Introduction
The potential for enhanced therapeutic responses to
combination DNMTi and HDACi EGT is predicated on evidence from
basic molecular studies identifying interaction of cellular
methylation and acetylation machinery in the transcriptional
regulation of gene expression (1,2).
Subsequent in vitro and in vivo studies, particularly
involving hematological malignancies, have identified enhanced
regulation of gene expression and responses to combination EGT
lending support to basic molecular studies (3–8).
Clinical studies evaluating the efficacy of
combination EGT have, in part, contributed to establishing the
molecular mechanisms for the observed therapeutic effects of EGT
although have failed to consistently demonstrate changes in early
global and gene specific methylation or acetylation status
correlate with clinical effect (9–12).
Given the duration of EGT either alone e.g., AZA or in combination
with HDACi frequently requires several cycles to determine evidence
of clinical benefit a significant need exists for identification of
molecular markers for determination of early response to
(combination) EGT in order to facilitate accurate selection of
patients for continued epigenetic treatment, avoidance of
unnecessary cost and exposure to side effects in patients unlikely
to respond, and to avert delays in provision of more appropriate
treatments where possible.
Our previous in vitro studies and recent
clinical report investigating the effects of combination AZA and
HDACi EGT identifies modulation of expression of genes implicated
in the pathogenesis of MDS and AML (8,13,14).
We sought to characterize the in vitro expression of genes
implicated in the pathogenesis of MDS/AML and potentially involved
in the transcriptional response to the specific combination of AZA
and LBH589 EGT in HL-60 cells. In addition we evaluated the
temporal expression of genes identified as being regulated from our
in vitro studies in peripheral blood mononuclear cells
(PBMCs) from trial patients with high-risk MDS and AML receiving
combination AZA and LBH589 EGT and correlated these findings with
subsequent clinical responses, predominately observed after a
minimum of 3 cycles of therapy (14), with a view to identification of
potential early molecular markers of combination EGT treatment
response.
Materials and methods
Cell culture
HL-60 cells were cultured in RPMI-1640 (Gibco BRL)
containing 10% heat inactivated fetal calf serum and kept in a 5%
CO2 incubator at 37°C. Agents: azacitidine (AZA), the
kind gift of Celgene, Australia and panobinostat (LBH589), the kind
gift of Novartis, Australia, were added to plates for 48 h. AZA was
dissolved in H2O with 0.2% acetic acid and used at a
final concentration of 1.0 μM. LBH589 was dissolved in PBS with 1%
DMSO and used at a final concentration of 20 nM.
Growth inhibition assay
HL-60 cells in log phase were plated at a density of
0.2×106 in 10 ml of medium. Cells were harvested at 48
h. Cell viability was assessed using 0.4% trypan blue staining
immediately after culture. Black staining cells were considered as
non-viable cells, and unstained bright cells as viable. All
experiments were repeated 3 times with averages displayed
graphically.
Normal healthy control and clinical trial
patient samples RNA extraction
TRIzol was used to extract RNA from PBMC fractions
from healthy donors (n=7) or day 25 samples from patients in a
recently completed phase Ib/II clinical trial (14). The clinical trial consisted of a
5-day schedule of AZA followed by LBH589 in high-risk MDS or AML.
AZA 75 mg/m2 was injected subcutaneously on days 1–5.
LBH589 was administered orally 3 times a week (Mon/Wed/Fri)
starting on day 5 for 7 doses of each 28-day cycle. Between January
2010 and January 2012, 40 evaluable patients were enrolled. There
were 30 patients with AML and 10 patients with high-risk MDS, 23
patients (Table I) had data
enabling evaluation of molecular marker expression and correlation
with treatment response determined either 1, 3 or 6 months after
treatment commencement. Treatment responses were defined according
to International Working Group criteria for AML and MDS (15).
 | Table IClinical and molecular response
correlation. |
Table I
Clinical and molecular response
correlation.
| Patient | Age/sex | Diagnosis | Best response | NUR77 mRNA
inductiona | p21 mRNA
inductionb |
|---|
| 1 | 68 M | MDS | Marrow CR | Yes | NA |
| 2 | 70 M | AML | PR | Yes | Yes |
| 3 | 72 F | MDS | CR | Yes | No |
| 4 | 61 M | MDS | Resistant | No | NA |
| 6 | 63 F | MDS | CR | Yes | NA |
| 7 | 58 M | AML | Resistant | No | No |
| 8 | 75 M | AML | Resistant | No | No |
| 9 | 72 M | AML | CR | No | Yes |
| 11 | 73 M | AML | Resistant | No | NA |
| 12 | 67 F | MDS | PR | Yes | NA |
| 13 | 79 M | MDS | SD | Yes | NA |
| 16 | 72 M | MDS | Resistant | No | No |
| 17 | 60 F | AML | Resistant | No | Yes |
| 18 | 67 F | MDS | PR | Yes | Yes |
| 19 | 80 M | AML | Resistant | No | Yes |
| 22 | 78 M | AML | CR | Yes | Yes |
| 23 | 69 M | AML | Resistant | No | No |
| 24 | 72 M | AML | PR | No | Yes |
| 26 | 75 M | MDS | PD | Yes | No |
| 28 | 69 M | AML | PR | No | Yes |
| 31 | 56 M | AML | Resistant | No | No |
| 33 | 62 F | MDS | Marrow CR | Yes | No |
| 34 | 70 F | AML | Resistant | Yes | No |
PCR
Semi-quantitative reverse
transcription-PCR (RT-PCR)
RT-PCR was performed on total TRIzol extracted RNA
from HL-60 cells untreated or treated for 48 h with AZA or LBH589
or a combination of AZA and LBH589. The primers used for
p21WAF/CIP1 were: forward, 5′-ATT AGC AGC GGA ACA AGG
AGT CAG CAT-3′; and reverse, 5′-CTG TGA AAG ACA CAG AAC AGT ACA GGG
T-3′. The primers used for Bcl-xL were: forward, 5′-TTG GAC AAT GGA
CTG GTT GA-3′; and reverse, 5′-GTA GAG TGG ATG GTC AGT G-3′. The
primers used for caspase-3 were: forward, 5′-GCA GCA AAC CTC AGG
GAA AC-3′; and reverse, 5′-TGT CGGCAT ACT GTT TCA GCA-3′. The human
β-actin gene was used as an internal control. The forward primer
for β-actin was 5′-GAC AGG ATG CAG AAG GAG ATT ACT-3′ and the
reverse primer was 5′-TGA TCC ACA TCT GCT GGA AGG T-3′.
Real-time PCR
Reaction volumes of 20 μl contained SYBR Green 1
buffer and forward and reverse primers for target genes. The
primers used for Bcl-xL were: forward, 5′-GGC TGG GAT ACT TTT GTG
GA-3′; and reverse, 5′-GTA GAG TGG ATG GTC AGT G-3′. The primers
used for caspase-3 were: forward, 5′-CAG TGG AGG CCG ACT TCT TG-3′;
and reverse, 5′-TGT CGG CAT ACT GTT TCA GCA-3′. The primers used
for p15INK4B were: forward, 5′-AGT CAA CCG TTT CGG GAG
GC-3′; and reverse, 5′-ACC ACC AGC GTG TCC AGG AAG-3′. The primers
used for p21WAF/CIP1 were: forward, 5′-TGG ACC TGT CAC
TGT CTT GT-3′; and reverse, 5′-TCC TGT GGG CGG ATT AG-3′. The
primers used for Nor-1 were: forward, 5′-GTC CTC AGA CTT TCC
ATC AGG T-3′; and reverse, 5′-GAT CAG TAA ATC CCG GAA TCC-3′. The
primers used for NUR77 were: forward, 5′-GCT GCA GAA TGA CTC
CAC C-3′; and reverse, 5′-ACA GCA GCA CTG GGC TTA-3′. The primers
used for β-actin were forward, 5′-GAC AGG ATG CAG AAG GAG ATT
ACT-3′; and reverse, 5′-TGA TCC ACA TCT GCT GGA AGG T-3′. Each PCR
run also included wells of no template control (NTC). A melting
point dissociation curve generated by the instrument was used to
confirm that only a single product was present. The fluorescence
data were quantitated using the threshold cycle (Ct) value. Data
were normalized to β-actin and presented as the mean-fold change.
PCR of each patient sample was completed a minimum of two times to
ensure consistency (8).
Statistical methods
The effects of AZA and LBH589 alone and in
combination on HL-60 cell growth, p15INK4B,
p21WAF1/CIP1, caspase-3, Bcl-xL, NUR77 and Nor-1
mRNA expression were assessed by Student’s t-test and analysis of
variance (ANOVA). Data were expressed as means ± SEM and p<0.05
was considered statistically significant.
Results
AZA and LBH589 alone and in combination
inhibits HL-60 cell growth
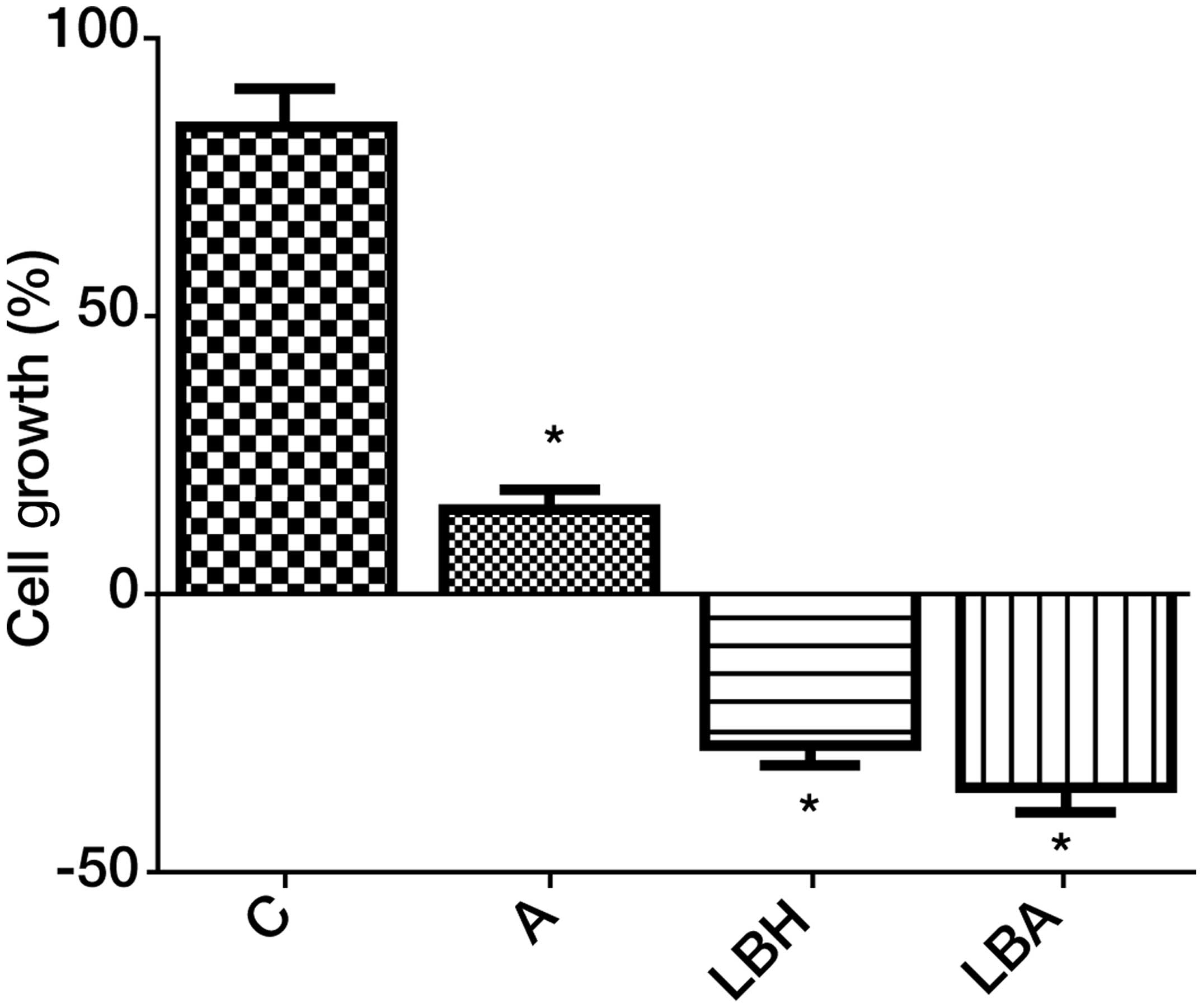
We examined the effects of AZA and LBH589 alone or
in combination on the growth of the human leukemia cell line HL-60.
Cells were exposed to AZA (1.0 μM) and/or LBH589 (20 nM) for ≤48 h.
Treatment doses were selected based on inhibition of cell growth
concentrations identified in previous in vitro studies
(3,4,7). AZA
or LBH589 treatment alone significantly inhibited HL-60 cell growth
in vitro with LBH589 being the more potent agent. LBH589 in
combination with AZA demonstrated a non-significant increase in
inhibition of cell growth over AZA or LBH589 treatment alone
(Fig. 1).
Effect of AZA and LBH589 on expression of
Bcl-xL, caspase-3, Nor-1, NUR77, p15INK4B and
p21WAF1/CIP1 mRNA in HL-60 cells
As we have previously identified a combination of
other hydroxamate HDACi’s and AZA as producing modulation of
several genes implicated in the pathogenesis of MDS including the
putative tumor suppressor gene NUR77 (8,13,16)
we were interested in determining the effect of the hydroxamate
HDACi LBH589 in combination with AZA on gene expression in HL-60
cells. LBH589 or AZA alone increased p21WAF1/CIP1 and
caspase-3 expression with LBH589 being the more potent agent
(Fig. 2A). The combination of AZA
with LBH589 demonstrated a non-significant increase in
p21WAF1/CIP expression over single agent treatment
(Fig. 2A). LBH589 or AZA alone
significantly decreased expression of Bcl-xL expression and the
combination of AZA with LBH589 significantly decreased Bcl-xL
expression over single agent treatment (Fig. 2A).
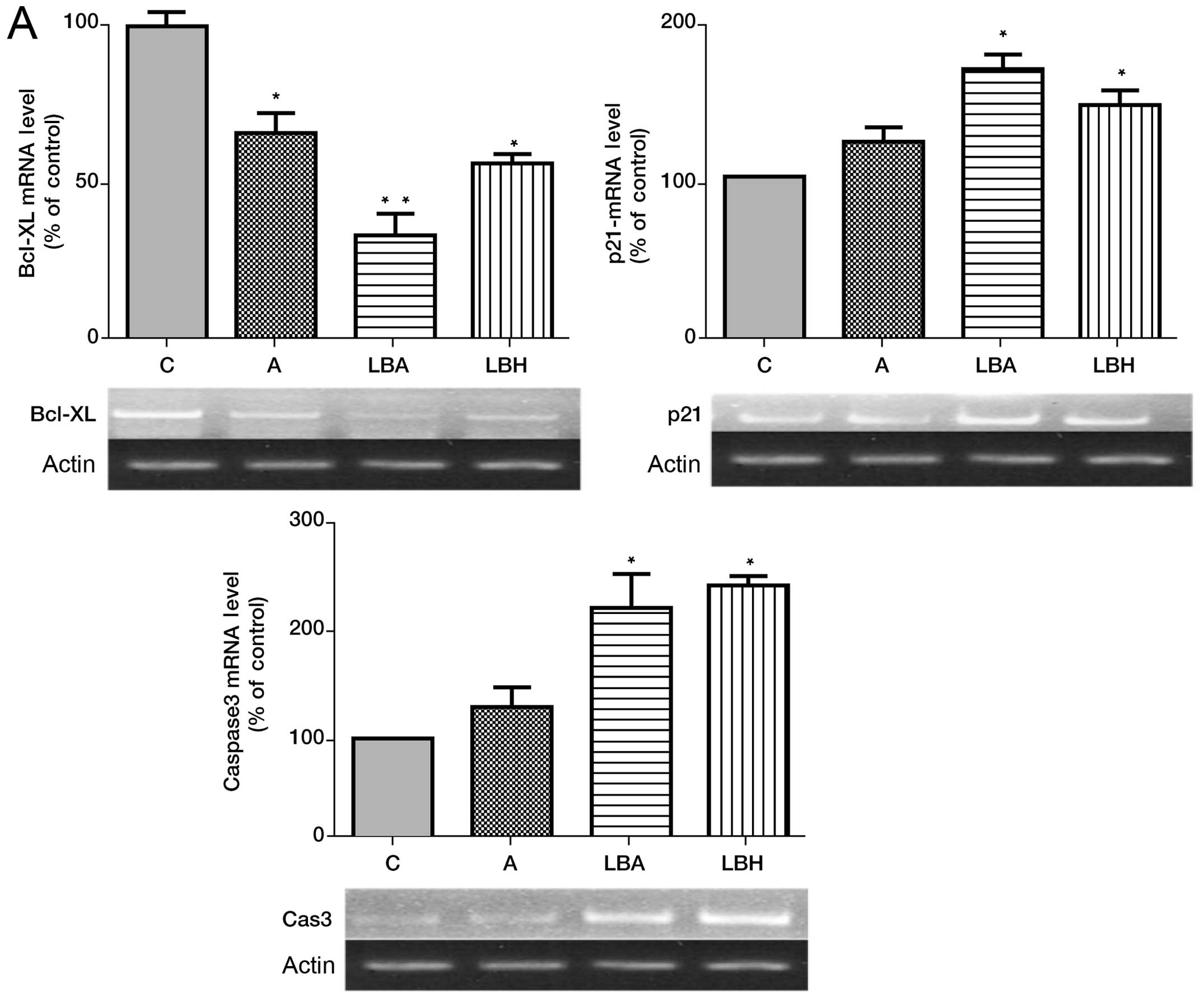 | Figure 2Effects of AZA and/or LBH589 alone
and in combination on Bcl-xL, caspase-3, Nor-1, NUR77,
p15INK4B and p21WAF1/CIP1 mRNA in HL-60
cells. (A) Effect of AZA and/or LBH589 on Bcl-xL, caspase-3 and
p21WAF1/CIP1 mRNA expression in HL-60 cells. (B) Effect
of AZA and/or LBH589 on Nor-1, NUR77, p15INK4B
mRNA expression in HL-60 cells. C, HL-60 cells without treatment;
A, cells treated with 1.0 μM 5-azacitidine; LBH, cells treated with
20 nM LBH589; LBA, cells treated with 20 nM LBH589 + 1.0 μM AZA.
*p<0.05 vs C, **p<0.05 LBA vs LBH or
AZA, and C, n=3. |
Real-time PCR analysis demonstrated AZA, and LBH589
alone significantly increased expression of the novel tumor
suppressor genes, NUR77, Nor-1 and p15INK4B with
LBH589 demonstrating a more potent single agent effect in relation
to induction of NUR77 expression (Fig. 2B). The combination of AZA with
LBH589 identified a non-significant increase in
p15INK4B, NUR77 and Nor-1 expression over single
agent treatment (Fig. 2B).
Effect of AZA and LBH589 on the
expression of Bcl-xL, caspase-3, Nor-1, NUR77, p15INK4B
and p21WAF1/CIP1 mRNA in patient samples
Analysis of gene expression in patient samples prior
to treatment (screening) demonstrated a significant reduction in
NUR77 and p21WAF1/CIP1 mRNA expression compared
to healthy controls (Fig. 3A).
NUR77 and p21WAF1/CIP1 mRNA expression levels
were similar between treatment non-responders and responders at
screening (Fig. 3B). Post
treatment (day 25, first cycle) demonstrated a significant increase
in expression of both NUR77, and p21WAF1/CIP1
ranging from 1.5- to 6-fold over screening levels (Fig. 3C). Importantly a significant
increase in NUR77, and p21WAF1/CIP1 (Table I and Fig. 3C) together with a trend to increase
in p15INK4B expression was observed in treatment
responders compared to non-responders (Fig. 3C).
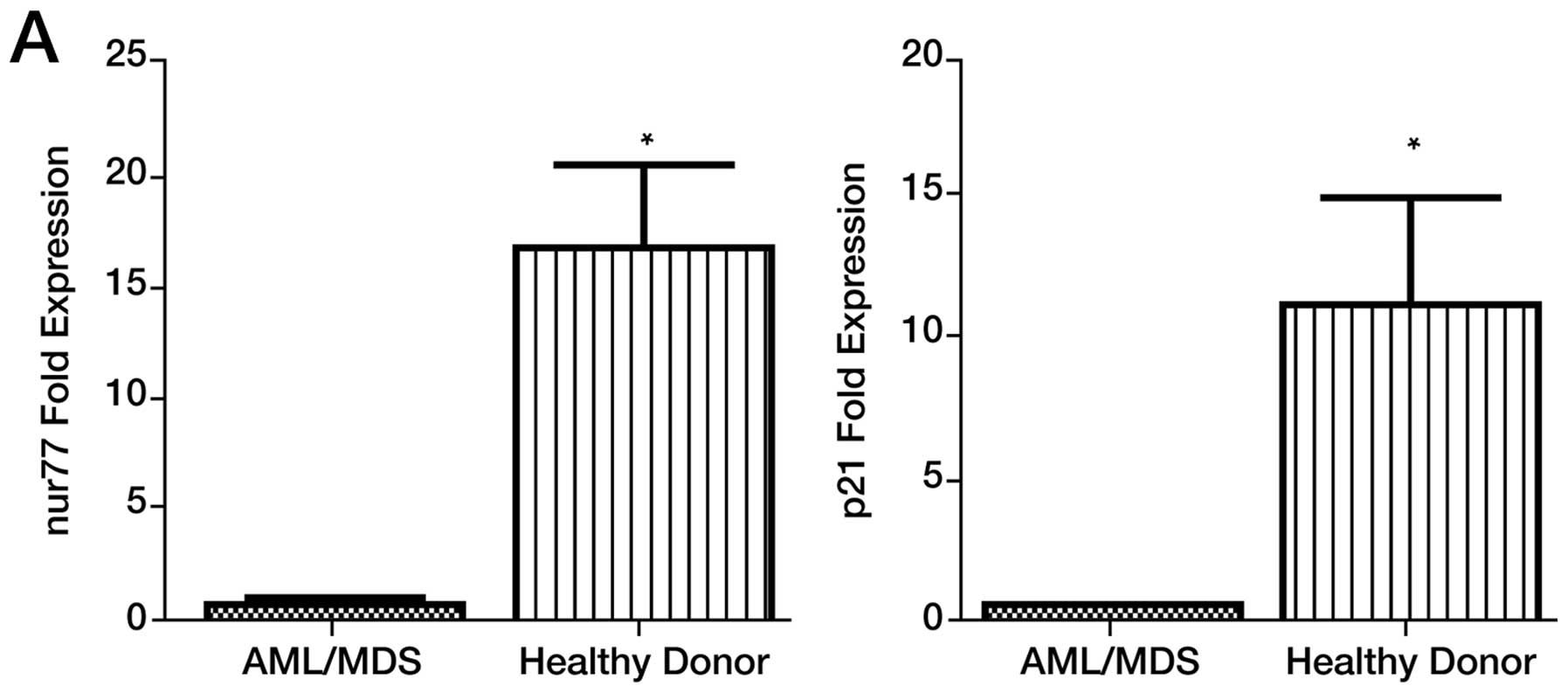 | Figure 3Effects of combination LBH589 and AZA
on Bcl-xL, caspase-3, Nor-1, NUR77, p15INK4B and
p21WAF1/CIP1 expression and correlation with clinical
response in PBMC’s. (A) Pre-treatment patient and healthy control
NUR77 and p21WAF/CIP1 mRNA expression levels. (B)
Pre-treatment NUR77 and p21WAF/CIP1 mRNA
expression levels for responders and non-responders. (C)
Correlation of gene expression with clinical response to epigenetic
treatment, day 25 mRNA expression levels for NUR77,
p21WAF/CIP1, p15INK4B, caspase-3, Bcl-xL and
NOR-1 correlated with clinical response to treatment
determined at 1, 3 and 6 months. n=23, p<0.05 deemed
significant. Responses were defined according to International
Working Group criteria for AML and MDS (15). |
Discussion
Novel epigenetic treatments for myelodysplastic
syndrome (MDS) including DNA methyl transferase inhibitors (DNMTi)
and potentially histone deacetylase inhibitors (HDACi) are able to
improve survival (17,18). However, not all patients respond
and treatment regimens are often lengthy with responses frequently
only observed after several cycles of therapy (17,18).
In order to reduce unnecessary treatment exposure, associated
complications and delays in commencement of other potentially
effective therapies a significant demand exists for molecular
markers to improve early prediction of response to epigenetic
therapy, particularly as baseline and early treatment epigenetic
modifications including methylation and acetylation status may not
inform on this issue (9,10,17,18).
LBH589 is a novel cinnamic hydroxamic acid analog
with established antitumor activity in pre-clinical models
(19). LBH589 has demonstrated
limited single-agent activity in advanced acute leukemia (20). Interestingly our in vitro
observations identified LBH589 as a more potent single agent than
AZA with the combination of LBH589 and AZA demonstrating a
non-significant increase in potency in inhibition of cell growth
over single agent treatment. Single agent treatment demonstrated
induction of p15 INK4B, p21WAF1/CIP1,
NUR77, Nor1 and caspase-3 together with suppression of
Bcl-xl expression with LBH589 demonstrating greater potency and a
non-significant trend to enhanced modulation of gene expression
with combination treatment.
Having identified significant in vitro
modulation of several genes implicated in the pathogenesis of
MDS/AML we were interested to identify temporal expression profiles
for these genes in PBMC samples from high-risk MDS/AML patients
receiving combination AZA and LBH589 therapy in our recently
reported phase 1b/II clinical trial (14). Analysis of NUR77 and
p21WAF1/CIP1 expression in patients compared with
healthy controls identified significantly reduced expression of
both NUR77 and p21WAF1/CIP1 compared with healthy
controls as has been previously demonstrated (16,21).
Screening levels of both NUR77 and p21WAF1/CIP1
demonstrated no significant correlation with clinical response to
therapy (Fig. 3B) similar to
several previous studies undertaking analysis of other epigenetic
markers in response to single or combination EGT (18). However determination of
NUR77 and p21WAF1/CIP1 expression levels,
compared with other genes implicated in the pathogenesis of
MDS/AML, from early treatment time-points (day 25), demonstrated a
significant correlation with clinical response (Fig. 3C and Table I). Whilst our observation is not
unprecedented, for example Link and colleagues demonstrated a
statistically significant concordance between response to AZA
therapy and induction of p53-inducible-ribonucleotide-reductase
(p53R2) (22) these findings were
only noted post several cycles of therapy. Identification of
NUR77 as a primary inhibitor of leukemogenesis (16), upregulation of NUR77 in
primary AML cells and leukemic stems cells in response to
epigenetic treatment (23) and
early response of NUR77 expression to EGT together with
strong correlation with clinical response suggests the
identification of a potentially novel molecular marker of early
response to combination EGT in the setting of MDS/AML. Unlike other
potential indirect molecular markers of EGT treatment response
e.g., global or gene specific, methylation/acetylation status the
delineation of the critical molecular involvement of NUR77
in the leukemogenesis process, its ubiquitously reduced expression
in AML/MDS compared to healthy controls and its early upregulation
and correlation with clinical response posits a compelling argument
for an early molecular marker of response to combination EGT in
MDS/AML. Interestingly, whilst in vitro expression levels of
the related orphan nuclear receptor Nor-1 were significantly
upregulated by combination EGT and previous in vivo studies
have demonstrated downpregulation of Nor-1 in patient leukemic
blasts (16) no significant
correlation between clinical response and early Nor-1 expression
was observed in our study suggesting that NUR77 has a more
central role in the early and subsequent clinical response to
combination EGT.
Our in vitro and in vivo observations
identified significant modulation of expression of apoptosis
machinery molecules including Bcl-xL and caspase-3, previously
identified as important in mediating the therapeutic effects of EGT
(24), although neither
demonstrated a significant correlation with clinical response
(Figs. 1–3). NUR77 has also been identified
to play a critical upstream role in mitochondrial-mediated
apoptotic events translocating from nucleus to mitochondria,
binding Bcl-2 and releasing cytochrome c with resultant
induction of apoptosis (25,26).
The critical molecular and upstream role of NUR77 in the
leukemogenic process as opposed to the effector functions of Bcl-xL
and caspase-3 may potentially explain why EGT-mediated restitution
of NUR77 and not Bcl-xL and caspase-3 expression levels is
associated with subsequent clinical response.
Our results also identified a trend toward early
upregulation of p15INK4B and clinical response. The
effects of AZA alone and in combination with HDACi on
p15INK4B expression are well documented and have been
thought pivotal in the therapeutic activity of this agent in MDS
(8,13,27–29).
Indeed early studies showed that promoter methylation of the
p15INK4B gene was associated with disease progression in
MDS and that the p15INK4B promoter was demethylated
during treatment with AZA at an early time-point (28). However, baseline and early
treatment methylation status has subsequently been demonstrated not
to predict response to epigenetic therapy (9,10)
and early re-expression of p15INK4B is not associated
with clinical response (30,31).
Together these results suggest whilst p15INK4B
expression may be modulated in response to EGT it is unlikely to
serve as a robust molecular marker of early epigenetic treatment
response and subsequent clinical response.
In conclusion, our observations identify potential
molecular mechanisms associated with combination AZA and LBH589
treatment in in vitro and in vivo settings and posit
an argument for prospective clinical investigation of orphan
nuclear receptor NUR77 and cyclin-dependent kinase inhibitor
p21WAF1/CIP1 as potential molecular markers of early
response to combination AZA and HDACi EGT in the setting of MDS and
AML.
References
|
1
|
Feinberg AP and Tycko B: The history of
cancer epigenetics. Nat Rev Cancer. 4:143–153. 2004. View Article : Google Scholar
|
|
2
|
Bolden JE, Peart MJ and Johnstone RW:
Anticancer activities of histone deacetylase inhibitors. Nat Rev
Drug Discov. 5:769–784. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cameron EE, Bachman KE, Myohanen S, Herman
JG and Baylin SB: Synergy of demethylation and histone deacetylase
inhibition in the re-expression of genes silenced in cancer. Nat
Genet. 21:103–107. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gore SD, Baylin S and Sugar E: Combined
DNA methyltransferase and histone deacetylase inhibition in the
treatment of myeloid neoplasms. Cancer Res. 66:6361–6369. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Griffiths EA and Gore SD: DNA
methyltransferase and histone deacetylase inhibitors in the
treatment of myelodysplastic syndromes. Semin Hematol. 45:23–30.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang H, Hoshino K, Sanchez-Gonzalez B,
Kantarjian H and Garcia-Manero G: Antileukemia activity of the
combination of 5-aza-2′-deoxycytidine with valproic acid. Leuk Res.
29:739–748. 2005.PubMed/NCBI
|
|
7
|
Shaker S, Bernstein M, Momparler LF and
Momparler RL: Preclinical evaluation of antineoplastic activity of
inhibitors of DNA methylation (5-aza-2′-deoxycytidine) and histone
deacetylation (trichostatin A, depsipeptide) in combination against
myeloid leukemic cells. Leuk Res. 27:437–444. 2003.
|
|
8
|
Liu HB, Mayes PA, Perlmutter P, McKendrick
JJ and Dear AE: The anti-leukemic effect and molecular mechanisms
of novel hydroxamate and benzamide histone deacetylase inhibitors
with 5-aza-cytidine. Int J Oncol. 38:1421–1425. 2011.
|
|
9
|
Shen L, Kantarjian H, Guo Y, Lin E, Shan
J, Huang X, Berry D, Ahmed S, Zhu W, Pierce S, Kondo Y, Oki Y,
Jelinek J, Saba H, Estey E and Issa JP: DNA methylation predicts
survival and response to therapy in patients with myelodysplastic
syndromes. J Clin Oncol. 28:605–613. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fandy TE, Herman JG, Kerns P, Jiemjit A,
Sugar EA, Choi SH, Yang AS, Aucott T, Dauses T, Odchimar-Reissig R,
Licht J, McConnell MJ, Nasrallah C, Kim MK, Zhang W, Sun Y, Murgo
A, Espinoza-Delgado I, Oteiza K, Owoeye I, Silverman LR, Gore SD
and Carraway HE: Early epigenetic changes and DNA damage do not
predict clinical response in an overlapping schedule of
5-azacytidine and entinostat in patients with myeloid malignancies.
Blood. 114:2764–2773. 2009. View Article : Google Scholar
|
|
11
|
Prince HM, Bishton MJ and Harrison SJ:
Clinical studies of histone deacetylase inhibitors. Clin Cancer
Res. 15:3958–3969. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lane AA and Chabner BA: Histone
deacetylase inhibitors in cancer therapy. J Clin Oncol.
27:5459–5468. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu HB, Voso MT, Gumiero D, Duong J,
McKendrick JJ and Dear AE: The anti-leukemic effect of a novel
histone deacetylase inhibitor MCT-1 and 5-aza-cytidine involves
augmentation of Nur77 and inhibition of MMP-9 expression. Int J
Oncol. 34:573–579. 2009.PubMed/NCBI
|
|
14
|
Tan P, Wei A, Mithraprabhu S, Cummings N,
Liu H, Perugini M, Reed K, Avery S, Patil S, Walker P, Mollee P,
Grigg A, D’Andrea R, Dear A and Spencer A: Dual epigenetic
targeting with panobinostat and azacitidine in acute myeloid
leukemia and high-risk myelodysplastic syndrome. Blood Cancer J.
4:e1702014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cheson BD, Greenberg PL, Bennett JM, et
al: Clinical application and proposal for modification of the
International Working Group (IWG) response criteria in
myelodysplasia. Blood. 108:419–425. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mullican SE, Zhang S, Konopleva M, Ruvolo
V, Andreeff M, Millbrant J and Conneely OM: Abrogation of nuclear
receptors Nr4a3 and Nr4a1 leads to development of acute myeloid
leukemia. Nat Med. 13:730–735. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fenaux P, Mufti GJ, Hellstrom-Lindberg E,
et al: Efficacy of azacitidine compared with that of conventional
care regimens in the treatment of higher-risk myelodysplastic
syndromes: a randomised, open-label, phase III study. Lancet Oncol.
10:223–232. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tan PT and Wei AH: The epigenomics
revolution in myelodysplasia: a clinico-pathological perspective.
Pathology. 243:536–546. 2011.PubMed/NCBI
|
|
19
|
Matthews GM, Lefebure M, Doyle MA, Shortt
J, Ellul J, Chesi M, Banks KM, Vidacs E, Faulkner D, Atadja P,
Bergsagel PL and Johnstone RW: Preclinical screening of histone
deacetylase inhibitors combined with ABT-737, rhTRAIL/MD5-1 or
5-azacytidine using syngeneic Vk*MYC multiple myeloma. Cell Death
Dis. 4:e7982013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Giles F, Fischer T, Cortes J,
Garcia-Manero G, Beck J, Ravandi F, Masson E, Rae P, Laird G,
Sharma S, Kantarjian H, Dugan M, Albitar M and Bhalla K: A phase 1
study of intravenous LBH589, a novel cinnamic hydroxamic acid
analogue histone deacetylase inhibitor, in patients with refractory
hematologic malignancies. Clin Cancer Res. 12:4628–4635. 2006.
View Article : Google Scholar
|
|
21
|
Xiao Y, Wang J, Song H, Zou P, Zhou D and
Liu L: CD34+ cells from patients with myelodysplastic
syndrome present different p21 dependent premature senescence. Leuk
Res. 37:333–340. 2013.
|
|
22
|
Link PA, Baer MR, James SR, Jones DA and
Karpf AR: p53-inducible ribonucleotide reductase (p53R2/RRM2B) is a
DNA hypomethylation-independent decitabine gene target that
correlates with clinical response in myelodysplastic syndrome/acute
myelogenous leukemia. Cancer Res. 68:9358–9366. 2008. View Article : Google Scholar
|
|
23
|
Zhou L, Ruvolo VR, McQueen T, Chen W,
Samudio IJ, Conneely O, Konopleva M and Andreeff M: HDAC inhibition
by SNDX-275 (Entinostat) restores expression of silenced
leukemia-associated transcription factors Nur77 and Nor1 and of key
pro-apoptotic proteins in AML. Leukemia. 27:1358–1368. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sonnemann J, Hartwig M, Plath A, Saravana
Kumar K, Muller C and Beck JF: Histone deacetylase inhibitors
require caspase activity to induce apoptosis in lung and prostate
carcinoma cells. Cancer Lett. 232:148–160. 2006. View Article : Google Scholar
|
|
25
|
Ekert PG and Vaux DL: The mitochondrial
death squad: hardened killers or innocent bystanders? Curr Opin
Cell Biol. 17:626–630. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lin B, Kolluri SK, Lin F, Liu W, Han YH,
Cao X, Dawson MI, Reed JC and Zhang XK: Conversion of Bcl-2 from
protector to killer by interaction with nuclear orphan receptor
Nur77/TR3. Cell. 116:527–540. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Uchida T, Kinoshita T, Nagai H, Nakahara
Y, Saito H, Hotta T and Murate T: Hypermethylation of the
p15INK4B gene in myelodysplastic syndromes. Blood.
90:1403–1409. 1997.
|
|
28
|
Daskalakis M, Nguyen TT, Nguyen C,
Guldberg P, Kohler G, Wijermans P, Jones PA and Lübbert M:
Demethylation of a hypermethylated P15/INK4B gene in patients with
myelodysplastic syndrome by 5-Aza-2′-deoxycytidine (decitabine)
treatment. Blood. 100:2957–2964. 2002.PubMed/NCBI
|
|
29
|
Hitomi T, Matsuzaki Y, Yokota T, Takaoka Y
and Sakai T: p15(INK4B) in HDAC inhibitor-induced growth arrest.
FEBS Lett. 554:347–350. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Soriano AO, Yang H, Faderl S, Estrov Z,
Giles F, Ravandi F, Cortes J, Wierda WG, Ouzounian S, Quezada A,
Pierce S, Estey EH, Issa JP, Kantarjian HM and Garcia-Manero G:
Safety and clinical activity of the combination of 5-azacytidine,
valproic acid, and all-trans retinoic acid in acute myeloid
leukemia and myelodysplastic syndrome. Blood. 110:2302–2308. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Garcia-Manero G, Kantarjian HM,
Sanchez-Gonzalez B, Yang H, Rosner G, Verstovsek S, Rytting M,
Wierda WG, Ravandi F, Koller C, Xiao L, Faderl S, Estrov Z, Cortes
J, O’brien S, Estey E, Bueso-Ramos C, Fiorentino J, Jabbour E and
Issa JP: Phase 1/2 study of the combination of
5-aza-2′-deoxycytidine with valproic acid in patients with
leukemia. Blood. 108:3271–3279. 2006.PubMed/NCBI
|