEK/ERK inhibition halts the
expression of malignant phenotype in rhabdomyosarcoma and in non
muscle-derived human tumors. Mol Cancer. 5:312006. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Lowry OH, Rosebrough NJ, Farr AL and
Randall RJ: Protein measurement with the Folin phenol reagent. J
Biol Chem. 193:265–275. 1951.PubMed/NCBI
|
|
33.
|
Fukazawa H, Mizuno S and Uehara Y: A
microplate assay for quantitation of anchorage-independent growth
of transformed cells. Anal Biochem. 228:83–90. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Ciccarelli C, Marampon F, Scoglio A, Mauro
A, Giacinti C, De Cesaris P and Zani BM: p21WAF1 expression induced
by MEK/ERK pathway activation or inhibition correlates with growth
arrest, myogenic differentiation and onco-phenotype reversal in
rhabdomyosarcoma cells. Mol Cancer. 4:412005. View Article : Google Scholar
|
|
35.
|
Wilson WR and Hay MP: Targeting hypoxia in
cancer therapy. Nat Rev Cancer. 11:393–410. 2011. View Article : Google Scholar
|
|
36.
|
Jensen RL: Hypoxia in the tumorigenesis of
gliomas and as a potential target for therapeutic measures.
Neurosurg Focus. 20:E242006. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Gupta AK, Bakanauskas VJ, Cerniglia GJ,
Cheng Y, Bernhard EJ, Muschel RJ and McKenna WG: The Ras radiation
resistance pathway. Cancer Res. 61:4278–4282. 2001.PubMed/NCBI
|
|
38.
|
Vordermark D, Katzer A, Baier K, Kraft P
and Flentje M: Cell-type-specific association of hypoxia-inducible
factor-1alpha (HIF-1alpha) protein accumulation and radiobiologic
tumor hypoxia. Int J Radiat Oncol Biol Phys. 58:1242–1250. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Williams KJ, Telfer BA, Xenaki D, Sheridan
MR, Desbaillets I, Peters HJ, Honess D, Harris AL, Dachs GU, van
der Kogel A and Stratford IJ: Enhanced response to radiotherapy in
tumors deficient in the function of hypoxia-inducible factor-1.
Radiother Oncol. 75:89–98. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Dai S, Huang ML, Hsu CY and Chao KS:
Inhibition of hypoxia inducible factor 1alpha causes
oxygen-independent cytotoxicity and induces p53 independent
apoptosis in glioblastoma cells. Int J Radiat Oncol Biol Phys.
55:1027–1036. 2003. View Article : Google Scholar
|
|
41.
|
Sasabe E, Zhou X, Li D, Oku N, Yamamoto T
and Osaki T: The involvement of hypoxia-inducible factor-1alpha in
the susceptibility to gamma-rays and chemotherapeutic drugs of oral
squamous cell carcinoma cells. Int J Cancer. 120:268–277. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Pore N, Gupta AK, Cerniglia GJ, Jiang Z,
Bernard EJ, Evans SM, Koch CJ, Hahn SM and Maity A: Nelfinavir
down-regulates hypoxia-inducible factor 1alpha and VEGF expression
and increases tumor oxygenation: implications for radiotherapy.
Cancer Res. 66:9252–9259. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Tian X, Chen G, Xing H, Weng D, Guo Y and
Ma D: The relationship between the down-regulation of DNA-PKcs or
Ku70 and the chemosensitization in human cervical carcinoma cell
line HeLa. Oncol Rep. 18:927–32. 2007.PubMed/NCBI
|
|
44.
|
Chang HW, Kim SY, Yi SL, Son SH, Song do
Y, Moon SY, Kim JH, Choi EK, Ahn SD, Shin SS, Lee KK and Lee SW:
Expression of Ku80 correlates with sensitivities to radiation in
cancer cell lines of the head and neck. Oral Oncol. 42:979–986.
2006. View Article : Google Scholar : PubMed/NCBI
|