Introduction
Hepatocellular carcinoma (HCC) is one of the most
fatal tumors worldwide, particularly in Sub-Saharan Africa and
Southeastern Asia (1). In recent
years, the incidence of HCC in China has increased (2). The major risk factors for the
development of HCC include infection by hepatitis B and C viruses,
exposure to aflatoxin B1, and cirrhosis of any etiology (3). Liver resection and transplantation
are currently regarded the most effective treatments; however, the
postoperative survival rate is only 30–40% at 5 years (4). In addition, most patients with
advanced HCC are rejected for treatment because of indications and
contraindications of surgery. Therefore, there is an urgent need to
advance our understanding of hepatocarcinogenesis and explore novel
effective therapeutic strategies.
Family with sequence similarity 189, member B
(FAM189B), also called COTE1, maps to chromosome 1q21 and is widely
expressed in heart, brain, placenta, lung, liver, skeletal muscle,
kidney, and pancreas (5,6). The COTE1 gene was originally
identified by Winfield et al, who found that COTE1 is
located near the gene for the lysosomal enzyme glucosylceramidase,
deficiency of which is associated with Gaucher disease (6). Alternative splicing of COTE1 results
in multiple transcript variants: 1, 2, and 3. Variant 1 represents
the longest transcript and encodes the longest protein (isoform a).
Variants 2 and 3 lack an in-frame portion of the 5′ coding region
compared with variant 1, and the resulting proteins (isoforms b and
c) are shorter than isoform a (National Center for Biotechnology
Information Reference Sequences). The COTE1 protein contains 669
amino acids with two potential N-glycosylation sites, a leucine
zipper, and multiple potential phosphorylation sites and
N-myristoylation sites (6). Recent
data showed that COTE1 contains a predicted four-transmembrane
domain, suggesting that it might reside in a membrane-bound
subcellular organelle such as the Golgi (7). Kallin et al found that
expression of COTE1 correlated with activation of endogenous
SREBP-1 (sterol-regulatory element binding protein) in
vitro, and speculated that it plays a role in lipid metabolism
(8). Moreover, the protein has
been identified as a potential binding partner of a WW
domain-containing protein that is involved in tumor suppression
(7,9).
In a previous study, we showed for the first time
that COTE1 is markedly upregulated in HCC clinical specimens
compared with adjacent non-cancerous livers (10). In the present study we verified
upregulation of COTE1 in 42 of 80 paired HCC specimens and 11 of 15
HCC cell lines. These findings indicate that COTE1 may represent a
new potential oncogene. Subsequent experiments showed that COTE1
contributed to cell growth and colony formation in vitro,
and tumorigenesis in vivo. Furthermore, COTE1 was found to
physically interact with the tumor suppressor WW domain-containing
oxidoreductase (WWOX), blocking its tyrosine phosphorylation and
thereby suppressing WWOX-mediated endogenous apoptosis and cell
cycle arrest.
Materials and methods
Tissue specimens
Eighty pairs of clinical specimens were obtained
from patients with HCC who were hospitalized in the First
Affiliated Hospital of Nanjing Medical University with informed
consent. Adjacent non-tumor tissues were excised 2 cm from the edge
of the primary focus. Both HCC specimens and adjacent non-tumor
tissues were immediately stored in liquid nitrogen after excision
and confirmed by pathological examination. The protocols for
investigations involving humans and animals were approved by the
Institutional Animal Care and Use Committee at Nanjing Medical
University.
Liver cancer cell lines
Human hepatocellular carcinoma cell lines (QGY-7703,
Focus, Hep3B, HepG2, HepG2.2.15, Huh7, LM3, LM6, MHCC-H, MHCC-L,
PLC/PRF/5, SK-hep-s, SNU-398, WRL-68, and YY-8103) obtained from
the Chinese National Human Genome Center at Shanghai were used in
this study. All cell lines were propagated at 37°C in a 5%
CO2 humidified incubator in Dulbecco’s modified Eagle’s
medium (DMEM) supplemented with 10% fetal bovine serum, penicillin
(50 U/ml), and streptomycin (50 μg/ml).
Semiquantitative reverse
transcription-polymerase chain reaction (RT-PCR) and quantitative
real-time PCR
Total RNA was extracted from clinical samples or
cell lines using TRIzol solution (Invitrogen) according to the
manufacturer’s protocol and reverse-transcribed into cDNA using a
M-MLV reverse transcriptase kit (Promega). Primers used in
semiquantitative RT-PCR and quantitative real-time PCR were as
follows: COTE1, 5′-GGGCTCTGACCTAGGCTTCT-3′ (forward) and
5′-ACAGAAGCTCTCCCAGTCCA-3′ (reverse); β-actin (loading control):
5′-AGAGCCTCGCCTTTGCCGATCC-3′ (forward) and
5′-CTGGGCCTCGTCGCCCACATA-3′ (reverse). All primers were synthesized
by Shanghai Biosune Co. Ltd.
Immunofluorescence assay
HCC cells grown on polylysine-treated slides were
washed twice with phosphate-buffered saline (PBS), fixed with 4%
paraformaldehyde on ice for 30 min, and blocked with 5% BSA at room
temperature. Cells were stained with primary antibody [goat
anti-COTE1 antibody (1:50) or mouse anti-WWOX antibody (1:50),
Santa Cruz Biotechnology, CA, USA] at 4°C overnight, followed by
incubation with secondary antibody [Cy5-conjugated anti-mouse
secondary antibody (1:200, red); Cy3-conjugated anti-goat antibody
(1:200, green), Molecular Probes Inc., Eugene, OR, USA] at room
temperature for 30 min. After rinsing three times with
PBS-Tween-20, nuclei were stained with 4,6-diamidino-2-phenylindole
(DAPI) and the cells were analyzed by inverted fluorescence
microscopy.
siRNA preparation
Two siRNAs against COTE1 were designed using the web
server of Invitrogen Co. and chemically synthesized by Shanghai
GenePharma Co. The commonly used negative control (NC) siRNA
supplied by Qiagen was used as a control. The sense and antisense
sequences of human COTE1 were as follows: siRNA-2852 sequence:
5′-GUAUGUAAGCCUUCAAUAAdTdT-3′ (sense) and 5′-UUA
UUGAAGGCUUACAUACdTdT-3′ (antisense); siRNA-3129 sequence:
5′-AGCUCUUAACAGUAUGUAAdTdT-3′ (sense) and
5′-UUACAUACUGUUAAGAGCUdTdT-3′ (antisense).
Construction of COTE1 shRNA plasmid and
COTE1 expression vector
To construct RNAi plasmid, the shRNA expression
cassette containing sequence identical to the siRNA2852/3129
sequence of the target gene was inserted into the expression
plasmid pSUPER containing the polymerase-III H1-RNA gene promoter.
Negative control oligos served as controls. For the construction of
COTE1 recombinant plasmid, the COTE1 open reading frame was
amplified from a human liver cDNA library (Genbank: NM_006589.2)
using nested PCR and inserted into pcDNA3.1B-FLAG-GFP (Chinese
National Human Genome Center, Shanghai). The sequences of the
shRNAs and primers used for COTE1 expression vector construction
are shown in Table I.
 | Table ISequences of shRNAs. |
Table I
Sequences of shRNAs.
| Sequences of
shRNAs |
| COTE1-sh2852 |
| Sense |
GATCCCCGTATGTAAGCCTTCAATAATTCAAGAGATTATTGAAGGCTTACATACTTTTTGGAAA |
| Antisense |
AGCTTTTCCAAAAAGTATGTAAGCCTTCAATAATCTCTTGAATTATTGAAGGCTTACATACGGG |
| COTE1-sh3129 |
| Sense |
GATCCCCAGCTCTTAACAGTATGTAATTCAAGAGATTACATACTGTTAAGAGCTTTTTTGGAAA |
| Antisense |
AGCTTTTCCAAAAAAGCTCTTAACAGTATGTAATCTCTTGAATTACATACTGTTAAGAGCTGGG |
|
| Sequences of
primers used for COTE1 expression vector construction |
| COTE1-nest-out |
| Forward |
GGGTGGAGAGGAGAAAGGAC |
| Reverse |
CAGTGCTATAAGAAGGGGCATC |
| COTE1-nest-in |
| Forward |
CGGGATCCACGAGCCCAGTCTCCCGGCTG |
| Reverse |
CGGAATTCATGATGCCCTCGCCTAGTGACTCCAGCCGC |
Cell transfection
RNAi and plasmid transfections were performed using
Lipofectamine 2000 (Invitrogen) according to the manufacturer’s
instructions at a cell density of 30–50% and 80–90%,
respectively.
Cell proliferation and colony
formation
For the cell proliferation assay, cells were plated
in 96-well plates and cultured for 24 h to a density of 30–50%
before transient transfection. The cell counting kit-8 (CCK-8;
Dojindo Labs.) was used to measure cell viability according to the
manufacturer’s instructions. Briefly, 90 μl DMEM free medium plus
10 μl CCK-8 solution was added to the 96-well plate. After
incubation at 37°C for 1 h, the absorbance at 450 nm was measured.
Three replicate wells were tested per assay condition, and each
experiment was repeated at least three times.
For the colony formation assay, plasmid-transfected
cells were plated in 100 mm plate and cultured for 2–3 weeks until
visible colonies were present. Cells were cultured in media
containing fetal bovine serum and G418 (Life Technologies, Inc.) at
a final concentration of 0.6–1 mg/ml. Colonies were stained with
Coomassie Brillant Blue R-250 (CBBR-250) for 1 h. All experiments
were repeated independently at least three times.
For the soft agar colony formation assay, 3000 cells
were plated in each well of a 24-well plate containing 1% base agar
and 0.5% top agar and cultured at 37°C for 2–3 weeks. Colonies were
counted under a dissecting microscope. All experiments were
repeated independently at least three times.
Tumor xenograft model
To establish the tumor xenograft model, we first
generated stable transfected cell lines. Briefly, Focus and WRL-68
HCC cells were transfected with pcDNA3.1B-cote1-Flag and
pSUPER-shRNA2852, respectively, and grown in the presence of G418
(0.6–1 mg/ml) for 2–3 weeks. Visible colonies were picked and
transferred into 96-well plates with DMEM++ medium and G418. Stable
transfected cells in the 96-well plate were digested and
successively transferred into 24-well plates, 6-well plates, and
100-mm plates. Stable cells were injected subcutaneously into both
flanks of nude mice (male BALB/c, 4–6 weeks-old). Tumor volume for
each mouse was determined by measuring in two dimensions and
calculated as: tumor volume = length × (width)2/2. The
tumor tissues were formalin-fixed and paraffin-embedded for
immunohistochemistry.
Immunohistochemical staining
COTE1 expression in tissues of HCC specimens or nude
mouse tumor xenografts was determined by immunohistochemistry.
Briefly, formalin-fixed samples were paraffin-embedded and cut into
4-μm sections. Slides were incubated with goat anti-COTE1
polyclonal antibody (1:50; Santa Cruz Biotechnology) or normal goat
IgG as a negative control at 4°C overnight. For detection,
MaxVision™ HRP-Polymer anti-Goat IHC Kit (Maixin Bio. Ltd., China)
was used according to the manufacturer’s protocol. Stained slides
were observed under a light microscope.
Flow cytometric analysis of cell cycle
and apoptosis
Transfected cells were harvested at different time
points, fixed in cold 70% ethanol, washed, rehydrated in PBS, and
stained with propidium iodide (PI) binding buffer (10 mg/ml RNase A
and 10 μg/ml PI) for 30 min at room temperature. DNA content of the
cells was analyzed using a FACSCalibur flow cytometer
(Becton-Dickinson) with collection of 10,000 events. Analyses of
cell cycle and apoptotic cells (sub-G1 population) were performed
using Becton-Dickinson FACScan.
Terminal deoxyribonucleotide transferase
(TdT)-mediated dUTP nick-end labeling (TUNEL) staining
Cells were fixed in 4% methanol-free formaldehyde
solution in PBS (pH 7.4) for 25 min at 4°C, washed with PBS for 10
min at room temperature, and permeabilized in 0.2% Triton X-100
solution in PBS for further 5 min. After equilibration for 10 min,
the cells were incubated with rTdT (Promega) and observed under a
fluorescence microscope. A nucleus with bright green fluorescent
staining was recorded as a TUNEL-positive event (11).
Western blot analysis
Cell lysates were prepared in cold lysis buffer
containing 25 mmol/l Tris-Cl (pH 7.5), 5 mmol/l EDTA, 1% sodium
dodecyl sulfate, and protease inhibitor cocktail (Sigma). After
boiling for 5 min, samples were separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and transferred onto a
polyvinylidene difluoride membrane, which was blocked in 5%
blocking buffer for 2 h at room temperature. The membrane was
incubated with primary antibody in PBS-Tween-20 (0.1% Tween-20 in
PBS) at 4°C overnight and with secondary antibody at room
temperature for 1 h. Antibodies used in this study were: goat
anti-COTE1 (1:200; Santa Cruz Biotechnology), mouse anti-WWOX
(1:200; Santa Cruz Biotechnology), rabbit anti-WWOX (phospho Y33,
phospho Y287, 1:500; Abcam, UK), mouse anti-flag (1:200; Santa Cruz
Biotechnology), rabbit anti-cyclin D1, E1 (1:500; Abcam), rabbit
anti-p53 (p53 and phospho S46, 1:500; Abcam), rabbit anti-Bcl-2
(1:500; Abcam), rabbit anti-caspase 3, 9 (1:500; Abcam), and
anti-β-actin (1:500; Santa Cruz Biotechnology). Proteins were
detected using the Odyssey Infared Imaging System (Li-COR).
Co-immunoprecipitation
Cells transfected with pcDNA3.1-COTE1-Flag were
resuspended in 1 ml lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl,
1.0% Triton X-100, 1 mM EDTA, and protease inhibitor cocktail).
Immunoprecipitation of lysates was conducted using anti-Flag
antibody (1:100; Santa Cruz Biotechnology), followed by
immunoblotting with antibodies against WWOX (1:200; Santa Cruz
Biotechnology) or COTE1 (1:100; Santa Cruz Biotechnology). Lysate
of cells transfected with empty vector (pcDNA3.1) served as a
control (12).
Statistical analysis
All quantitative data were recorded as means ±
standard deviation (SD). Differences between two groups were
assessed by Student’s t-test (two-tailed) using GraphPad PRISM 5
software. Comparisons among multiple groups were performed by
one-way analysis of variance and least significant difference
t-test. Categorical data were evaluated by the χ2 test.
In all tests, p<0.05 was considered statistically
significant.
Results
Overexpression of COTE1 and its
association with malignancy of HCC
Previous gene microarray analysis performed in our
laboratory showed that COTE1 was upregulated in 11 paired
HCC tissues (data not shown). To confirm these findings, we
performed RT-PCR and quantitative PCR to measure the mRNA
expression level of COTE1 in 80 paired HCC clinical specimens
relative to the levels in corresponding adjacent non-cancerous
liver. The results clearly showed upregulation of COTE1 mRNA
in HCC specimens (Fig. 1A). To
evaluate the protein level of COTE1 in HCC, we performed
immunochemical staining with a specific antibody against COTE1 in
another 80 matched samples. In non-HCC tissues 24/80 (30%) showed
no or weak (+/−) positive staining with the rest showed no
staining, whereas 37/80 (46.25%) of cancer specimens showed
positive staining: seven mildly positive, 17 moderately positive,
and 13 strongly positive (Fig.
1B). We also evaluated the expression pattern of COTE1 in HCC
cell lines by RT-PCR and found that COTE1 was highly expressed in
11 of 15 HCC cells relative to expression in normal human adult
liver tissue (Fig. 1C). Together,
these data showed that COTE1 is upregulated in HCC, confirming the
results of the gene microarray analysis.
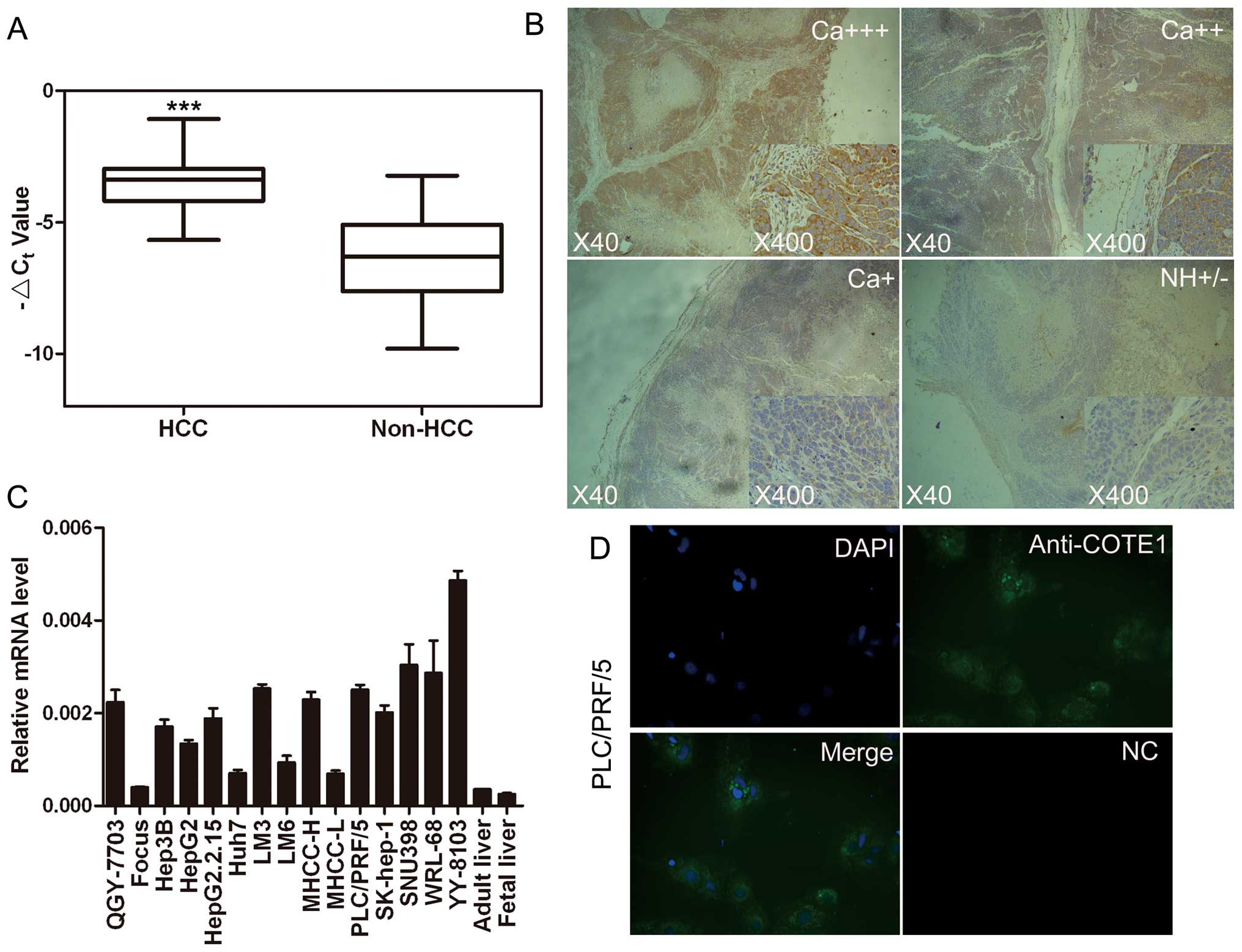 | Figure 1Expression pattern of COTE1 in HCC
tissue specimens and cell lines. (A) Comparison of COTE1 mRNA
expression in 80 paired HCC and non-HCC specimens by real-time PCR.
The line within each box represents the median ΔCt
value; the upper and lower edges of each box represent the 75th and
25th percentile, respectively; the upper and lower bars indicate
the highest and lowest values detected, respectively. For each
sample, the relative COTE1 mRNA level was normalized to that of
β-actin (***p<0.001, Student’s t-test). (B)
Representative immunohistochemical staining of 80 paired HCC
specimens and noncancerous tissue using anti-COTE1 antibody. The
nuclei were counterstained with hematoxylin. The bottom right panel
is enlarged from the center region of the middle image. Original
magnification, ×40 (middle); ×400 (bottom right). Ca+++,
cancerous specimen with strongly positive COTE1 expression;
Ca++, cancerous specimen with moderately positive COTE1
expression; Ca+, cancerous specimen with mildly positive
COTE1 expression; NH, non-HCC tissue with no or weakly positive
expression of COTE1. (C) Expression of COTE1 in 15 HCC cell lines,
healthy adult liver, and fetal liver was evaluated by real-time
PCR, including β-actin as an internal control. (D) Cytoplasmic
localization of endogenous COTE1 (green) in PLC/PRF/5 cells
detected by immunofluorescent staining. The nuclei were stained
with DAPI. Original magnification, ×400. |
To investigate the relationship between COTE1
expression and HCC clinical features, we further analyzed the
results of RT-PCR in the 80 HCC specimens. The clinical
characteristics of the patients and tumors are summarized in
Table II. The prepared specimens
were grouped by gender (male or female), age (≥45 years or <45
years), etiology (HBV+ or HBV−), tumor size
(≥3 cm or <3 cm), metastasis (yes or no) and Edmondson grade
(I–II or III–IV). The resulting data showed that increased
transcriptional expression of COTE1 was statistically correlated
with the pathological tumor size and Edmondson grade (p<0.05).
However, no statistical correlation was found for the other
variables. Taken together, these data indicate that upregulation of
COTE1 contributes to HCC growth and poor differentiation.
| Table IIExpression of COTE1 versus clinical
features. |
Table II
Expression of COTE1 versus clinical
features.
| HCC parameters | No. of
patients | COTE1(+) (%) | COTE1(−) (%) | χ2 | p-value |
|---|
| Gender |
| Male | 69 | 36 (45.0) | 33 (41.25) | 0.021 | 0.884 |
| Female | 11 | 6 (7.5) | 5 (6.25) | | |
| Age (years) |
| ≥45 | 58 | 32 (40.0) | 26 (32.5) | 0.604 | 0.437 |
| <45 | 22 | 10 (12.5) | 12 (15.0) | | |
| Etiology |
|
HBV+ | 67 | 36 (45.0) | 31 (38.75) | 0.251 | 0.617 |
|
HBV− | 13 | 6 (7.5) | 7 (8.75) | | |
| Pathological size
(cm) |
| ≥3 | 25 | 18 (22.5) | 7 (6.75) | 5.545 | 0.019 |
| <3 | 55 | 24 (30.0) | 31 (38.75) | | |
| Metastasis |
| Yes | 36 | 24 (42.11) | 12 (21.05) | 0.132 | 0.716 |
| No | 21 | 13 (22.87) | 8 (14.04) | | |
| Edmondson
grade |
| I–II | 49 | 21 (26.25) | 28 (35.0) | 4.715 | 0.03 |
| III–IV | 31 | 21 (26.25) | 10 (12.5) | | |
COTE1 is localized in the cytoplasm and
cell membrane
To identify the subcellular localization of COTE1 in
HCC cells we performed immunofluorescence staining of endogenous
COTE1 in PLC/PRF/5 cells. The results showed localization of COTE1
predominantly in the cytoplasm and to a lesser extent in the cell
membrane of HCC cells (Fig.
1D).
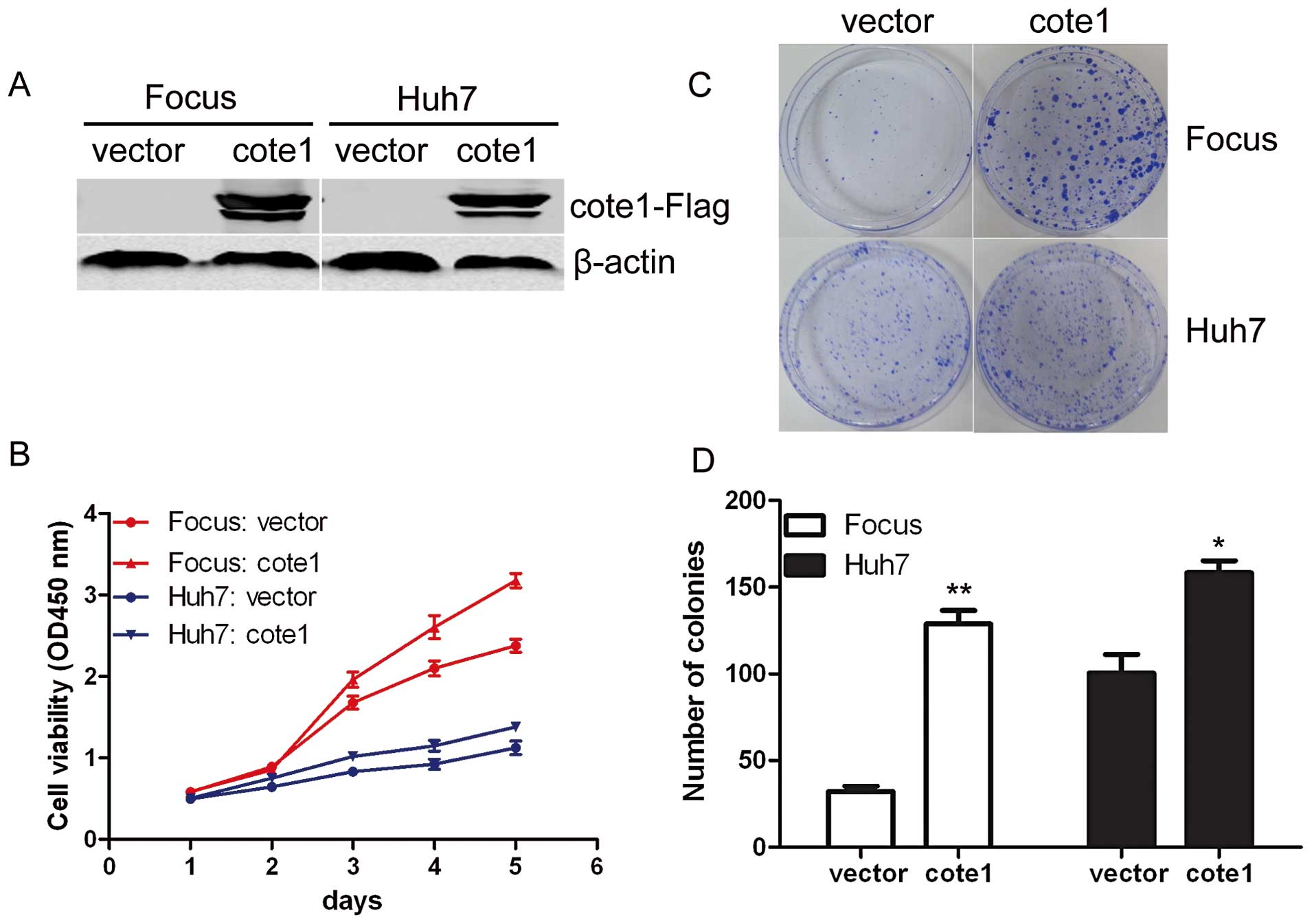
Exogenous COTE1 promotes cellular
proliferation and colony formation
To investigate whether COTE1 contributes to
hepatocarcinogenesis, we investigated the effect of COTE1 on cell
proliferation and colony formation. Based on the expression pattern
of COTE1 in HCC cell lines, recombinant pcDNA3.1-COTE1-Flag was
transiently transfected into Focus, Huh7, MHCC-LM6, and MHCC-L
cells, all of which express a relatively low level of COTE1
(Fig. 1C). The empty vector
pcDNA3.1-Flag was used as a control. Interestingly, cellular growth
of Focus and Huh7 cells was significantly promoted by exogenous
COTE1 compared with cells transfected with empty vector (Fig. 2A and B) but a similar response was
not observed in MHCC-LM6 and MHCC-L. To further investigate the
long-term effect of COTE1 on cell proliferation, transfected Focus
and Huh7 cells were cultured in G418 for 2–3 weeks and colony
formation was measured. Few colonies formed for cells transfected
with vector alone, whereas colonies were more visible and a greater
number of colonies formed in COTE1-overexpressing cells (Fig. 2C and D). These data suggest that
ectopic COTE1 expression selectively enhanced the viability of HCC
cells.
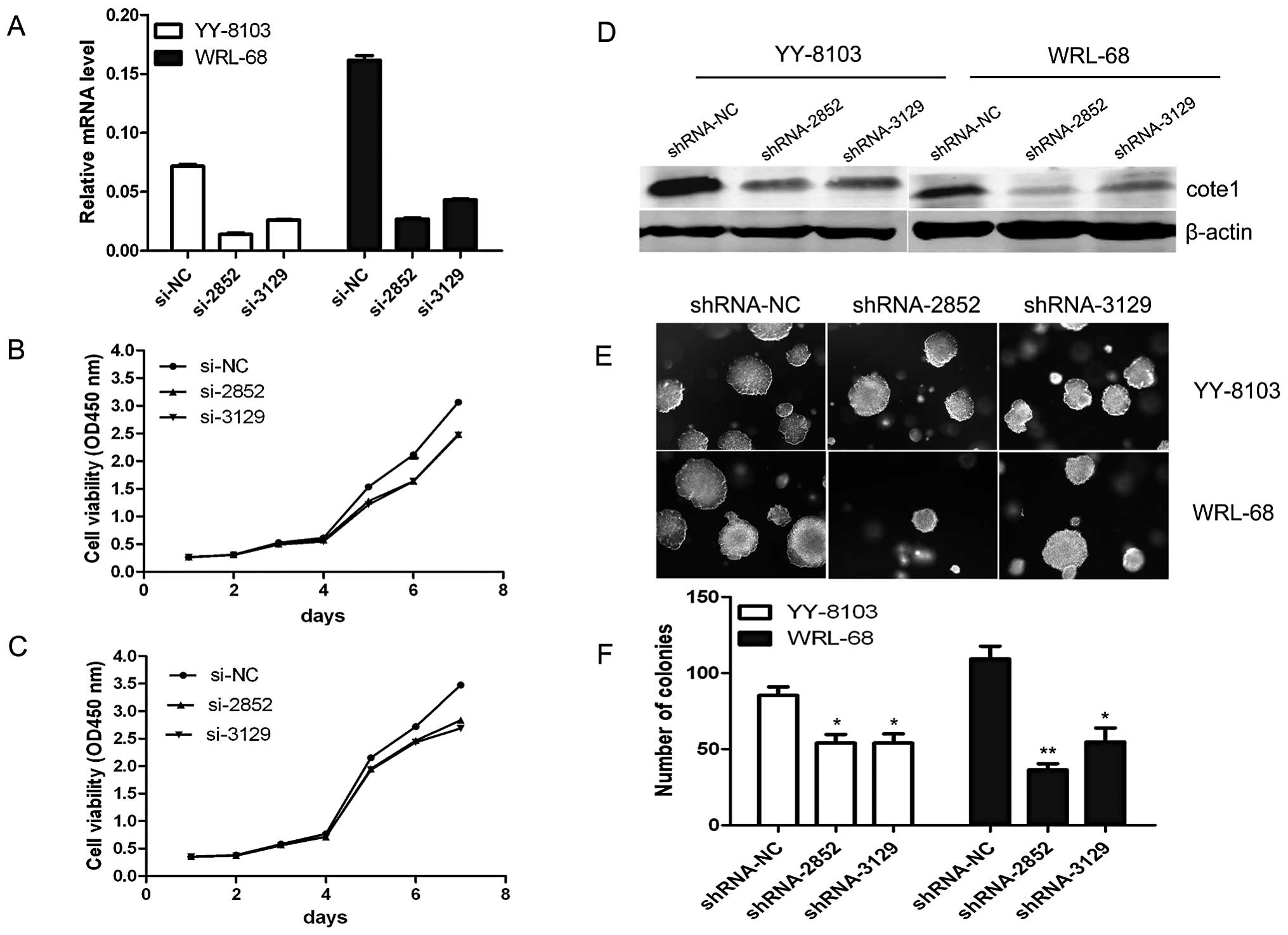
COTE1 knockdown inhibits cell growth and
soft agar colony formation of HCC cells
We further evaluated the effect of COTE1 on cellular
proliferation using YY-8103, WRL-68, PLC, and MHCC-97H cells, which
showed high COTE1 expression levels (Fig. 1C). These cells were transfected
with two chemically synthesized siRNAs that target COTE1,
siRNA-2852 and siRNA-3129. As expected, endogenous COTE1 expression
was efficiently knocked down in all cell lines. Proliferation was
significantly inhibited in YY-8103 and WRL-68 cells transfected
with siRNAs, but not in PLC or MHCC-97H cells (Fig. 3A-C). To further test the effect of
COTE1 on cellular growth, we performed a soft agar colony formation
assay, which more closely imitates in vivo growth. Given the
short half-life of siRNAs, we used shRNAs derived from recombinant
pSUPER vector for these experiments. YY-8103 and WRL-68 cells were
transfected with shRNA and cultured in soft agar for 2–3 weeks. As
expected, cells transfected with shRNA-2852/3129 produced fewer
colonies than those transfected with shRNA-NC plasmid (Fig. 3D-F). These collective data indicate
that endogenous COTE1 plays an essential role in cellular
proliferation and colony formation of HCC cells.
COTE1 contributes to the tumorigenicity
of HCC in vivo
To assess the effect of COTE1 on HCC cell
proliferation in vivo, stable COTE1-transfected Focus or
WRL-68 cells were implanted into one flank of nude mice and
negative control cells were injected into the other flank. As
expected, transfection with pcDNA3.1B-COTE1-Flag markedly enhanced
tumorigenicity of cells compared with the controls (Fig. 4A and D, upper), whereas shRNA2852
suppressed tumorigenicity (Fig. 4B and
D, lower). Immunohistochemical analysis confirmed elevated
COTE1 expression in the tumors formed by COTE1-transfected cells,
and reduced COTE1 expression in tumors formed by cells transfected
with COTE1 shRNA (Fig. 4C).
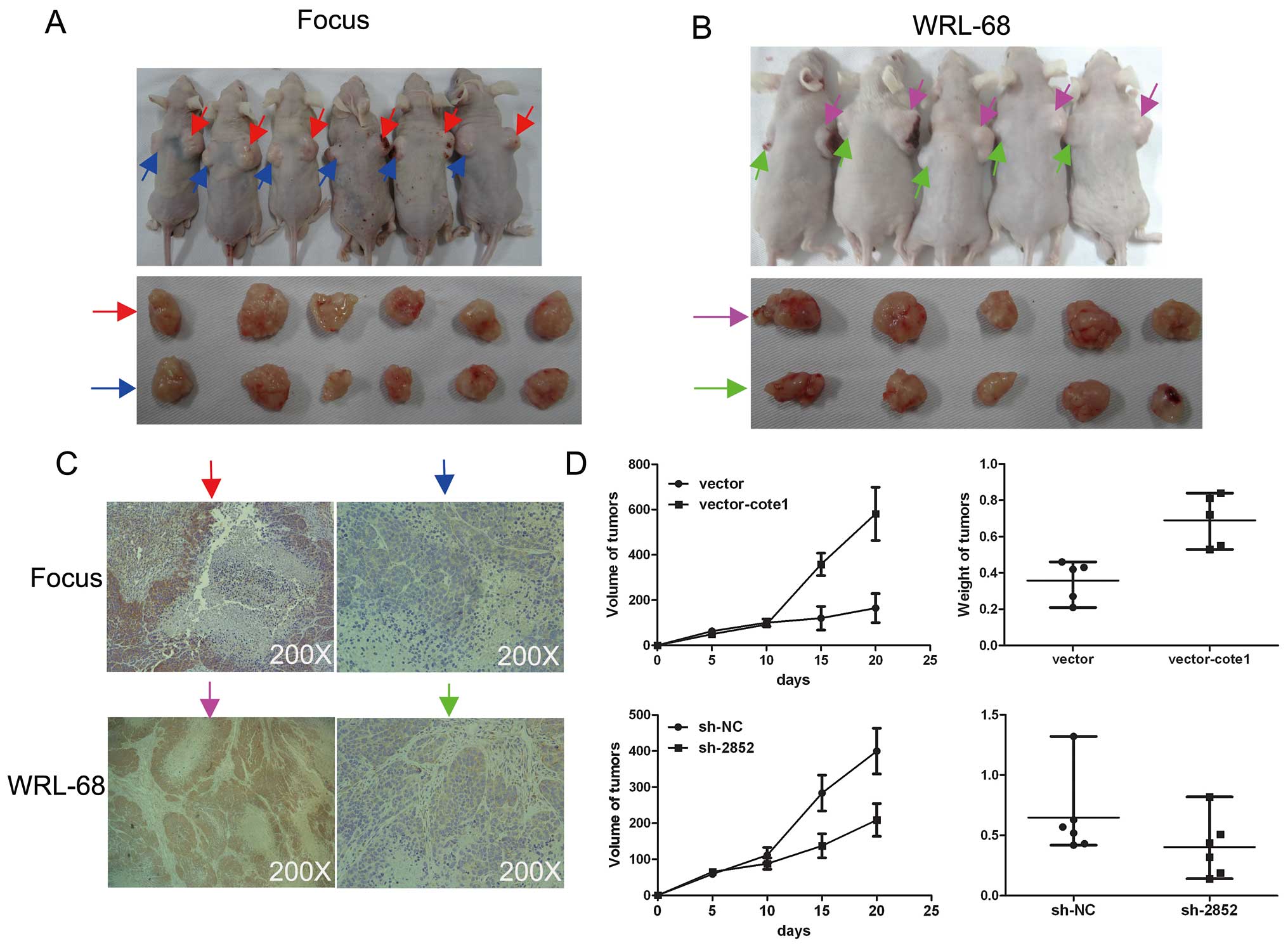 | Figure 4COTE1 promotes tumorigenicity of HCC
cells. (A, B) Two stable transfected cell lines, Focus transfected
with pcDNA3.1B-COTE1-Flag and WRL-68 transfected with shRNA-2852,
were injected subcutaneously into mice. Overexpression of COTE1
enhanced tumorigenicity of Focus cells (A, upper), whereas gene
silencing of COTE1 inhibited tumorigenicity of WRL-68 cells (B,
upper). All xenograft tumors were removed and photographed after
sacrificing the mice (A and B, lower). Red arrows, vector-COTE1;
blue arrows, vector only; pink arrows, shRNA-NC; green arrows,
shRNA-2852. (C) Immunohistochemical staining to verify the
expression levels of COTE1 in xenografts. Focus cell tumors
transfected with pcDNA3.1B-COTE1-Flag expressed high levels of
COTE1, whereas WRL-68 tumors transfected with shRNA-2852 showed low
levels of COTE1 expression. Original magnification, ×200. (D)
Growth of Focus (top left) and WRL-68 (bottom left) tumors was
monitored for 5 days by measuring the diameter (mean ± SD). Tumor
weight of Focus (top right) and WRL-68 (bottom right) tumors was
measured on the 20th day. Each scatter plot shows the weight of a
given xenograft tumor, where the lines represent the median with
interquartile range. |
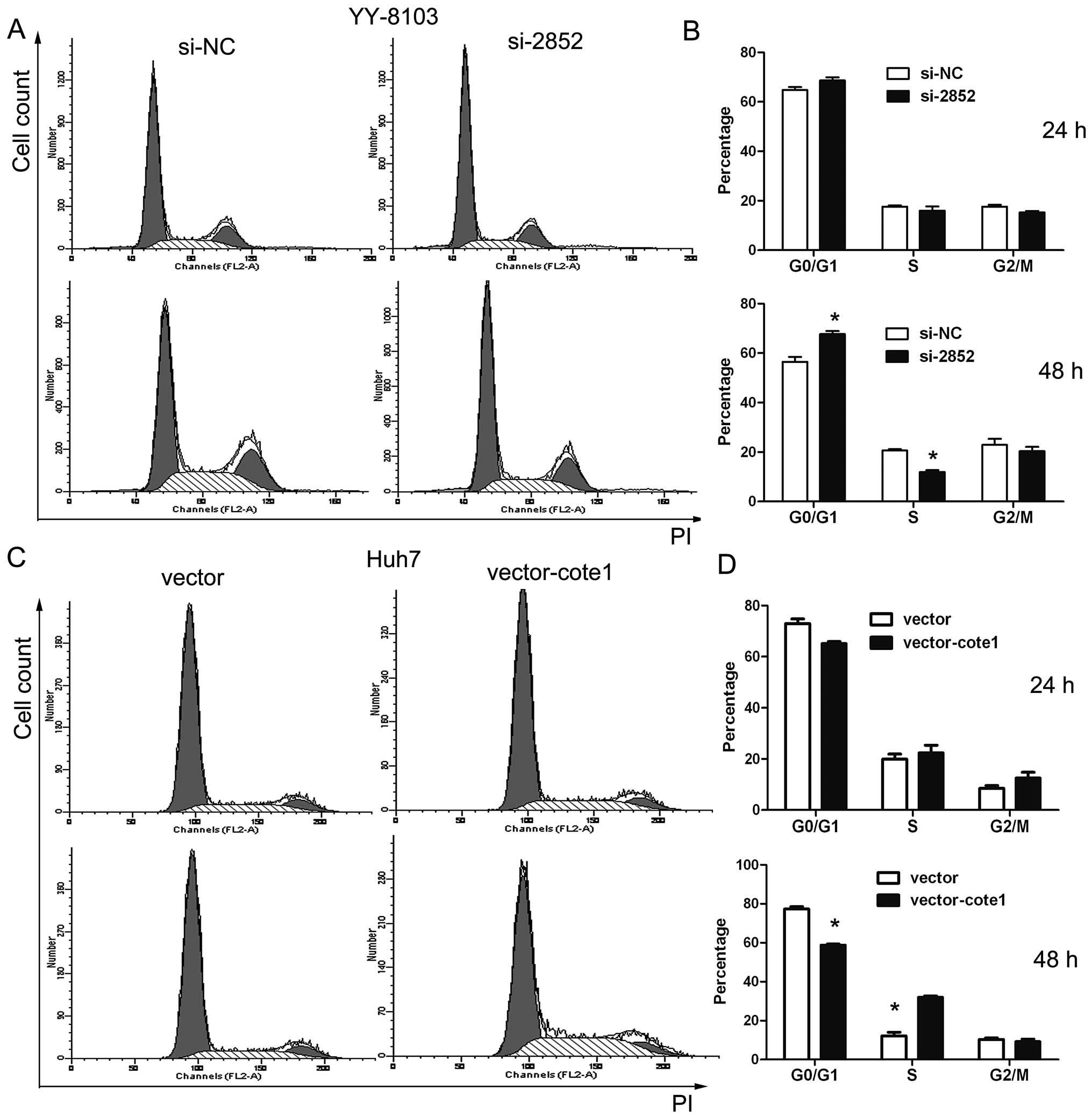
Role of COTE1 in cell cycle regulation
and apoptosis of HCC
We first performed flow cytometry to evaluate cell
cycle distribution of Focus, Huh7, YY-8103, and WRL-68 cells 24 and
48 h after transfection with siRNA2852. G0/G1 phase arrest was
obvious in YY-8103 cells 48 h after transfection (Fig. 5A and B). In contrast, G1- to
S-phase transition was evident in Huh7 cells at 24 h and reached a
peak at 48 h post-transfection (Fig.
5C and D).
We next performed flow cytometry and TUNEL assays to
determine the effect of COTE1 on apoptosis under reduced serum
conditions. First, we investigated the apoptosis of YY-8103 and
WRL-68 cells at different time points after shRNA transfection.
Compared with the control group, COTE1 inhibition resulted in a
larger percentage of sub-G1 cells in WRL-68, but not YY-8103 cells,
reaching a peak at 72 h (Fig. 6A).
To obtain further evidence of apoptosis, we performed the TUNEL
assay in WRL-68 cells at 72 h post-transfection and obtained
results consistent with those of flow cytometry (Fig. 6B and C). The effect of COTE1
upregulation on apoptosis was examined by treatment of
COTE1-overexpressing Focus and Huh7 cells with doxorubicin. As
shown in Fig. 6D and E,
doxorubicin (5 μM, 24 h) treatment resulted in a larger proportion
of apoptotic cells in Focus cells transfected with empty vector
compared with the COTE1 overexpressing cells.
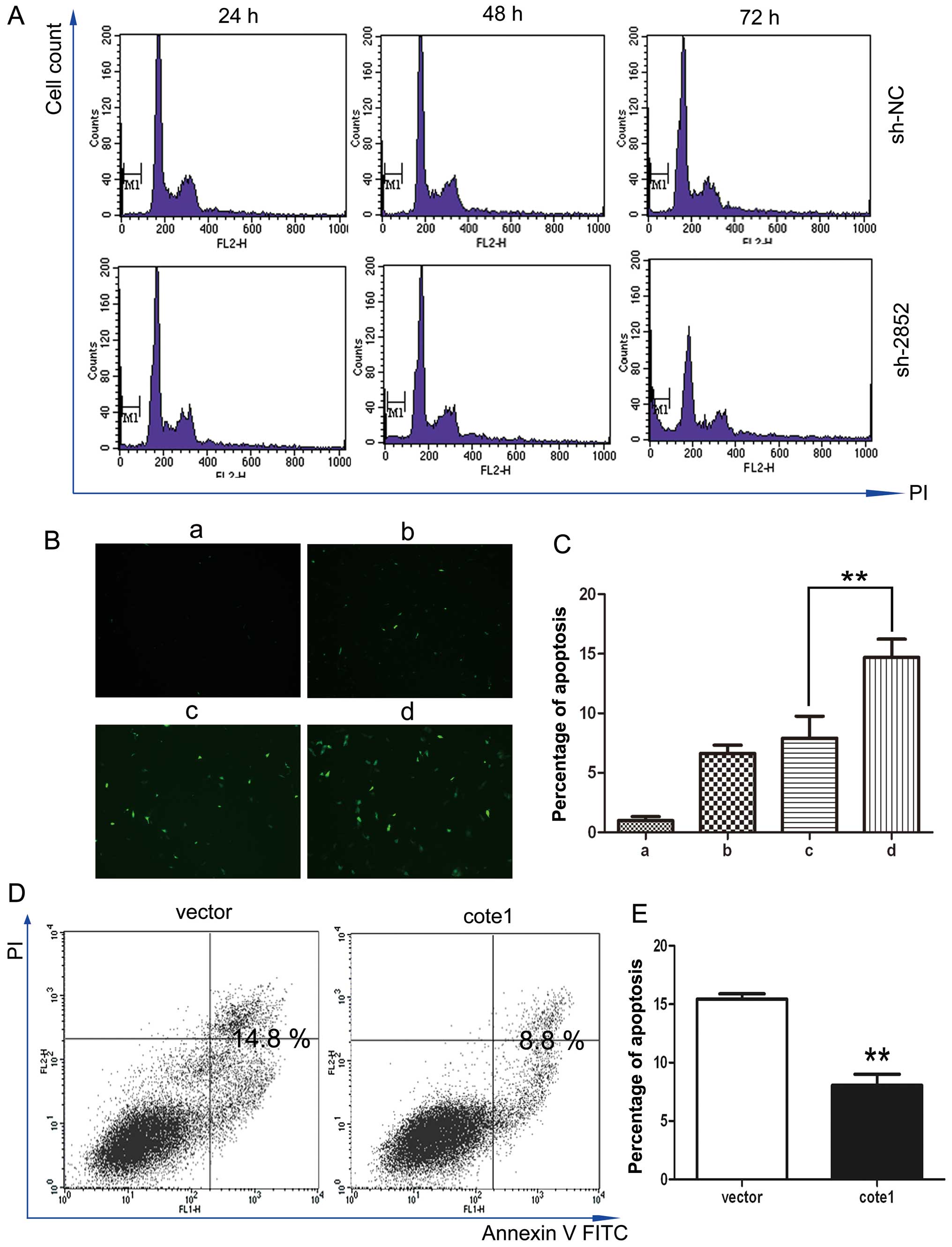 | Figure 6COTE1 mediated apoptosis in HCC
cells. (A, B, and C) Apoptosis in WRL-68 cells transfected with
shRNA at 24, 48 and 72 h post-transfection. Cell apoptosis was
assessed by flow cytometry (A) and TUNEL (B). Compared with the
control group, the sub-diploid peak and the percentage of
TUNEL-positive nuclei were significantly higher in cells
transfected with COTE1-shRNA2852 (E, **p<0.01). a,
null; b, lipofectamine 2000; c, lipofectamine 2000 + shNC; d,
lipofectamine 2000 + sh2852. (D, E) Apoptosis in Focus cells
transfected with vector alone or COTE1 and treated with doxorubicin
(5 μM, 24 h) analyzed by FACS with Annexin V/PI labeling. Data
shown in the histogram (E) are the means of triplicate
determinations (± SD; **p<0.01). |
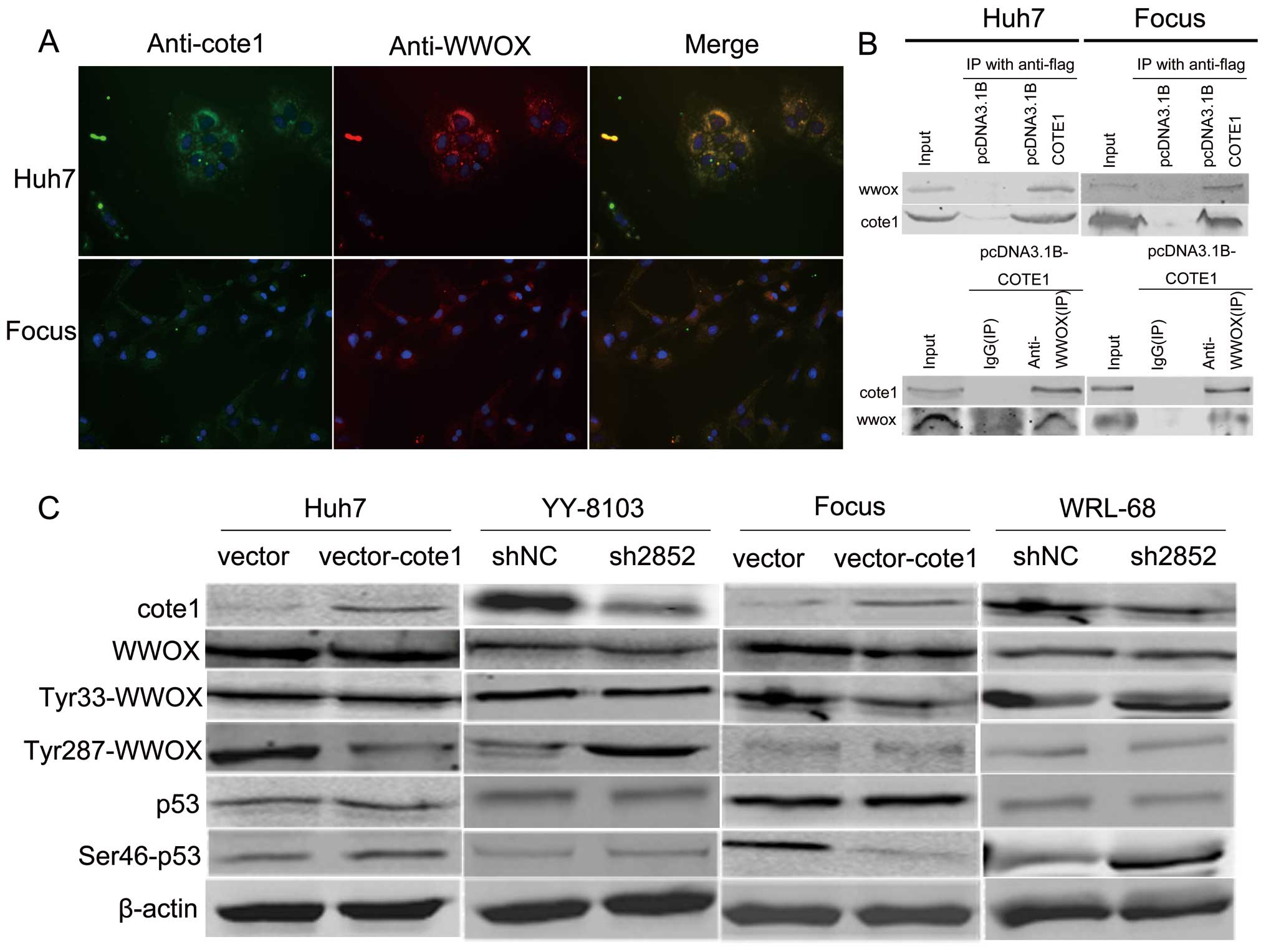
COTE1 regulates the WWOX signaling
pathway via inhibition of tyrosine phosphorylation
The data presented above suggest that COTE1
functions as a novel oncogene in HCC, since COTE1 inhibition
obviously suppresses cell proliferation via cell cycle arrest and
apoptosis. Considering previous reports that COTE1 binds to the WW
domain of WWOX in vitro (7,9), we
investigated whether the effect of COTE1 on cell proliferation was
mediated by WWOX. First, we carried out co-localization experiments
to determine whether COTE1 might interact with WWOX, and observed
COTE1-WWOX co-localization in the cytoplasm of Focus and Huh7 cells
by fluorescence microscopy (Fig.
7A). Next, we performed co-immunoprecipitation (co-IP) assays
to determine whether COTE1 and WWOX physically interact in Focus
and Huh7 cells transfected with pcDNA3.1-COTE1-Flag. The mutual
co-IP data indicated that COTE1 physically associates with WWOX
(Fig. 7B). To explore the effect
of COTE1 on WWOX, we measured tyrosine phosphorylation of WWOX
(Tyr33 and Tyr287) in transfected Focus, Huh7, YY-8103, and WRL-68
cells. Our data showed that the level of pTyr33 was decreased in
Focus cells and increased in WRL-68; whereas the level of pTyr287
was reduced in Huh7 cells and increased in YY-8103 (Fig. 7C). We also measured p-p53 (Ser46),
which participates in WWOX-mediated apoptosis, and found that p-p53
levels were suppressed in Focus cells, and increased in WRL-68
(Fig. 7C).
COTE1-pWWOX mediates mitochondrial
apoptosis and cell cycle regulation
To validate the hypothesis that COTE1-pWWOX mediates
mitochondrial apoptosis in HCC cells, we performed western blot
analysis to measure the expression of molecules involved in the
mitochondrial apoptosis pathway: Bax, Bcl-2, caspase-9, and
caspase-3. Our data showed that upregulation of pWWOX (Tyr33) by
COTE1 knockdown induced p53 (Ser46)-mediated endogenous apoptosis
of WRL-68 cells and, conversely, Tyr33 dephosphorylation of WWOX by
COTE1 overexpression rendered Focus cells resistant to p53
(Ser46)-mediated apoptosis (Fig.
8A).
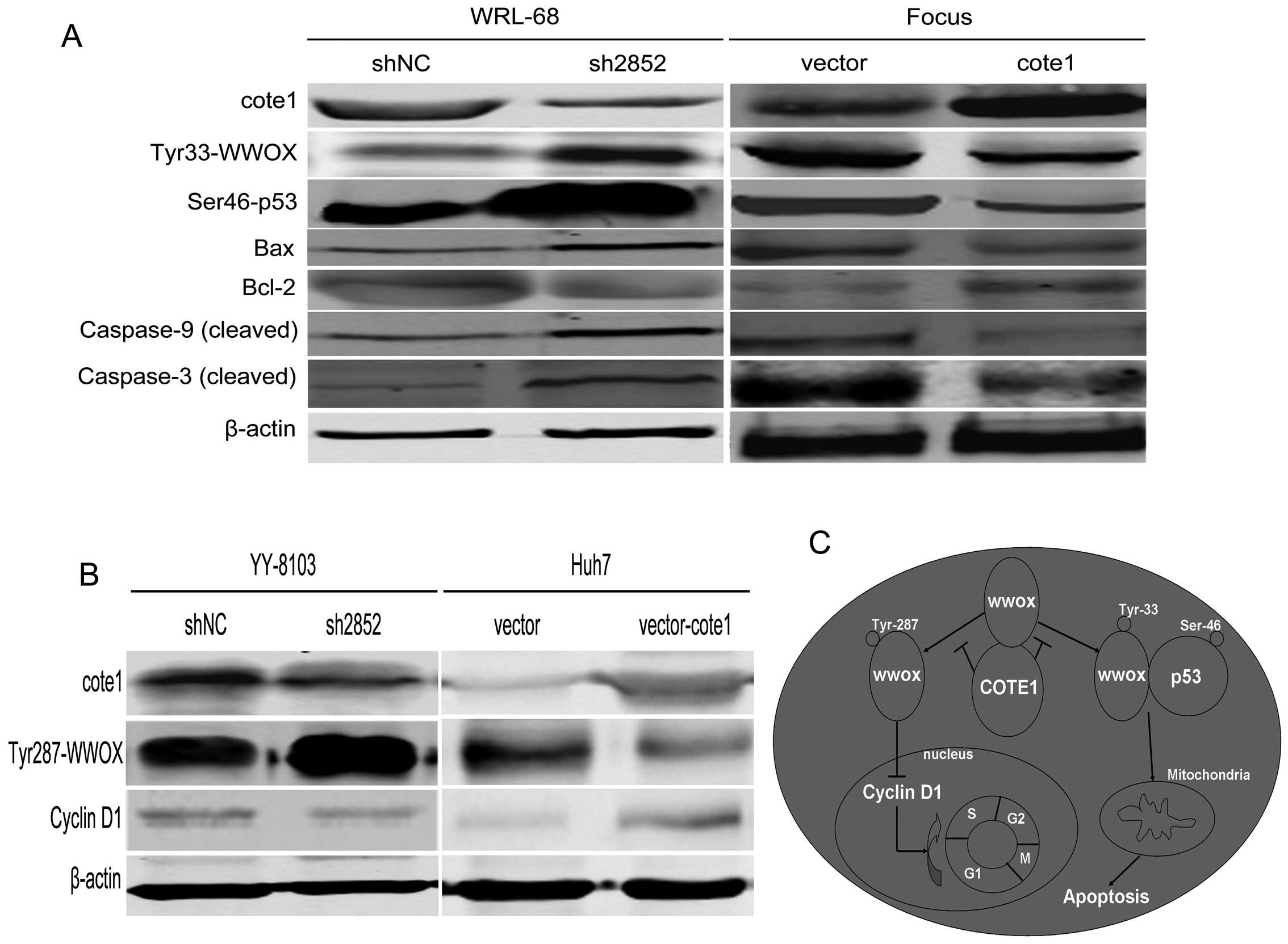 | Figure 8COTE1-pWWOX mediates cell cycle
regulation and mitochondrial apoptosis of HCC cells. (A) Western
blot analysis of endogenous apoptosis-associated downstream
molecules, Bax, Bcl-2, caspase-9, and caspase-3, in COTE1 silenced
WRL-68 cells and COTE1 overexpressing Focus cells. β-actin was used
as a loading control. (B) Western blot analysis of cyclin D1
expression in COTE1 knockdown YY-8103 and COTE1 overexpressing Huh7
cells. β-actin was used as a loading control. (C) A hypothetical
model of the functions of COTE1 in HCC via regulation of the WWOX
signaling pathway. Arrows represent activation, whereas bars
represent inhibition. In physiological conditions, COTE1 inhibits
the tyrosine phosphorylation of WWOX by physically associating with
WWOX, and thus blocks the WWOX signaling pathway. Upregulation of
COTE1 in HCC may contribute to oncogenesis and tumor progression
through multiple mechanisms. On the one hand, dephosphorylation of
Tyr33 of WWOX by COTE1 results in decreased formation of
WWOXTyr33-p53Ser46 complex, which in turn leads to mitochondrial
apoptosis resistance. On the other hand, overexpression of COTE1
causes downregulation of WWOX Tyr287, which may negatively regulate
the expression of cyclin D1 leading to cell cycle progression
through promotion of the G1− to S-phase transition. |
We then measured expression of cyclin D1 and cyclin
E1, both of which function as positive regulators in promoting G1-
to S-phase transition, in Huh7 and YY-8103 cells and found that
expression of cyclin D1, but not cyclin E1, negatively correlated
with COTE1 expression. However, there was a positive correlation
between cyclin D1 and pWWOX (Tyr287) (Fig. 8B). These data suggest that
COTE1-pWWOX (Tyr287)-cyclin D1 mediate cell cycle regulation in
HCC.
In conclusion, the above findings indicate that
COTE1 regulates the WWOX-p53-mediated apoptosis pathway through
Tyr33 dephosphorylation and participates in WWOX-induced cell cycle
progression via Tyr287 dephosphorylation (Fig. 8C).
Discussion
Most cancer cells contain chromosomes that are
broken, truncated, deleted, amplified, or translocated to other
chromosomes. Such chromosomal abnormalities may lead to the
inactivation of tumor suppressor genes or the activation of
oncogenes via amplification (13).
A previous study showed a high incidence of C1q copy number gain in
HCC (60–80%) (14). Many
cancer-related genes that are located at 1q12–q22, such as JTB,
SHC1, CCT3, and COPA, have been shown to be up-regulated in HCC
(15). COTE1, a novel potential
oncogene that was identified by our laboratory, is located at
chromosome 1q 21. Thus, we hypothesized that the COTE1 gene could
be a candidate HCC-specific molecular marker.
The biological functions of COTE1, especially in
cancers, remain unclear. In the present study, we showed that COTE1
was upregulated in HCC cancer tissue compared with adjacent normal
liver tissue, and statistically correlated with tumor size and
differentiation. In addition, high expression of COTE1 was observed
in HCC cell lines. These findings implied that COTE1 could function
as an oncogene in HCC. We also showed that overexpression of COTE1
promoted proliferation of Focus and Huh7 cells in vivo and
in vitro. In contrast, gene silencing of COTE1 reduced cell
viability of YY-8103 and WRL-68 cells in vivo and in
vitro. Together, these data suggest that COTE1 does indeed play
an important role in HCC neoplasia. We subsequently revealed that
knockdown of COTE1 induced cell cycle arrest in YY-8103 cells and
apoptosis in WRL-68 cells. These data indicate that the oncogenic
role of COTE1 in HCC is potentially mediated through regulation of
the cell cycle and apoptosis.
COTE1 appears to participate in apoptosis regulation
by direct physical association with the tumor suppressor WWOX and
modulation of WWOX tyrosine phosphorylation. The WW domains of WWOX
interact with a growing list of interesting proteins (16–18);
for example, WWOX participates in TRADD (TNF receptor-associated
death domain protein)-mediated cell death and mitochondrial
apoptosis (19–23). WWOX is inactivated in a range of
tumor cells, and its decreased activity correlates with the
malignancy of human HCC and other tumors (24–28).
Phosphorylation of WWOX at Tyr33 and subsequent phosphorylation of
other focal apoptosis complex-associated proteins, such as p53, are
required for mitochondrial apoptosis (29,30).
WWOX is typically localized in the mitochondria, nucleus, and Golgi
(31,32), and is released from mitochondria
during the mitochondrial membrane permeability transition, when it
translocates to the nucleus and cooperates with p53 to mediate
apoptosis (29,30,33).
Surprisingly, our data indicated co-localization and
co-immunoprecipitation of COTE1 and WWOX in HCC cells. We also
found that COTE1 knockdown via RNAi stimulated the WWOX tyrosine
phosphorylation cascade and apoptosis-associated downstream
signaling pathways. This suggests that COTE1 contributes to HCC
tumorigenesis by regulating the WWOX-p53-mediated endogenous
apoptosis pathway through tyrosine-33 dephosphorylation of
WWOX.
WWOX is also known to play an important role in the
regulation of cell cycle progression (29,34,35).
It was previously shown that WWOX inhibits cell cycle progression
(29) and that its expression
levels are negatively correlated with the expression of cyclin D1
and E1 (35). Cyclin D1 and E1 are
well-documented important regulators that promote the G1- to
S-phase transition of the cell cycle and function as oncogenes in
many cancers, including HCC (36–40).
Here, we confirmed the negative correlation between COTE1 and
cyclin D1 expression in Huh7 and YY-8103 cells, and further
demonstrated that Tyr287 phosphorylation of WWOX is modulated by
COTE1 in Huh7 and YY-8103 cells. However, no change in WWOX
expression was detected. We speculate that phosphorylation of WWOX
at Tyr287 modulates cell cycle progression by regulating the
expression of cyclin D1. In addition, WWOX could be activated by
phosphorylation of other sites, including Tyr61, Tyr293, and Ser14
residues (29,30,41),
that were not investigated in this study. Furthermore, the key
domain of COTE1 responsible for tyrosine phosphorylation of WWOX
remains unknown, and is worthy of in-depth research. The diverse
ways in which COTE1 appears to contribute to HCC proliferation via
different pathways encourages us to further explore the role of
COTE1 in other HCC cell lines in future studies.
In conclusion, COTE1 contributes to neoplasia of HCC
via WWOX-p53-mediated apoptosis and WWOX-cyclin D1 associated cell
cycle delay regulation (Fig. 8C).
Based on these findings, COTE1 may represent a new target for HCC
gene therapy.
Acknowledgments
The authors thank Qing Deng for kindly providing us
with the pSUPER-GFP and pcDNA3.1B-Flag-hrGFP plasmids and Qun Wang
for her excellent technical assistance. This work was supported by
the National Natural Science Foundation, China (grant no.
81170415).
References
|
1
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar
|
|
2
|
Wang JS, Huang T, Su J, et al:
Hepatocellular carcinoma and aflatoxin exposure in Zhuqing Village,
Fusui County, People’s Republic of China. Cancer Epidemiol
Biomarkers Prev. 10:143–146. 2001.PubMed/NCBI
|
|
3
|
Coleman WB: Mechanisms of human
hepatocarcinogenesis. Curr Mol Med. 3:573–588. 2003. View Article : Google Scholar
|
|
4
|
Aravalli RN, Steer CJ and Cressman EN:
Molecular mechanisms of hepatocellular carcinoma. Hepatology.
48:2047–2063. 2008. View Article : Google Scholar
|
|
5
|
Yu W, Andersson B, Worley KC, et al:
Large-scale concatenation cDNA sequencing. Genome Res. 7:353–358.
1997.PubMed/NCBI
|
|
6
|
Winfield SL, Tayebi N, Martin BM, Ginns EI
and Sidransky E: Identification of three additional genes
contiguous to the glucocerebrosidase locus on chromosome 1q21:
implications for Gaucher disease. Genome Res. 7:1020–1026.
1997.PubMed/NCBI
|
|
7
|
Ludes-Meyers JH, Kil H, Bednarek AK, Drake
J, Bedford MT and Aldaz CM: WWOX binds the specific proline-rich
ligand PPXY: identification of candidate interacting proteins.
Oncogene. 23:5049–5055. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kallin A, Johannessen LE, Cani PD, et al:
SREBP-1 regulates the expression of heme oxygenase 1 and the
phosphatidylinositol-3 kinase regulatory subunit p55γ. J Lipid Res.
48:1628–1636. 2007.PubMed/NCBI
|
|
9
|
Abdeen SK, Salah Z, Maly B, et al: Wwox
inactivation enhances mammary tumorigenesis. Oncogene.
30:3900–3906. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang H, Huang CJ, Tian Y, Wang YP, Han ZG
and Li XC: Ectopic overexpression of COTE1 promotes cellular
invasion of hepatocellular carcinoma. Asian Pac J Cancer Prev.
13:5799–5804. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ren F, Wu H, Lei Y, et al: Quantitative
proteomics identification of phosphoglycerate mutase 1 as a novel
therapeutic target in hepatocellular carcinoma. Mol Cancer.
9:812010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang J, Zheng DL, Qin FS, et al: Genetic
and epigenetic silencing of SCARA5 may contribute to human
hepatocellular carcinoma by activating FAK signaling. J Clin
Invest. 120:223–241. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim TM, Yim SH, Shin SH, et al: Clinical
implication of recurrent copy number alterations in hepatocellular
carcinoma and putative oncogenes in recurrent gains on 1q. Int J
Cancer. 123:2808–2815. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wong N, Lam WC, Lai PB, Pang E, Lau WY and
Johnson PJ: Hypomethylation of chromosome 1 heterochromatin DNA
correlates with q-arm copy gain in human hepatocellular carcinoma.
Am J Pathol. 159:465–471. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wong N, Chan A, Lee SW, et al: Positional
mapping for amplified DNA sequences on 1q21–q22 in hepatocellular
carcinoma indicates candidate genes overexpression. J Hepatol.
38:298–306. 2003.PubMed/NCBI
|
|
16
|
Aqeilan RI, Donati V, Gaudio E, et al:
Association of Wwox with ErbB4 in breast cancer. Cancer Res.
67:9330–9336. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Del Mare S, Salah Z and Aqeilan RI: WWOX:
its genomics, partners, and functions. J Cell Biochem. 108:737–745.
2009.PubMed/NCBI
|
|
18
|
Kurek KC, Del Mare S, Salah Z, et al:
Frequent attenuation of the WWOX tumor suppressor in osteosarcoma
is associated with increased tumorigenicity and aberrant RUNX2
expression. Cancer Res. 70:5577–5586. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chang NS, Pratt N, Heath J, Schultz L,
Sleve D, Carey GB and Zevotek N: Hyaluronidase induction of a WW
domain-containing oxidoreductase that enhances tumor necrosis
factor cytotoxicity. J Biol Chem. 276:3361–3370. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Aqeilan RI, Donati V, Palamarchuk A,
Trapasso F, et al: WW domain-containing proteins, WWOX and YAP,
compete for interaction with ErbB-4 and modulate its
transcriptional function. Cancer Res. 65:6764–6772. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aderca I, Moser CD, Veerasamy M, et al:
The JNK inhibitor SP600129 enhances apoptosis of HCC cells induced
by the tumor suppressor WWOX. J Hepatol. 49:373–383. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hong Q, Sze CI, Lin SR, et al: Complement
C1q activates tumor suppressor WWOX to induce apoptosis in prostate
cancer cells. PLoS One. 4:e57552009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Teng CC, Yang YT, Chen YC, Kuo YM and Sze
CI: Role of WWOX/WOX1 in Alzheimer’s disease pathology and in cell
death signaling. Front Biosci (Elite Ed). 4:1951–1965. 2012.
|
|
24
|
Park SW, Ludes-Meyers J, Zimonjic DB,
Durkin ME, Popescu NC and Aldaz CM: Frequent downregulation and
loss of WWOX gene expression in human hepatocellular carcinoma. Br
J Cancer. 91:753–759. 2004.PubMed/NCBI
|
|
25
|
Nunez MI, Ludes-Meyers J, Abba MC, et al:
Frequent loss of WWOX expression in breast cancer: correlation with
estrogen receptor status. Breast Cancer Res Treat. 89:99–105. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Aqeilan RI and Croce CM: WWOX in
biological control and tumorigenesis. J Cell Physiol. 212:307–310.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ramos D, Abba M, López-Guerrero JA, et al:
Low levels of WWOX protein immunoexpression correlate with tumour
grade and a less favourable outcome in patients with urinary
bladder tumours. Histopathology. 52:831–839. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gourley C, Paige AJ, Taylor KJ, et al:
WWOX gene expression abolishes ovarian cancer tumorigenicity in
vivo and decreases attachment to fibronectin via integrin alpha3.
Cancer Res. 69:4835–4842. 2009. View Article : Google Scholar
|
|
29
|
Chang NS, Doherty J and Ensign A: JNK1
physically interacts with WW domain-containing oxidoreductase
(WOX1) and inhibits WOX1-mediated apoptosis. J Biol Chem.
278:9195–9202. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen ST, Chuang JI, Cheng CL, Hsu LJ and
Chang NS: Light-induced retinal damage involves tyrosine33
phosphorylation, mitochondrial and nuclear translocation of WW
domain-containing oxidoreductase in vivo. Neuroscience.
130:397–407. 2005. View Article : Google Scholar
|
|
31
|
Yang G, Zhang G, Pittelkow MR, Ramoni M
and Tsao H: Expression profiling of UVB response in melanocytes
identifies a set of p53-target genes. J Invest Dermatol.
126:2490–2506. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gaudio E, Palamarchuk A, Palumbo T,
Trapasso F, Pekarsky Y, Croce CM and Aqeilan RI: Physical
association with WWOX suppresses c-Jun transcriptional activity.
Cancer Res. 66:11585–11589. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chang NS, Doherty J, Ensign A, Schultz L,
Hsu LJ and Hong Q: WOX1 is essential for tumor necrosis factor-, UV
light-, staurosporine-, and p53-mediated cell death, and its
tyrosine 33-phosphorylated form binds and stabilizes serine
46-phosphorylated p53. J Biol Chem. 280:43100–43108. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Salah Z, Aqeilan R and Huebner K: WWOX
gene and gene product: tumor suppression through specific protein
interactions. Future Oncol. 6:249–259. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Płuciennik E, Nowakowska M, Wujcicka WI,
Sitkiewicz A, Kazanowska B, Zielińska E and Bednarek AK: Genetic
alterations of WWOX in Wilms’ tumor are involved in its
carcinogenesis. Oncol Rep. 28:1417–1422. 2012.PubMed/NCBI
|
|
36
|
Nishida N, Fukuda Y, Komeda T, et al:
Amplification and overexpression of the cyclin D1 gene in
aggressive human hepatocellular carcinoma. Cancer Res.
54:3107–3110. 1994.PubMed/NCBI
|
|
37
|
Deane NG, Parker MA, Aramandla R, et al:
Hepatocellular carcinoma results from chronic cyclin D1
overexpression in transgenic mice. Cancer Res. 61:5389–5395.
2001.PubMed/NCBI
|
|
38
|
Tashiro E, Tsuchiya A and Imoto M:
Functions of cyclin D1 as an oncogene and regulation of cyclin D1
expression. Cancer Sci. 98:629–635. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Geng Y, Yu Q, Sicinska E, Das M, et al:
Cyclin E ablation in the mouse. Cell. 114:431–443. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hwang HC and Clurman BE: Cyclin E in
normal and neoplastic cell cycles. Oncogene. 24:2776–2786. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chang JY, He RY, Lin HP, et al: Signaling
from membrane receptors to tumor suppressor WW domain-containing
oxidoreductase. Exp Biol Med. 235:796–804. 2010. View Article : Google Scholar
|