Introduction
Thyroid cancer is the most common cancer of the
endocrine system. In the United States, differentiated thyroid
cancer (DTC) is the sixth most common cancer among women and the
eight most common cancer overall (http://seer.cancer.gov/statfacts/html/thyro.html).
Most DTCs respond favorably to conventional therapy such as
thyroidectomy and radioactive iodine (RAI) therapy. However, a
significant proportion of patients with DTC develop
life-threatening RAI-refractory disease that is usually resistant
to cytotoxic chemotherapy (1–4).
Because of this poor response to cytotoxic chemotherapy, new
molecular-targeted therapies are being developed for the treatment
of DTC patients who require systemic therapy (3,5). In
this context, protein kinase inhibitors targeting the
RAS-RAF-MEK-ERK (MAPK) signaling pathway have been investigated
intensively and BRAF inhibitors have shown beneficial effects in
clinical trials (6–8). However, the development of resistance
to kinase inhibitors is an emerging problem for clinicians and
patients, and the design of strategies to overcome this resistance
is a primary concern of researchers (9,10).
Mammalian sirtuin 1 (Sirt1) belongs to a highly
conserved family of nicotinamide adenosine dinucleotide-dependent
(NAD+-dependent) protein deacetylases and is widely
expressed in most mammalian organs (11). The deacetylase activity of Sirt1
and its role in the regulation of several stress-induced
transcription factors such as p53, heat shock transcription factor
1 (HSF1), nuclear factor κB (NF-κB), peroxisome
proliferator-activated receptor γ, coactivator 1α (PGC-1α) and the
forkhead box O (FOXO) family of transcription factors have been
studied extensively (12–16). The activation of Sirt1 in response
to stress may therefore be an evolutionarily conserved process to
drive cellular homeostasis related to oxidative stress and
apoptosis (17). Sirt1 activity is
modulated by calorie restriction (CR), HuR, NAD+ and
active regulator of Sirt1 (AROS); in turn, the deacetylase activity
of Sirt1 promotes stress adaptation responses including DNA repair
and anti-apoptotic effects in response to genotoxic stress
(18–23). However, its dynamic role in the
regulation of cytoprotective effects and apoptosis suggests that
Sirt1 acts both as an oncogene and a tumor suppressor (24,25).
In the present study, we investigated the
cytoprotective effects of Sirt1 in thyroid cancer cells exposed to
genotoxic stress induced by etoposide treatment, which can cause
DNA damage by preventing the re-ligation of DNA strands and
promoting DNA strand breaks (26).
Our results showed that the etoposide-induced differential
expression of Sirt1 is cell type-specific and related to the
resistance of thyroid cancer cells to etoposide-induced cell death.
Our data also indicated that Bax and p21 are signature molecules
possibly associated with the cytoprotective effects of Sirt1.
Materials and methods
Tissue specimens and immunohistochemical
analysis
Thyroid tissue specimens were obtained from 50
patients with papillary thyroid cancer (PTC) who underwent surgery
from 2010 to 2013 at the Yonsei Cancer Center, Yonsei University
College of Medicine (Seoul, Korea). All protocols were approved by
the institutional review board and written informed consent was
obtained from all subjects. Immunohistochemical (IHC) staining for
Sirt1 and Sirt3 was performed in PTC samples and matched normal
tissues. Tissue sections were incubated with primary antibodies
against Sirt1 (sc-15404, Santa Cruz Biotechnology, Santa Cruz, CA,
USA) and Sirt3 (sc-99143, Santa Cruz Biotechnology).
Cell lines and materials
TPC-1 (papillary thyroid cancer cell line) and
FTC-133 (follicular thyroid cancer cell line) cells were cultured
in DMEM (Sigma, St. Louis, MO, USA) supplemented with 10% FBS. FRO
(undifferentiated/anaplastic thyroid cancer cell line) cells were
cultured in RPMI-1640 (Sigma) with 10% FBS. Etoposide (Sigma) was
used at 20 μM for the indicated times.
Immunoblot analysis
Cells were lysed in lysis buffer, and cell lysates
were separated using SDS-polyacrylamide gel electrophoresis.
Proteins were transferred to nitrocellulose (NS) membranes
(Amersham Biosciences, Freiburg, Germany), which were blocked with
5% skim milk and incubated with the indicated primary antibodies
overnight at 4°C. After washing, the membranes were incubated with
secondary antibodies for 1 h at room temperature. The
immunoreactive bands were visualized using peroxidase-conjugated
secondary antibodies (Phototope-HRP Western Blot Detection Kit, New
England Biolabs, Beverly, MA, USA). The primary antibodies used in
this study were obtained from Santa Cruz Biotechnology (Santa Cruz,
CA, USA) and were as follows: anti-Sirt1 (sc-15404), anti-Sirt3
(sc-365175), and anti-actin (sc-1616).
Immunofluorescence staining
Cells were plated at 1×105 cells/well on
coverslips in 6-well plates. After 3 days, cells were fixed and
permeabilized using conventional methods. Then, the cells were
incubated with anti-Sirt1 (sc-15404, Santa Cruz Biotechnology) and
anti-p21 (sc-397, Santa Cruz Biotechnology) antibodies at a 1:100
dilution in 3% bovine serum albumin for 24 h at 4°C. After washing,
cells were incubated with goat anti-rabbit IgG H&L (Alexa
Fluor® 488) (ab150077, Cambridge, MA, USA). After
washing, the cells on the coverslips were mounted on glass slides
using mounting medium (Sigma-Aldrich) and observed using a
laser-scanning confocal microscope (Carl Zeiss AG, Oberkochen,
Germany). All experiments were performed in triplicate and were
repeated at least three times.
Apoptosis detection
Apoptosis was assessed using the PE Annexin V
Apoptosis Detection Kit I (BD Biosciences, Warsaw, Poland)
according to the manufacturer’s protocol. Briefly, cells were
washed twice with cold PBS and re-suspended in 1× binding buffer at
a concentration of 1×106 cells/ml. Then, 100 μl of the
solution (1×105 cells) was transferred to a 5-ml culture
tube, treated with 5 μl of FITC Annexin V and 5 μl of propidium
iodide (PI), and incubated for 15 min at RT (25°C) in the dark.
After adding 400 μl of 1× binding buffer to each tube, flow
cytometry analysis was performed. Data were analyzed using a BD
FACSVerse system and BD FACSuite software (BD Biosciences).
MTT
(3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide)
assay
Cell viability was assessed by the MTT dye
conversion assay. After treatment with etoposide, MTT (25 μl of 5
mg/ml MTT in sterile PBS) was added to 100 μl of a cell suspension
and incubated for 2 h at 37°C. After the reaction was stopped, the
cells were lysed by addition of 100 μl lysis buffer. Cell lysates
were incubated at 37°C overnight to allow cell lysis and dye
solubilization. The OD was read at 595 nm using a THERMOmax
microplate reader (Molecular Devices, Menlo Park, CA, USA). Data
are expressed as a percentage of vehicle-treated (DMSO) control
values and are the result of three independent experiments, each
performed in triplicate.
RNA isolation and real-time PCR
Total RNA was extracted using TRIzol (Invitrogen,
Carlsbad, CA, USA) and complementary DNA (cDNA) was prepared from
total RNA using M-MLV Reverse Transcriptase (Invitrogen) and
oligo-dT primers (Promega, Madison, WI, USA). Quantitative RT-PCR
(qRT-PCR) was performed using cDNA, a QuantiTect SYBR®
Green RT-PCR kits (Qiagen, Valencia, CA, USA), and the following
primers: Bax, 5′-CCC GAG AGG TCT TTT TCC GAG-3′ and 5′-CCA GCC CAT
GAT GGT TCT GAT-3′; Bcl-xL, 5′-GAG CTG GTG GTT GAC TTT CTC-3′ and
5′-TCC ATC TCC GAT TCA GTC CCT-3′; p53, 5′-CAG CAC ATG ACG GAG GTT
GT-3′ and 5′-TCA TCC AAA TAC TCC ACA CGC-3′; p21, 5′-TGT CCG TCA
GAA CCC ATG C-3′ and 5′-AAA GTC GAA GTT CCA TCG CTC-3′; and GAPDH,
5′-GGA GCG AGA TCC CTC CAA AAT-3′ and 5′-GGC TGT TGT CAT ACT TCT
CAT GG-3′. Relative expression was measured using the Applied
Biosystems® StepOne™ Real-Time PCR system (Foster City,
CA, USA). qRT-PCR experiments were performed in triplicate and
repeated three times.
Public data and statistical analysis
Analysis of gene expression using public repository
data was performed using the Human Protein Atlas (http://www.proteinatlas.org/), BioGPS (http://biogps.org/#goto=welcome), NCBI Gene
Expression Omnibus (GEO) profiles (http://www.ncbi.nlm.nih.gov/geoprofiles), and
GeneNetwork (a free scientific web resource, http://www.genenetwork.org/). Statistical analysis was
performed using GraphPad Prism (GraphPad Software, Inc., CA, USA).
Comparisons were performed with the Mann-Whitney U test. Data are
expressed as the mean ± SEM, *p<0.05,
**p<0.01, ***p<0.001. All reported
p-values are two-sided.
Results
Sirt1 is differentially expressed in
human papillary thyroid carcinoma
To compare the expression patterns of Sirt1 in
normal and thyroid cancer tissues, we performed IHC using
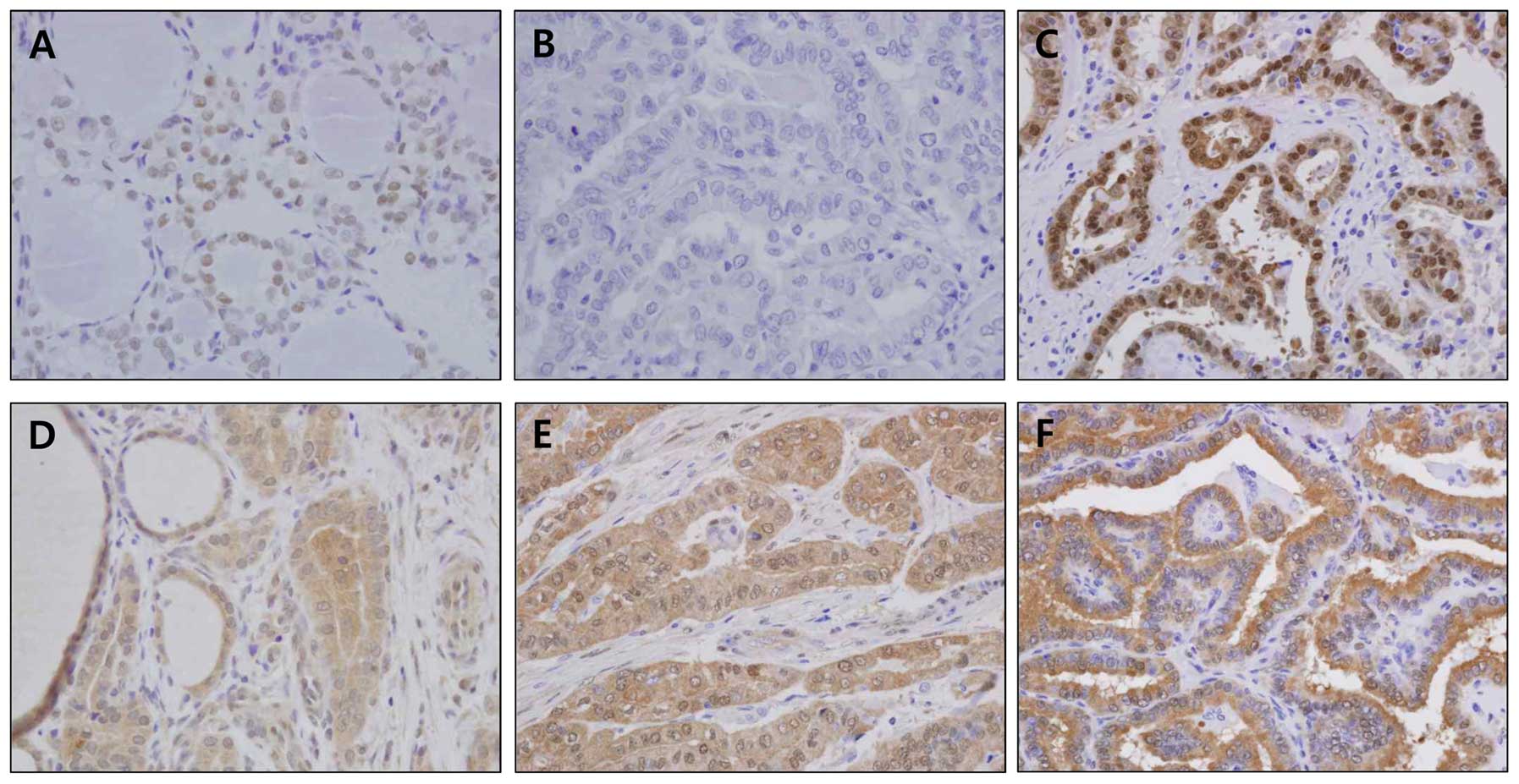
anti-Sirt1 and anti-Sirt3 antibodies. As shown in Fig. 1A, Sirt1 expression in the nuclei of
normal follicular cells was variable. In the PTC samples, Sirt1
expression was also variable, and expression ranged from no
staining (Fig. 1B) to strong
nuclear staining (Fig. 1C). In the
case of Sirt3, variable expression levels were observed in normal
and PTC tissues (Fig. 1D–F). To
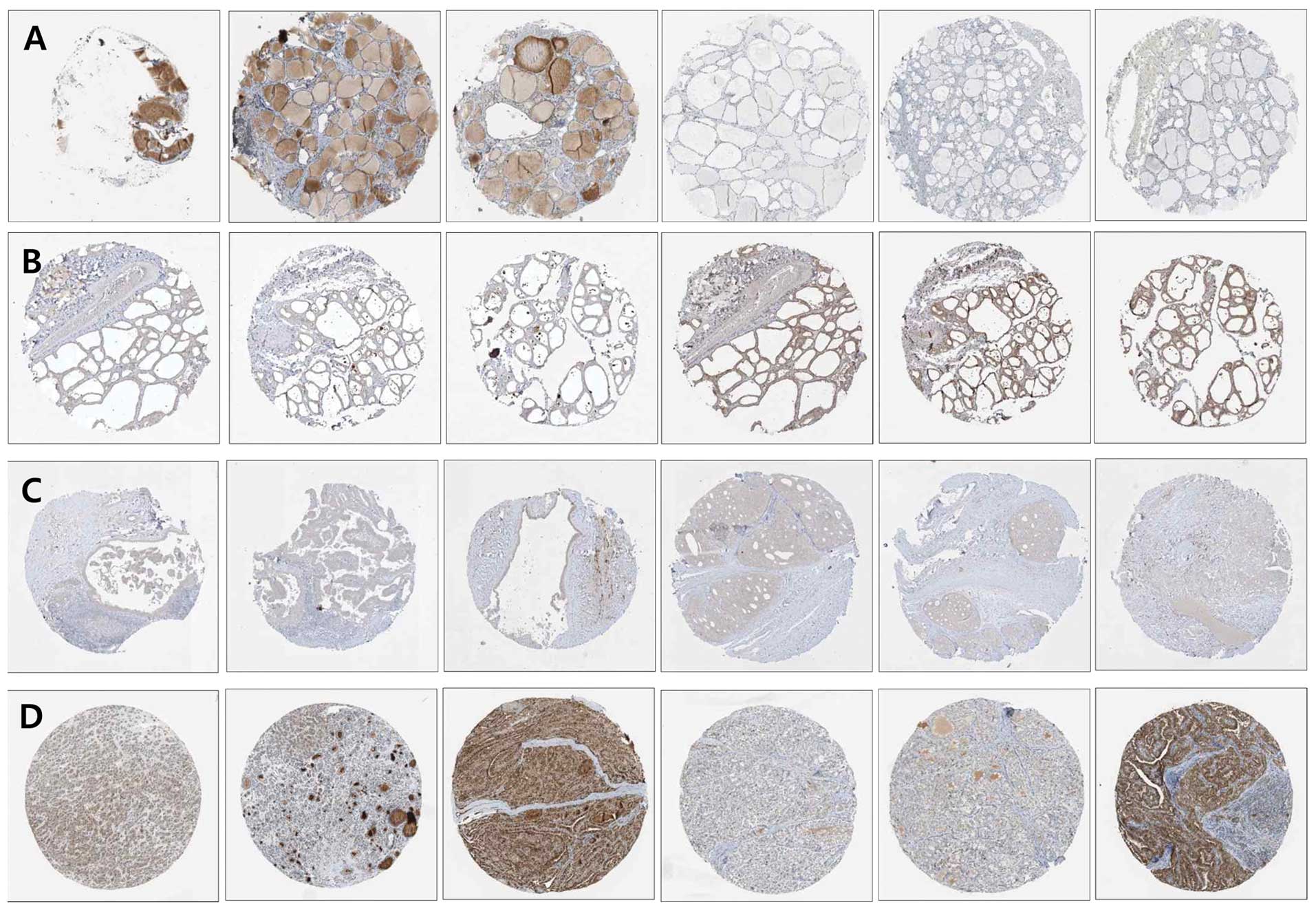
support these results, we conducted a search of public gene
expression repositories such as the Human Protein Atlas (http://www.proteinatlas.org/), BioGPS (http://biogps.org/#goto=welcome) and NCBI Gene
Expression Omnibus (GEO) profiles (http://www.ncbi.nlm.nih.gov/geoprofiles). The Human
Protein Atlas showed heterogeneous expression patterns of Sirt1 in
normal thyroid and papillary thyroid cancer tissues with remarkable
variation from faint to strong expression (Fig. 2A and B). Consistent with the
pattern of Sirt1 expression, Sirt3 showed heterogeneous and
remarkably variable expression in normal and papillary thyroid
cancers tissues (Fig. 2C and D).
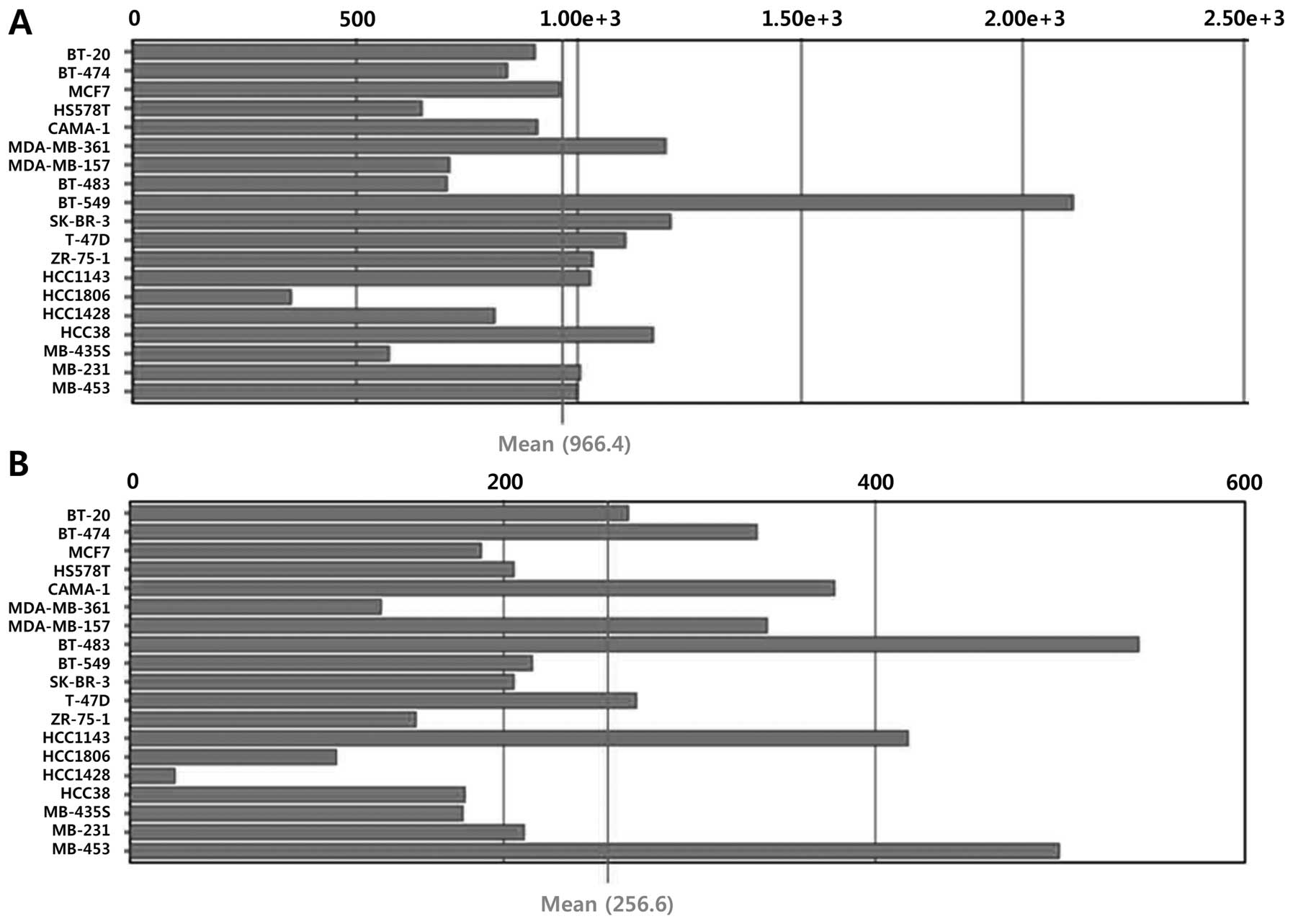
Because stress-induced changes in Sirt1 levels are regulated not
only at the transcriptional level but also by modulation of RNA
stability and post-translational control, the mRNA expression
levels of Sirt1 and Sirt3 were assessed using BioGPS and NCBI GEO
profiles. No data on the mRNA expression profiles of Sirt1 and
Sirt3 in thyroid cancers were found in BioGPS; however, variable
mRNA expression of Sirt1 and Sirt3 was observed in breast cancer
cell lines (Fig. 3). In line with
the BioGPS profiles, we consistently found variable Sirt1 and Sirt3
mRNA expression profiles in normal thyroid follicular cells and
papillary thyroid cancer cells in the NCBI GEO profiles (Fig. 4). Taken together, our analysis
using public repositories indicated that Sirt1 and Sirt3 mRNA and
protein expression may be regulated in a cell type- or
context-dependent manner.
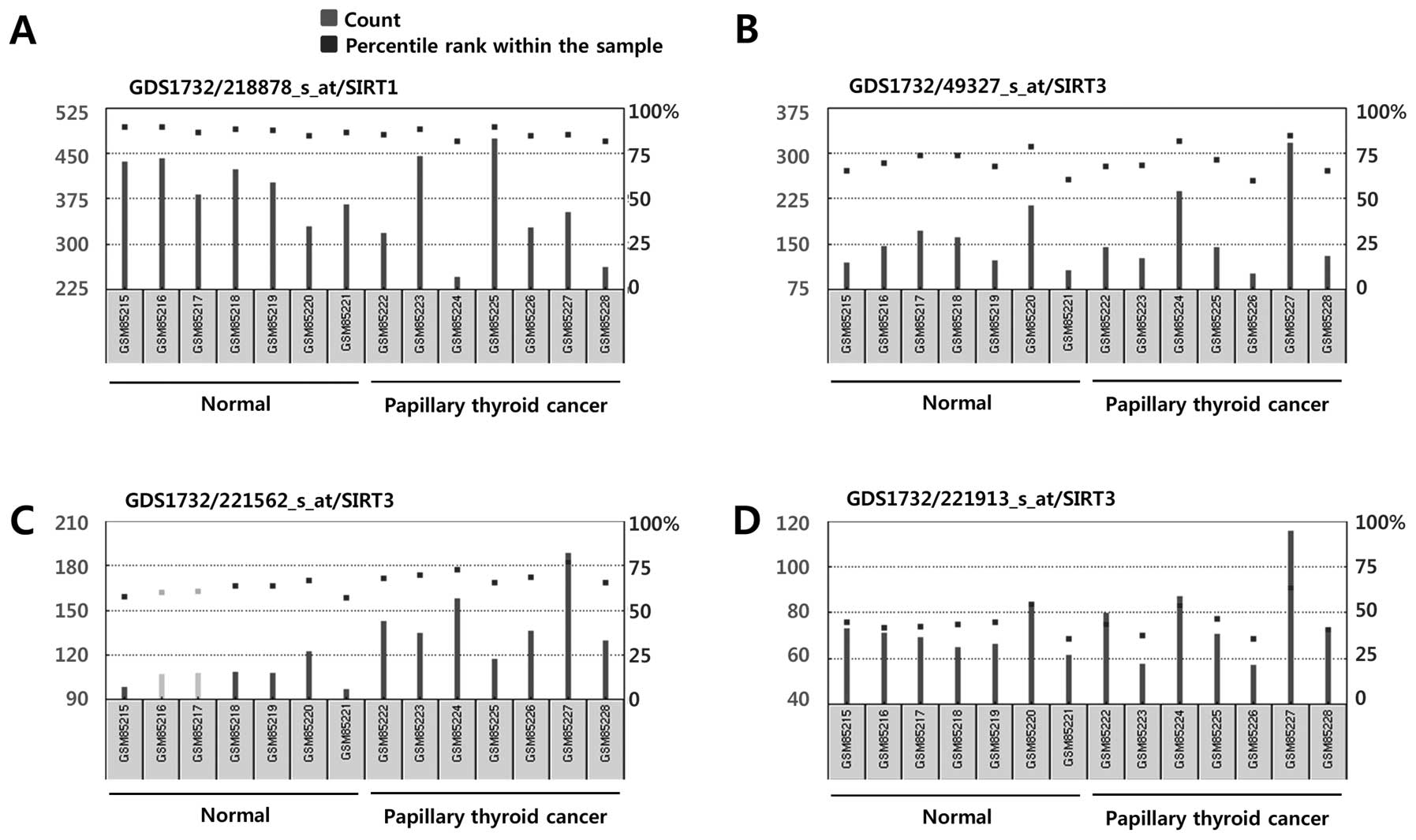 | Figure 4mRNA expression of Sirt1 (A) and
Sirt3 (B–D) in human thyroid follicular cells and papillary thyroid
cancer cells. Data were obtained from NCBI Gene Expression Omnibus
(GEO) profiles (http://www.ncbi.nlm.nih.gov/geoprofiles). (A) Sirt1,
Reporter: GPL570, 218878_s_at (ID_REF), GDS1732, 23411 (Gene ID),
NM_012238. (B) Sirt3, Reporter: GPL570, 49327_at (ID_REF), GDS1732,
23410 (Gene ID), AI492888. (C) Sirt3, Reporter: GPL570, 221562_s_at
(ID_REF), GDS1732, 23410 (Gene ID), AF083108. (D) Sirt3, Reporter:
GPL570, 221913_at (ID_REF), GDS1732, 23410 (Gene ID), AI492888. |
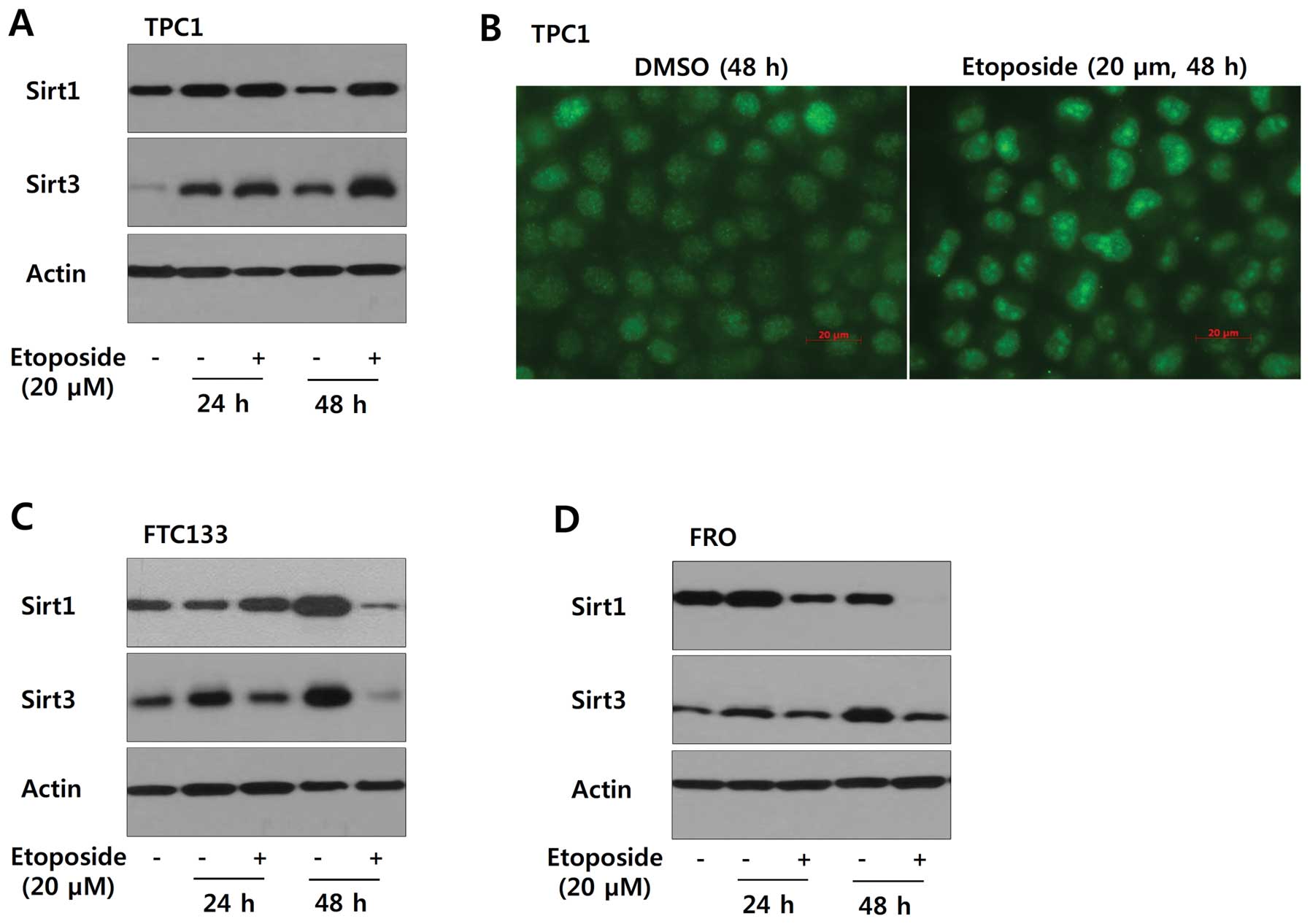
The induction of Sirt1 expression by
etoposide occurs in a cell type-specific manner in thyroid cancer
cell lines
Based on the variable expression of Sirt1 and Sirt3
in papillary thyroid cancer, we examined whether etoposide-induced
genotoxic stress has a differential effect on the expression of
Sirt1 and Sirt3 in a cell type-specific manner. As shown in
Fig. 5A and B, TPC1 cells treated
with etoposide (20 μM) for 24 or 48 h showed increased expression
of Sirt1 and Sirt3. However, Sirt1 and Sirt3 were downregulated in
FTC133 and FRO cells under the same experimental conditions
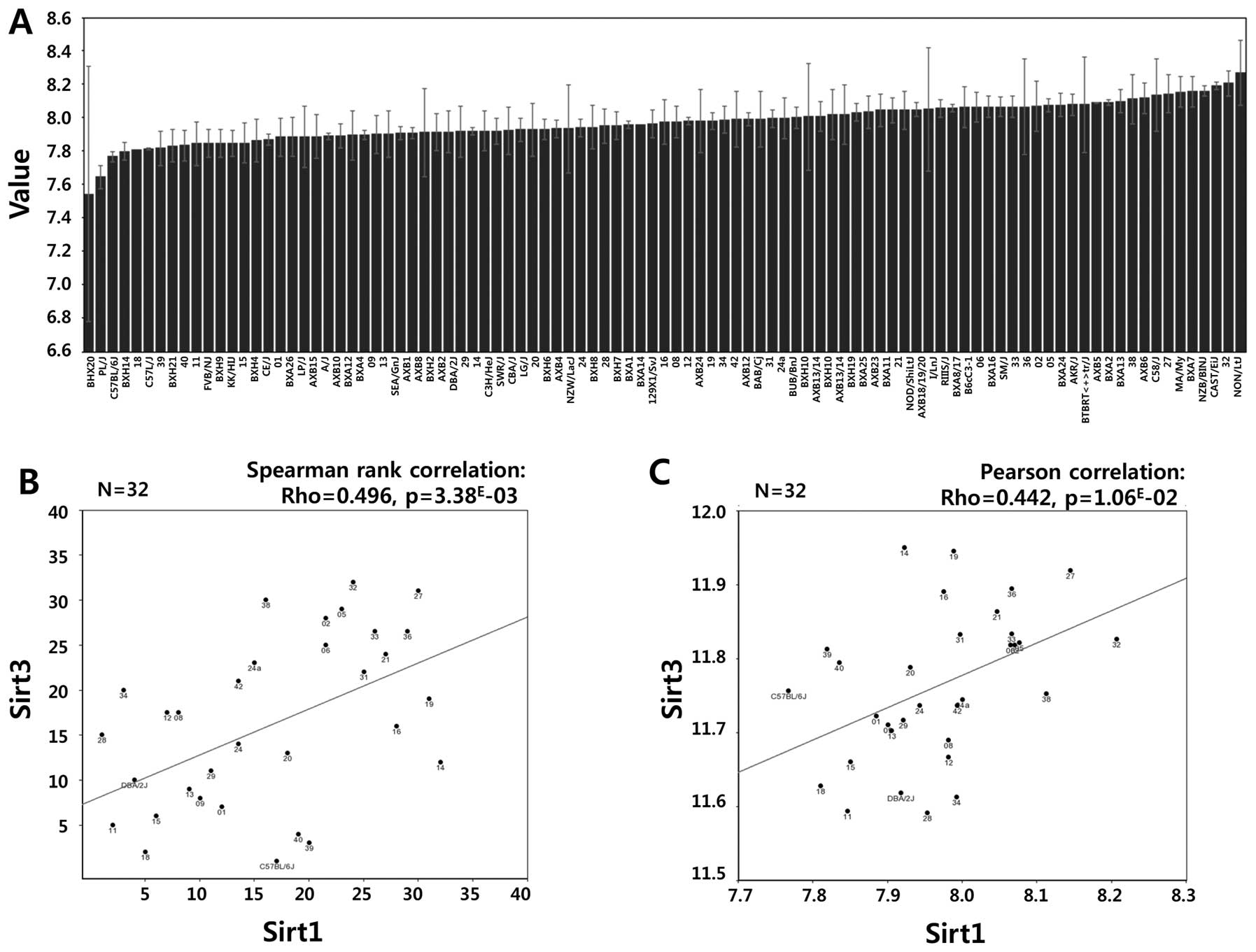
(Fig. 5C and D). Consistent with
our data, a strong positive correlation between Sirt1 and Sirt3
mRNA expression was observed in the GSE16780 UCLA Hybrid MDP Liver
Affy HT M430A (Sep11) RMA Database from GeneNetwork (http://www.genenetwork.org/) (Fig. 6, Spearman’s rank correlation:
Rho=0.496, p=3.38E-03; Pearson’s correlation: Rho=0.442,
p=1.06E-02). In fact, Sirt3 possesses stress responsive
deacetylase activity similar to that of Sirt1, protecting cells
from genotoxic and oxidative stress-mediated cell death. Combined
with data from the public repository, the differential induction of
Sirt1 and Sirt3 in TPC1, FTC133 and FRO cells confirmed that Sirt1
and Sirt3 might function cooperatively in a cell type- or
context-dependent manner.
Sirt1 induction is related to
etoposide-induced cell death
To support our results suggesting the cell type- or
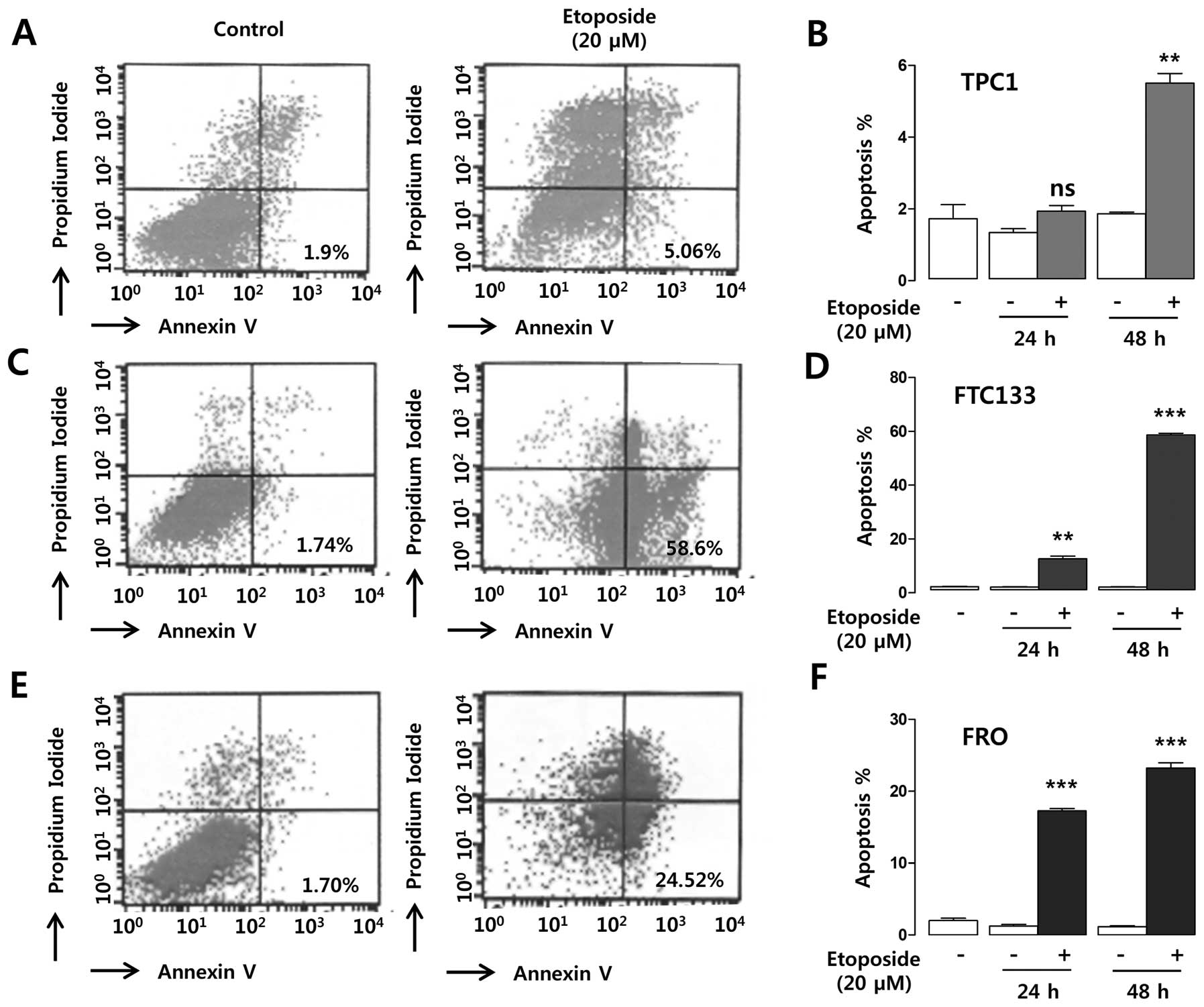
context-dependent action of Sirt1 and Sirt3, we analyzed apoptotic
cell death induced by etoposide in TPC1, FTC133 and FRO cells using
Annexin V flow cytometric analysis. As shown in Fig. 7A, TPC1 cells showed a minimal
increase of apoptosis (5.06%) in response to etoposide (20 μM)
treatment for 48 h compared to the untreated controls (1.9%),
whereas 24-h etoposide treatment had no significant effect on the
rate of apoptosis (Fig. 7B). By
contrast, a dramatic increase of apoptotic cell death (58.6%) was
observed in response to etoposide for 48 h in FTC133 cells compared
to the untreated controls (1.74%, Fig.
7C), and the effect was statistically significant after 24 h of
treatment (Fig. 7D). FRO cells
also showed a dramatic increase of apoptotic cell death (1.70 vs.
24.52%) after 48 h of etoposide treatment (Fig. 7E), and this effect was
statistically significant after 24 h of treatment (Fig. 7F). To verify the flow cytometry
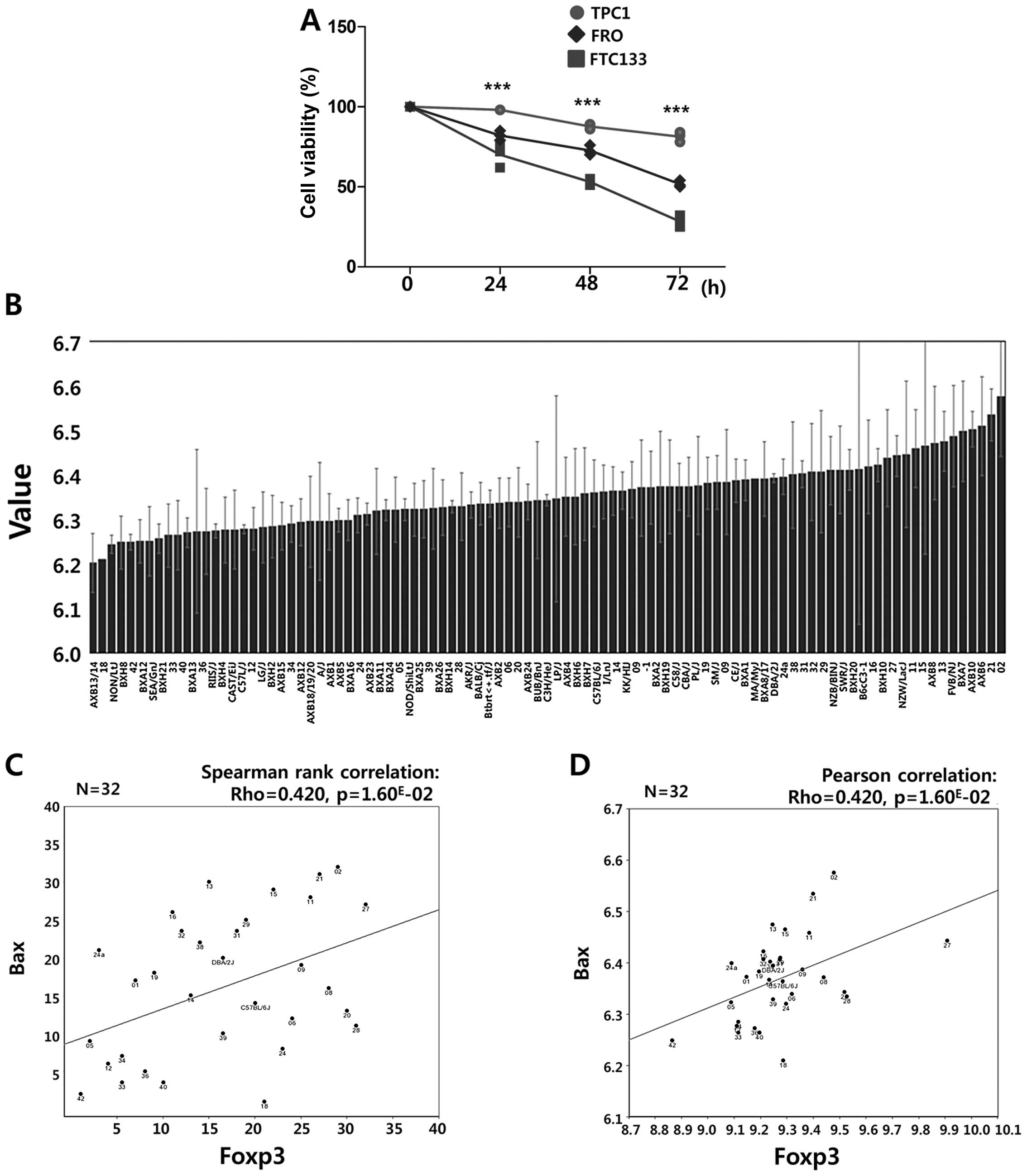
results, cell viability was assessed by MTT assay after exposure to
etoposide (20 μM) for the indicated times. Consistent with the flow
cytometry data, the reduction of enzymatic activity of
NAD(P)H-dependent cellular oxido-reductase was lower in TPC1 cells
than in FTC133 and FRO cells (Fig.
8A). Taken together, these results suggest that the higher
induction of Sirt1 and Sirt3 in TPC1 cells may confer increased
resistance against etoposide-induced genotoxic stress, as observed
by reduced apoptosis and increased cell viability compared to cells
with low Sirt1 and Sirt3 induction.
cDNA microarray data using BXD mice show
a correlation of Bax and p21 with Sirt1 activation
To gain insight into the molecular mechanisms via
which Sirt1 and Sirt3 contribute to resistance against
etoposide-induced genotoxic stress, we reviewed the literature and
analyzed public gene expression repositories. A recent study
suggested that Sirt1 deacetylates and negatively regulates the
forkhead box protein P3 (Foxp3) transcription factor (27). In addition, Ex-527, a Sirt1
inhibitor, enhanced Foxp3 expression during ex vivo Treg
expansion (27). In line with this
recent report, we investigated the correlations between Foxp3
expression and apoptosis-related molecules on GeneNetwork.
Interestingly, we found that the mRNA expression of Foxp3 showed a
positive relationship with Bax in the GSE16780 UCLA Hybrid MDP
Liver Affy HT M430A (Sep11) RMA Database (Fig. 8, Spearman’s rank correlation:
Rho=0.420, p=1.60E-02; Pearson’s correlation: Rho=0.453,
p=8.49E-03), suggesting that a Sirt1-Foxp3-Bax signaling
pathway contributes to resistance to etoposide-induced genotoxic
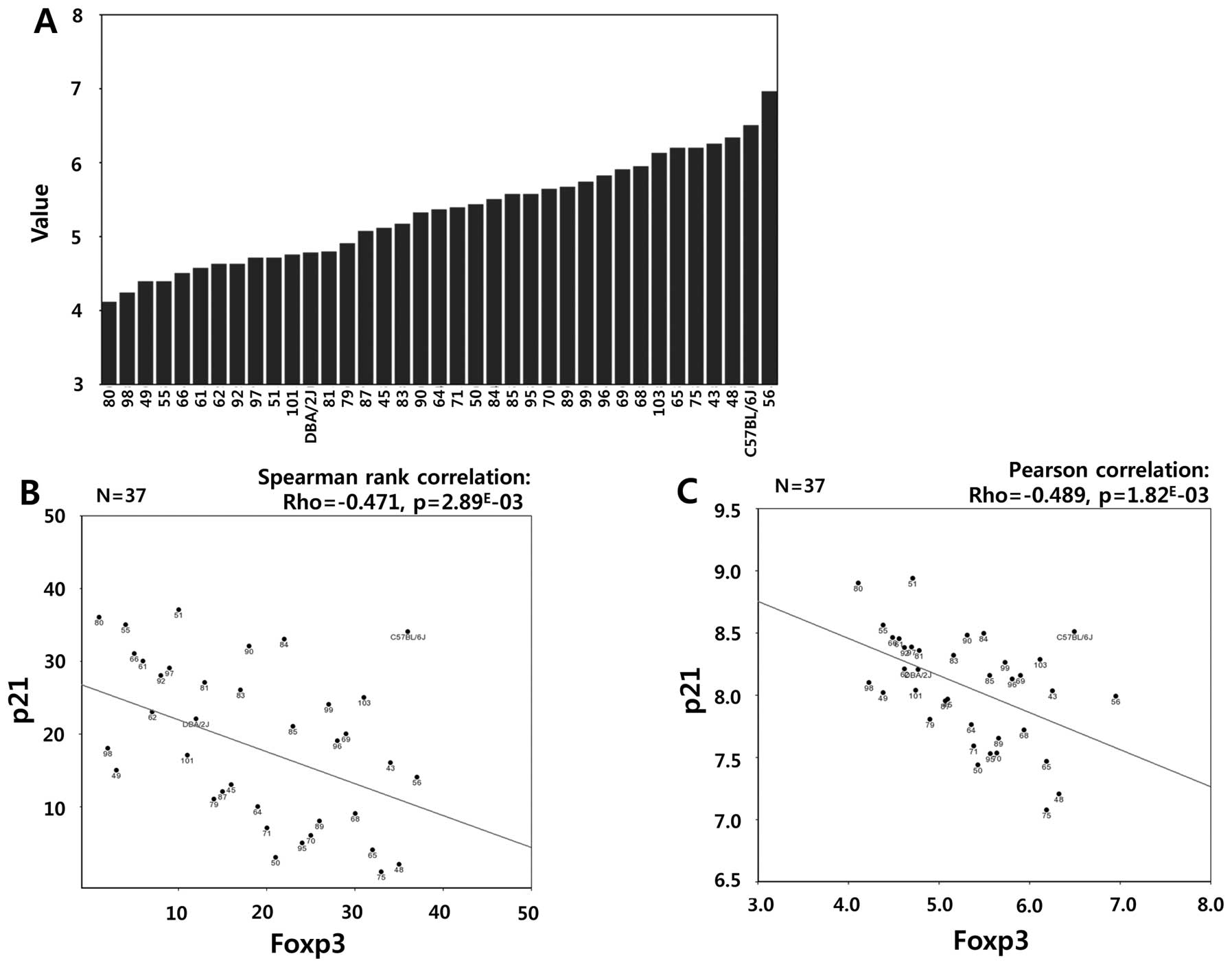
stress by decreasing Bax expression. In addition, a statistically
significant negative correlation between Foxp3 and p21
[cyclin-dependent kinase inhibitor 1A (CDKN1A), Cip1] was also
found in EPFL/LISP BXD CD Brown Adipose Affy Mouse Gene 2.0 ST Exon
Level (Oct13) RMA (Fig. 9,
Spearman’s rank correlation: Rho=−0.471, p=2.89E-03;
Pearson’s correlation: Rho=−0.489, p=1.82E-03). Taken
together, these results suggest that Sirt1-Foxp3-Bax/p21 signaling
might generate a cytoprotective effect in TPC1 cells exposed to
etoposide treatment.
The differential expression of Bcl-2
family proteins in TPC1 cells is correlated with Sirt1
To gather further evidence in support of the
proposed mechanism of resistance to etoposide in TPC1 cells, we
performed qRT-PCR for Bax and p21. In addition, we also included
another Bcl-2 family protein, Bcl-xL, and p53, which is the first
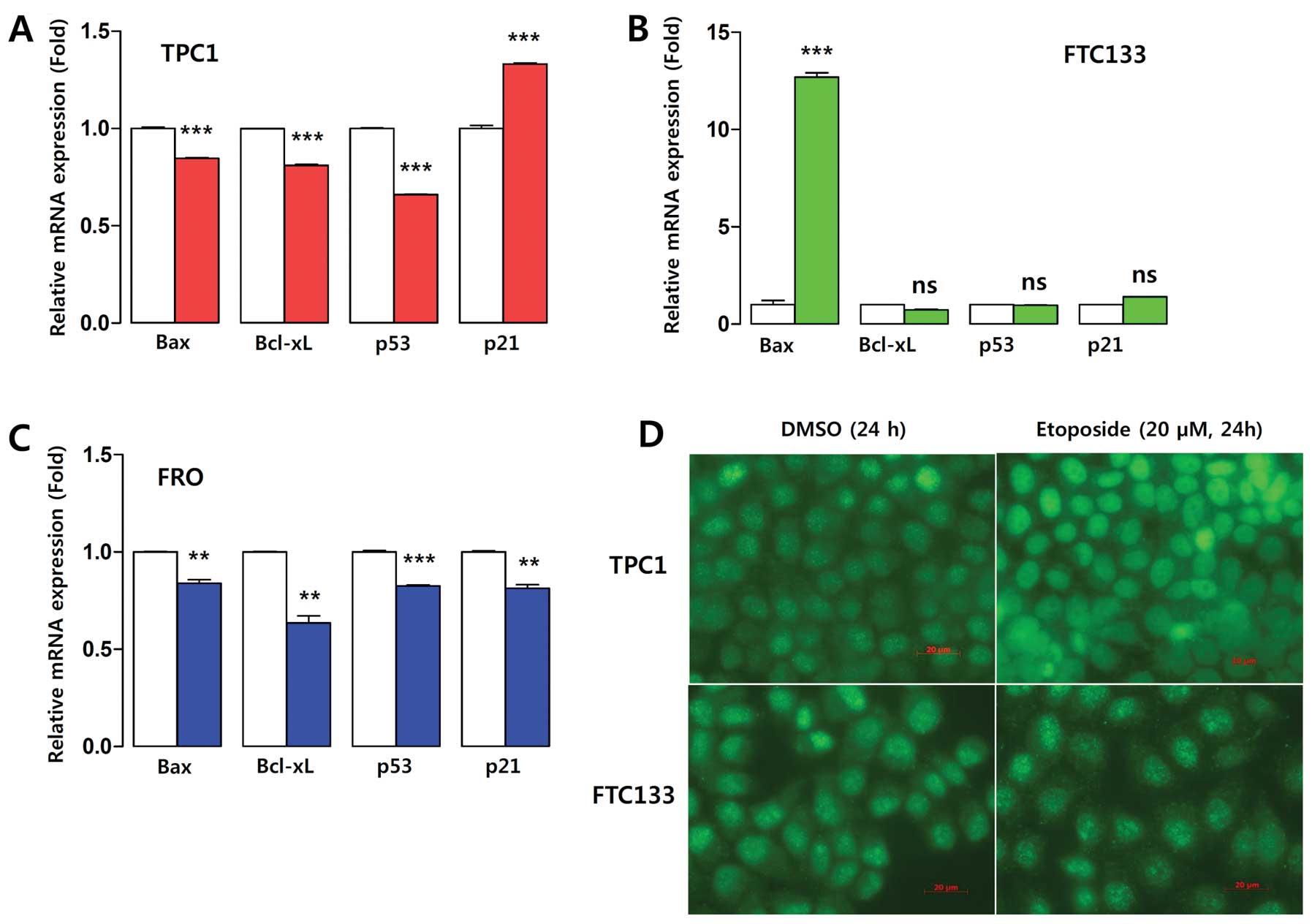
known non-histone target of Sirt1. Etoposide treatment
significantly decreased Bax mRNA levels in TPC1 cells (Fig. 10A), whereas it remarkably
upregulated Bax expression in FRO cells (Fig. 10C). Bcl-xL and p53 showed
decreased mRNA expression in the three cell lines after etoposide
treatment (Fig. 10A–C). p21
expression was significantly upregulated in TPC1 cells and
downregulated in FRO cells, suggesting a cytoprotective effect for
p21 in TPC1 cells (Fig. 10A, C and
D). The downregulation of Bax and upregulation of p21 in TPC1
cells suggests that the higher induction of Sirt1 confers increased
resistance to etoposide-induced apoptosis via Sirt1-Foxp3-Bax/p21
signaling.
Discussion
The use of selective RAF/MEK small molecule kinase
inhibitors as monotherapy has been examined in clinical trials
(28,29). The US Food and Drug Administration
expanded the approved uses of Nexavar (Sorafenib)® to
the treatment of late-stage (metastatic) DTC. However, the
development of resistance against RAF inhibitors is an emerging
obstacle to the treatment of RAI-refractory thyroid cancers
(9,30) and the development of novel
therapeutic drugs. To overcome these limitations, significant
efforts have been directed to improving our understanding of the
mechanisms underlying thyroid carcinogenesis. For example, protein
interactions between RAF paralogs have been suggested as a possible
drug resistance mechanism (31–33).
Point mutations in RAS or MEK are known to generate MEK/ERK signal
propagation in response to selective RAF kinase inhibitors
(34). Mitogen-activated protein
kinase kinase kinase 8 [MAP3K8, cancer Osaka thyroid (COT)]
overexpression is another proposed mechanism of drug resistance
(35). Ligand-specific receptor
tyrosine kinase activation is a druggable target in RAI-refractory
thyroid cancers (36–38). Recently, MEK-ERK independent
signaling pathways involved in thyroid carcinogenesis have been
investigated (39–42).
The sirtuin family, which includes seven members
(SIRT1-SIRT7), has emerged as an important regulator of diverse
physiologic or pathologic events including life-span extension,
age-related disorders and cancer (19). However, studies have suggested that
Sirt1 can act as either a tumor suppressor or promoter depending on
its targets in specific signaling pathways or in specific cancers,
and its role therefore remains unclear (24,25).
In the present study, we first investigated whether
Sirt1 expression could be used as a molecular marker to
differentiate thyroid cancers from normal thyroid cells.
Unfortunately, the IHC data showed heterogenous expression of Sirt1
and Sirt3, which prevented us using the expression of these
proteins to discriminate between cancer cells and normal cells.
Data from the Human Protein Atlas (HPA), a scientific research
program that aims to explore the whole human proteome using an
antibody-based approach, showed heterogeneous expression of Sirt1
and Sirt3 in normal follicular cells and papillary thyroid cancer
cells (43). Consistently, BioGPS
and NCBI Gene Expression Omnibus (GEO) profiles also indicated
heterogeneous expression of Sirt1 and Sirt3 in normal follicular
cells and papillary thyroid cancer cells, as well as breast cancer
cell lines. Additionally, western blot analysis indicated variable
baseline Sirt1 and Sirt3 protein levels in TPC1 (papillary), FTC133
(follicular) and FRO (anaplastic) cells (data not shown). The cell
type-dependent differential induction of Sirt1 and Sirt3 expression
by etoposide suggested that Sirt1 and Sirt3 induction in thyroid
cancer cell lines is related to the response to drug-induced
genotoxic stress. Consistent with this hypothesis, TPC1 cells,
which have the highest induction of the three cell lines, were more
resistant to etoposide treatment than FTC133 and FRO cells, as
demonstrated by Annexin V apoptosis and MTT assays.
To identify the signaling pathway mediating the
resistance to drug-induced genotoxic stress, we performed qRT-PCR
for Bcl2 family proteins such as pro-apoptotic Bax and
antiapoptotic Bcl-xL. The expression of p21, a cyclin-dependent
kinase inhibitor that is induced by p53-dependent and -independent
mechanisms in response to stress, was also assessed by qRT-PCR
(44). Our results showed that
etoposide downregulated pro-apoptotic Bcl2 family proteins and p53
and upregulated p21 expression in TPC1 cells. Previous studies
suggested that p21 suppresses tumor development by inhibiting cell
cycle progression in response to various stimuli. Additionally,
several biochemical and genetic studies have indicated that p21
acts as a master effector of multiple tumor suppressor pathways
that are independent of the classical p53 pathway (44). Despite its known anti-proliferative
role and its ability to promote cellular senescence, recent studies
have suggested that, under certain conditions, p21 can promote
cellular proliferation and oncogenicity (45). Consequently, p21 is often
dysregulated in human cancers, although it has been shown to act as
a tumor suppressor or oncogene depending on the cellular context
and conditions (45,46). Recent studies also suggested that
p21 suppresses the induction of pro-apoptotic genes by MYC and E2F1
through direct binding and inhibition of their transactivation
functions (47). In addition, p21
plays an important role in modulating DNA repair processes by
inhibiting cell cycle progression and allowing DNA repair to
proceed while inhibiting apoptosis. Furthermore, p21 can compete
for PCNA binding with several PCNA-reliant proteins involved
directly in DNA repair processes (48).
To the best of our knowledge, a direct relationship
between Sirt1 and Bax or p21 independently from the p53 pathway has
not been investigated to date. However, Foxp3, which is
deacetylated and degraded by Sirt1, showed a statistically
significant strong positive correlation with Bax and negative
correlation with p21 in our analysis using GeneNetwork. These
results suggested that a Sirt1-Foxp3 signaling axis may be a
signature molecular event in TPC1 cells associated with resistance
against etoposide-induced genotoxic stress (49).
In conclusion, we showed that Sirt1 and Sirt3 are
expressed at different levels in different cell and tissue samples.
Furthermore, the induction of Sirt1 and Sirt3 expression differed
among thyroid cancer cell lines and was correlated with survival
under conditions of genotoxic stress. Our results suggest the
involvement of a Sirt1-Foxp3 signaling pathway in the resistance of
thyroid cancer cells against genotoxic stress. The role of p21 in
cells exposed to genotoxic stress should be addressed in future
studies to identify novel therapeutic targets for the treatment of
RAI-refractory thyroid cancer.
Acknowledgements
This study was supported by a faculty research grant
of Yonsei University College of Medicine (6-2014-0056).
References
|
1
|
Lang BH, Wong KP, Cheung CY, Wan KY and Lo
CY: Evaluating the prognostic factors associated with
cancer-specific survival of differentiated thyroid carcinoma
presenting with distant metastasis. Ann Surg Oncol. 20:1329–1335.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lin JD, Huang MJ, Juang JH, et al: Factors
related to the survival of papillary and follicular thyroid
carcinoma patients with distant metastases. Thyroid. 9:1227–1235.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sabra MM, Dominguez JM, Grewal RK, et al:
Clinical outcomes and molecular profile of differentiated thyroid
cancers with radioiodine-avid distant metastases. J Clin Endocrinol
Metab. 98:E829–E836. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Droz JP, Schlumberger M, Rougier P, Ghosn
M, Gardet P and Parmentier C: Chemotherapy in metastatic
nonanaplastic thyroid cancer: experience at the Institut
Gustave-Roussy. Tumori. 76:480–483. 1990.PubMed/NCBI
|
|
5
|
Schlumberger M: Target therapies for
radioiodine refractory advanced thyroid tumors. J Endocrinol
Invest. 35:40–44. 2012.PubMed/NCBI
|
|
6
|
Salvatore G, De Falco V, Salerno P, et al:
BRAF is a therapeutic target in aggressive thyroid carcinoma. Clin
Cancer Res. 12:1623–1629. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xing M: BRAF mutation in papillary thyroid
cancer: pathogenic role, molecular bases, and clinical
implications. Endocr Rev. 28:742–762. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu D, Hu S, Hou P, Jiang D, Condouris S
and Xing M: Suppression of BRAF/MEK/MAP kinase pathway restores
expression of iodide-metabolizing genes in thyroid cells expressing
the V600E BRAF mutant. Clin Cancer Res. 13:1341–1349. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lito P, Rosen N and Solit DB: Tumor
adaptation and resistance to RAF inhibitors. Nat Med. 19:1401–1409.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Montero-Conde C, Ruiz-Llorente S,
Dominguez JM, et al: Relief of feedback inhibition of HER3
transcription by RAF and MEK inhibitors attenuates their antitumor
effects in BRAF-mutant thyroid carcinomas. Cancer Discov.
3:520–533. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Haigis MC and Guarente LP: Mammalian
sirtuins - emerging roles in physiology, aging, and calorie
restriction. Genes Dev. 20:2913–2921. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Luo J, Nikolaev AY, Imai S, et al:
Negative control of p53 by Sir2alpha promotes cell survival under
stress. Cell. 107:137–148. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Westerheide SD, Anckar J, Stevens SM Jr,
Sistonen L and Morimoto RI: Stress-inducible regulation of heat
shock factor 1 by the deacetylase SIRT1. Science. 323:1063–1066.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rodgers JT, Lerin C, Haas W, Gygi SP,
Spiegelman BM and Puigserver P: Nutrient control of glucose
homeostasis through a complex of PGC-1alpha and SIRT1. Nature.
434:113–118. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rodgers JT, Lerin C, Gerhart-Hines Z and
Puigserver P: Metabolic adaptations through the PGC-1 alpha and
SIRT1 pathways. FEBS Lett. 582:46–53. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Brunet A, Sweeney LB, Sturgill JF, et al:
Stress-dependent regulation of FOXO transcription factors by the
SIRT1 deacetylase. Science. 303:2011–2015. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brachmann CB, Sherman JM, Devine SE,
Cameron EE, Pillus L and Boeke JD: The SIR2 gene family, conserved
from bacteria to humans, functions in silencing, cell cycle
progression, and chromosome stability. Genes Dev. 9:2888–2902.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bishop NA and Guarente L: Genetic links
between diet and lifespan: shared mechanisms from yeast to humans.
Nat Rev Genet. 8:835–844. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Raynes R, Brunquell J and Westerheide SD:
Stress inducibility of SIRT1 and its role in cytoprotection and
cancer. Genes Cancer. 4:172–182. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Verdin E: AROuSing SIRT1: identification
of a novel endogenous SIRT1 activator. Mol Cell. 28:354–356. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gorospe M and de Cabo R: AsSIRTing the DNA
damage response. Trends Cell Biol. 18:77–83. 2008. View Article : Google Scholar
|
|
22
|
Brennan CM and Steitz JA: HuR and mRNA
stability. Cell Mol Life Sci. 58:266–277. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lopez de Silanes I, Zhan M, Lal A, Yang X
and Gorospe M: Identification of a target RNA motif for RNA-binding
protein HuR. Proc Natl Acad Sci USA. 101:2987–2992. 2004.PubMed/NCBI
|
|
24
|
Song NY and Surh YJ: Janus-faced role of
SIRT1 in tumorigenesis. Ann NY Acad Sci. 1271:10–19. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bosch-Presegue L and Vaquero A: The dual
role of sirtuins in cancer. Genes Cancer. 2:648–662. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yuan J, Luo K, Liu T and Lou Z: Regulation
of SIRT1 activity by genotoxic stress. Genes Dev. 26:791–796. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kwon HS, Lim HW, Wu J, Schnolzer M, Verdin
E and Ott M: Three novel acetylation sites in the Foxp3
transcription factor regulate the suppressive activity of
regulatory T cells. J Immunol. 188:2712–2721. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bollag G, Tsai J, Zhang J, et al:
Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat
Rev Drug Discov. 11:873–886. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Turajlic S, Ali Z, Yousaf N and Larkin J:
Phase I/II RAF kinase inhibitors in cancer therapy. Expert Opin
Investig Drugs. 22:739–749. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Brilli L and Pacini F: Targeted therapy in
refractory thyroid cancer: current achievements and limitations.
Future Oncol. 7:657–668. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Poulikakos PI, Zhang C, Bollag G, Shokat
KM and Rosen N: RAF inhibitors transactivate RAF dimers and ERK
signalling in cells with wild-type BRAF. Nature. 464:427–430. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Poulikakos PI, Persaud Y, Janakiraman M,
et al: RAF inhibitor resistance is mediated by dimerization of
aberrantly spliced BRAF(V600E). Nature. 480:387–390. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Montagut C, Sharma SV, Shioda T, et al:
Elevated CRAF as a potential mechanism of acquired resistance to
BRAF inhibition in melanoma. Cancer Res. 68:4853–4861. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Greger JG, Eastman SD, Zhang V, et al:
Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome
acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib,
mediated by NRAS or MEK mutations. Mol Cancer Ther. 11:909–920.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Johannessen CM, Boehm JS, Kim SY, et al:
COT drives resistance to RAF inhibition through MAP kinase pathway
reactivation. Nature. 468:968–972. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Su F, Bradley WD, Wang Q, et al:
Resistance to selective BRAF inhibition can be mediated by modest
upstream pathway activation. Cancer Res. 72:969–978. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nazarian R, Shi H, Wang Q, et al:
Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or
N-RAS upregulation. Nature. 468:973–977. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Atefi M, von Euw E, Attar N, et al:
Reversing melanoma cross-resistance to BRAF and MEK inhibitors by
co-targeting the AKT/mTOR pathway. PLoS One. 6:e289732011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sanchez-Hernandez I, Baquero P, Calleros L
and Chiloeches A: Dual inhibition of (V600E)BRAF and the
PI3K/AKT/mTOR pathway cooperates to induce apoptosis in melanoma
cells through a MEK-independent mechanism. Cancer Lett.
314:244–255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vergani E, Vallacchi V, Frigerio S, et al:
Identification of MET and SRC activation in melanoma cell lines
showing primary resistance to PLX4032. Neoplasia. 13:1132–1142.
2011.PubMed/NCBI
|
|
41
|
Lee MH, Lee SE, Kim DW, et al:
Mitochondrial localization and regulation of BRAFV600E in thyroid
cancer: a clinically used RAF inhibitor is unable to block the
mitochondrial activities of BRAFV600E. J Clin Endocrinol Metab.
96:E19–E30. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lee SE, Lee JU, Lee MH, et al: RAF kinase
inhibitor-independent constitutive activation of Yes-associated
protein 1 promotes tumor progression in thyroid cancer.
Oncogenesis. 2:e552013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Uhlen M, Oksvold P, Fagerberg L, et al:
Towards a knowledge-based Human Protein Atlas. Nat Biotechnol.
28:1248–1250. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zuo S, Liu C, Wang J, et al: IGFBP-rP1
induces p21 expression through a p53-independent pathway, leading
to cellular senescence of MCF-7 breast cancer cells. J Cancer Res
Clin Oncol. 138:1045–1055. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Warfel NA and El-Deiry WS:
p21WAF1 and tumourigenesis: 20 years after. Curr Opin
Oncol. 25:52–58. 2013.PubMed/NCBI
|
|
46
|
Deng C, Zhang P, Harper JW, Elledge SJ and
Leder P: Mice lacking p21CIP1/WAF1 undergo normal
development, but are defective in G1 checkpoint control. Cell.
82:675–684. 1995.
|
|
47
|
Gartel AL and Tyner AL: Transcriptional
regulation of the p21((WAF1/CIP1)) gene. Exp Cell Res. 246:280–289.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Abbas T and Dutta A: p21 in cancer:
intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
van Loosdregt J, Brunen D, Fleskens V,
Pals CE, Lam EW and Coffer PJ: Rapid temporal control of Foxp3
protein degradation by sirtuin-1. PLoS One. 6:e190472011.PubMed/NCBI
|