1. Introduction
As a staple approach for cancer treatment, radiation
therapy plays a critical role in local disease control. When
combined with chemotherapy (i.e., chemoradiation), radiation
provides additional benefits, which give better disease control and
significantly improve cancer patient survival (1–3).
However, radioresistance and the presence of residual disease after
radiation therapy remain major problems that result in the loss of
the therapy effectiveness (4–7).
Currently, there is no clinical approach available either for
predicting the benefit of radiation therapy for individual cancer
patients or for radiosensitization of cancer cells. Thus, an
improved understanding of the mechanisms that promote cancer cell
survival after radiation could allow pharmacological strategies to
be developed to improve the efficacy of radiation therapy.
Radiation exposure induces numerous cellular
signaling pathways, which can lead to cellular responses including
apoptosis, cellular senescence and cell cycle checkpoint
activation/DNA repair (8). Among
the radiation-induced pro-survival signaling pathways, some are
involved in inducing cell cycle arrest and promoting DNA repair,
while others are engaged in suppressing apoptosis induction
(9,10). These pathways act synergistically
to protect cancer cells from the cytotoxic effects of radiation,
ultimately leading to the development of radioresistance. This
review summarizes the signaling pathways that positively contribute
to cancer cell survival in response to ionizing radiation.
2. HER (also called ERBB or EGFR)
signaling
The HER family of receptor tyrosine kinases (RTKs)
consists of HER1, HER2, HER3 and HER4, which localize on the cell
membrane (11). HER RTKs share a
similar protein structure that contains an extracellular region
(ligand binding and dimerization domains), a transmembrane region
and an intracellular region (protein tyrosine kinase domain and
phosphorylation regulatory tail) (12). Among HER receptors, HER2 has no
known ligand and HER3 possesses very low kinase activity (12). Binding of ligands to the ligand
binding domain of HER1, HER3 and HER4 results in homo- or
hetero-dimerization of the receptors followed by
trans-phosphorylation of several tyrosines in the c-terminal
regulatory tail of the receptor (12). The phosphorylated tyrosines form
docking sites for downstream adaptors and signal transducers,
activating downstream signaling pathways including PI3K/AKT,
RAS/RAF/MEK/ERK, phospholipase C-γ/protein kinase C and JAK/STAT
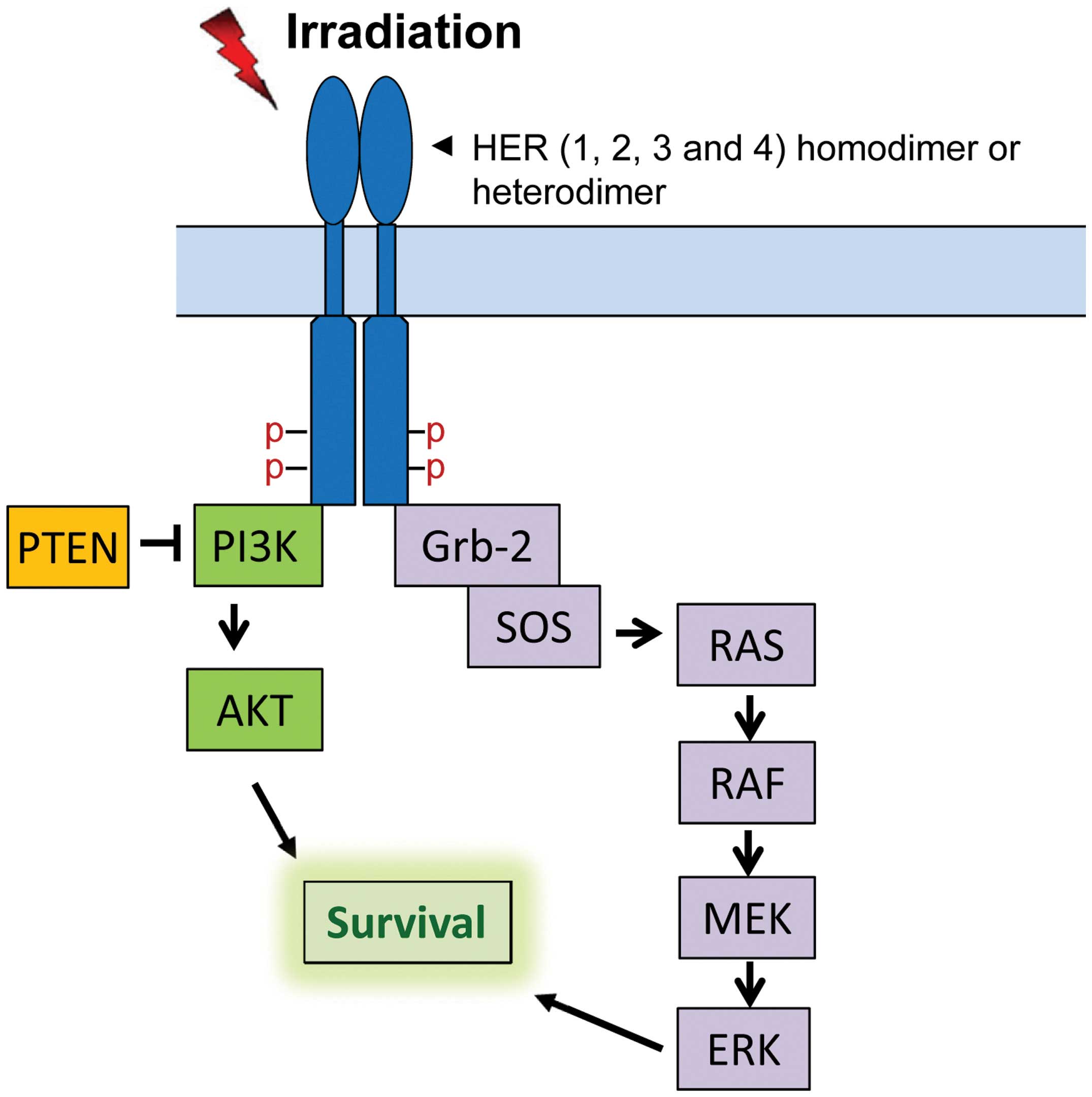
pathways (13,14). Among those pathways, PI3K/AKT and
RAS/RAF/MEK/ERK cascades have been shown to play important roles in
cell survival after radiation (Fig.
1) (15).
An increase in HER1 phosphorylation, indicative of
HER activation, following ionizing radiation has been reported
previously (16–18). Our most recent study in human
breast cancer cells demonstrates that ionizing radiation results in
an increase in phosphorylation of not only HER1, but also HER2,
HER3 and HER4 (19). Although the
mechanisms responsible for this phosphorylation of HER receptors
has not yet been determined, previous studies have shown that
receptor protein tyrosine phosphatases (PTPs), which suppress HER
RTK phosphorylation, can effectively be inhibited by reactive
oxygen/nitrogen species (ROS/RNS) through oxidation (20). Previous studies have also
demonstrated that radiation induces ROS/RNS production via a
mitochondria-dependent mechanism (21). Thus, the ROS/RNS production in
response to radiation could lead to the inhibition of PTPs,
resulting in the activation of HER RTKs. Future studies will be
needed to examine this possibility for the activation of HER RTKs
following radiation.
Inhibition of HER RTKs has been shown to increase
the radiosensitivity of cancer cells. Inhibition of HER RTKs by HER
pan-inhibitor CI-1033 notably enhances the radiosensitivity of
human colon carcinoma cells both in vitro and in vivo
(22), while HER1 inhibition by
gefitinib and HER2 inhibition by herceptin, respectively,
radiosensitizes EGFR amplified glioma cells and breast cancer cells
(23,24). Generally, the pro-survival function
of HER receptors involves at least two possible mechanisms. The
first mechanism is based on the capability of HER receptors to
activate AKT and ERK1/2 signaling, which play critical roles in
suppressing apoptosis (15).
Another possible mechanism for the pro-survival function of HER
receptors is through their regulation of the cell cycle checkpoint
response and DNA repair. In our recent study, we found that HER2
activation following radiation is necessary for the activation of
the G2/M cell cycle checkpoint response (19). In addition, HER1 has been reported
to promote the activation of DNA-dependent protein kinase (DNA-PK),
which plays an essential role in the NHEJ-mediated repair of DNA
double-strand breaks (DSBs) (25,26).
3. Extracellular signal-regulated kinase
(ERK1/2) pathway
In a wide variety of cell types, ionizing radiation
induces rapid activation of MAPK family members, including ERK1/2,
JNK and p38 (27,28). Among those, radiation-induced
ERK1/2 signaling activation has been shown to play an important
role in promoting cell survival in response to radiation (29–31).
Following radiation, ERK1/2 is activated through
dual tyrosine and threonine phosphorylation by MEK1/2 and the
activation, in turn, leads to the phosphorylation/activation of
over 160 substrates (32). Some of
these substrates are transcription factors that regulate the
expression of genes encoding for anti-apoptotic proteins (27,32).
The best characterized antiapoptotic transcription factors targeted
by ERK1/2 signaling are the cyclic AMP-responsive element binding
protein (CREB) and CAAT/enhancer binding protein β (C/EBP-β). In
response to radiation, ERK1/2 phosphorylates/activates
p90rsk kinase, which in turn activates CREB and C/EBP-β,
thereby inducing the expression of a number of anti-apoptotic
proteins including Bcl-xL, Mcl-1 and c-FLIPs (33–35).
In addition, ERK1/2 can directly phosphorylate and inhibit a number
of pro-apoptotic proteins, including Bad, Bim and caspase 9
(36–39). Thus, by increasing the
expression/activity of anti-apoptotic proteins and inhibiting the
activity of pro-apoptotic proteins, the net effect of the
radiation-induced ERK1/2 signaling activation is the suppression of
apoptosis in irradiated cells.
Studies from our group and others have demonstrated
that ERK1/2 signaling activation after radiation is essential for
activation of the G2/M cell cycle checkpoint in response to
radiation (29,31,40–42).
Radiation-induced ERK1/2 signaling is required for the activation
of key regulators of the G2 checkpoint, most notably ATR and BRCA1
(31,42). ERK1/2 signaling also plays an
important role in promoting DNA repair. Radiation-induced ERK1/2
signaling has been associated with the transcriptional upregulation
of genes involved in DNA repair, such as ERCC1, XRCC1 and
XPC (43,44). Furthermore, ERK1/2 signaling can
activate DNA-PK, which plays a critical role in NHEJ-mediated DSB
repair, and PARP-1, which recognizes single-stranded DNA breaks
(SSBs) on the damaged DNA (44–47).
Also, ERK1/2 signaling functions as a positive regulator of ataxia
telangiectasia mutated (ATM)-dependent homologous recombination
(HR) DSB repair (48). Thus, by
promoting G2/M cell cycle checkpoint activation and increasing DNA
repair, ERK1/2 signaling positively regulates cancer cell survival
following radiation. Consistent with these observations, an
increasing number of studies demonstrate that constitutive
activation of Ras results in an increase in the radioresistance of
cancer cells, whereas inhibition of MEK or ERK leads to the
radiosensitization of cancer cells (29,40,41,49).
While the exact mechanisms responsible for the
activation of ERK1/2 signaling by radiation has not yet been
clearly elucidated, several signaling mechanisms have been proposed
to be involved in this activation. As demonstrated by us and
others, the rapid activation of HER family receptors following
ionizing radiation contributes to ERK1/2 signaling activation in
cancer cells of the breast and lung (17). Furthermore, this role of HER
receptors involves Ras GTPase. An activation of Ras in response to
HER receptor activation (mainly HER1 and HER2) has been
demonstrated and ectopic expression of Ras-N17 dominant negative
mutant abolishes the ERK1/2 activation by radiation (50,51).
Via recruitment of Grb-2 to the activated HER receptors, Grb-2
becomes activated and forms a complex with SOS protein, which
triggers the activation of Ras/Raf/MEK/ERK signaling (Fig. 1) (50,51).
Furthermore, the activated Ras can induce HER1-ligand production,
which, through an autocrine feedback loop, further activates HER1
and then Ras/Raf/MEK/ERK signaling (52,53).
Another mechanism implicated in radiation-induced ERK1/2 activation
involves the tumor suppressor BRCA1. Studies from our laboratory
show that decreasing BRCA1 expression in breast cancer cells using
shRNA markedly diminishes the activation of ERK1/2 signaling after
radiation (42). Conversely,
inhibition of ERK1/2 signaling using pharmacological inhibitors or
siRNA also results in the destabilization of BRCA1 protein in
irradiated breast cancer cells (42). These results suggest a positive
feedback loop involving ERK1/2 and BRCA1 in response to ionizing
radiation. Lastly, the DNA damage sensor ATM has also been
implicated in radiation-induced ERK1/2 activation (48). ERK1/2 activation following
radiation has been shown to require ATM, as ATM inhibition
partially blocks the radiation-induced ERK1/2 activation (48). Conversely, inhibition of ERK1/2
signaling can also attenuate radiation-induced ATM phosphorylation,
as well as the recruitment of ATM to DNA damage foci (48). These studies suggest another
positive feedback loop in the radiation response, this time
involving ATM and ERK1/2. Collectively, these studies indicate that
the activation of ERK1/2 signaling in response to radiation is
regulated by multiple inter-regulated signaling pathways.
4. AKT signaling pathway
The AKT signaling pathway plays a vital role in cell
survival. Aberrant activation of this signaling cascade has been
detected in various types of malignancies and is associated with
tumorigenesis (54). AKT functions
as a pro-survival factor by inhibiting apoptotic signal cascades
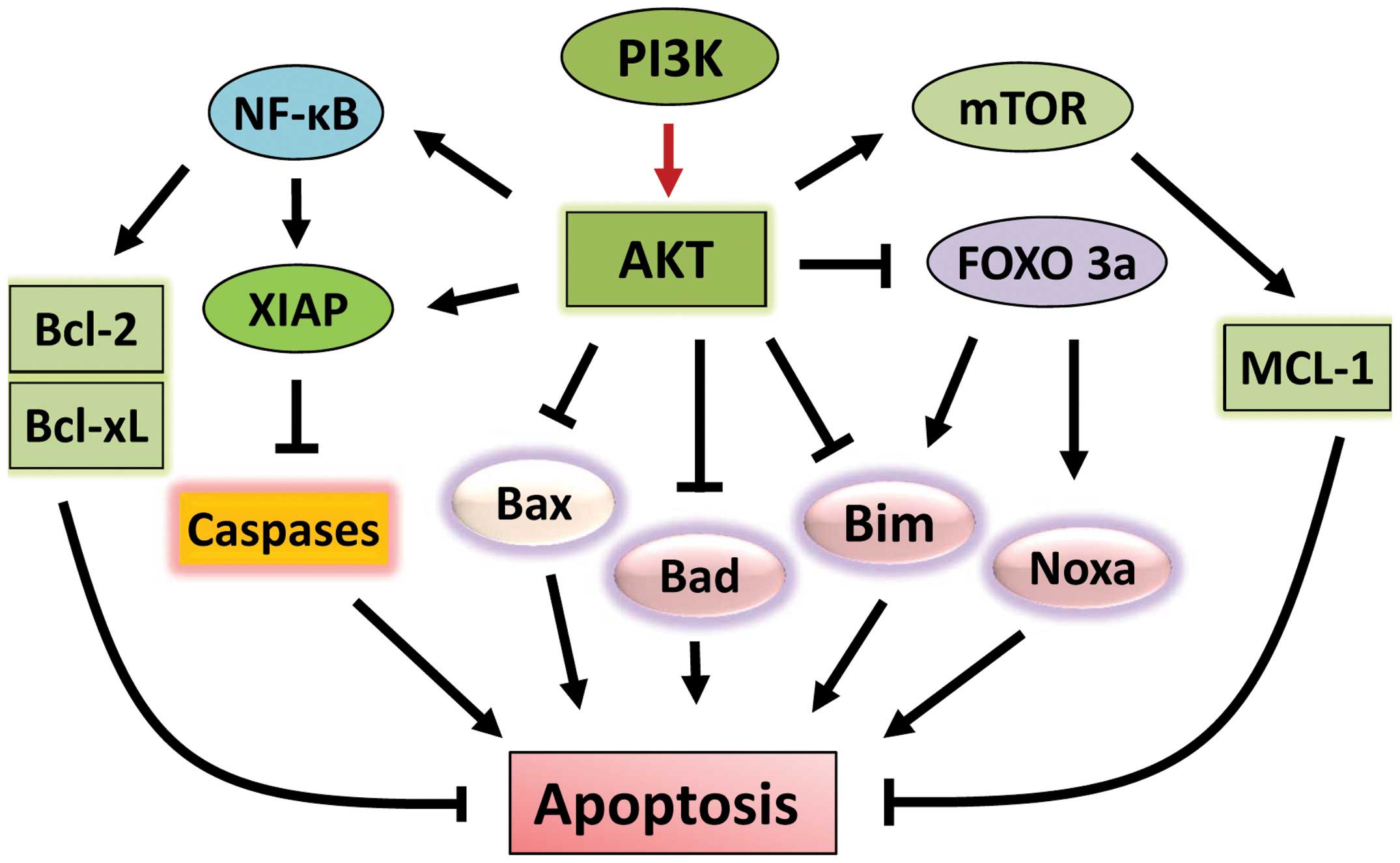
and activating pro-survival signaling pathways (Fig. 2). Upon activation, AKT
phosphorylates and inhibits a number of pro-apoptotic Bcl-2 family
members, including Bad, Bax and Bim (55–57).
Furthermore, through direct inhibition and exclusion of
proapoptotic transcription factor FOXO3a (Forkhead box O3), AKT
also suppresses the expressions of the pro-apoptotic factors Bim
and Noxa (58–61).
AKT also positively regulates anti-apoptotic
pathways (Fig. 2). AKT induces
activation of NF-κB transcription factor, which promotes the
transcription of a wide range of anti-apoptotic genes, in
particular BCL-2 and BCL-XL (62). Furthermore, AKT
phosphorylates/activates pro-survival protein XIAP (X-linked
inhibitor of apoptosis protein), resulting in an increase of
binding of XIAP to caspases 3, 7 and 9 and inhibition of these
caspases, the activities of which are essential for apoptosis
induction (63). Another key
pro-survival pathway targeted by AKT is the mTOR signaling pathway.
AKT phosphorylates and activates mTOR kinase, leading to the
phosphorylation/activation of anti-apoptotic protein Mcl-1
(64,65). Furthermore, AKT negatively
regulates hypoxia-induced apoptosis. Following radiation therapy,
hypoxia is often induced in tissues by radiation and can lead to
apoptosis in the injured tissue (66,67).
The hypoxia-induced apoptosis requires glycogen synthase kinase
(GSK) to activate the mitochondria-dependent death signaling
pathway (67,68). However, AKT activation following
radiation can inhibits the activity of GSK through phosphorylation,
resulting in an activation of glycolysis and glucose transport that
suppresses apoptosis induction (69). Lastly, AKT is directly involved in
the activation of the catalytic subunit of DNA-PK after radiation,
promoting NHEJ-mediated DSB repair that increases cell survival
(70). These studies establish a
pro-survival role for AKT mediated signaling pathways in the
response of cancer cells to radiation.
Activation of the PI3K/AKT signaling pathway in
response to ionizing radiation has been widely observed (15). A likely mechanism for this
activation involves HER RTKs. Upon activation of HER RTKs, the
phosphorylated tyrosines in the carboxyl-terminal regulatory tail
of HER3 can form six docking sites for recruitment of the p85
adaptor subunit of phosphatidylinositol 3-kinase (PI3K) (71). Subsequently, the p110 catalytic
subunit of PI3K phosphorylates phosphatidylinositol-4,5-biphosphate
(PIP2) to generate phosphatidylinositol (3,4,5)-triphosphate (PIP3), which then leads
to the membrane recruitment and activation of proteins that contain
a phospholipid-binding (PH) domain, such as
phosphoinositide-dependent kinase (PDK) 1 (72). The activated PDK1 phosphorylates
AKT-Thr308 and leads to the initial AKT activation (72). The full-activation of AKT requires
further phosphorylation of its Ser473 by PDK2 (72). Furthermore, mutant K-Ras also
positively contributes to the activation of PI3K-AKT signaling in
response to radiation, which is through its activation of autocrine
production of EGFR ligands (73,74).
The pro-survival function of PI3K/AKT signaling is
expected to positively contribute to the radioresistance of tumor
cells. Indeed, an increasing number of studies indicate that
inhibition of PI3K/AKT signaling by either pharmacological
inhibitors or genetic approaches leads to an enhancement of
radiosensitivity of cancer cells both in vitro and in
vivo (75–77). Furthermore, the increase in
radiosensitivity by PI3K/AKT inhibition involves both the
diminution of DNA repair and an enhancement of apoptosis induction
(70,75,76,78,79).
On the other hand, in some cell-based models, inhibition of
PI3K/AKT has been shown to have little effect on radiosensitivity
(29,80–83),
suggesting an involvement of PI3K/AKT-independent mechanisms in the
regulation of radiosensitivity.
5. Cell cycle checkpoint signaling
In response to DNA damage, cell cycle checkpoints
often become activated to block cell cycle progression, allowing
time for cells to repair the damage (84). Depending on the phase of the cell
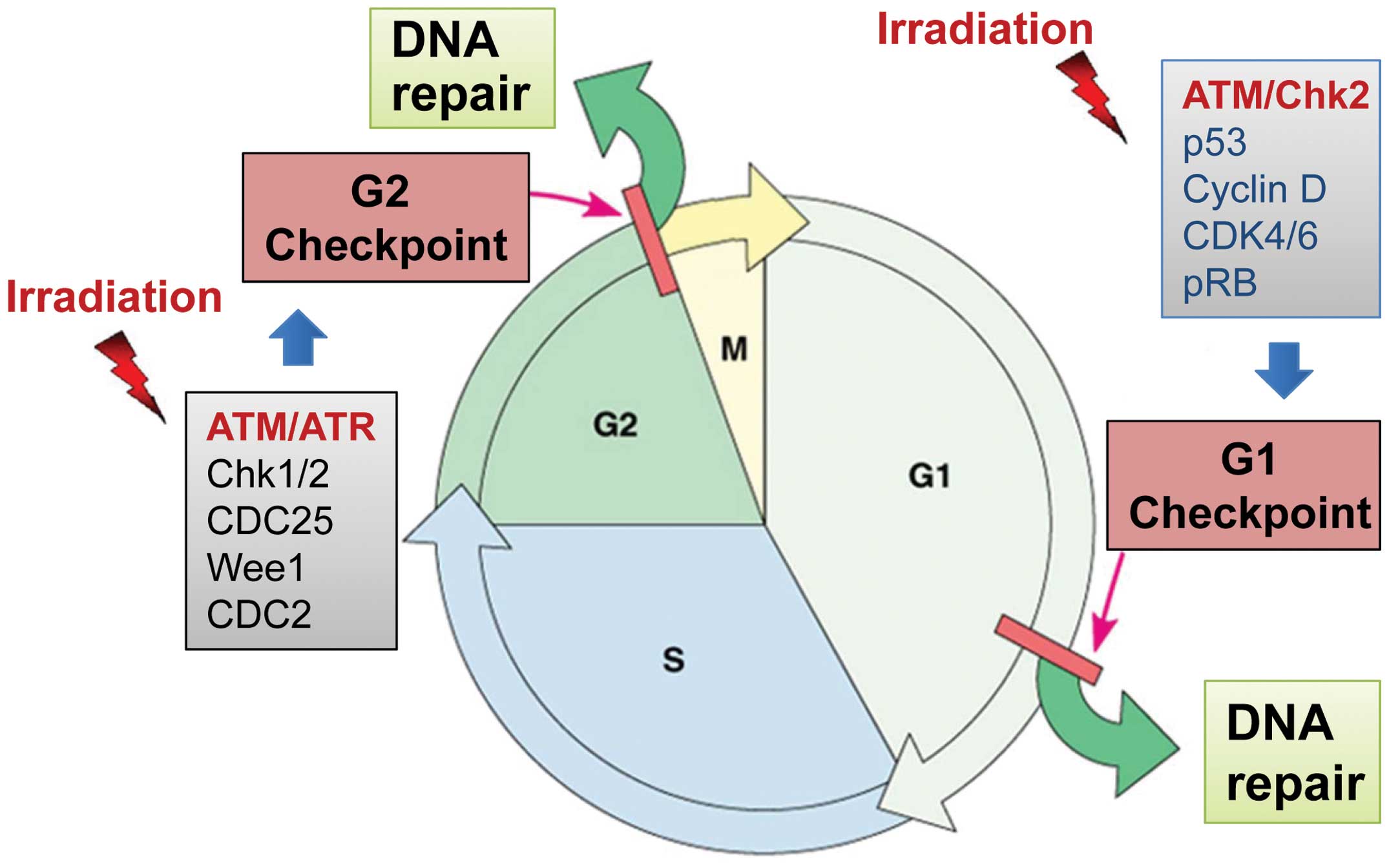
cycle at which the damage is sensed, the cells can be blocked at
the G1/S or G2/M border of the cell cycle (Fig. 3) (84). If the damage is irreversible or the
cell cycle checkpoint is dysfunctional, apoptosis may be triggered
to eliminate the injured cells (84). Thus, properly functioning cell
cycle checkpoints promote cell survival by counteracting the
cytotoxicity of DNA damage.
The G1/S transition is controlled by the activity of
Cdk4/6 kinases coupled with Cyclin D, the activities of which are
predominantly regulated by the p53/p21 pathway (80). The G2/M border is tightly
controlled by the Cdc2/Cyclin B complex, whose activity is required
for the G2/M transition of the cell cycle (85). The G1 checkpoint is defective in
most cancer cells, commonly due to mutations/alterations of key
regulators of the G1 checkpoint (p53, Cyclin D, etc.) (80), whereas activation of the G2
checkpoint is rarely impaired in cancer cells, as this checkpoint
operates primarily via a p53-independent mechanism (Fig. 3) (86). In fact, in cancer cells lacking a
functional G1 checkpoint, abrogation of the G2 checkpoint often
sensitizes the cells to radiation (87).
Previous studies identified Cdc2-Y15 as a vital site
involved in G2 checkpoint control in response to radiation
(88). Cdc2-Y15 is phosphorylated
in response to radiation exposure and this phosphorylation is
maintained during radiation-induced G2/M arrest (88–90).
Cdc2-Y15 is phosphorylated by the Wee1 and Myt1 kinases (91,92)
and dephosphorylated by the Cdc25 dual-specificity phosphatases
(93).
ATM- and ATR-mediated signaling pathways play
essential roles in the radiation-induced cell cycle checkpoint
responses (84). In response to
radiation-induced DNA-damage, ATM and ATR kinases are rapidly
activated, which, in turn, activate their respective downstream
targets, including p53 as well as the Chk1 and Chk2 kinases
(Fig. 3) (84). Activation of Chk1 and Chk2 results
in phosphorylation of Cdc25, leading to the subcellular
sequestration, degradation and/or inhibition of the Cdc25 that
normally activates Cdc2/Cyclin B at the G2/M boundary (94). Chk2 can also phosphorylate
p53-Ser20 to induce stabilization of the p53 protein following
radiation (84). Activation of p53
by ATM, ATR and Chk2 kinases leads to the induction of p21 protein,
which can directly inhibit the activity of the Cdc2/Cyclin B
complex (84).
In summary, radiation-induced cell cycle checkpoint
signaling pathways promote cell cycle arrest, which, in turn,
contributes positively to cell survival in response to
radiation.
6. DNA repair pathways
The cytotoxicity caused by ionizing radiation is
mainly the result of DNA damage. Radiation induces several forms of
DNA damage, which include SSBs, DSBs, sugar and base modifications
and DNA-protein crosslinks (95,96).
Among these, DSBs are not only a dominant form of damage caused by
ionizing radiation (97,98), but also is the most deadly form of
DNA damage, as unrepaired DSBs can lead to lethality of cells
(97,99).
In response to ionizing radiation, the activation of
the phosphoinositide 3-kinase-related kinases (PIKKs), including
ATM, ATR and DNA-PK, transduces and amplifies the DNA-damage
signal, triggering the assembly of DNA repair apparatuses at the
damaged sites and initiating DNA repair (10). A DSB is repaired by one of two
competing mechanisms: non-homologous end joining repair (NHEJ) and
homologous recombination (HR) (10), with both mechanisms regulated by
PIKKs. With no sequence homology required, NHEJ rejoins the free
ends in a process that commonly produces errors at the point of
junction (100). Each of the two
ends is recognized by the Ku70/Ku80 heterodimer, which then
recruits DNA-PK (100). Once
formed, these complexes bring the ends together for further
processing and ligation by DNA ligase IV (100). In contrast to NHEJ, HR repairs
DSBs accurately and with very high fidelity (100). This process operates during the S
and G2 phases and repairs DSBs taking advantage of sequence
information present in the intact sister chromatid (100). Radiation also creates SSBs,
mainly through base oxidation driven by ROS/RNS (98). The repair of this type of damage
uses base excision repair, which removes the damaged base using a
DNA glycosylase and AP endonuclease and then fills up the nick
through the actions of DNA polymerases and DNA ligase (101). Consequently, successful DNA
repairs promote cell survival in response to radiation, whereas a
failure to repair the DNA damage enhances the cytotoxic effect of
radiation, leading to lethality of the cells.
7. Conclusion
As a standard of care, radiation therapy plays an
important role in cancer therapy. However, radiation resistance
remains a major obstacle that limits the efficacy of radiation
therapy for cancer treatment. In order to improve the efficacy of
radiation therapy, it is necessary that we fully understand the
signaling network that drives cancer cells to overcome
radiation-induced cytotoxicity, leading to survival. As discussed
above, the lethal cytotoxicity caused by ionizing radiation is
mainly the result of DSBs. However, radiation also simultaneously
induces multiple signaling pathways that can protect cells from the
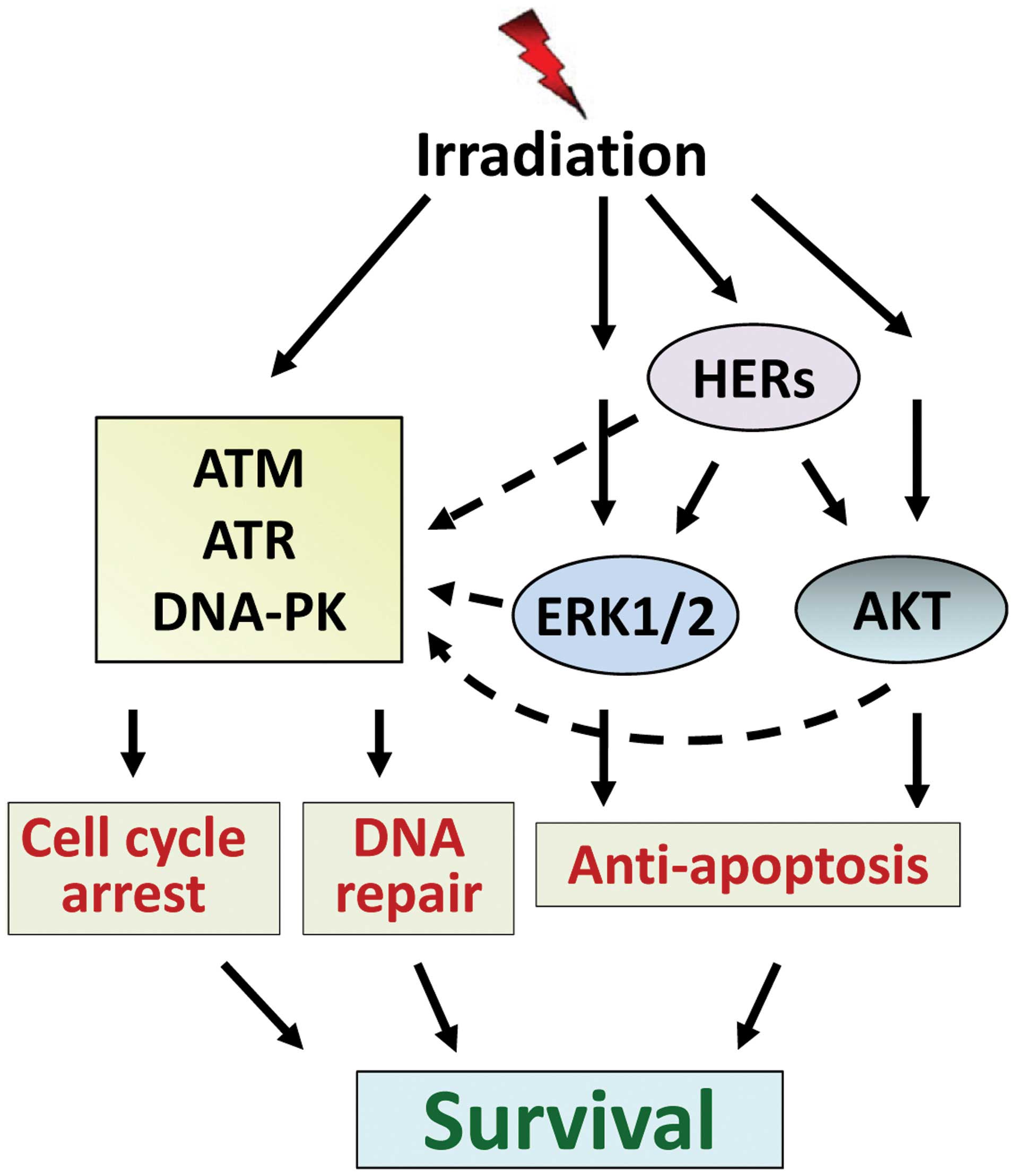
cytotoxic effect of radiation. Among these, signalings mediated by
HER receptors, ERK1/2 and AKT prevent the irradiated cells from
undergoing apoptosis induction, while signalings mediated by ATM,
ATR and DNA-PK drive cells into cycle arrest and initiate DNA
repair. In addition, HER ERK1/2 and AKT signaling also positively
regulate the cell cycle checkpoint response and DNA repair
machinery. Consequently, these signaling pathways act conjointly to
rescue the cells from radiation-induced injury and promote survival
(Fig. 4). To overcome radiation
therapy resistance, future research should focus on the development
of pharmacological approaches to block the activation of these
pro-survival signaling pathways in irradiated cells.
Acknowledgements
This study was supported by a Nebraska DHHS-LB506
grant 2010-40 to Y.Y. and NCI SPORE grant (P50 CA127297) to
M.M.O.
References
|
1
|
Pignon JP, le Maitre A, Maillard E and
Bourhis J: Meta-analysis of chemotherapy in head and neck cancer
(MACH-NC): an update on 93 randomised trials and 17,346 patients.
Radiother Oncol. 92:4–14. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ramnath N, Dilling TJ, Harris LJ, et al:
Treatment of stage III non-small cell lung cancer: diagnosis and
management of lung cancer, 3rd ed: American College of Chest
Physicians evidence-based clinical practice guidelines. Chest.
143:eS314–eS340. 2013. View Article : Google Scholar
|
|
3
|
Wilkinson-Ryan I, Binder PS,
Pourabolghasem S, et al: Concomitant chemotherapy and radiation for
the treatment of advanced-stage endometrial cancer. Gynecol Oncol.
134:24–28. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Johnstone RW, Ruefli AA and Lowe SW:
Apoptosis: a link between cancer genetics and chemotherapy. Cell.
108:153–164. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Milas L, Raju U, Liao Z and Ajani J:
Targeting molecular determinants of tumor chemo-radioresistance.
Semin Oncol. 32:S78–S81. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bernier J: Current state-of-the-art for
concurrent chemoradiation. Semin Radiat Oncol. 19:3–10. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ghiam AF, Spayne J and Lee J: Current
challenges and future perspectives of radiotherapy for locally
advanced breast cancer. Curr Opin Support Palliat Care. 8:46–52.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gewirtz DA: Growth arrest and cell death
in the breast tumor cell in response to ionizing radiation and
chemotherapeutic agents which induce DNA damage. Breast Cancer Res
Treat. 62:223–235. 2000. View Article : Google Scholar
|
|
9
|
Hawkins AJ, Golding SE, Khalil A and
Valerie K: DNA double-strand break-induced pro-survival signaling.
Radiother Oncol. 101:13–17. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Raleigh DR and Haas-Kogan DA: Molecular
targets and mechanisms of radiosensitization using DNA damage
response pathways. Future Oncol. 9:219–233. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Navolanic PM, Steelman LS and McCubrey JA:
EGFR family signaling and its association with breast cancer
development and resistance to chemotherapy (Review). Int J Oncol.
22:237–252. 2003.PubMed/NCBI
|
|
12
|
Linggi B and Carpenter G: ErbB receptors:
new insights on mechanisms and biology. Trends Cell Biol.
16:649–656. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Arteaga Carlos L and Engelman Jeffrey A:
ERBB receptors: from oncogene discovery to basic science to
mechanism-based cancer therapeutics. Cancer Cell. 25:282–303.
2014.PubMed/NCBI
|
|
14
|
Rexer BN and Arteaga CL: Intrinsic and
acquired resistance to HER2-targeted therapies in HER2
gene-amplified breast cancer: mechanisms and clinical implications.
Crit Rev Oncog. 17:1–16. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Valerie K, Yacoub A, Hagan MP, et al:
Radiation-induced cell signaling: inside-out and outside-in. Mol
Cancer Ther. 6:789–801. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Goldkorn T, Balaban N, Shannon M and
Matsukuma K: EGF receptor phosphorylation is affected by ionizing
radiation. Biochim Biophys Acta. 1358:289–299. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee H-C, An S, Lee H, et al: Activation of
epidermal growth factor receptor and its downstream signaling
pathway by nitric oxide in response to ionizing radiation. Mol
Cancer Res. 6:996–1002. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kiyozuka M, Akimoto T, Fukutome M, Motegi
A and Mitsuhashi N: Radiation-induced dimer formation of EGFR:
implications for the radiosensitizing effect of cetuximab.
Anticancer Res. 33:4337–4346. 2013.PubMed/NCBI
|
|
19
|
Yan Y, Hein AL, Greer PM, et al: A novel
function of HER2/Neu in the activation of G2/M checkpoint in
response to [gamma]-irradiation. Oncogene. June 9–2014.(Epub ahead
of print). View Article : Google Scholar
|
|
20
|
Meng TC, Fukada T and Tonks NK: Reversible
oxidation and inactivation of protein tyrosine phosphatases in
vivo. Mol Cell. 9:387–399. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Leach JK, Van Tuyle G, Lin PS,
Schmidt-Ullrich R and Mikkelsen RB: Ionizing radiation-induced,
mitochondria-dependent generation of reactive oxygen/nitrogen.
Cancer Res. 61:3894–3901. 2001.PubMed/NCBI
|
|
22
|
Nyati MK, Maheshwari D, Hanasoge S, et al:
Radiosensitization by Pan ErbB Inhibitor CI-1033 in vitro and in
vivo. Clin Cancer Res. 10:691–700. 2004. View Article : Google Scholar
|
|
23
|
Liang K, Lu Y, Jin W, Ang KK, Milas L and
Fan Z: Sensitization of breast cancer cells to radiation by
trastuzumab. Mol Cancer Ther. 2:1113–1120. 2003.PubMed/NCBI
|
|
24
|
Geoerger B, Gaspar N, Opolon P, et al:
EGFR tyrosine kinase inhibition radiosensitizes and induces
apoptosis in malignant glioma and childhood ependymoma xenografts.
Int J Cancer. 123:209–216. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dittmann K, Mayer C, Fehrenbacher B, et
al: Radiation-induced epidermal growth factor receptor nuclear
import is linked to activation of DNA-dependent protein kinase. J
Biol Chem. 280:31182–31189. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dittmann K, Mayer C and Rodemann HP:
Inhibition of radiation-induced EGFR nuclear import by C225
(Cetuximab) suppresses DNA-PK activity. Radiother Oncol.
76:157–161. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dent P, Yacoub A, Fisher PB, Hagan MP and
Grant S: MAPK pathways in radiation responses. Oncogene.
22:5885–5896. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cui W, Yazlovitskaya EM, Mayo MS, Pelling
JC and Persons DL: Cisplatin-induced response of c-jun N-terminal
kinase 1 and extracellular signal-regulated protein kinases 1 and 2
in a series of cisplatin-resistant ovarian carcinoma cell lines.
Mol Carcinog. 29:219–228. 2000. View Article : Google Scholar
|
|
29
|
Abbott DW and Holt JT: Mitogen-activated
protein kinase kinase 2 activation is essential for progression
through the G2/M checkpoint arrest in cells exposed to ionizing
radiation. J Biol Chem. 274:2732–2742. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tang D, Wu D, Hirao A, et al: ERK
activation mediates cell cycle arrest and apoptosis after DNA
damage independently of p53. J Biol Chem. 277:12710–12717. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yan Y, Black CP and Cowan KH:
Irradiation-induced G2/M checkpoint response requires ERK1/2
activation. Oncogene. 26:4689–4698. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Munshi A and Ramesh R: Mitogen-activated
protein kinases and their role in radiation response. Genes Cancer.
4:401–408. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Boucher MJ, Morisset J, Vachon PH, Reed
JC, Laine J and Rivard N: MEK/ERK signaling pathway regulates the
expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of
human pancreatic cancer cells. J Cell Biochem. 79:355–369. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Aoudjit F and Vuori K: Matrix attachment
regulates Fas-induced apoptosis in endothelial cells: a role for
c-flip and implications for anoikis. J Cell Biol. 152:633–643.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jost M, Huggett TM, Kari C, Boise LH and
Rodeck U: Epidermal growth factor receptor-dependent control of
keratinocyte survival and Bcl-xL expression through a MEK-dependent
pathway. J Biol Chem. 276:6320–6326. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bonni A, Brunet A, West AE, Datta SR,
Takasu MA and Greenberg ME: Cell survival promoted by the Ras-MAPK
signaling pathway by transcription-dependent and -independent
mechanisms. Science. 286:1358–1362. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Clark CJ, McDade DM, O’Shaughnessy CT and
Morris BJ: Contrasting roles of neuronal Msk1 and Rsk2 in Bad
phosphorylation and feedback regulation of Erk signalling. J
Neurochem. 102:1024–1034. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ewings KE, Hadfield-Moorhouse K, Wiggins
CM, et al: ERK1/2-dependent phosphorylation of BimEL promotes its
rapid dissociation from Mcl-1 and Bcl-xL. EMBO J. 26:2856–2867.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Allan LA, Morrice N, Brady S, Magee G,
Pathak S and Clarke PR: Inhibition of caspase-9 through
phosphorylation at Thr 125 by ERK MAPK. Nat Cell Biol. 5:647–654.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tamamoto T, Ohnishi K, Takahashi A, et al:
Correlation between gamma-ray-induced G2 arrest and radioresistance
in two human cancer cells. Int J Radiat Oncol Biol Phys.
44:905–909. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fritz G, Brachetti C and Kaina B:
Lovastatin causes sensitization of HeLa cells to ionizing
radiation-induced apoptosis by the abrogation of G2 blockage. Int J
Radiat Biol. 79:601–610. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yan Y, Black CP, Cao PT, et al:
Gamma-irradiation-induced DNA damage checkpoint activation involves
feedback regulation between extracellular signal-regulated kinase
1/2 and BRCA1. Cancer Res. 68:5113–5121. 2008. View Article : Google Scholar
|
|
43
|
Yacoub A, McKinstry R, Hinman D, Chung T,
Dent P and Hagan MP: Epidermal growth factor and ionizing radiation
up-regulate the DNA repair genes XRCC1 and ERCC1 in DU145 and LNCaP
prostate carcinoma through MAPK signaling. Radiat Res. 159:439–452.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Golding SE, Morgan RN, Adams BR, Hawkins
AJ, Povirk LF and Valerie K: Pro-survival AKT and ERK signaling
from EGFR and mutant EGFRvIII enhances DNA double-strand break
repair in human glioma cells. Cancer Biol Ther. 8:730–738. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wei F, Yan J, Tang D, et al: Inhibition of
ERK activation enhances the repair of double-stranded breaks via
non-homologous end joining by increasing DNA-PKcs activation.
Biochim Biophys Acta. 1833.90–100. 2013.PubMed/NCBI
|
|
46
|
Cohen-Armon M: PARP-1 activation in the
ERK signaling pathway. Trends Pharmacol Sci. 28:556–560. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cohen-Armon M, Visochek L, Rozensal D, et
al: DNA-independent PARP-1 activation by phosphorylated ERK2
increases Elk1 activity: a link to histone acetylation. Mol Cell.
25:297–308. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Golding SE, Rosenberg E, Neill S, Dent P,
Povirk LF and Valerie K: Extracellular signal-related kinase
positively regulates ataxia telangiectasia mutated, homologous
recombination repair, and the DNA damage response. Cancer Res.
67:1046–1053. 2007. View Article : Google Scholar
|
|
49
|
Yan Y, Greer PM, Cao PT, Kolb RH and Cowan
KH: RAC1 GTPase plays an important role in gamma-irradiation
induced G2/M checkpoint activation. Breast Cancer Res. 14:R602012.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sasaoka T, Langlois WJ, Leitner JW,
Draznin B and Olefsky JM: The signaling pathway coupling epidermal
growth factor receptors to activation of p21ras. J Biol Chem.
269:32621–32625. 1994.PubMed/NCBI
|
|
51
|
Janes PW, Daly RJ, deFazio A and
Sutherland RL: Activation of the Ras signalling pathway in human
breast cancer cells overexpressing erbB-2. Oncogene. 9:3601–3608.
1994.PubMed/NCBI
|
|
52
|
Dent P, Reardon DB, Park JS, et al:
Radiation-induced release of transforming growth factor alpha
activates the epidermal growth factor receptor and
mitogen-activated protein kinase pathway in carcinoma cells,
leading to increased proliferation and protection from
radiation-induced cell death. Mol Biol Cell. 10:2493–2506.
1999.
|
|
53
|
Hagan M, Wang L, Hanley JR, Park JS and
Dent P: Ionizing radiation-induced mitogen-activated protein (MAP)
kinase activation in DU145 prostate carcinoma cells: MAP kinase
inhibition enhances radiation-induced cell killing and G2/M-phase
arrest. Radiat Res. 153:371–383. 2000. View Article : Google Scholar
|
|
54
|
Polivka J Jr and Janku F: Molecular
targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol
Ther. 142:164–175. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yamaguchi H and Wang HG: The protein
kinase PKB/Akt regulates cell survival and apoptosis by inhibiting
Bax conformational change. Oncogene. 20:7779–7786. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gardai SJ, Hildeman DA, Frankel SK, et al:
Phosphorylation of Bax Ser184 by Akt regulates its activity and
apoptosis in neutrophils. J Biol Chem. 279:21085–21095. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Qi XJ, Wildey GM and Howe PH: Evidence
that Ser87 of BimEL is phosphorylated by Akt and regulates BimEL
apoptotic function. J Biol Chem. 281:813–823. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Engström M, Karlsson R and Jönsson J-I:
Inactivation of the forkhead transcription factor FoxO3 is
essential for PKB-mediated survival of hematopoietic progenitor
cells by kit ligand. Exp Hematol. 31:316–323. 2003.PubMed/NCBI
|
|
59
|
Yang JY, Xia W and Hu MC: Ionizing
radiation activates expression of FOXO3a, Fas ligand, and Bim, and
induces cell apoptosis. Int J Oncol. 29:643–648. 2006.PubMed/NCBI
|
|
60
|
Obexer P, Geiger K, Ambros PF, Meister B
and Ausserlechner MJ: FKHRL1-mediated expression of Noxa and Bim
induces apoptosis via the mitochondria in neuroblastoma cells. Cell
Death Differ. 14:534–547. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Jang S-W, Yang S-J, Srinivasan S and Ye K:
Akt phosphorylates MstI and prevents its proteolytic activation,
blocking FOXO3 phosphorylation and nuclear translocation. J Biol
Chem. 282:30836–30844. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Ozes ON, Mayo LD, Gustin JA, Pfeffer SR,
Pfeffer LM and Donner DB: NF-kappaB activation by tumour necrosis
factor requires the Akt serine-threonine kinase. Nature. 401:82–85.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Dan HC, Sun M, Kaneko S, et al: Akt
phosphorylation and stabilization of X-linked inhibitor of
apoptosis protein (XIAP). J Biol Chem. 279:5405–5412. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Shaw RJ and Cantley LC: Ras, PI(3)K and
mTOR signalling controls tumour cell growth. Nature. 441:424–430.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Fumarola C, Bonelli MA, Petronini PG and
Alfieri RR: Targeting PI3K/AKT/mTOR pathway in non small cell lung
cancer. Biochem Pharmacol. 90:197–207. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Fleckenstein K, Zgonjanin L, Chen L, et
al: Temporal onset of hypoxia and oxidative stress after pulmonary
irradiation. Int J Radiat Oncol Biol Phys. 68:196–204. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Sendoel A and Hengartner MO: Apoptotic
cell death under hypoxia. Physiology. 29:168–176. 2014. View Article : Google Scholar
|
|
68
|
King TD, Bijur GN and Jope RS: Caspase-3
activation induced by inhibition of mitochondrial complex I is
facilitated by glycogen synthase kinase-3β and attenuated by
lithium. Brain Res. 919:106–114. 2001.
|
|
69
|
Loberg RD, Vesely E and Brosius FC:
Enhanced glycogen synthase kinase-3β activity mediates
hypoxia-induced apoptosis of vascular smooth muscle cells and is
prevented by glucose transport and metabolism. J Biol Chem.
277:41667–41673. 2002.
|
|
70
|
Toulany M, Kehlbach R, Florczak U, et al:
Targeting of AKT1 enhances radiation toxicity of human tumor cells
by inhibiting DNA-PKcs-dependent DNA double-strand break repair.
Mol Cancer Ther. 7:1772–1781. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Soltoff SP, Carraway KL III, Prigent SA,
Gullick WG and Cantley LC: ErbB3 is involved in activation of
phosphatidylinositol 3-kinase by epidermal growth factor. Int J
Radiat Biol. 14:3550–3558. 1994.PubMed/NCBI
|
|
72
|
Marone R, Cmiljanovic V, Giese B and
Wymann MP: Targeting phosphoinositide 3-kinase - moving towards
therapy. Biochim Biophys Acta. 1784:159–185. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Toulany M, Baumann M and Rodemann HP:
Stimulated PI3K-AKT signaling mediated through ligand or
radiation-induced EGFR depends indirectly, but not directly, on
constitutive K-Ras activity. Mol Cancer Res. 5:863–872. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Minjgee M, Toulany M, Kehlbach R, Giehl K
and Rodemann HP: K-RAS(V12) induces autocrine production of EGFR
ligands and mediates radioresistance through EGFR-dependent Akt
signaling and activation of DNA-PKcs. Int J Radiat Oncol Biol Phys.
81:1506–1514. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Toulany M, Lee K-J, Fattah KR, et al: Akt
promotes post-irradiation survival of human tumor cells through
initiation, progression, and termination of DNA-PKcs-dependent DNA
double-strand break repair. Mol Cancer Res. 10:945–957. 2012.
View Article : Google Scholar
|
|
76
|
Sahlberg SH, Gustafsson AS, Pendekanti PN,
Glimelius B and Stenerlow B: The influence of AKT isoforms on
radiation sensitivity and DNA repair in colon cancer cell lines.
Tumour Biol. 35:3525–3534. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Shimura T, Kakuda S, Ochiai Y, Kuwahara Y,
Takai Y and Fukumoto M: Targeting the AKT/GSK3β/cyclin D1/Cdk4
survival signaling pathway for eradication of tumor radioresistance
acquired by fractionated radiotherapy. Int J Radiat Oncol Biol
Phys. 80:540–548. 2011.
|
|
78
|
Kim IA, Bae SS, Fernandes A, et al:
Selective inhibition of Ras, phosphoinositide 3 kinase, and Akt
isoforms increases the radiosensitivity of human carcinoma cell
lines. Cancer Res. 65:7902–7910. 2005.PubMed/NCBI
|
|
79
|
Contessa JN, Hampton J, Lammering G, et
al: Ionizing radiation activates Erb-B receptor dependent Akt and
p70 S6 kinase signaling in carcinoma cells. Oncogene. 21:4032–4041.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Kastan MB, Onyekwere O, Sidransky D,
Vogelstein B and Craig RW: Participation of p53 protein in the
cellular response to DNA damage. Cancer Res. 51:6304–6311.
1991.PubMed/NCBI
|
|
81
|
Shonai T, Adachi M, Sakata K, et al:
MEK/ERK pathway protects ionizing radiation-induced loss of
mitochondrial membrane potential and cell death in lymphocytic
leukemia cells. Cell Death Differ. 9:963–971. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Lee YJ, Soh JW, Jeoung DI, et al: PKC
epsilon-mediated ERK1/2 activation involved in radiation-induced
cell death in NIH3T3 cells. Biochim Biophys Acta. 1593:219–229.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Dai X-F, Ding J, Zhang R-G, Ren J-H, Ma
C-MC and Wu G: Radiosensitivity enhancement of human hepatocellular
carcinoma cell line SMMC-7721 by sorafenib through the MEK/ERK
signal pathway. Int J Radiat Biol. 89:12013.PubMed/NCBI
|
|
84
|
Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K
and Linn S: Molecular mechanisms of mammalian DNA repair and the
DNA damage checkpoints. Annu Rev Biochem. 73:39–85. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Smits VA and Medema RH: Checking out the
G(2)/M transition. Biochim Biophys Acta. 1519:1–12. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
O’Connell MJ and Cimprich KA: G2 damage
checkpoints: what is the turn-on? J Cell Sci. 118:1–6.
2005.PubMed/NCBI
|
|
87
|
Chen T, Stephens PA, Middleton FK and
Curtin NJ: Targeting the S and G2 checkpoint to treat cancer. Drug
Discov Today. 17:194–202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Kharbanda S, Saleem A, Datta R, Yuan ZM,
Weichselbaum R and Kufe D: Ionizing radiation induces rapid
tyrosine phosphorylation of p34cdc2. Cancer Res. 54:1412–1414.
1994.PubMed/NCBI
|
|
89
|
Rhind N, Furnari B and Russell P: Cdc2
tyrosine phosphorylation is required for the DNA damage checkpoint
in fission yeast. Genes Dev. 11:504–511. 1997. View Article : Google Scholar
|
|
90
|
O’Connell MJ, Raleigh JM, Verkade HM and
Nurse P: Chk1 is a wee1 kinase in the G2 DNA damage checkpoint
inhibiting cdc2 by Y15 phosphorylation. EMBO J. 16:545–554.
1997.PubMed/NCBI
|
|
91
|
Lundgren K, Walworth N, Booher R, Dembski
M, Kirschner M and Beach D: mik1 and wee1 cooperate in the
inhibitory tyrosine phosphorylation of cdc2. Cell. 64:1111–1122.
1991. View Article : Google Scholar
|
|
92
|
Parker LL, Atherton-Fessler S and
Piwnica-Worms H: p107wee1 is a dual-specificity kinase
that phosphorylates p34cdc2 on tyrosine 15. Proc Natl Acad Sci USA.
89:2917–2921. 1992.PubMed/NCBI
|
|
93
|
Bulavin DV, Higashimoto Y, Demidenko ZN,
et al: Dual phosphorylation controls Cdc25 phosphatases and mitotic
entry. Nat Cell Biol. 5:545–551. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
94
|
Kastan MB and Bartek J: Cell-cycle
checkpoints and cancer. Nature. 432:316–323. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Nikjoo H, O’Neill P, Wilson WE and
Goodhead DT: Computational approach for determining the spectrum of
DNA damage induced by ionizing radiation. Radiat Res. 156:577–583.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Yu H: Typical cell signaling response to
ionizing radiation: DNA damage and extranuclear damage. Chin J
Cancer Res. 24:83–89. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Ward JF: DNA damage as the cause of
ionizing radiation-induced gene activation. Radiat Res.
138:S85–S88. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Haddy N, Tartier L, Koscielny S, et al:
Repair of ionizing radiation-induced DNA damage and risk of second
cancer in childhood cancer survivors. Carcinogenesis. Apr
19–2014.Epub ahead of print.
|
|
99
|
Huhn D, Bolck HA and Sartori AA: Targeting
DNA double-strand break signalling and repair: recent advances in
cancer therapy. Swiss Med Wkly. 143:w138372013.PubMed/NCBI
|
|
100
|
Hosoya N and Miyagawa K: Targeting DNA
damage response in cancer therapy. Cancer Sci. 105:370–388. 2014.
View Article : Google Scholar
|
|
101
|
Iyama T and Wilson DM III: DNA repair
mechanisms in dividing and non-dividing cells. DNA Repair.
12:620–636. 2013. View Article : Google Scholar : PubMed/NCBI
|