Introduction
Spindle cell tumors are clinically heterogeneous
neoplasms with similar morphology. The term is descriptive and
based on the tumor cells’ long and slender microscopic appearance.
The tumor may be a true sarcoma (connective tissue cancer) or only
sarcomatoid which means a tumor that looks like a sarcoma under the
microscope (http://www.cancer.org/index). Spindle cell sarcomas
can occur anywhere. They are more common in adults and are treated
primarily with surgery to which is often added adjuvant or
neoadjuvant radiation (1).
Sub-classification of spindle cell sarcomas requires integration of
histology, clinicopathological parameters, immunohistochemistry,
cytogenetics (including fluorescence in situ hybridization)
and/or molecular genetics. When no identifiable differentiation can
be made the diagnosis is undifferentiated spindle cell sarcoma by
exclusion (2). Other tumors are
subdivided depending on the differentiation they show. The latter
group includes leiomyosarcoma (smooth muscle differentiation),
malignant peripheral nerve sheath tumor, fibrosarcoma (fibroblastic
differentiation) and myofibroblastic sarcoma (myofibroblastic
differentiation) among others. In the subgroup with uncertain
differentiation, synovial sarcoma is the most common (1). Cytogenetic and molecular genetic
analyses of spindle cell sarcomas have led to the recognition of
several distinct chromosomal translocations and fusion genes that
characterize specific tumor types. For example, synovial sarcomas
carry the translocation t(X;18)(p11;q11) in more than 95% of the
cases. It results in rearrangement of the SS18 gene in 18q11
with one of the SSX1, SSX2 or SSX4 genes in
Xp11 creating an SS18-SSX1, SS18-SSX2 or
SS18-SSX4 chimeric gene (3). A subset of inflammatory
myofibroblastic tumor, a neoplasm composed of myofibroblastic
spindle cells and infiltrating inflammatory cells, harbors clonal
chromosomal rearrangements involving chromosome band 2p23. These
rearrangements target the ALK gene, serving as the
3′-partner in fusions with various translocation partners and
activating ALK tyrosine kinase function (3). Dermatofibrosarcoma protuberans,
another subtype of spindle cell sarcoma, is characterized
cytogenetically either by supernumerary ring chromosomes (60% of
all examined tumors) or by the translocation t(17;22)(q22;q13)
(http://cgap.nci.nih.gov/Chromosomes/Mitelman). Either
event results in the formation of a COL1A1-PDGFB fusion gene
in which PDGFB exon 1 is deleted and replaced by a variable
segment of the COL1A1 gene (4). Solitary fibrous tumors are
distinguished by their hemangiopericytoma-like branching vascular
pattern, positivity for CD34 by immunohistochemistry and
NAB2-STAT6 fusion (2,5,6).
Further sub-classification of spindle cell sarcomas may in the
future be of value but is currently difficult. Eyden et al
(7) presented five ‘diagnostically
problematic spindle cell sarcomas’ which lacked defined line of
differentiation by light microscopy and showed different clinical
courses. In a correlation study between clinicopathological
features and karyotypes in spindle cell sarcomas, Fletcher et
al (8) reported three
unclassified spindle cell sarcomas, two of which showed a normal
karyotype whereas the third had a complex karyotype with multiple
aberrations. Alaggio et al (9) described two spindle cell tumors with
EWSR1-WT1 fusion gene and favorable prognosis. According to
the authors, these tumors could represent ‘an unrecognized subgroup
of tumors with spindle cell morphology, bearing the same
translocation as desmoplastic small round cell tumor, but
characterized by a more favorable clinical course’ (9). Lestou et al (10) reported a case of spindle cell
sarcoma with a complex karyotype and ring chromosomes. Further
analysis with multicolor fluorescence in situ hybridization
(FISH) probes showed amplification of chromosome 18 and
co-amplification of 12p11 and 12q13~q22 in the ring chromosomes.
Recently, Nord et al (11)
reported a spindle cell sarcoma of the heart with ring chromosome,
amplification of the MDM2 gene and homozygous deletion of
CDKN2A (11).
In the present study, we describe a spindle cell
sarcoma with ring chromosome composed of chromosome 12 material,
several fusion genes mapping to 12q, and amplification of
MDM2.
Materials and methods
Ethics statement
The study was approved by the Regional Committee for
Medical Research Ethics (Regional komité for medisinsk
forskningsetikk Sør-Øst, Norge, http://helseforskning.etikkom.no).
Case report
The patient was a 69-year-old woman who presented
with right-sided chest pain and reduced general condition. Clinical
and radiological work-up revealed a large tumor in the lower lobe
of the right lung. No metastases were detected. After a needle
biopsy, bilobectomy was performed and the tumor was removed with
adequate margins. Macroscopic examination showed a well demarcated
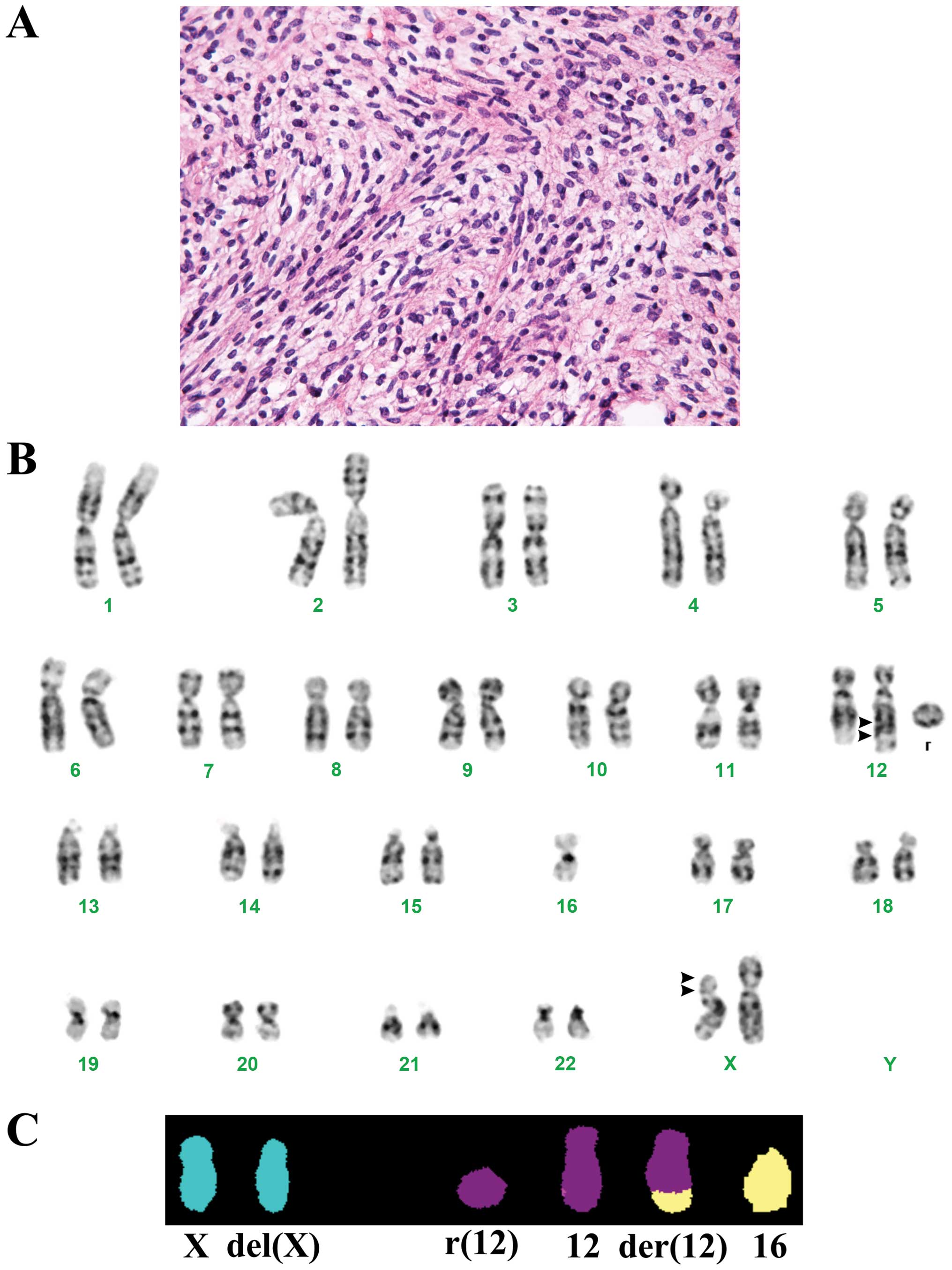
11 cm large tumor. Microscopic examination disclosed a cellular
tumor with spindle cells and some round cells with slight atypia
surrounded by loose intercellular substance and some collagen
(Fig. 1A). No atypical lipoblasts
or pleomorphic cells were seen. There was no necrosis. The mitotic
count was 8/10HPF and the French malignancy grade 2 (2). Immunohistochemical analysis showed
negative staining for AE1/AE3, CK5/6, EMA, CD31, CD34, CD45, CD117,
CD99, actin, smooth-muscle actin, desmin, h-caldesmon, calretinin,
HM B-45 and protein S-100. RT-PCR was negative for gene fusions
consistent with synovial sarcoma. The morphological picture and
immunohistochemical findings were consistent with a spindle cell
sarcoma not otherwise specified. No adjuvant therapy was
administered. Two-and-a-half years after primary surgery a systemic
relapse was diagnosed, with soft tissue metastases in the
retroperitoneum and the large adductor muscle of the left thigh, a
solitary lung metastasis in the right upper lobe, a tumor in the
pancreatic head and bone metastases in the 7th lumbar vertebra and
the left pubic bone. A core needle biopsy of the tumor in the left
thigh confirmed the diagnosis of metastatic spindle cell sarcoma.
No radiotherapy or systemic treatment was given, and the patient
died 15 months later of metastatic disease.
G-banding, karyotyping and multicolor
fluorescence in situ hybridization (mFISH)
A sample from the surgically removed primary tumor
was mechanically and enzymatically disaggregated and short-term
cultured as described elsewhere (12). The culture was harvested and the
chromosomes G-banded using Wright stain. The subsequent cytogenetic
analysis and karyotype description followed the recommendations of
the ISCN (13). For multicolor
FISH the 24XCyte Human Multicolor FISH Probe kit was used
(MetaSystems-international, http://www.metasystems-international.com/) according
to the manufacturer’s protocol. Fluorescent signals were captured
and analyzed using the CytoVision system (Leica Biosystems,
Newcastle, UK).
High-throughput paired-end
RNA-sequencing
Tumor tissue adjacent to that used for cytogenetic
analysis and histological examination had been frozen and stored at
−80°C. Total RNA was extracted from both primary tumor and the
metastasis in the left thigh using TRIzol reagent according to the
manufacturer’s instructions (Life Technologies, Oslo, Norway) and
its quality was checked by Experion Automated Electrophoresis
System (Bio-Rad Laboratories, Oslo, Norway). Three micrograms of
total RNA from the primary tumor were sent for high-throughput
paired-end RNA-sequencing at the Genomics Core Facility, The
Norwegian Radium Hospital (http://genomics.no/oslo/). The RNA was sequenced using
an Illumina HiSeq 2500 instrument and the Illumina software
pipeline was used to process image data into raw sequencing data.
Only sequence reads marked as ‘passed filtering’ were used in the
downstream data analysis. A total of 120 million reads were
obtained. The software FusionMap (release date 2012-04-16) and the
associated pre-built Human B37 and RefGene from the FusionMap
website (http://www.omicsoft.com/fusionmap/) were used for the
discovery of fusion transcripts (14).
Polymerase chain reaction
The primers used for PCR amplification and
sequencing are listed in Table I.
For Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR), 1 μg
of total RNA from both primary tumor and the metastasis was
reverse-transcribed in a 20-μl reaction volume using iScript
Advanced cDNA Synthesis kit for RT-qPCR according to the
manufacturer’s instructions (Bio-Rad). The cDNA was diluted to 50
μl and 2 μl were used as templates in subsequent PCR assays. The 25
μl PCR-volume contained 12.5 μl of Premix Taq (Takara Bio
Europe/SAS, Saint-Germain-en-Laye, France), 2 μl of diluted cDNA
and 0.4 μM of each of the forward and reverse primers. The PCRs
were run on a C-1000 Thermal cycler (Bio-Rad). The PCR conditions
were: an initial denaturation at 94°C for 30 sec followed by 35
cycles of 7 sec at 98°C and 2 min at 68°C, and a final extension
for 5 min at 68°C. The following primer sets were used to detect
the fusion transcripts: PTGES3-416F1/PTPRB-1610R1 for
(PTGES3-PTPRB), HM GA2-982F1/DYRK2-733R1 for
(HMGA2-DYRK2), TMBIM4-25F1/MSRB3-501R for
(TMBIM4-MSRB3) and USP15-655F1/CNTN1-413R for
(USP15-CNTN1).
 | Table IPrimers used for PCR amplification
and sequencing. |
Table I
Primers used for PCR amplification
and sequencing.
| Oligo name | Sequence
(5′→3′) |
|---|
| PTGES3-416F1 | CTC GG A GG A AGT
GAT AAT TTT AAG C |
| PTPRB-1610R1 | CTG TAG CCA TGT ATT
TTC GTC CAG |
| HMGA2-982F1 | CAA GAG TCC CTC TAA
AGC AGC TCA |
| DYRK2-733R1 | CCG TTT TGC CCA CTG
TTG TAA G |
| DYRK2-709R1 | GCC CAT TTG GTT GTG
TCG TGA GCA C |
| DYRK2-651R | TTG AAC CTG GAT CTG
TCC GTG AGC G |
| TMBIM4-25F1 | AGG AGG CGG TTG CGG
TTA GTG |
| MSRB3-501R | TTT CCA CCC TGT GCA
TCC CAT AG |
| USP15-655F1 | CCA GAC AGC ACC ATT
CAG GAT GC |
| CNTN1-413R | CTG GCT CGT GCC CTA
CAG TTG AGT |
|
PTGES3-Int4-F1New | CCT TGG TCA GAA ACG
GAG CTG GTC AA |
|
PTGES3-Int4-F2New | AAT GCT TGC TGC ACT
CCA GCC TGG |
| PTPRB-1602R | CCA TGT ATT TTC GTC
CAG GCA CCA GG |
| PTPRB-1490R | TCC CAT TCT GCT ACA
GG GTC TGC C |
| HMGA2-Int4-F1 | CCT CTG CAC TGT TGG
CAA GAG CAG C |
| HMGA2-Int4-F2 | CGA TTG AGC GTC ATG
GCT GTG CC |
| TMBIM4-55F1New | CGG TAG GGG TGC TGT
TGC CAT CAT G |
|
TMBIM4-121F1New | CGA CTT CAA CTA TGG
CAG CAG CGT GG |
| MSRB3-350R1New | TGC AGT CTC ACT GTG
CTT CCA CAG CTG A |
| MSRB3-261R1New | ACC ACG GTG GCC ATC
TGG CTT ATG TC |
For the detection of the genomic hybrid fragments,
extra-long PCR (XL PCR) was carried out using the XL PCR kit
(Applied Biosystems, Life Technologies). XL PCR was performed in 50
μl of 1:3 diluted 3.3X XL buffer II, 1.1 mM Mg(OAc)2,
0.2 mM of each dNTP, 1 unit of rTth DNA polymerase, XL, 0.4 μM of
each of the forward and reverse primers (Table I) and 300 ng tumor DNA (or 2 μl of
the first PCR in nested PCR). After an initial denaturation for 1
min at 94°C, 32 cycles of 15 sec at 94°C and 10 min at 68°C were
run followed by a final extension for 10 min at 72°C. For the
detection of genomic PTGES3-PTPTRB fusion DNA fragment, the
primers PTGES3-Int4-F1New and PTTRB-1602R were used. For possible
genomic HMGA2-DYRK2 and TMBIM4-MSRB3, nested XL PCR
was performed. For genomic HMGA2-DYRK2 amplification, the
primers HMG A2-Int4-F1/DYRK2-709R1 and HMG A2-Int4-F2/DYRK2-651R
were used for first and nested PCR, respectively. For genomic
TMBIM4-MSRB3 amplification, the primers
TMBIM4-55F1New/MSRB3-350R1New and TMBIM4-121F1New/MSRB3-261R1New
were used for first and nested PCR, respectively.
Four microliters of the PCR products were stained
with GelRed (Biotium, Hayward, CA, USA), analyzed by
electrophoresis through 1.0% agarose gel and photographed. The
amplified fragments were excised from the gel, purified using the
Qiagen gel extraction kit (Qiagen), and direct sequencing (Sanger
sequencing) was performed using the light run sequencing service of
GATC Biotech (http://www.gatc-biotech.com/en/sanger-services/lightrun-sequencing.html).
The BLAST software (http://blast.ncbi.nlm.nih.gov/Blast.cgi) was used for
computer analysis of sequence data.
MDM2 amplification
Genomic tumor DNA was extracted from both primary
tumor and metastasis paraffin-embedded samples using QIAamp DNA
FFPE Tissue kit as recommended by the manufacturer (Qiagen, Hilden,
Germany). Real-time PCR was performed using the ABI
PRISM® 7900HT Sequence Detection System (Life
Technologies, Carlsbad, CA). Primers and probes for MDM2 (on
12q15), CDK4 (on 12q14.1) and ALB (on 4q13.3, reference
gene) were as described by Hostein et al (15). PCR amplification was performed in
duplicates in 25 μl reaction volume containing 12 ng of genomic
DNA, 300 nM of each primer, 100 nM of the probe and Ix TaqMan
Universal PCR Master Mix (Life Technologies). For the amplification
of ALB, MgCl2 was added to a final concentration
of 400 nM. The gene copy numbers were calculated from standard
curves constructed by amplification of normal blood DNA of the
target genes MDM2 and CDK4 and the reference gene
ALB. The level of amplification was determined as copy
number of target gene (MDM2 or CDK4)/copy number of
reference gene (ALB). A pool of six paraffin-embedded
lipomas (confirmed by karyotyping) was used as normal control and
the osteosarcoma cell line SJSAI (ATCC CRL-2098) as a positive
control cell line.
Results
The G-banding and M-FISH analyses yielded the
karyotype 46,X,del(X)(p?11p?22), der(12)(12pter→12q?22::12q?15→q?
22::16p11→16pter),−16,+r(12)[23]
(Fig. 1B and C). Using the
FusionMap on the raw RNA sequencing data, approximately 500
potential fusion genes were found (data not shown). The
PTGES3-PTPRB, HMGA2-DYRK2, USP15-CNTN1 and
TMBIM4-MSRB3 fusion transcripts were ranked 1st with 60 seed
counts, 3rd with 27 seed counts, 4th with 26 seed counts and 5th
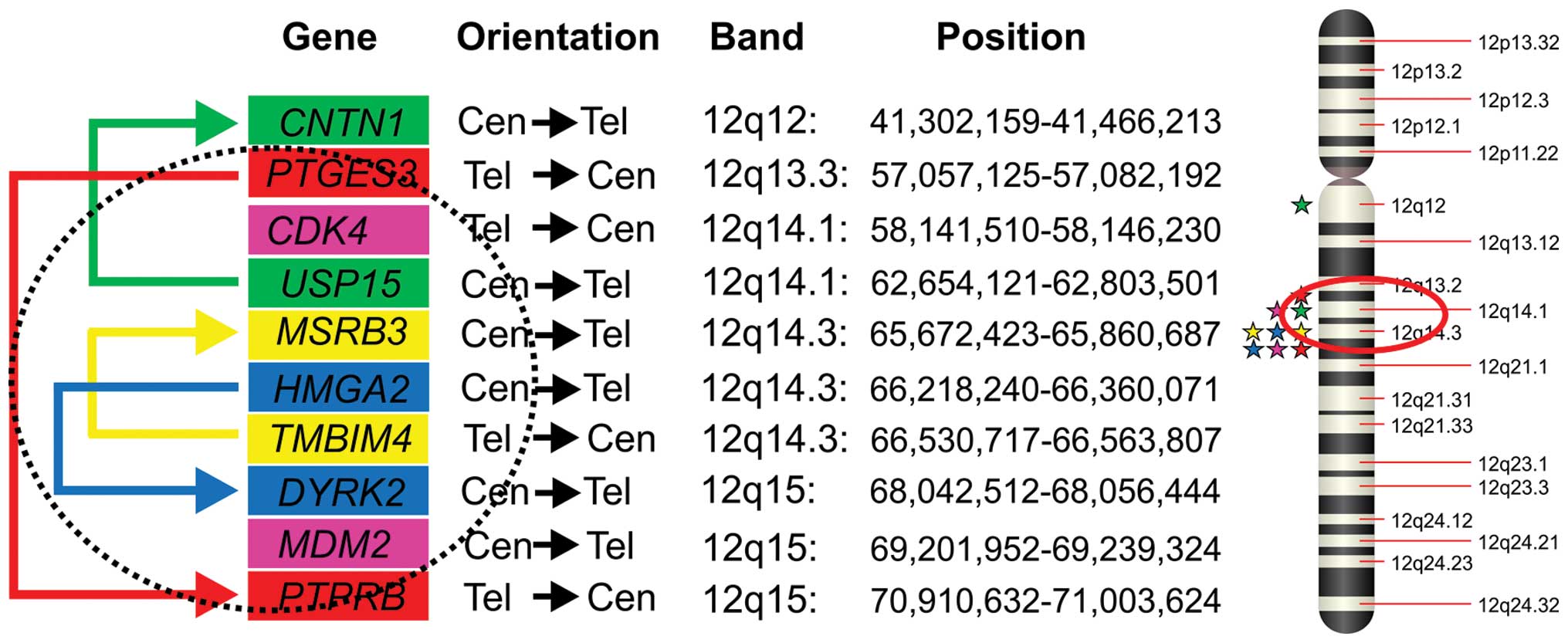
with 22 seed counts, respectively. We decided to investigate
further these fusion genes in materials obtained from both the
primary tumor and the metastasis because: i) all the partner genes
in the above-mentioned fusion transcripts map to the q arm of
chromosome 12 (Fig. 2) and ii) the
karyotype of the primary tumor had a ring chromosome composed of
chromosome 12 material and a translocation involving chromosomes 12
and 16. A potential read through sequence, C10orf68-CCDC7,
ranked 2nd with 50 seed counts, was not further studied.
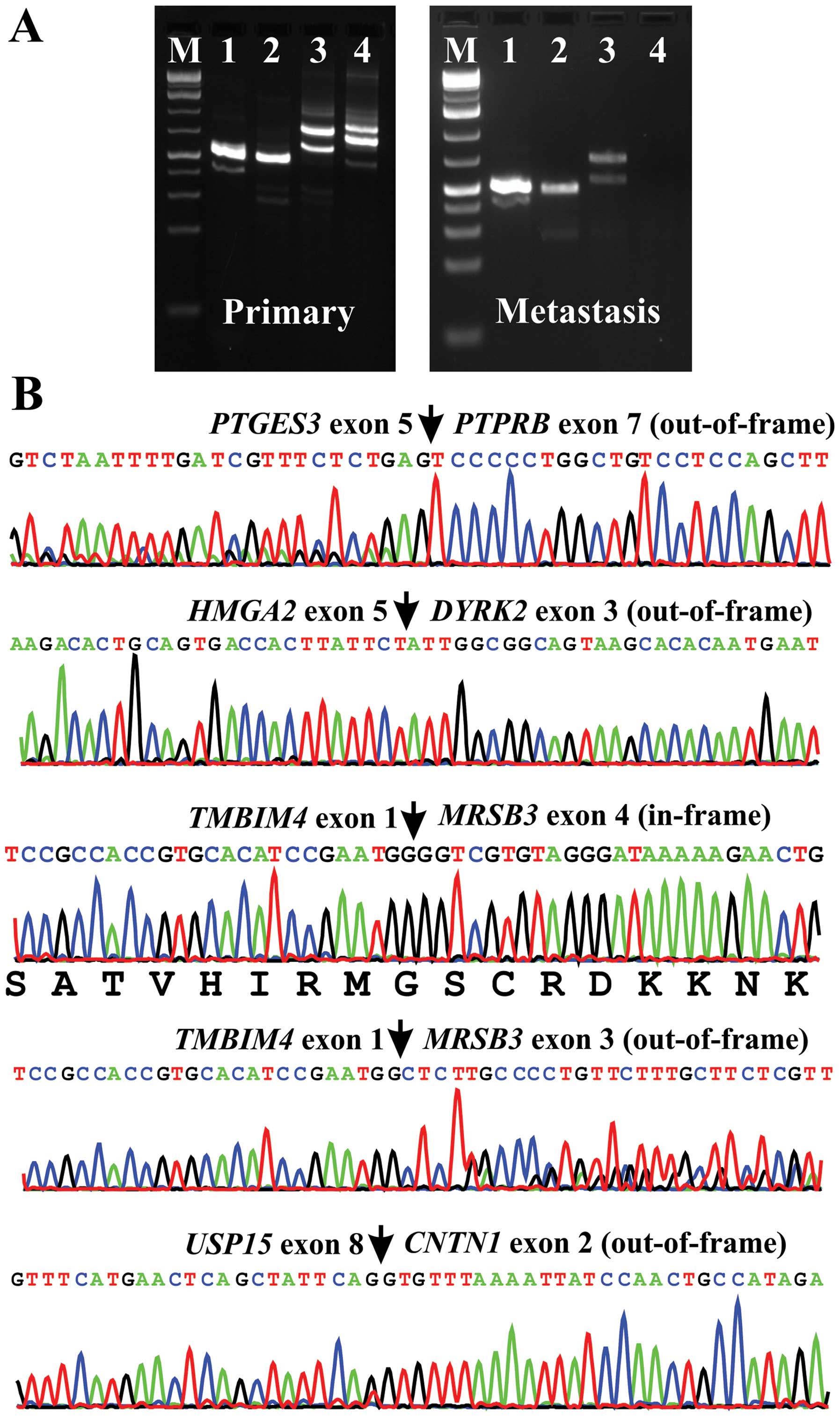
RT-PCR using cDNA from the primary tumor and
subsequent direct Sanger sequencing verified the presence of
PTGES3-PTPRB, HMGA2-DYRK2, TMBIM4-MSRB3 and
USP15-CNTN1 chimeric transcripts (Fig. 3). PCR analysis of the metastasis
amplified the fusion transcripts PTGES3-PTPRB,
HMGA2-DYRK2 and TMBIM4-MSRB3 but not
USP15-CNTN1 (Fig. 3A).
Sanger sequencing of the amplified products from the metastasis
showed that they were identical to those amplified from the primary
tumor.
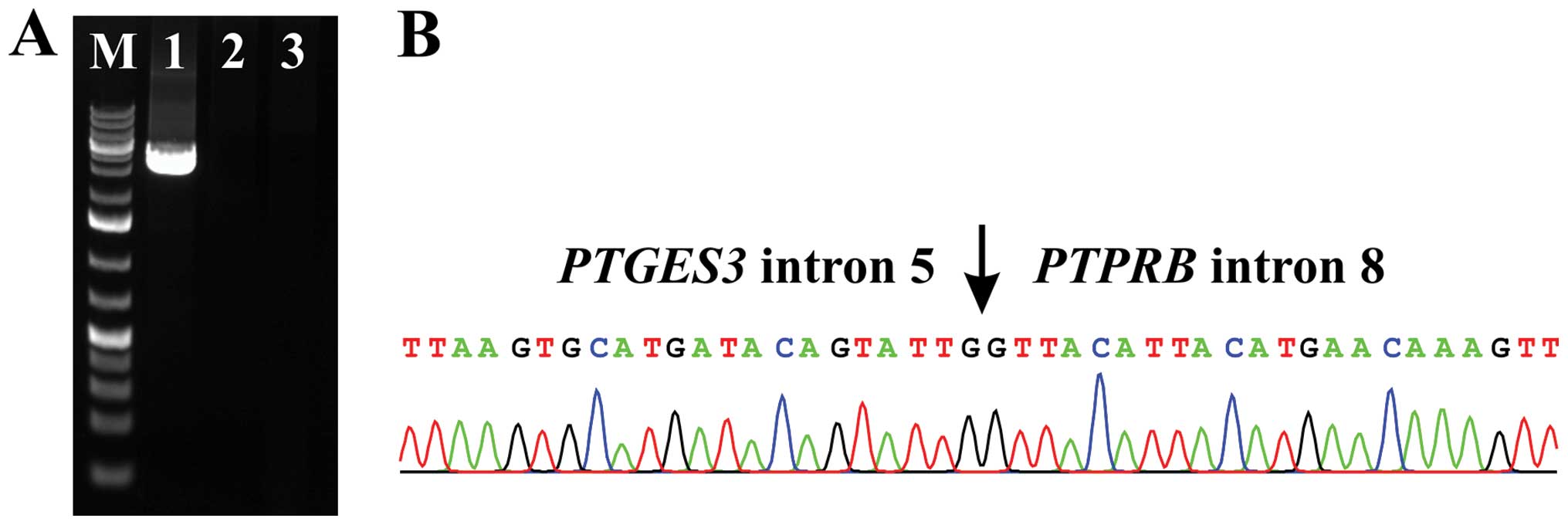
XL PCR using the primers PTGES3-Int4-F1New and
PTPRB-1602R amplified a genomic DNA fragment (Fig. 4A) that was a chimeric
PTGES3-PTPRB genomic DNA with the breakpoints located in
intron 5 of PTGES3 and intron 8 of PTPRB (Fig. 4B). XL PCR for amplification of
genomic breakpoints of HMGA2-DYRK2 and TMBIM4-MSRB3
with the primers given above did not amplify any products (Fig. 4A). XL PCR with additional primer
sets failed to amplify genomic hybrid HMGA2-DYRK2 and
TMBIM4-MSRB3 fragments (data not shown).
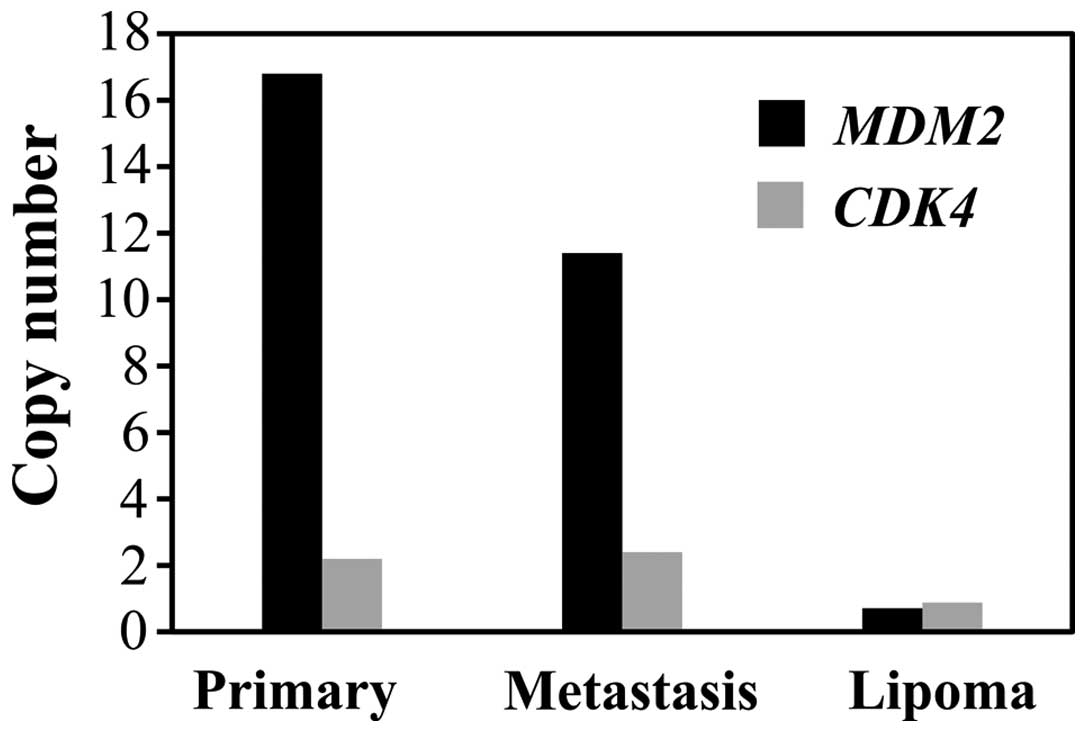
In the primary tumor, the copy number of MDM2
was 17.2 whereas the copy number of CDK4 was 2.2 (Fig. 5). In the metastasis, the copy
number was 11.4 and 2.4 for MDM2 and CDK4,
respectively. The gene copy number (mean levels) of lipoma was 0.8
and 0.9 for MDM2 and CDK4, respectively.
Discussion
The spindle cell sarcoma we describe had a ring
chromosome composed of material from chromosome 12. The molecular
studies of primary tumor and metastatic cells showed both
amplification of MDM2 and several fusion genes (four -
PTGES3-PTPRB, HMGA2-DYRK2, TMBIM4-MSRB3 and
USP15-CNTN1 in the primary tumor, whereas the first three,
but not USP15-CNTN1 were present also in the metastasis),
all of them located on 12q (Fig.
2). This suggests that USP15-CNTN1 was not expressed or
was absent in the metastasis. MDM2 amplification and the
fusion transcripts PTGES3-PTPRB, HMGA2-DYRK2,
TMBIM4-MSRB3 were found both in the primary tumor and the
metastasis examined, showing that the same clone set up these two
tumor lesions. Which molecular change is the primary, or most
primary, cannot be deduced from the data, but a role in initiation
and/or progression is possible and indeed likely. The multiple
copies of MDM2 are not only correlated with the occurrence
of ring chromosomes (16) but are
located on the rings (17,18). It is reasonable to assume that in
the present tumor both MDM2 amplification and the fusion
genes PTGES3-PTPRB, HMGA2-DYRK2, TMBIM4-MSRB3
and USP15-CNTN1 are located on the ring chromosome (Fig. 2) although lack of suitable material
prevented us from determining this with certainty. The absence of
USP15-CNTN1 in the metastasis could reflect the instability
of ring chromosomes. In the course of tumor progression, the
ring(s) may break and re-join to form new rings. Thus, the rings in
daughter cells can be different from each other and from the ring
of the mother cell (19).
With exception of HMGA2 and its multiple
fusion genes in neoplasia (http://cgap.nci.nih.gov/Chromosomes/MSearchForm), none
of the other three fusion genes we detected have been reported
before in cancer (http://cgap.nci.nih.gov/Chromosomes/MSearchForm). The
fusion transcripts are all out-of frame except one of the two
alternative splicing transcripts of TMBIM4-MRSB3 (Fig. 3). This suggests that the fusions
play a role in the expression and/or regulation of genes by
swapping promoter or 3′-untranslated region (UTR) and not by the
formation of fusion proteins.
In HMGA2-DYRK2 the fusion of HMGA2
with DYRK2 occurs at exon 5, 80 bp after the stop codon
(position 1221 in sequence with accession number NM_003483.4) and
exon 3 of DYRK2 (position 601 in sequence with accession
number NM_006482.2 or the position 452 of sequence with accession
number NM_003583). Thus, only one of the eight Let-a target
sites of the HMGA2 3′-UTR is present in the
HMGA2-DYRK2 fusion transcript (the one in position 1169 in
sequence with accession number NM_003483.4) (20). DYRK2 belongs to a family of protein
kinases whose members are presumed to be involved in cellular
growth and/or development. The family is defined by structural
similarity of their kinase domains and their capacity to
autophosphorylate on tyrosine residues. DYRK2 has demonstrated
tyrosine autophosphorylation and catalyzed phosphorylation of
histones H3 and H2B in vitro (http://www.ncbi.nlm.nih.gov/nuccore/Nm_006482). In the
HMGA2-DYRK2 fusion transcript, the part of DYRK2
coding the entire isoform 1 of DYRK2 protein (accession number
NP_003574.1) is also present. It is therefore possible that the
entire protein-encoding moiety of DYRK2 is expressed either
alone or as part of a bicistronic transcript encoding both HM GA2
and DYRK2 proteins under the influence of the HMGA2
promoter. Bicistronic transcripts encoding two independent proteins
have been reported in humans (21,22),
and a bicistronic CCND1-TROP2 mRNA chimera was even shown to
have an oncogenic role in human cancer (23). The chimeric CCND1-TROP2
mRNA, which independently translates both the cyclin D1 and TROP2
proteins, was isolated from human ovarian and mammary cancer cells
and was found to be expressed in gastrointestinal, ovarian and
endometrial tumors (23).
In PTGES3-PTPRB, the fusion of PTGES3
with PTPRB occurs on exon 5 (position 670 in the sequence
with accession number NM_006601.5) and exon 7 of PTPRB
(position 1427 in the sequence with accession number NM_002837.4).
PTGES3 functions as a chaperone which is required for proper
function of the glucocorticoid and other steroid receptors
(http://en.wikipedia.org/wiki/PTGES3).
PTPRB is a member of the protein tyrosine phosphatase (PTP) family.
PTPs are signaling molecules that regulate a variety of cellular
processes including cell growth, differentiation, mitotic cycle and
oncogenic transformation. This PTP belongs to the receptor type PTP
and contains an extracellular domain, a single transmembrane
segment, and an intracytoplasmic catalytic domain (http://en.wikipedia.org/wiki/PTPRB). The chimeric
PTGES3-PTPRB transcript contains the part of PTGES3
which codes for the putative Hsp90 binding site (conserved domain
cd00237) and the part of PTPRB which catalyzes the
dephosphorylation of phosphotyrosine peptides (conserved domain
cd00047). The translation of a hypothetical bicistronic
PTGES3-PTPRB fusion transcript would result in the
production of two independent PTGES3 and PTPRB proteins with the
above-mentioned functional domains.
The MRSB3 gene codes for a protein which
catalyzes the reduction of methionine sulphoxide to methionine
(http://www.ncbi.nlm.nih.gov/gene/253827). This enzyme
acts as a monomer and requires zinc as a cofactor. Four transcript
variants have been identified encoding two different isoforms of
the MRSB3 protein. Transcript variant 1 (accession number
NM_198080) encodes the longer isoform 1 of MRSB3 and is found in
the endoplasmic reticulum (ER), whereas transcript variants 2, 3
and 4 encode the same isoform 2 variant of MRSB3 which has shorter
and distinct N-terminus compared to isoform 1 and is found
primarily in mitochondria (24).
The result of TMBIM4-MRSB3 fusion is that the
expression of MRSB3 comes under the control of the
TMBIM4 promoter. In the out-of-frame TMBIM4-MRSB3
fusion transcript in which exon 1 of TMBIM4 (nt 173 in the
sequence with accession number NM_016056.2) is fused to exon 3 of
MSRB3 (nt 388 in the sequence with accession number
NM_001193460.1), the result would be the production of the isoform
2 variant of MRSB3 which is localized to mitochondria. In the
in-frame TMBIM4-MRSB3 fusion transcript in which exon 1 of
TMBIM4 (nt 173 in the sequence with accession number
NM_016056.2) is fused to exon 4 of MSRB3 (nt 515 in the
sequence with accession number NM_001193460.1), the result would be
a chimeric TMBIM4-MRSB3 protein in which the first 32 amino acids
of MRSB3 (MSPRRTLPRPLSLCLSLCLCLCLAAA LG SAQS) are replaced by the
first 32 amino acids of TMBIM4
(MADPDPRYPRSSIEDDFNYGSSVASATVHIRM).
In a recent study, Nord et al (11) divided ring chromosomes in
neoplastic cells into those with MDM2 amplification and
those without MDM2 amplification. The amplification of
MDM2 is associated with co-amplification of a variety of
potential driver oncogenes. Here we show that MDM2
amplification may be associated with a variety of fusion genes. The
sequence in which the various genetic events occur cannot be
determined. The fact that both MDM2 amplification and the
fusion genes are found in both the primary tumor and metastasis
indicate, however, that both play a role in tumorigenesis.
Acknowledgements
The authors thank Kristin Andersen for her technical
help. This study was supported by grants from the Norwegian Cancer
Society.
References
|
1
|
Collini P, Sorensen PH, Patel S, et al:
Sarcomas with spindle cell morphology. Semin Oncol. 36:324–337.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fletcher CD, Bridge JA, Hogendoorn P and
Mertens F: WHO Classification of Tumours of Soft Tissue and Bone.
4th edition. IARC; Lyon: 2013
|
|
3
|
Heim S and Mitelman F: Cancer
Cytogenetics. 3rd edition. Wiley-Blackwell; New York: 2009
|
|
4
|
Sirvent N, Maire G and Pedeutour F:
Genetics of dermatofibrosarcoma protuberans family of tumors: from
ring chromosomes to tyrosine kinase inhibitor treatment. Genes
Chromosomes Cancer. 37:1–19. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chmielecki J, Crago AM, Rosenberg M, et
al: Whole-exome sequencing identifies a recurrent NAB2-STAT6 fusion
in solitary fibrous tumors. Nat Genet. 45:131–132. 2013. View Article : Google Scholar
|
|
6
|
Robinson DR, Wu YM, Kalyana-Sundaram S, et
al: Identification of recurrent NAB2-STAT6 gene fusions in solitary
fibrous tumor by integrative sequencing. Nat Genet. 45:180–185.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Eyden BP, Banerjee SS, Harris M and Mene
A: A study of spindle cell sarcomas showing myofibroblastic
differentiation. Ultrastruct Pathol. 15:367–378. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fletcher CD, Dal Cin P, de Wever I, et al:
Correlation between clinicopathological features and karyotype in
spindle cell sarcomas. A report of 130 cases from the CHAMP study
group. Am J Pathol. 154:1841–1847. 1999. View Article : Google Scholar
|
|
9
|
Alaggio R, Rosolen A, Sartori F, et al:
Spindle cell tumor with EWS-WT1 transcript and a favorable clinical
course: a variant of DSCT, a variant of leiomyosarcoma, or a new
entity? Report of 2 pediatric cases. Am J Surg Pathol. 31:454–459.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lestou VS, O’Connell JX, Ludkovski O,
Gosling H, Lesack D and Horsman DE: Coamplification of 12p11 and
12q13 approximately q22 in multiple ring chromosomes in a spindle
cell sarcoma resolved by novel multicolor fluorescence in situ
hybridization analysis. Cancer Genet Cytogenet. 139:44–47. 2002.
View Article : Google Scholar
|
|
11
|
Nord KH, Macchia G, Tayebwa J, et al:
Integrative genome and transcriptome analyses reveal two distinct
types of ring chromosome in soft tissue sarcomas. Hum Mol Genet.
23:878–888. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mandahl N: Methods in solid tumour
cytogenetics. Human Cytogenetics: Malignancy and Acquired
Abnormalities. Rooney DE: Oxford University Press; New York: pp.
165–203. 2001
|
|
13
|
Schaffer LG, Slovak ML and Campbell LJ:
ISCN 2009: An International System for Human Cytogenetic
Nomenclature. Karger; Basel: 2009
|
|
14
|
Ge H, Liu K, Juan T, Fang F, Newman M and
Hoeck W: FusionMap: detecting fusion genes from next-generation
sequencing data at base-pair resolution. Bioinformatics.
27:1922–1928. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hostein I, Pelmus M, Aurias A, Pedeutour
F, Mathoulin-Pelissier S and Coindre JM: Evaluation of MDM2 and
CDK4 amplification by real-time PCR on paraffin wax-embedded
material: a potential tool for the diagnosis of atypical lipomatous
tumours/well-differentiated liposarcomas. J Pathol. 202:95–102.
2004. View Article : Google Scholar
|
|
16
|
Nilbert M, Rydholm A, Willen H, Mitelman F
and Mandahl N: MDM2 gene amplification correlates with ring
chromosome in soft tissue tumors. Genes Chromosomes Cancer.
9:261–265. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gisselsson D, Höglund M, Mertens F,
Mitelman F and Mandahl N: Chromosomal organization of amplified
chromosome 12 sequences in mesenchymal tumors detected by
fluorescence in situ hybridization. Genes Chromosomes Cancer.
23:203–212. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pedeutour F, Forus A, Coindre JM, et al:
Structure of the supernumerary ring and giant rod chromosomes in
adipose tissue tumors. Genes Chromosomes Cancer. 24:30–41. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gisselsson D, Pettersson L, Höglund M, et
al: Chromosomal breakage-fusion-bridge events cause genetic
intratumor heterogeneity. Proc Natl Acad Sci USA. 97:5357–5362.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee YS and Dutta A: The tumor suppressor
microRNA let-7 represses the HM GA2 oncogene. Genes Dev.
21:1025–1030. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gray TA, Saitoh S and Nicholls RD: An
imprinted, mammalian bicistronic transcript encodes two independent
proteins. Proc Natl Acad Sci USA. 96:5616–5621. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ritchie H and Wang LH: A mammalian
bicistronic transcript encoding two dentin-specific proteins.
Biochem Biophys Res Commun. 231:425–428. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guerra E, Trerotola M, Dell’Arciprete R,
et al: A bicistronic CYCLIN D1-TROP2 mRNA chimera demonstrates a
novel oncogenic mechanism in human cancer. Cancer Res.
68:8113–8121. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim HY and Gladyshev VN: Methionine
sulfoxide reduction in mammals: characterization of
methionine-R-sulfoxide reductases. Mol Biol Cell. 15:1055–1064.
2004. View Article : Google Scholar : PubMed/NCBI
|