Introduction
Gastric cancer (GC) is the second leading cause of
cancer death in the world, and especially in Asian countries,
including China (1,2). Conventional treatment modalities
including surgery, radiotherapy and chemotherapy play a role mainly
in patients at early stage. However, these modalities are far from
satisfactory for patients with advanced gastric cancer, with
considerable treatment-associated toxicity and dismal overall
survival time (3,4). With better understanding of the
biology and underlying molecular mechanism of carcinogenesis, new
therapeutic approaches are needed for advanced gastric cancer
treatment.
Increasing evidence demonstrates that PI3K signaling
pathway regulates various cellular processes including
proliferation, cell cycle progression, apoptosis, migration and
metabolism (5,6). The dysregulation of PI3K/AKT pathway
has been recently found to be involved in the pathogenesis of
various human cancers such as gastric, colon, breast, pancreatic
and prostate cancer (7). PI3K
catalyzes the phosphorylated of 3-hydroxyl position of PIP2
(phosphatidylinositol 4,5-diphosphate) to PIP3
(phosphatidylinositol 3,4,5-triphosphate) (8,9).
PI3Ks are grouped into three classes (І, II and Ш) with regulatory
subunit for each, based on their respective structural
characteristics and substrate specificity (10,11).
The most extensively studied PI3Ks are class І PI3Ks, especially
class ІA PI3Ks which are composed of heterodimers of a p85
regulatory subunit and a p110 catalytic subunit (12). The dysregulation of p110α has been
observed in many human cancers such as gastric, colon, ovarian,
hepatocellular and breast carcinoma (13–16).
The Akt family, also well known as protein kinase B,
is one of the major downstream mediators of the PI3K pathway. Akt
plays very important roles in various cellular functions such as
cell cycle progression, proliferation, apoptosis, migration and
angiogenesis (17). Akt gene
amplification has been observed in a number of human cancers such
as gastric, breast and ovarian cancer. In addition to
amplification, recent studies have shown that elevation of Akt
activities is associated with a poor prognosis in human cancers.
Whereas tumor suppressor phosphatase and tensin homologue (PTEN)
negatively regulates the PI3K signals (18). The control of cell growth by
PI3K/Akt pathway via regulating cell proliferation, cell cycle
progression and apoptosis implicates a crucial role of this pathway
in carcinogenesis and cancer development. Therefore, PI3K is a
potential target for cancer prevention and therapy. Inhibition of
PI3K/Akt pathway in gastric cancer seems to be a promising strategy
for the treatment.
Cell cycle progression is promoted by the activity
of phase-specific kinase complexes composed of cyclins and
cyclin-dependent kinases (19). It
consists of four distinct phases: G1 phase, S phase
(synthesis), G2 phase and M phase (mitosis). From
G2 phase to M phase, it requires the activation of
cyclin B/CDK1, which push the cells through the G2
checkpoint and are regulated by CDC25C (20).
There is ample evidence that cancer is characterized
by uncontrolled cellular growth and proliferation, and therefore
inducing cancer cells into apoptosis is one of the important
therapeutic intervention approaches in cancer (21,22).
Apoptosis is characterized by a number of morphological and
biochemical features, such as cell shrinkage, nuclear DNA
fragmentation and membrane blebbing (23,24).
Apoptosis is mediated through two main routes the death receptor
pathway (extrinsic) and the mitochondrial pathway (intrinsic). In
the death receptor pathway, Fas/CD95 combines with its ligand FasL
recruits procaspase-8 and activates downstream effectors caspase-3
and/or caspase-7 (25). Upon
activation of the mitochondrial pathways, the change of
mitochondrial membrane results in release of cytochrome c, and
caspase-9 activation. In turn, activated caspase-9 leads to
cleavage of the executioner caspase-3 and caspase-7, and finally
results in chromatin condensation, DNA laddering and formation of
apoptotic bodies (26).
Quinazoline derivatives have been reported to
possess a wide range of therapeutic activities including anticancer
(27), anti-inflammation (28), anti-bacterial (29), antihypertension (30). Recently, some quinazoline
derivatives have been reported to have antitumor effects in several
human tumor cell lines by our research group (31,32).
With a goal of developing a more effective derivative, a series of
quinazoline derivatives were synthesized and screened. Among them,
methyl
4-([6-chloro-2-([1′-methyl-(1,4′-bipiperidin)-4-yl]amino)quinazolin-4-yl]amino)benzoate
(WYK431) displayed the most potent antitumor activity and induced
apoptosis in vitro. However, molecular mechanisms of action
underlying WYK431 against cancer remain unknown. Therefore,
investigating the molecular mechanisms of WYK431 is urgent for the
development of WYK431 as a potential anticancer agent. In this
study, we demonstrate that WYK431 inhibits BGC823 cells
proliferation, induces G2/M arrest and apoptosis though
intrinsic apoptotic pathway in vitro and suppresses tumor
growth in vivo.
Materials and methods
Materials
DMSO,3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
(MTT), propidium iodide (PI), Triton X-100 and rhodamine-123
(Rh-123) were purchased from Sigma Chemical Co. (St. Louis, MO,
USA). The Annexin V-FITC apoptosis detection kit was purchased from
KoradBio (Beijing, China). The primary antibodies against
caspase-3, caspase-8, caspase-9, Bcl-2, Bax, Akt, p-Akt (Ser473),
CDK1, Cyclin B1, CDC25C, p85 and p110α were purchased from Cell
Signaling Technology (Beverly, MA, USA). Antibodies against
cytochrome c and β-actin were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). TUNEL (terminal
deoxynucleotidyl transferase mediated dUTP nick end-labeling) assay
kit was purchase from Millipore (CA, USA). RPMI-1640 or DMEM were
obtained from Gibco BRL Co. (Grand Island, NE, USA). All of the
chemicals employed in this study were culture grade and analytic
purity.
WYK431 was synthesized and supplied as a yellow
solid by Institute of Materiel Medica, Chinese Academy of Medical
Sciences and Peking Union Medical College. Its structural formula
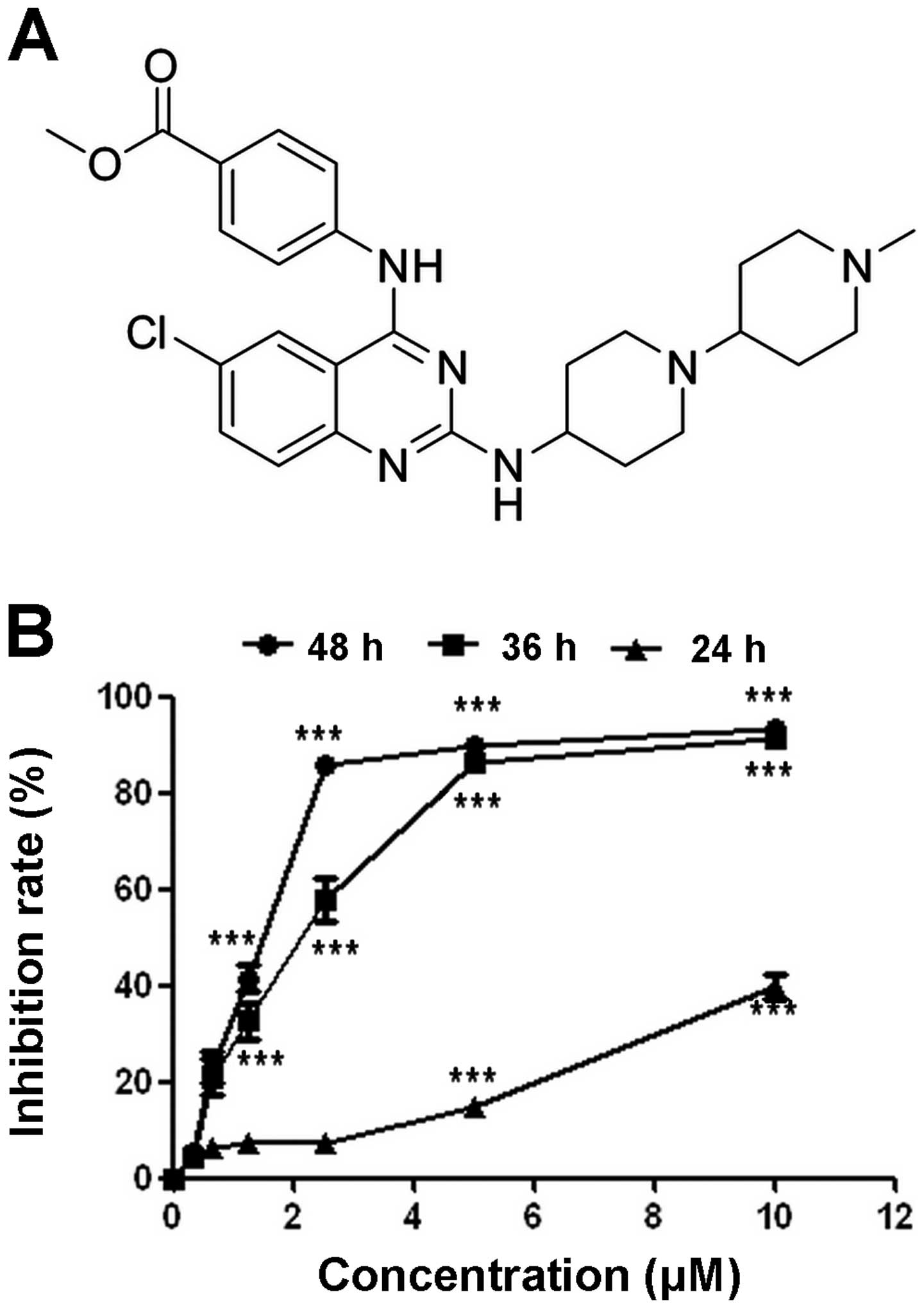
is shown in Fig. 1A. WYK431 was
dissolved in dimethyl sulfoxide (DMSO) to give a 5 mM stock
solution, which was stored at 4°C and diluted with the relevant
medium for the in vitro experiments. For the in vivo
studies, WYK431 was solubilized in 20% PEG (Beijing Chemical Works,
Beijing, China) to yield stock solutions of 2, 1 and 0.5 mg/ml, and
stored at −20°C.
Cell culture
The following human carcinoma cell lines were
obtained from American Type Culture Collection (ATCC, Manassas, VA,
USA): MCF7 cells; HepG2 cells; A2780 cells; A549 cells; HCT 116 and
T47D. BGC823 and BGC803 purchased from Cell Culture Center of
Institute of Basic Medical Sciences, Chinese Academy of Medical
Sciences. Cells were cultured in DMEM or RPMI-1640 media containing
10% FBS and 1% antibiotics (penicillin and streptomycin) under
humidified conditions with 5% CO2 at 37°C.
Cell viability assay
MTT assay was used to measure the cell viability
after WYK431 treatment. Briefly, the cells (3×103
cells/well) were seeded in a 96-well plate and cultured for 24 h.
After WYK431 treatment, the drug containing medium was removed and
replaced by fresh medium. MTT solution (100 μl of 0.5 mg/ml) was
added per well and incubated for another 4 h at 37°C, then the
supernatant fluid was removed and DMSO was added 150 μl to each
well. The plates were gently agitated until the color reaction was
uniform. The OD570 was measured using a microplate reader (Wellscan
MK3, Labsystems Dragon), and the IC50 values were
detected.
Cell cycle analysis
Cells were seeded in 6-well plates at the density of
2×105/well and cultured for 24 h, followed by indicated
concentrations WYK431 treatment for 24 h. Then, the cells were
incubated with 1 ml of a solution containing 50 μg/ml propidium
iodide (PI) and 0.1% Triton X-100 for 10 min in the dark and then
immediately analyzed by flow cytometer.
Morphological analysis after DAPI
staining
To investigate the apoptosis induction effect of
WYK431, morphological analysis by DAPI staining was performed.
Briefly, BGC823 cells (1×105) were seeded in 6-well
plates for 24 h, followed by WYK431 treatment for 24 h. After fixed
with 70% of ethanol, the cells were rinsed with PBS. Then cells
were examined with an inverted fluorescence microscopy after
staining with DAPI.
Apoptosis analysis by flow cytometry
(FCM)
To further confirm the apoptosis-induced effect by
WYK431, we analyzed the percentage of the early apoptotic cells
using an Annexin V-FITC apoptosis detection kit according to the
manufacturer’s instructions. Briefly, cells were seeded in 6-well
plates at the density of 2×105/well and cultured for 24
h, followed by WYK431 treatment for 36 h, and then collected and
washed with cold PBS twice. Cells were stained with an Annexin
V-FITC apoptosis detection kit following the manufacturer’s
instructions and then immediately examined by a flow cytometer.
Analysis of the mitochondrial membrane
potential
The mitochondrial transmembrane potential (ΔΨm) of
cells treated with WYK431 or medium alone was measured using the
rhodamine 123 (Rh123) as previously described (33). Cells were seeded in 6-well plates
at the density of 2×105/well and cultured for 24 h.
After WYK431 treatment for 24 h, the cells were incubated with 5
μg/ml Rh123 at 37°C in the dark for 30 min. Then fluorescence
emitted from the Rh123 was measured by FCM.
Western blot analysis
Western blot analysis was performed as previously
described (34). Briefly, the
cells were treated with WYK431 for various concentrations and lysed
with protein lysis buffer. Protein concentration was determined
with Bio-Rad Protein Assay kit (Bio-Rad Laboratories), dissolved in
5X SDS sample buffer and denatured. The samples were separated
according to molecular weight on 8–15% sodium dodecyl
sulfate-polyacrylamide (SDS-PAGE) gel and electroblotted onto a
polyvinylidene difluoride (PVDF) membrane. Membranes were blocked
for 1 h in 5% dried milk in TBST at room temperature with rotation.
Then membranes were incubated overnight at 4°C with the respective
primary antibodies diluted in blocking buffer. The membrane blots
were washed thrice for ~10 min with TBST and incubated with
appropriate horseradish peroxidase-conjugated species-specific
antibody diluted in blocking buffer for 2 h at room temperature
with rotation. After 3 additional washes, the immunoreactive bands
were visualized using the enhanced chemiluminescence method.
Analysis of cytochrome c in cytosolic and
mitochondria extracts
For detection of cytochrome c, the cytosolic and
mitochondria fractions were prepared as described previously
(35). Briefly, BGC823 cells were
treated with WYK431 at various concentrations for 24 h and then
harvested by centrifugation at 800 × g at 4°C for 15 min. After
washing with ice-cold PBS for 3 times, the cell pellets were
resuspended in HEPES buffer (20 mM HEPES, 10 mM KCl, 1.5 mM
MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 0.1 mM PMSF, pH
7.5) containing 250 mM sucrose, homogenized with a homogenizer and
centrifugation at 800 × g at 4°C for 15 min. The supernatants were
centrifuged at 10000 × g for 15 min at 4°C, and the mitochondria
pellets were dissolved in SDS sample buffer (25 μl), subjected to
15% SDS polyacrylamide gel electrophoresis and analyzed by
immunoblotting with monoclonal antibodies against cytochrome c
(Santa Cruz Biotechnology). Aliquots of supernatant were also
analyzed for cytochrome c expression by western blotting.
Immunofluorescence staining
To assess whether WYK431 induces cytochrome c
release from mitochondria to cytosol, immunofluorescence staining
was performed, and the cells were treated with 5 μM WYK431 for 24
h. To identify the location of cytochrome c, the cells were fixed
with freshly prepared buffer containing 4% formaldehyde for 15 min
at 37°C. After fixation, the cells were permeabilized with buffer
containing 2% Triton X-100 for 10 min at room temperature. The
cells were blocked with 5% goat serum in PBS and incubated
overnight with the anti-cytochrome c monoclonal antibody (diluted
1:100 in 5% goat serum, Santa Cruz Biotechnology). After three
washes, cells were incubated with Alexa Fluor 488® goat
anti-mouse IgG (H+L) (diluted 1:100 in 5% goat serum, Molecular
Probe) for 1 h at room temperature in the dark followed by rinsing
the cells three times in PBS for 5 min each. In addition, the cells
were stained with DAPI for 5 min in the dark. The coverslips were
mounted onto the slides using the Dako® Fluorescence
Mounting Medium (Dako, Glostrup, Denmark) and fluorescence images
were recorded using fluorescence microscopy.
Tumor xenograft experiments
Seven-week-old BALB/c athymic nude mice (Beijing
Animal Center, Beijing, China) were established as a xenograft
tumor model of BGC-823 in present study. BGC-823 cells
(1×107) were inoculated in the dorsal area of mice.
After 3 weeks of growth, the tumors were chopped into 3×3×3
mm3 pieces and implanted by subcutaneous injection in
the dorsum of the mice with a gange trocar. When tumors grew to
approximately 100 mm3, the nude mice bearing a tumor
were randomly assigned into four groups (6 per group) and injected
intraperitoneally with the following treatments, respectively: i)
20 mg/kg WYK431, ii) 10 mg/kg WYK431, iii) 5 mg/kg WYK431, iv)
Vehicle. The systemic therapy started on day 7 and was repeated
every three days for 12 days. Tumor volumes were assessed by
bilateral Vernier caliper measurement every three days and
calculated according to the following equation: [tumor volume =
(width)2 × length/2)]. Body weight was measured every
three days and clinical symptoms, including mortality, body weight,
movement and gross findings, were observed daily. The mice were
fasted overnight after the last administration and sacrificed.
Blood was collected for hematological analysis using a Nihon Kohden
MEK-5216K automatic hematology analyzer. All experiments were
carried out on animals in compliance with the guidelines set by the
institute’s Animal Care and Use Committee.
TUNEL detection
The analysis of apoptotic cells in tumor tissue was
conducted by TUNEL staining by an apoptotic cell detection kit
(Minipore). Images of the sections were taken by a fluorescence
microscope.
Statistical analysis
The data are presented as the means ± SE.
Statistical analysis was performed using SPSS 13.0 software
(Chicago, IL, USA). Statistical significance of the difference
between groups was analyzed by one-way analysis of variance.
P<0.05 was considered statistically significant.
Results
Effects of WYK431 on cell viability
The effect of WYK431 on cell viability was detected
using MTT assay. Our results showed WYK431 markedly reduced the
viability of a panel of cancer cell lines with IC50
ranged from 2 to 10 μM (Table I).
BGC8-23 cell line was the most sensitive to WYK431 treatment.
Furthermore, we investigated the time-and concentration-dependent
effects of WYK431 on BGC823 cell viability. As shown in Fig. 1B, the IC50 was 1.9±0.3
and 2.24±0.4 after WYK431 treatment for 48 and 36 h, respectively.
The results indicated that WYK431 inhibited the proliferation of
BGC823 cells in a time- and concentration-dependent manner.
 | Table IThe effects of 431 on tumor cells
viability. |
Table I
The effects of 431 on tumor cells
viability.
| Cell line | Cell type | IC50
(μM) |
|---|
| BGC823 | Human gastric
carcinoma cell line | 2.16 |
| BGC803 | Human gastric
carcinoma cell line | 2.59 |
| A549 | Human lung
adenocarcinoma cell line | 3.12 |
|
HepG2 | Human
hepatoblastoma cell line | 6.66 |
| A2780 | Human ovarian
carcinoma cell line | 7.22 |
| T47D | Human breast
adenocarcinoma cell line | 4.73 |
| MCF7 | Human breast
adenocarcinoma cell line | 4.68 |
| HCT116 | Human colorectal
carcinoma cell line | >10 |
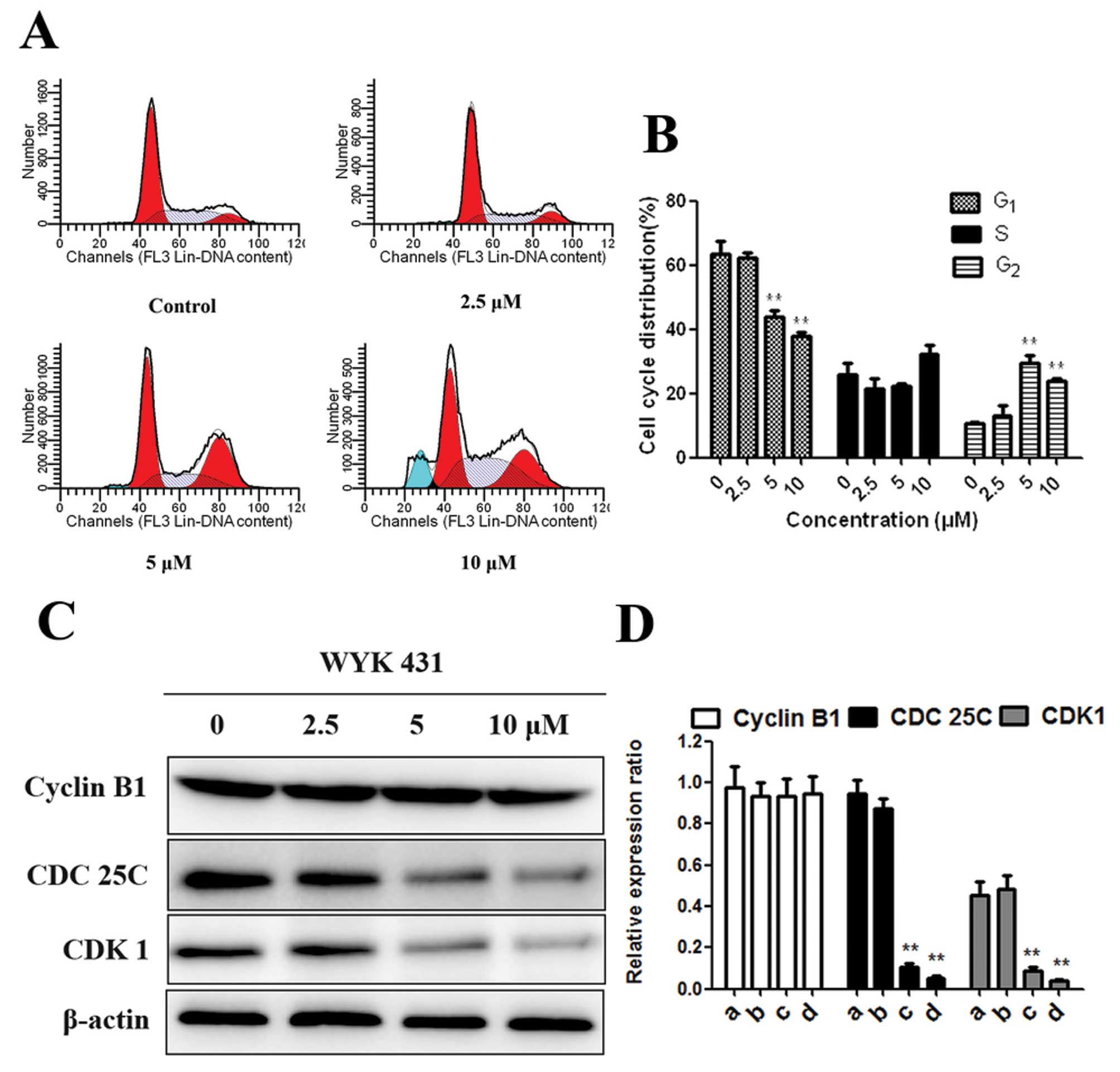
WYK431 treatment induces G2/M
phase arrest in BGC823 cells
To investigate the mechanism responsible for
WYK431-induced inhibition of cell growth, flow cytometric analysis
was performed after treatment of BGC823 with indicated
concentrations of WYK431 for 24 h. As can be seen in Fig. 2A, cells treated with 5 μM WYK431
increased cells population at G2/M phase compared with
control cells. In addition, at 10 μM of WYK431, the G2/M
phase population started to decrease, but the sub-G1
phase population slightly increased (Fig. 2A and B). To further elucidate the
mechanism of 431-induced cell cycle arrest at G2/M
phase, the expression of CDK1, CDC25C and Cyclin B1 all of which
participated in G2/M phases were detected by western
blot assay. It was observed that the expression of CDK1 and CDC25C
were significantly decreased at 5 and 10 μM, whereas the expression
of Cyclin B1 was not changed after treatment of BGC823 cells with
WYK431 for 24 h (Fig. 2C and D).
These results indicated that WYK431-induced G2/M phase
arrest related to the expression levels of CDK1 and CDC25C.
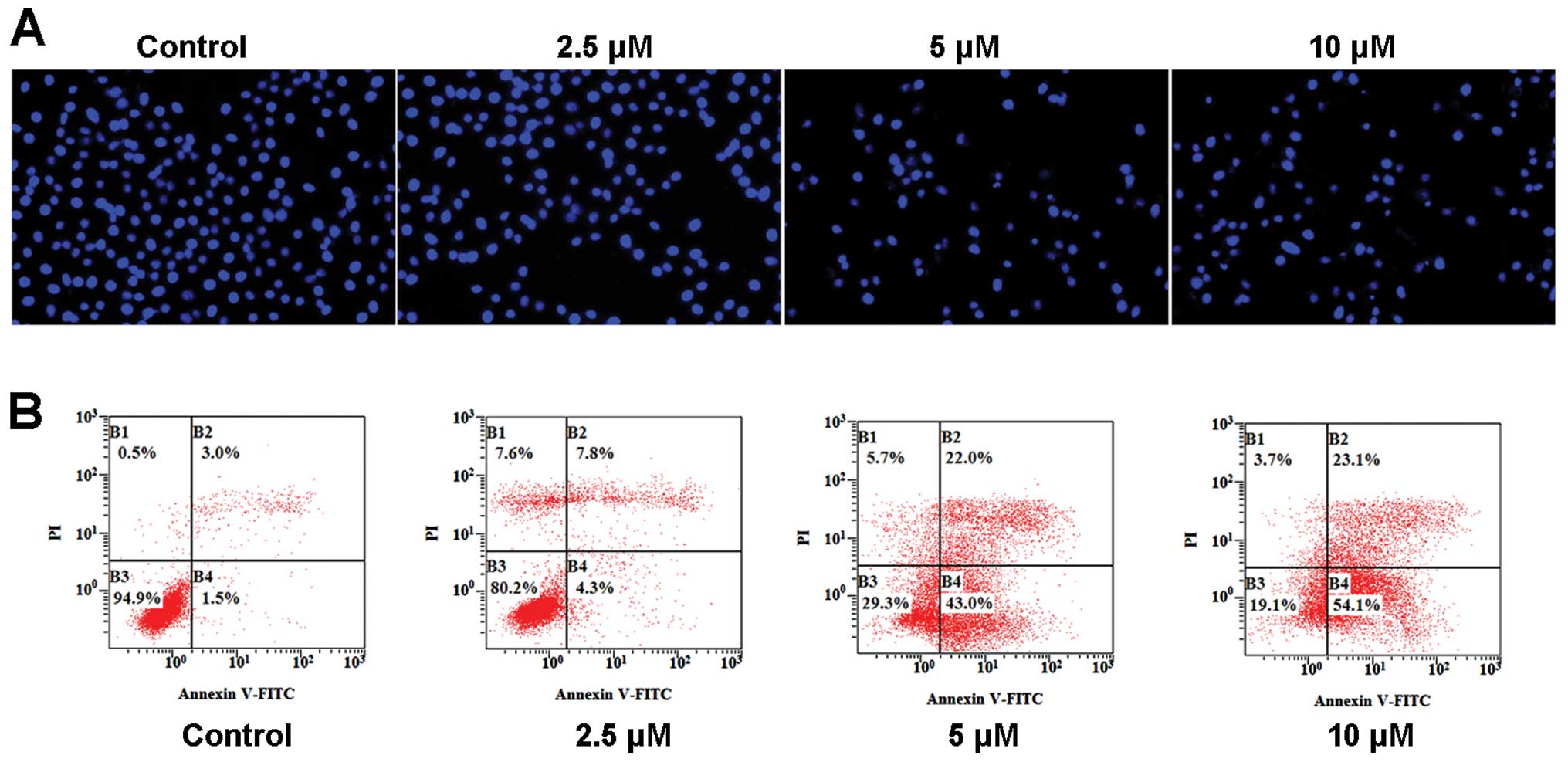
WYK431 treatment induces apoptosis in
BGC823 cells
We explored whether treatment of BGC823 cells with
WYK431 resulted in morphological changes. Fluorescence microscopic
examination of DAPI stained cells was performed. Treatment with
WYK431 showed features of apoptotic cells such as a bright blue
fluorescent condensed nuclei and apoptotic bodies (Fig. 3A).
Furthermore, apoptosis was measured using an Annexin
V-FITC apoptosis detection kit via flow cytometry. This combination
allows the differentiation among viable cells
(AV−/PI−), early phase apoptotic cells
(AV+/PI−), late phase apoptotic cells
(AV+/PI+) and necrotic cells
(AV−/PI+). After treatment with 2.5, 5 and 10
μM WYK431 for 36 h, the percentages of apoptotic cells was 12.1, 65
and 77.2%, respectively (Fig. 3B).
These results indicated that WYK431 could induce apoptosis and
death in BGC823 cells in a dose-dependent manner in vitro
(Table II).
| Table IIQuantification of apoptosis by
Annexin V-FITC/PI staining. |
Table II
Quantification of apoptosis by
Annexin V-FITC/PI staining.
| Treatment (36
h) | Percentage ±
standard deviation |
|---|
|
|---|
|
AV−/PI− |
AV+/PI− |
AV+/PI+ |
AV−/PI+ |
|---|
| Control | 94.0±2.5 | 1.0±0.3 | 2.5±0.7 | 2.5±0.6 |
| 2.5 μM | 83.7±5.8a | 3.8±1.4 | 7.0±1.6a | 5.5±1.4 |
| 5 μM | 27.4±5.2b | 45.1±4.2b | 20.4±3.7b | 7.1±2.0 |
| 10 μM | 18.5±1.7b | 53.7±2.1b | 24.5±2.4b | 3.3±0.7 |
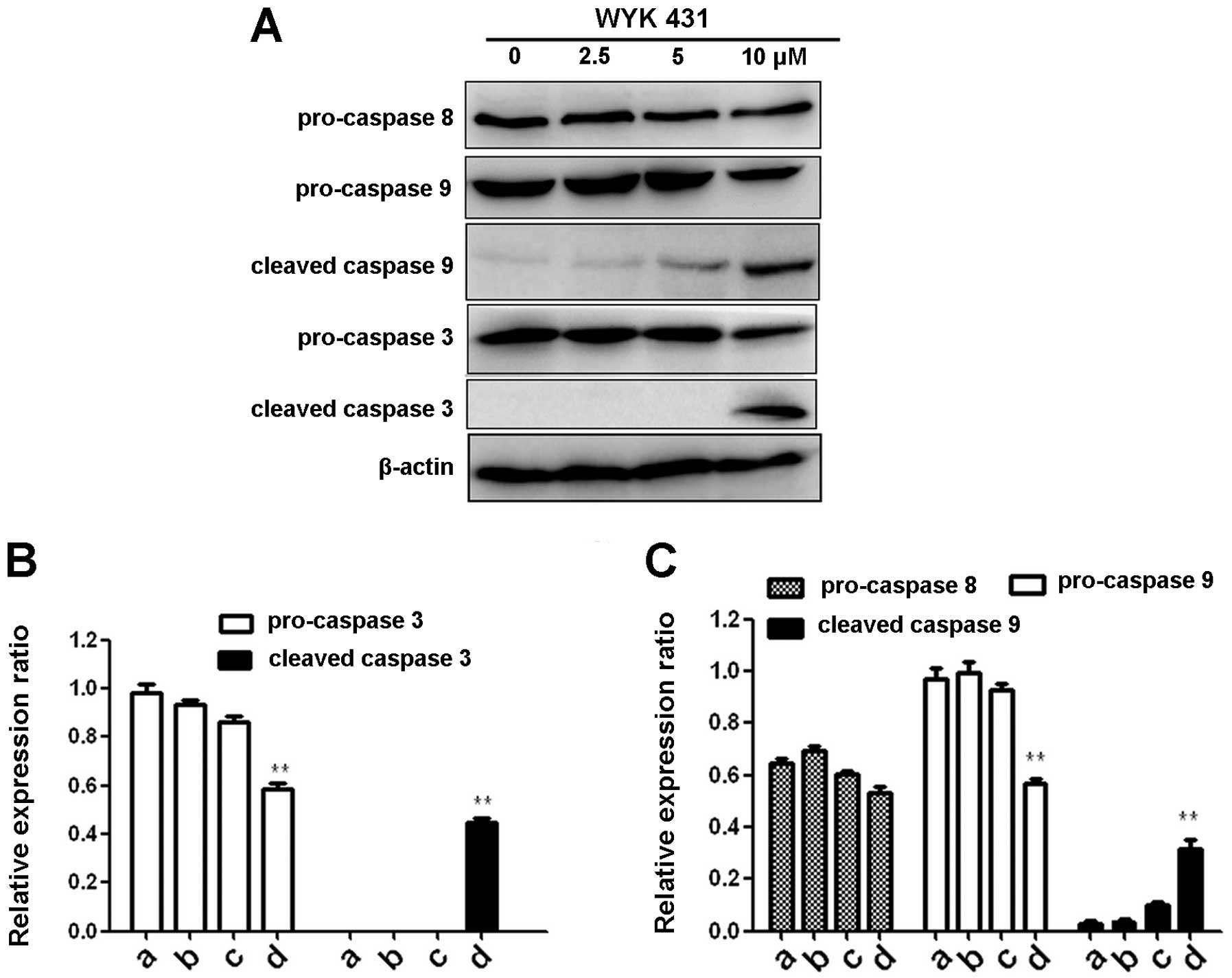
WYK431 treatment induces apoptosis via
the intrinsic pathway
To further confirm the induction of apoptosis with
WYK431 treatment, we examined caspase-3, caspase-8 and caspase-9
expression levels in BGC823 cells after WYK431 treatment for 24 h
by western blot analysis. As shown in Fig. 4, procaspase-3, and procaspase-9
decreased significantly after WYK431 exposure for 24 h in BGC823
cells and the levels of cleaved caspase-3 and -9 increased, but no
change of caspase-8 was observed. According to the results, we
propose that WYK431 triggers apoptosis trough the intrinsic
pathway.
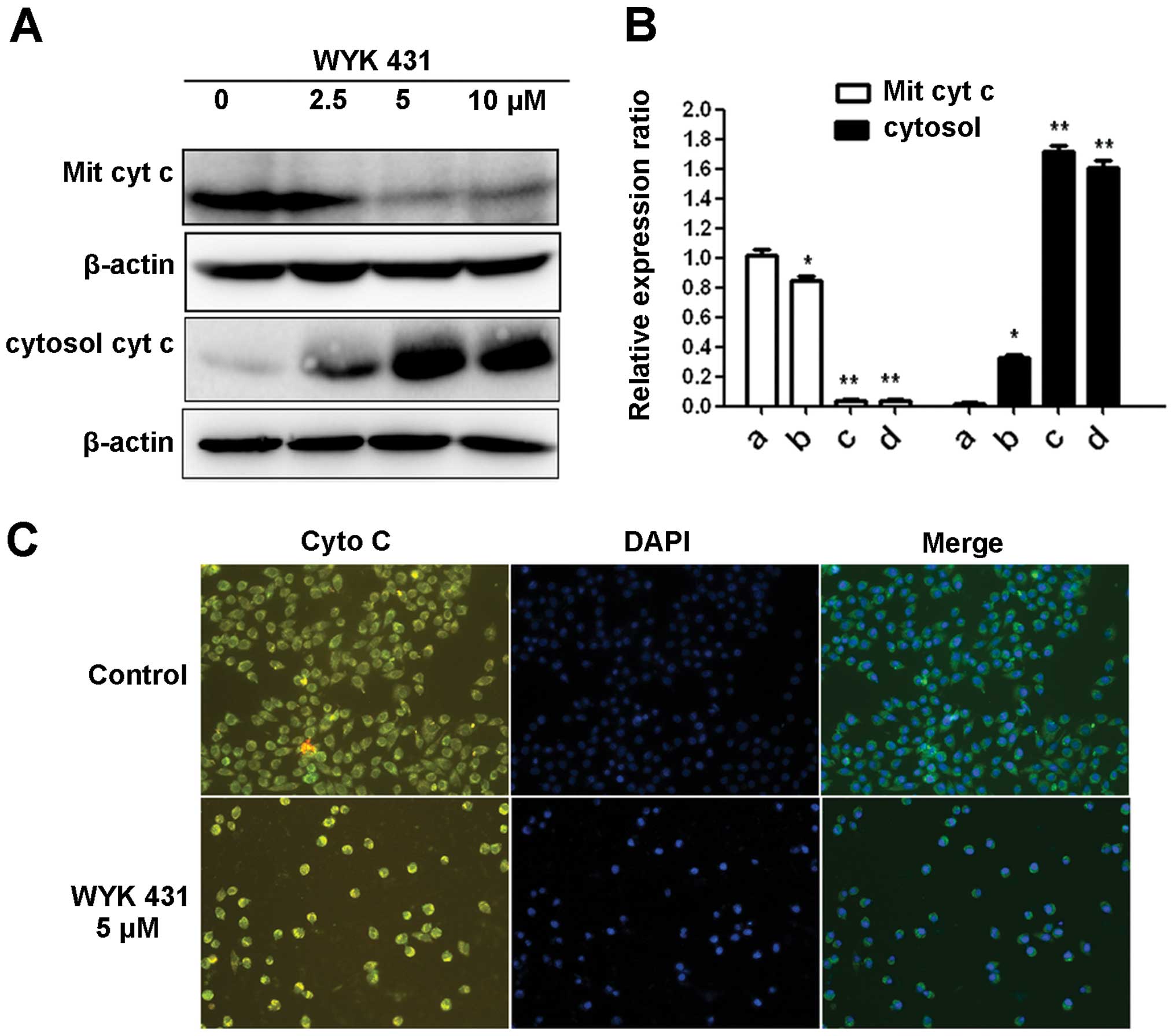
WYK431 induces cytochrome c released
Previously, it has been reported that during
mitochondrion-mediated apoptotic pathway, the release of cytochrome
c from mitochondria into cytosol promotes caspase activation
(36). Therefore, after treatment
of BGC823 cells with WYK431 at indicated concentrations for 24 h,
cytochrome c was detected in cytosolic fraction
concentration-dependently, whereas a decreased level of cytochrome
c was detected in mitochondria fraction (Fig. 5A and B). In addition, the release
of cytochrome c from mitochondria into cytosol was visualized in
WYK431-treated cells by fluorescence microscopy (Fig. 5C). Overall, these results indicated
that WYK431 induced the release of cytochrome c from mitochondria
into cytosol in BGC823 cells.
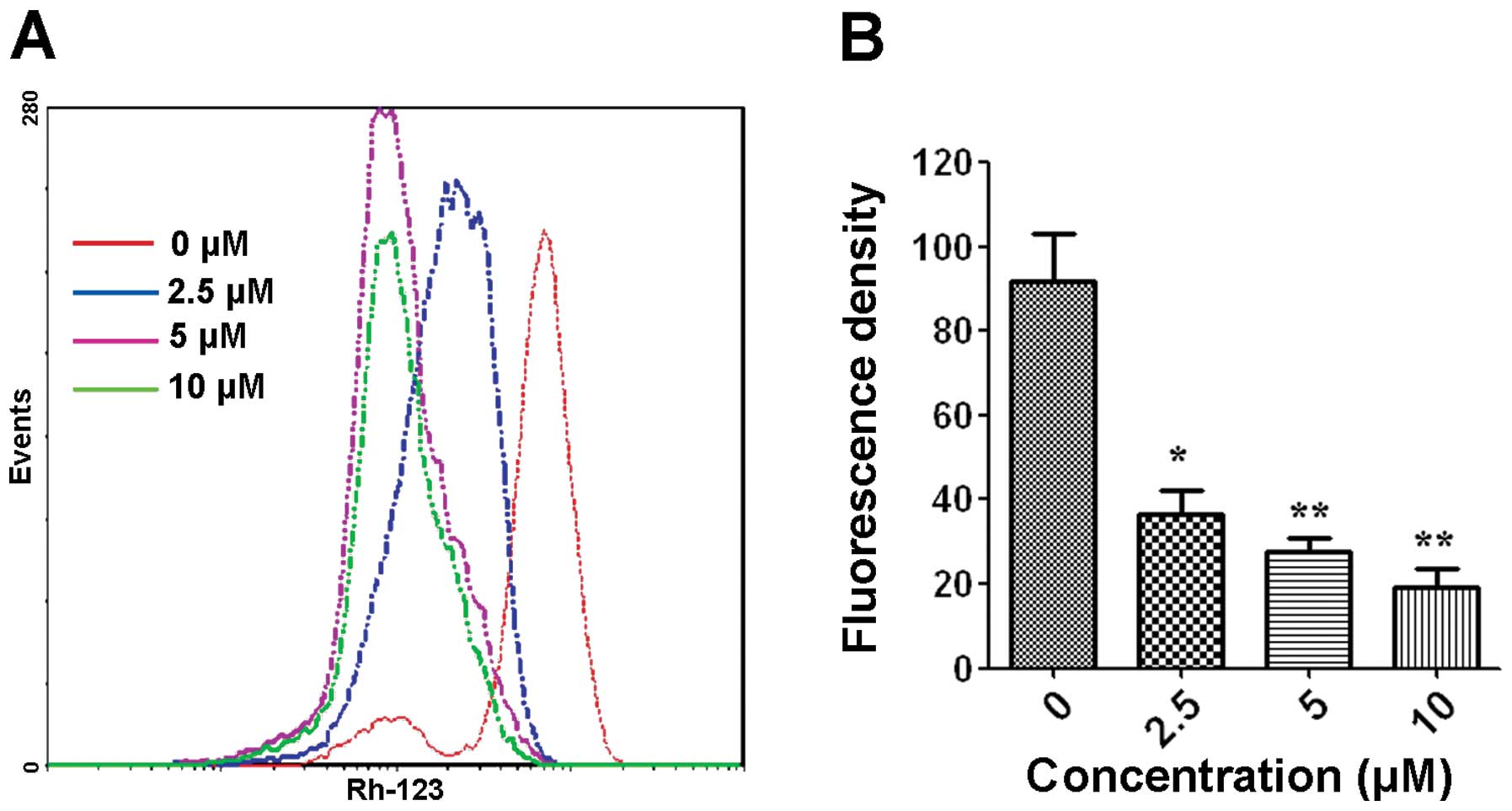
Effects of WYK431 on mitochondrial
transmembrane potential
A key step in the intrinsic apoptotic pathway is the
disruption of the mitochondrial membrane, which lead to loss
mitochondrial transmembrane potential ΔΨm (37). It is an early event coinciding with
caspase activation. To gain further insight into the mechanism
underlying apoptosis induced by WYK431, we investigated whether
WYK431 promotes mitochondrial disruption. Therefore, ΔΨm was
examined by flow cytometry using the cationic lipophilic green
fluorochrome rhodamine 123. Mitochondrial membrane permeability
disruption is associated with a lack of rhodamine 123 retention and
a decrease in fluorescence in intrinsic apoptosis pathway. Fig. 6 showed that WYK431 triggered the
loss of mitochondrial transmembrane potential in BGC823 cells.
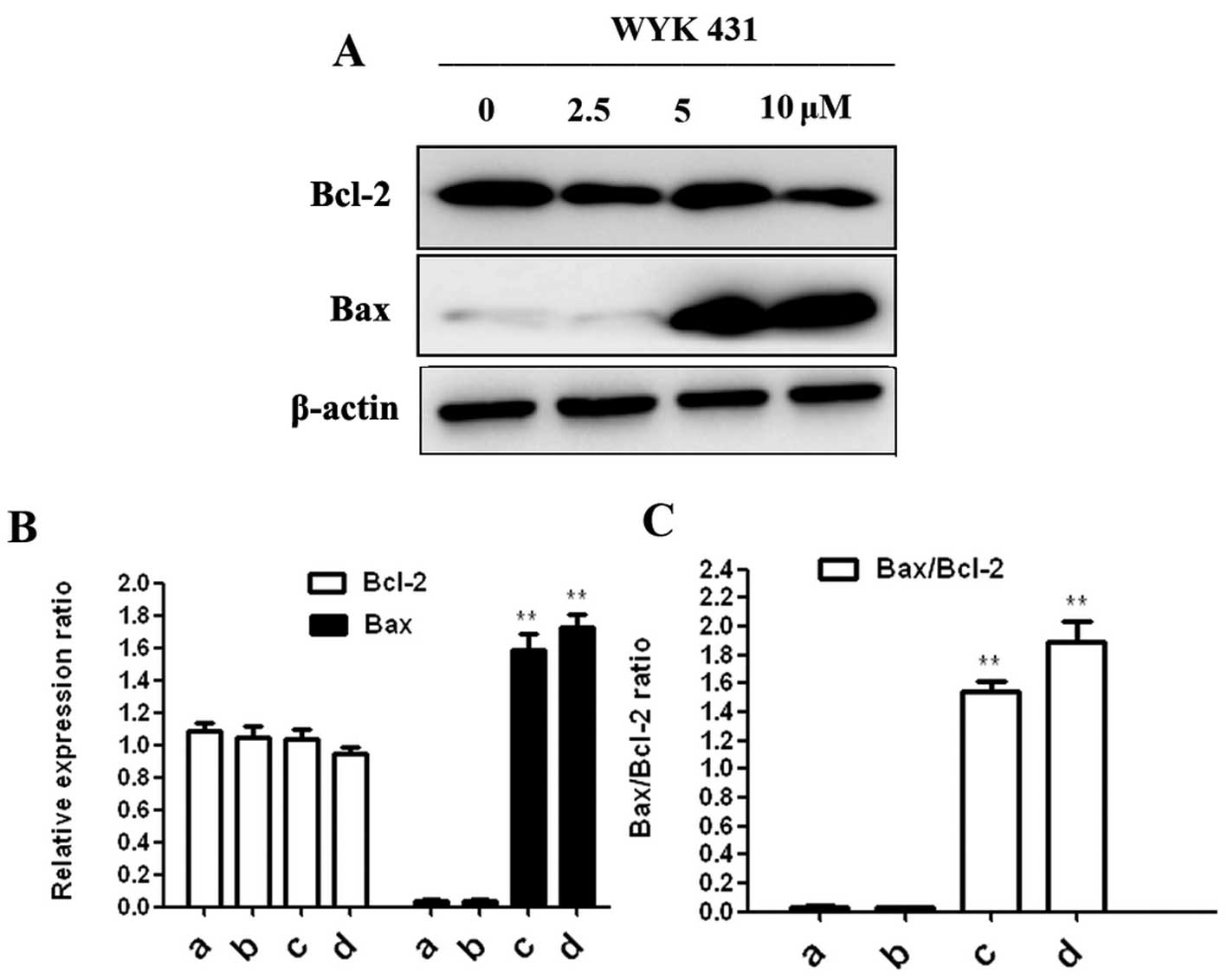
Effect of WYK431 on Bcl-2 and Bax
expression
We measured Bcl-2 and Bax expression in BGC823 cells
after WYK treatment for 24 h. Fig.
7 shows that WYK431 treatment led to an increased level of Bax
in BGC823 cells, but no significant change of Bcl-2 was observed.
The result was coincident with the result of cytochrome c release.
Collectively, we propose that WYK431 promotes apoptosis through the
intrinsic pathway.
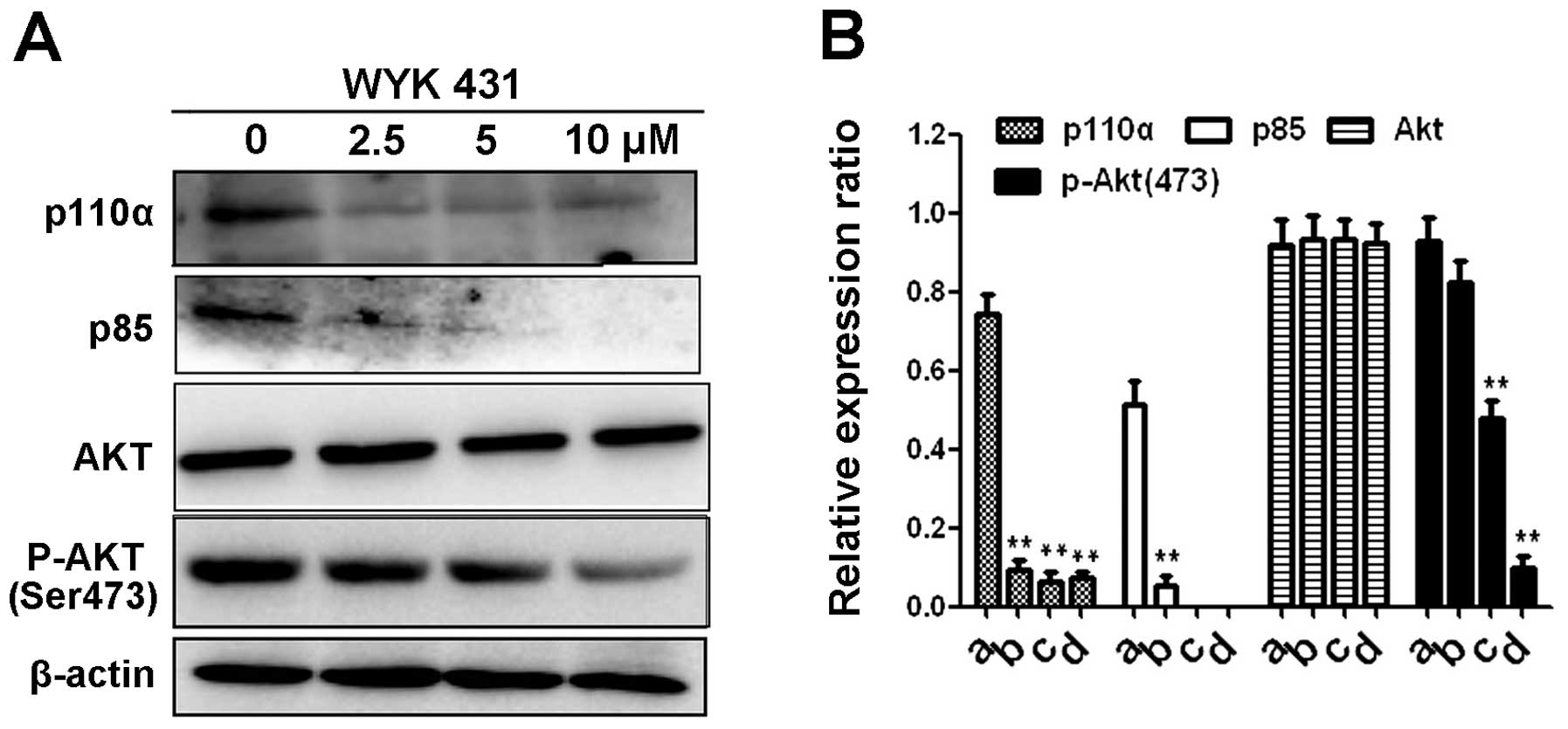
Effects of WYK431 on the PI3K/Akt
signaling pathways
To investigate the effect of WYK431 on PI3K/Akt
pathway, the expression level of PI3K and its downstream components
in BGC823 cells after WYK431 treatment for 8 h were detected by
western blot analysis. Our data indicated that exposure of BGC823
cells to WYK431 markedly decreased the expression levels of PI3K
p110α and p85 proteins (Fig. 8).
Akt, the downstream target of PI3K, plays a crucial role in
carcinogenesis and cancer development. Our result showed that the
expression of p-Akt (473), as the active form of Akt, substantially
decreased after WYK431 treatment. However, the level of total Akt
was unaffected by WYK431.
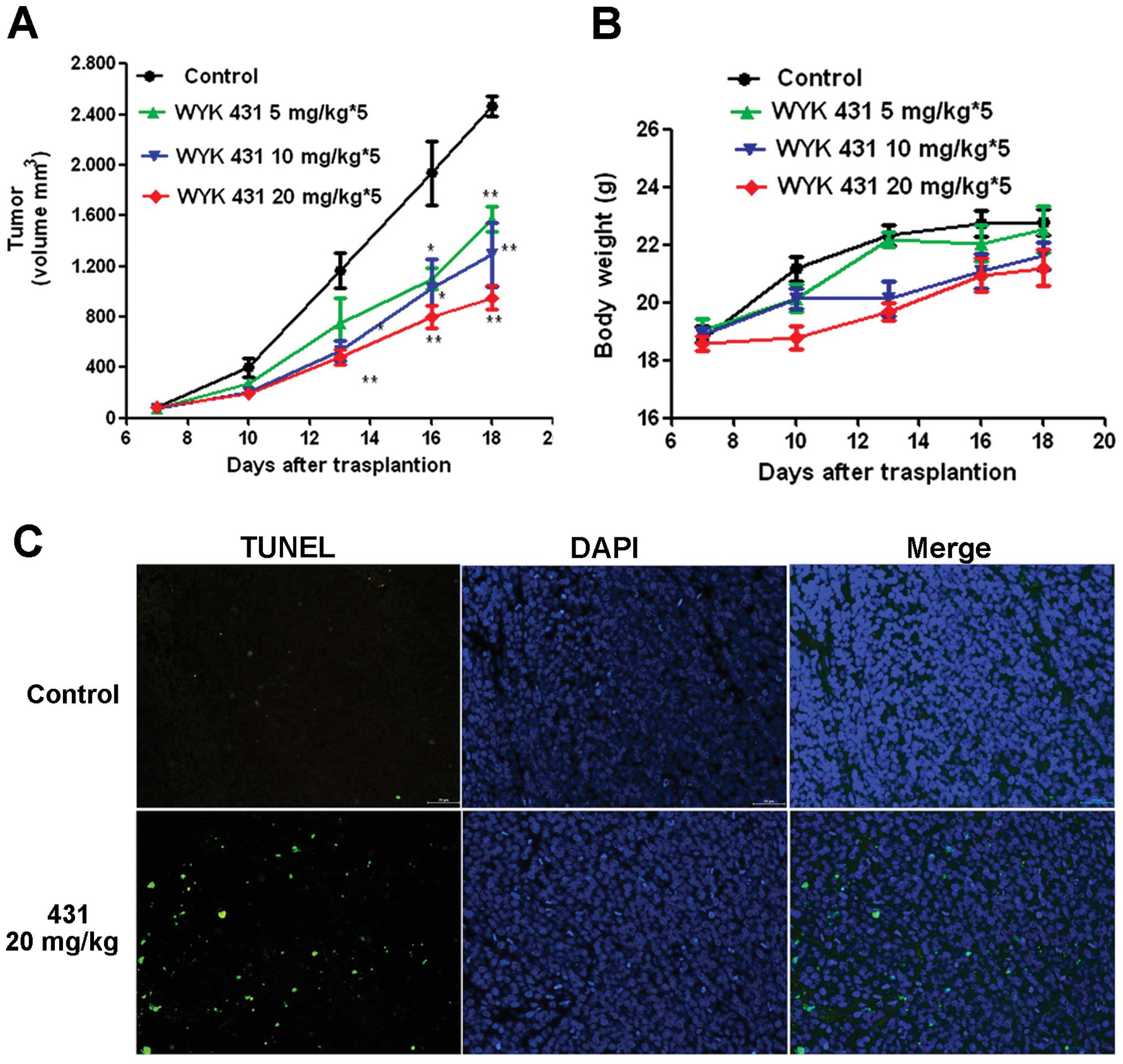
Antitumor activity of WYK431 in vivo
To evaluate the antitumor activity of WYK431 in
vivo, BGC823 bearing BALB/c nude mice were treated with
vehicle, 5, 10 and 20 mg/kg of WYK431, respectively. WYK431
exhibited a significant antitumor activity in inhibiting tumor
progression compared with vehicle-treated animals. As noted in
Fig. 9A, WYK431 administered at 5,
10 and 20 mg/kg of WYK431 suppressed tumors 28.4, 41.9, 63.6%,
compared with vehicle-treated animals (P<0.05). Moreover, WYK431
treatment was well tolerated, with only small effects on body
weight, which was consistent with the notion that WYK431 preferably
targets tumor cells and thus exhibited little toxicity (Fig. 9B). Hematological analysis showed
that WYK431 did not significantly decrease the number of white
blood corpuscles (Table
III).
| Table IIIThe final WBC parameters of different
groups. |
Table III
The final WBC parameters of different
groups.
| Types | Control | WYK431
5 mg/kg | WYK431
10 mg/kg | WYK431
20 mg/kg |
|---|
| LYM % | 8.6±1.2 | 6.4±3.2 | 8.0±6.0 | 10.1±5.4 |
| MON % | 4.9±2.6 | 3.7±3.1 | 5.4±5.9 | 8.4±7.3 |
| NEUT % | 73.2±4.9 | 77.5±3.7 | 73.3±8.1 | 69.5±7.2 |
| EOS % | 10.5±2.8 | 10.2±2.6 | 10.9±3.3 | 10.2±5.5 |
| BAS % | 2.9±1.2 | 2.2±0.7 | 2.4±0.8 | 1.7±0.5 |
Histological analysis by TUNEL
The tumors were removed and TUNEL assay was
performed. Five equal-sized fields were randomly chosen and
analyzed in tumor sections. We observed more TUNEL-positive cells
with dark green fluorescent staining in the treatment group,
indicating a significant increased apoptosis in the treatment group
compared with control group (Fig.
9C). These results suggest that WYK431 induced apoptosis of
tumor cells in vivo.
Discussion
Quinazoline derivatives possess significant
antitumor effects via inducing apoptosis and cell cycle arrest
(31,38). WYK431, a novel quinazoline
derivative, is a highly potent and broad spectrum anticancer
compound, and BGC823 cells were the most sensitive. To our
knowledge, this study is the first to demonstrate that WYK431
possesses significant antitumor activities through inducing
G2/M phase arrest and apoptosis by blocking the PI3K
signaling pathway in human gastric cancer cells in vitro.
Moreover, it inhibits tumor growth in vivo as well.
Hallmarks of cancer include an uncontrolled cell
cycle and resistance to apoptosis signals (39). Therefore, the induction of
apoptosis and cell cycle blockage are important cancer prevention
treatment goals. Cell cycle arrest is a critical mechanism of
anticancer drug suppressing cell proliferation (40). Cell cycle is promoted by a number
of proteins including cyclin dependent kinases (CDKs) and cyclins.
In G2 phase, cells synthesize cyclin A/cyclin B, which
can form a complex with CDK1 to promote entry into mitosis
(41). When the CDK1/cyclin B or
cyclin A complex initially forms, it is maintained in the inactive
form by phosphorylation at two sites, threonine 14 and tyrosine 15
(42). Cdc25C can dephosphorylate
Cdk1 at threonine 14 and tyrosine 15 and activate CDK1-cyclin B or
CDK1-cyclin A complex. The activated CDK1-cyclin B or CDK1-cyclin A
complex translocate into nucleus at the G2/M checkpoint
to promote cell entry into mitosis (43,44).
Therefore, we investigated the effect of WYK431 on the cell cycle.
Our result showed that treatment of BGC823 with WYK431 for 24 h
caused cells to accumulate in G2/M phase which was
associated with downregulation of CDK1 and Cdc25C proteins.
PI3K/AKT pathway has been implicated in cell cycle progression
(45). Inhibited PI3K/AKT pathway
regulated the expression of CDKs and cyclins, eventually causing
G2/M arrest (46–48).
In the present study, we observed that WYK431-mediated
downregulation of cell cycle regulatory molecular may be associated
with the decrease of p110α and p85. Further studies are required to
investigate the detailed mechanism of how PI3K/Akt pathway
regulates WYK431-induced G2/M arrest in BGC823
cells.
Apoptosis is the most common way that antitumor
agents induce cell death (49,50).
Hoechst staining and FCM analysis showed that WYK431 induced BGC823
cells apoptosis. Our study also demonstrated that WYK431 could
induce tumor cell apoptosis by TUNEL assay. In order to explore the
mechanism of apoptosis induced by WYK431, we examined the proteins
involved in apoptosis. Our results showed that caspase-3 was
activated by WYK431. There are two main pathway of apoptosis, the
death receptor (extrinsic) pathway and mitochondria (intrinsic)
pathway. Activation of caspase-3 is in either one of the pathways
(51). To determine the pathway of
apoptosis induced by WYK431, caspase-8 and caspase-9 involved in
the extrinsic and intrinsic pathway, respectively, was detected
(52). We found that caspase-9,
but not caspase-8 was activated by WYK431, indicating that WYK431
induces BGC823 cell apoptosis via intrinsic pathway. The disruption
of mitochondrial membrane potential and the release of cytochrome c
from mitochondria into cytoplasm are recognized as key step in the
mitochondria pathway.
Our results showed that the treatment of BGC823
cells with WYK431 led to a loss of ΔΨm and released cytochrome c
from mitochondria into cytoplasm, indicating an activation of the
intrinsic apoptosis pathway. The process is regulated by Bcl-2
family proteins which include proapoptotic members such as Bax and
anti-apoptotic members such as Bcl-2 (53). Bcl-2 family members located on the
mitochondrial membrane can alter the permeability of the
mitochondrial membrane and trigger the release of cytochrome c. To
further confirm the involvement of the mitochondrial pathway in
WYK431 induced apoptotic death, the expression of Bax and Bcl-2
were detected. The expression of Bax was increased in cells treated
with WYK431, whereas there was no change in Bcl-2 protein
expression. Thus, the ratio of Bax to Bcl-2 was altered. We assume
that upregulation of Bax may be involved in the release of
cytochrome c from mitochondria into cytosol after WYK431
treatment.
Many studies showed that inactivation of Akt by
dephosphorylation play a key role in tumor suspression (54,55).
Our results show that WYK431 could inhibit Akt phosphorylation. It
is well-established that the inhibition of PI3K/Akt signal pathway
can induce apoptosis of cancer cells (56). Our investigations revealed that
treatment with WYK431 remarkably decreased Akt phosphorylation and
promote cell apoptosis indicating that PI3K/Akt pathway is involved
in WYK431-induced apoptosis. These results suggest that PI3K/Akt is
involved in apoptosis induced by WYK431.
In conclusion, this is the first study reporting
that WYK431 exerts potential anti-proliferative effect against
BGC823 cells probably through cell cycle arrest and the
mitochondria apoptotic pathway. Our studies show that WYK431
treatment blocks the PI3K/Akt pathways. In addition, the in
vivo study showed that WYK431 can suppress tumor growth of
BGC823 cells without obvious toxicity, indicating that WYK431
provide chemical structure information that can be used for the
design and development of more effective derivatives.
Acknowledgements
We thank Ms. Furong Zhang and Hanze Yang for
technical assistance and Professor Bailing Xu for providing
compound 431. This study was supported by the National Natural
Science Foundation (Grant no. 81273380).
References
|
1
|
Kamangar F, Dores GM and Anderson WF:
Patterns of cancer incidence, mortality, and prevalence across five
continents: defining priorities to reduce cancer disparities in
different geographic regions of the world. J Clin Oncol.
24:2137–2150. 2006. View Article : Google Scholar
|
|
2
|
Crew KD and Neugut AI: Epidemiology of
gastric cancer. World J Gastroenterol. 12:354–362. 2006.
|
|
3
|
Wagner AD, Grothe W, Behl S, et al:
Chemotherapy for advanced gastric cancer. Cochrane Database Syst
Rev. 2:CD0040642005.
|
|
4
|
Van Cutsem E, Moiseyenko VM, Tjulandin S,
et al: Phase III study of docetaxel and cisplatin plus fluorouracil
compared with cisplatin and fluorouracil as first-line therapy for
advanced gastric cancer: a report of the V325 Study Group. J Clin
Oncol. 24:4991–4997. 2006.
|
|
5
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vanhaesebroeck B, Guillermet-Guibert J,
Graupera M and Bilanges B: The emerging mechanisms of
isoform-specific PI3K signalling. Nat Rev Mol Cell Biol.
11:329–341. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Paweletz CP, Charboneau L, Bichsel VE, et
al: Reverse phase protein microarrays which capture disease
progression show activation of pro-survival pathways at the cancer
invasion front. Oncogene. 20:1981–1989. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bellacosa A, Kumar CC, Di Cristofano A and
Testa JR: Activation of AKT kinases in cancer: implications for
therapeutic targeting. Adv Cancer Res. 94:29–86. 2005. View Article : Google Scholar
|
|
9
|
Testa JR and Bellacosa A: AKT plays a
central role in tumorigenesis. Proc Natl Acad Sci USA.
98:10983–10985. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu P, Cheng H, Roberts TM and Zhao JJ:
Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev
Drug Discov. 8:627–644. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Engelman JA, Luo J and Cantley LC: The
evolution of phosphatidylinositol 3-kinases as regulators of growth
and metabolism. Nat Rev Genet. 7:606–619. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee JW, Soung YH, Kim SY, et al: PIK3CA
gene is frequently mutated in breast carcinomas and hepatocellular
carcinomas. Oncogene. 24:1477–1480. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Campbell IG, Russell SE, Choong DY, et al:
Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer
Res. 64:7678–7681. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li VS, Wong CW, Chan TL, et al: Mutations
of PIK3CA in gastric adenocarcinoma. BMC Cancer. 5:292005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bachman KE, Argani P, Samuels Y, et al:
The PIK3CA gene is mutated with high frequency in human breast
cancers. Cancer Biol Ther. 3:772–775. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Duronio V, Scheid MP and Ettinger S:
Downstream signalling events regulated by phosphatidylinositol
3-kinase activity. Cell Signal. 10:233–239. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Blume-Jensen P and Hunter T: Oncogenic
kinase signalling. Nature. 411:355–365. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Johnson DG and Walker CL: Cyclins and cell
cycle checkpoints. Annu Rev Pharmacol Toxicol. 39:295–312. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Senderowicz AM and Sausville EA:
Preclinical and clinical development of cyclin-dependent kinase
modulators. J Natl Cancer Inst. 92:376–387. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kasibhatla S and Tseng B: Why target
apoptosis in cancer treatment? Mol Cancer Ther. 2:573–580.
2003.PubMed/NCBI
|
|
22
|
Jemal A, Siegel R, Ward E, et al: Cancer
statistics, 2008. CA Cancer J Clin. 58:71–96. 2008. View Article : Google Scholar
|
|
23
|
Plenchette S, Filomenko R, Logette E, et
al: Analyzing markers of apoptosis in vitro. Methods Mol Biol.
281:313–331. 2004.PubMed/NCBI
|
|
24
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sartorius U, Schmitz I and Krammer PH:
Molecular mechanisms of death-receptor-mediated apoptosis.
Chembiochem. 2:20–29. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jiang X and Wang X: Cytochrome c promotes
caspase-9 activation by inducing nucleotide binding to Apaf-1. J
Biol Chem. 275:31199–31203. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wakeling AE, Guy SP, Woodburn JR, et al:
ZD1839 (Iressa): an orally active inhibitor of epidermal growth
factor signaling with potential for cancer therapy. Cancer Res.
62:5749–5754. 2002.PubMed/NCBI
|
|
28
|
Baba A, Kawamura N, Makino H, Ohta Y,
Taketomi S and Sohda T: Studies on disease-modifying antirheumatic
drugs: synthesis of novel quinoline and quinazoline derivatives and
their anti-inflammatory effect. J Med Chem. 39:5176–5182. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rohini R, Muralidhar Reddy P, Shanker K,
Hu A and Ravinder V: Antimicrobial study of newly synthesized
6-substituted indolo[1,2-c]quinazolines. Eur J Med Chem.
45:1200–1205. 2010.PubMed/NCBI
|
|
30
|
Honkanen E, Pippuri A, Kairisalo P, Nore
P, Karppanen H and Paakkari I: Synthesis and antihypertensive
activity of some new quinazoline derivatives. J Med Chem.
26:1433–1438. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jin J, Zhu L-N, Zhang H-J, et al: XLN306
induces apoptosis in human breast carcinoma MX-1 cells. Acta Pharm
Sin B. 1:84–88. 2011. View Article : Google Scholar
|
|
32
|
Wang YK, Jin J, Zhu LN, et al: Synthesis
and antitumor activity of novel
2-(1-substituted-piperidin-4-ylamino)quinazolines as antitumor
agents. Yao Xue Xue Bao. 47:1164–1178. 2012.PubMed/NCBI
|
|
33
|
Scaduto RC Jr and Grotyohann LW:
Measurement of mitochondrial membrane potential using fluorescent
rhodamine derivatives. Biophys J. 76:469–477. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Konopleva M, Contractor R, Tsao T, et al:
Mechanisms of apoptosis sensitivity and resistance to the BH3
mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 10:375–388.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lewis JS, Meeke K, Osipo C, et al:
Intrinsic mechanism of estradiol-induced apoptosis in breast cancer
cells resistant to estrogen deprivation. J Natl Cancer Inst.
97:1746–1759. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nunez G, Benedict MA, Hu Y and Inohara N:
Caspases: the proteases of the apoptotic pathway. Oncogene.
17:3237–3245. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar
|
|
38
|
Cubedo E, Cordeu L, Bandres E, et al: New
symmetrical quinazoline derivatives selectively induce apoptosis in
human cancer cells. Cancer Biol Ther. 5:850–859. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shapiro GI and Harper JW: Anticancer drug
targets: cell cycle and checkpoint control. J Clin Invest.
104:1645–1653. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
von Bergwelt-Baildon MS, Kondo E,
Klein-Gonzalez N and Wendtner CM: The cyclins: a family of widely
expressed tumor antigens? Expert Rev Vaccines. 10:389–395.
2011.PubMed/NCBI
|
|
42
|
Goss VL, Cross JV, Ma K, Qian Y, Mola PW
and Templeton DJ: SAPK/JNK regulates cdc2/cyclin B kinase through
phosphorylation and inhibition of cdc25c. Cell Signal. 15:709–718.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Karlsson-Rosenthal C and Millar JB: Cdc25:
mechanisms of checkpoint inhibition and recovery. Trends Cell Biol.
16:285–292. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Galea CA, Wang Y, Sivakolundu SG and
Kriwacki RW: Regulation of cell division by intrinsically
unstructured proteins: intrinsic flexibility, modularity, and
signaling conduits. Biochemistry. 47:7598–7609. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chang F, Lee JT, Navolanic PM, et al:
Involvement of PI3K/Akt pathway in cell cycle progression,
apoptosis, and neoplastic transformation: a target for cancer
chemotherapy. Leukemia. 17:590–603. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang J, Wu A, Xu Y, Liu J and Qian X:
M(2)-A induces apoptosis and G(2)-M arrest via inhibiting PI3K/Akt
pathway in HL60 cells. Cancer Lett. 283:193–202. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yuan Q, Cai S, Zhang X, et al: A new
protoapigenone analog RY10-4 induces apoptosis and suppresses
invasion through the PI3K/Akt pathway in human breast cancer.
Cancer Lett. 324:210–220. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li Y, Zhang P, Qiu F, et al: Inactivation
of PI3K/Akt signaling mediates proliferation inhibition and
G2/M phase arrest induced by andrographolide in human
glioblastoma cells. Life Sci. 90:962–967. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Xu YZ, Zheng RL, Zhou Y, et al: Small
molecular anticancer agent SKLB703 induces apoptosis in human
hepatocellular carcinoma cells via the mitochondrial apoptotic
pathway in vitro and inhibits tumor growth in vivo. Cancer Lett.
313:44–53. 2011. View Article : Google Scholar
|
|
50
|
Sun B, Geng S, Huang X, et al: Coleusin
factor exerts cytotoxic activity by inducing
G0/G1 cell cycle arrest and apoptosis in
human gastric cancer BGC-823 cells. Cancer Lett. 301:95–105. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ferreira CG, Epping M, Kruyt FA and
Giaccone G: Apoptosis: target of cancer therapy. Clin Cancer Res.
8:2024–2034. 2002.PubMed/NCBI
|
|
52
|
Adams JM and Cory S: The Bcl-2 protein
family: arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Youle RJ and Strasser A: The BCL-2 protein
family: opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Brognard J, Clark AS, Ni Y and Dennis PA:
Akt/protein kinase B is constitutively active in non-small cell
lung cancer cells and promotes cellular survival and resistance to
chemotherapy and radiation. Cancer Res. 61:3986–3997.
2001.PubMed/NCBI
|
|
55
|
Martelli AM, Tazzari PL, Tabellini G, et
al: A new selective AKT pharmacological inhibitor reduces
resistance to chemotherapeutic drugs, TRAIL, all-trans-retinoic
acid, and ionizing radiation of human leukemia cells. Leukemia.
17:1794–1805. 2003. View Article : Google Scholar
|
|
56
|
Hussain AR, Al-Rasheed M, Manogaran PS, et
al: Curcumin induces apoptosis via inhibition of PI3’-kinase/AKT
pathway in acute T cell leukemias. Apoptosis. 11:245–254.
2006.PubMed/NCBI
|